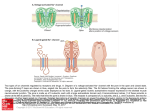
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
SCIENTIFIC REPORT SCK•CEN-BLG-990 04/MDC/P-48 Geochemistry of Boom Clay pore water at the Mol site Status 2004 M. De Craen, L. Wang, M. Van Geet and H. Moors September, 2004 SCK•CEN Boeretang 200 2400 Mol Belgium Waste & Disposal Department © SCK•CEN Belgian Nuclear Research Centre Boeretang 200 2400 Mol Belgium Phone +32 14 33 21 11 Fax +32 14 31 50 21 http://www.sckcen.be Contact: Knowledge Centre [email protected] RESTRICTED All property rights and copyright are reserved. Any communication or reproduction of this document, and any communication or use of its content without explicit authorization is prohibited. Any infringement to this rule is illegal and entitles to claim damages from the infringer, without prejudice to any other right in case of granting a patent or registration in the field of intellectual property. SCK•CEN, Boeretang 200, 2400 Mol, Belgium. SCIENTIFIC REPORT OF THE BELGIAN NUCLEAR RESEARCH CENTRE SCK•CEN-BLG-990 04/MDC/P-48 Geochemistry of Boom Clay pore water at the Mol site Status 2004 M. De Craen, L. Wang, M. Van Geet and H. Moors September, 2004 Status: Unclassified ISSN 1379-2407 SCK•CEN Boeretang 200 2400 Mol Belgium Waste & Disposal Department 3 Executive summary In Belgium, geological disposal in clay is the primary option for the final disposal of high-level radioactive waste and spent fuel. The Boom Clay is studied as the reference host rock for methodological studies on the geological disposal of radioactive waste. In many of these studies, an in-depth understanding of the Boom Clay pore water geochemistry is essential. The objective of this report is to evaluate the most reliable technique(s) to obtain representative pore water samples, to determine the variation of the pore water composition in the Boom Clay, to present a coherent geochemical model for explaining the origin of the Boom Clay pore water composition, and to propose a reference pore water composition to be used in the laboratory experiments and for speciation calculations and assessments of perturbation of the Boom Clay. It is important to mention that this report is not the result of an integrated study on the Boom Clay pore water composition. In fact, all the available information from previous studies is put together in this report. It is therefore considered to be a 'state of the art' report, status 2004. Pore water sampling is done in situ from various piezometers, or by the mechanical squeezing or leaching of clay cores in the laboratory. These three pore water sampling techniques are compared and evaluated. At the present time, piezometer water is considered to be the most representative for the in situ pore water. This is because piezometer waters experience minimum laboratory manipulations and therefore suffer minimum artefacts. Squeezed pore water is comparable to piezometer-derived water when considering the major ionic composition, but not for trace elements and organic matter. Squeezed pore water samples can thus be considered as representative for the in situ conditions, up to a certain degree. Comparing to the piezometer and squeezing techniques, batch leaching experiments provide comparable results for the major cation composition if the samples are carefully filtered. Due to the electrostatic properties of the Boom Clay, i.e., double layer phenomena, the leaching waters reveal a very different anion composition compared to the waters extracted from compacted clay using piezometers and squeezing techniques. A large data-set on pore water composition is available, however, because of the different sampling techniques, the different design of the piezometers, and the different filter materials used, it is not always unambiguous to interpret these data. The reliability and significance of these data have therefore been considered by statistical analyses and geochemical modelling. The statistical analysis of the available data at the Mol site (to about 40 m around the HADES URF) has shown that a vertical spatial variability (perpendicular to the bedding) is present within the Boom Clay pore water composition. This vertical variability shows no gradient, and is mostly influenced by the elements Na, Mg, Ca and Cl. The mechanism behind these variations in major cations is explained by cation exchange and calcite dissolution/precipitation. The ultimate cause of these chemical reactions is assumed to be due to the spatial variability in pCO2 and pH, although the reason of this is not yet understood. Nevertheless, if the assumed pCO2 variation exists, the pore water seemed to respond rapidly to reach a chemical equilibrium with the clay. Because transport in the Boom Clay is diffusion-controlled, 4 the spatial variability in the pore water composition can still be present, even on small scales. Due to this spatial variability, one single mean Boom Clay pore water composition at the Mol site cannot be given. However, a modelled reference composition, taking into account the current knowledge of Boom Clay mineralogy and calibrated towards a dataset including spatial variability, is provided. Boom Clay pore water is basically a NaHCO3 solution of 15 mM, containing about 115 mg C / l. The observed major cation concentrations can be explained by cation exchange and mineral dissolution/precipitation mechanisms. The current model assumes the equilibrium of calcite, siderite, pyrite, and chalcedony, and the cation exchange between Ca, Na, K, and Mg. The maximum redox potential Eh is about -270 mV; probably controlled by the equilibrium of pyrite and siderite under the in situ geochemical conditions. A lower redox potential is possible as the result of interactions involving natural organic matter mediated by biochemical processes. As mentioned before, this report is a 'putting together of all available information' on the Boom Clay pore water geochemistry. Comparison of the results of various studies was not always unambiguous, on the one hand because of the different design of the piezometers and filtermaterial, but on the other hand also because of the lack of uniformity in sampling conditions and analyses. Therefore, some recommendations are given for a systematic procedure of pore water sampling and analyses. Finally, some recommendations for further research on the pore water geochemistry are formulated. 5 Table of contents Executive summary List of abbreviations Introduction 1 2 Physical and mineralogical characteristics of Boom Clay .......... 13 1.1 Physical characteristics ................................................................................13 1.2 Mineralogical characteristics .......................................................................14 1.2.1 General mineralogical composition of Boom Clay .............................14 1.2.2 Clay-mineralogical composition of Boom Clay ..................................15 Boom Clay pore water sampling and analytical techniques....... 17 2.1 Pore water sampling techniques...................................................................17 2.1.1 Piezometers ..........................................................................................17 2.1.2 Squeezing .............................................................................................19 2.1.3 Leaching...............................................................................................21 2.2 Analytical techniques...................................................................................22 2.2.1 2.2.1.1 pH.....................................................................................................22 2.2.1.2 Eh ......................................................................................................24 2.2.1.3 pCO2 and other dissolved gasses .....................................................25 2.2.1.4 Electrolytic Conductivity (EC) ........................................................27 2.2.2 2.3 pH, Eh, pCO2, electrolytic conductivity and dissolved gases ..............22 Other techniques ..................................................................................28 2.2.2.1 The total organic carbon content (TOC) ..........................................28 2.2.2.2 UV/VIS spectrometry ......................................................................28 2.2.2.3 The cation concentration (Ca, Fe, K, Mg, Na, Si) ...........................28 2.2.2.4 The anion concentration (F-, Cl -, Br -, HPO42-, NO3-, SO4 2-) .........29 2.2.2.5 The trace element concentration ......................................................29 2.2.2.6 The stable isotope composition........................................................29 2.2.2.7 The radiochemical composition .......................................................30 Sample locations ..........................................................................................31 2.3.1 EG/BS piezometer ...............................................................................32 2.3.2 ARCHIMEDE piezometers .................................................................33 6 3 2.3.3 Spring 116............................................................................................34 2.3.4 ORPHEUS piezometer.........................................................................34 2.3.5 MORPHEUS piezometer .....................................................................35 Boom Clay pore water composition and characteristics............. 37 3.1 Chemical composition .................................................................................37 3.1.1 Major elements.....................................................................................37 3.1.2 Trace elements .....................................................................................42 3.1.3 pH, pCO2, and alkalinity of Boom Clay ..............................................44 3.1.3.1 pH of Boom Clay pore water ...........................................................45 3.1.3.2 Partial pressure of CO2 (g) in Boom Clay .......................................49 3.1.3.3 Alkalinity of Boom Clay pore water................................................54 3.1.3.4 Conclusions of pH/ pCO2.................................................................56 3.1.4 Redox processes and redox potential in Boom Clay ...........................56 3.1.4.1 Redox potential in Boom Clay.........................................................57 3.1.4.2 Redox capacity of Boom Clay pore water .......................................59 3.1.4.3 Conclusion of Boom Clay redox conditions....................................59 3.1.5 Electrolytic Conductivity (EC) ............................................................60 3.1.6 Dissolved organic carbon (DOC) and its effect on pore water composition..........................................................................................60 3.1.6.1 Presence of TOC in Boom Clay pore water.....................................60 3.1.6.2 TOC versus UV measurements........................................................63 3.1.6.3 Characteristics of the mobile organic matter in Boom Clay............64 3.1.7 3.2 Evaluation of extraction techniques and recommendations for water sampling and storage............................................................................68 Isotope geochemistry ...................................................................................70 3.2.1 Stable isotopes .....................................................................................70 3.2.2 Radioisotopes.......................................................................................72 3.3 3.2.2.1 U-Th isotopes...................................................................................72 3.2.2.2 14 C ....................................................................................................72 3.2.2.3 36 Cl ...................................................................................................72 Spatial variability .........................................................................................73 3.3.1 Vertical variability ...............................................................................73 3.3.2 Lateral variability.................................................................................74 3.4 Data quality ..................................................................................................77 3.4.1 Statistical Analysis...............................................................................77 7 3.4.1.1 Factors that might influence pore water composition......................77 3.4.1.2 Data and statistical techniques .........................................................78 3.4.1.3 Results of the statistical analyses.....................................................79 3.4.1.4 Effect of the filter material on the pore water composition .............82 3.4.1.5 Effect of the spatial variability on the pore water composition .......85 3.4.1.6 Conclusions of the statistical analyses.............................................88 3.4.2 4 Charge balance and equilibrium state of the pore water......................88 Model simulation of pore water chemistry................................... 91 4.1 Equilibrium model and water-rock interaction ............................................91 4.2 Computer code and thermodynamic database .............................................91 4.3 Mineral solubility and ion exchange............................................................92 4.4 Equilibrium model for the simulation of the pore water composition of Boom Clay...................................................................................................99 4.4.1 Mineral stability constants and ion exchange parameters..................100 4.4.2 Results of model simulations and discussions ...................................103 4.5 Concluding remarks ...................................................................................105 4.6 Future work needed to improve the model ................................................105 5 Reference Boom Clay pore water composition at the Mol site. 107 6 Conclusions .................................................................................... 111 7 Recommendations ......................................................................... 115 8 Acknowledgements ....................................................................... 117 9 References ...................................................................................... 119 10 Annexes .......................................................................................... 127 8 9 List of abbreviations ANC Acid Neutralising Capacity ANOVA ANalysis Of VAriance ARCHIMEDE Acquisition et Regulation de la Chimie des Eaux en Milieu Argileux pour le projet de Stockage de Déchets Radioactifs en Formation Géologique BCPW Boom Clay Pore Water BDT Below Drilling Table BSL Below Surface Level CA Carbo CEA Commissariat à l'Energie Atomique CEC Cation Exchange Capacity CERBERUS Control Experiments with Radiation of the Belgian Repository for Underground Storage CDT Canyon Diablo Troilite DOC Dissolved Organic Carbon EDZ Excavation Disturbed Zone EC Electrolitic Conductivity EG/BS Extension Gallery / Bottom Shaft Extra DOS depth versus the outside of the lining of the HADES URF FFFF Flow Field Flow Fractionation FT-IR Fourier Transform Infrared Spectroscopy HADES High Activity Disposal Experimental Site HLW High Level Waste HR-ICP-MS High Resolution - Inductively Coupled Plasma - Mass Spectrometry IAEA International Atomic Energy Agency IC Ion Chromatography ICP-AES Inductively Coupled Plasma - Atomic Emission Spectrometry ICP-MS Inductively Coupled Plasma - Mass Spectrometry Intra DOS depth versus the inside of the lining of the HADES URF ISE Ion Selective Electrode ISFET Ion Sensitive Field-Effect Transistor LLNL Lawrence Livermore National Library MANOVA Multivariate ANalysis Of Variance 10 MORPHEUS Mobile ORganic matter and Pore water extraction in the Hades Experimental Underground Site MWCO Molecular Weight Cutt-Off MWL Meteoric Water Line NEA Nuclear Energy Agency ORPHEUS Oxidation Reduction Potential and pH Experimental Underground Station PE Poly-Ethylene PEEK Poly-Ether-Ether-Keton PHYMOL PalaeoHYdrogeological study of the Mol site SBCPW Synthetic Boom Clay Pore Water SCK•CEN Belgian Nuclear Research Centre SBCW Synthetic Boom Clay Water SCW Synthetic Clay Water SG Sintered Glass SIC Synthetic Interstitial Clay water SICZH Synthetic Interstitial Clay water without (Zonder) Humus S/L Solid/Liquid ratio SMOW Standard Mean Ocean Water SPRING 116 Source Piezonest at RING 116 of the Test Drift gallery SS Stainless Steel ST SchumaTherm TAW Tweede Algemene Waterpassing TC Total Carbon TD Test Drift TDS Total Dissolved Salt TIC Total Inorganic Carbon TOC Total Organic Carbon TRANCOM Migration Case Study: TRANsport of Radionuclides due to Complexation with Organic Matter in Clay Formations URF Underground Research Facility UV/VIS Ultra Violet / Visible wavelenghts XRD X-Ray Diffraction YM3 3000 Molecular Weight Cutt-Off 11 Introduction In Belgium, geological disposal in clay is the primary option for the final disposal of high-level radioactive waste and spent fuel. The Boom Clay is studied as the reference host rock for methodological studies on the geological disposal of radioactive waste. This clay layer is present under the facilities of the SCK•CEN at Mol, at a depth of 190 to 293 m (Figure 0-1). Mol Figure 0-1. Location of Mol. Present-day outcrops of the Boom Clay in Belgium are indicated in black. To the north of the outcrops, the Boom Clay is present in the subsurface. In Mol, the Boom Clay is present at a depth of 190 to 293 m (Mol-1 borehole). In 1974, SCK•CEN started with the construction of the HADES underground research facility (URF), which was build at a depth of 223 m in the Boom Clay. HADES was designed to carry out experiments related to the disposal of radioactive waste. The current R&D programme is focussed on the feasibility and safety of HLW disposal in the Boom Clay. In this framework, a detailed characterisation of the clay is performed (mechanical, physico-chemical and hydrogeological properties, variability, role of organic matter, ...). In addition, high priority is given to the understanding of the basic phenomena which control the retention and/or mobility of radionuclides in the clay. Therefore, it is very important to characterise the pore water composition in the host rock. Previous studies, mainly involved with radionuclide migration studies, already showed the necessity to understand the pore water composition. Baeyens et al. (1985) studied the in situ physico-chemical characteristics of Boom Clay. This study mainly focussed on the pore water composition, the cation exchange capacity and specific surface of the clay. The study was performed on leached pore water samples. In the 12 frame of radionuclide migration studies, Henrion et al. (1985) sampled Boom Clay pore water from piezometers under anaerobic conditions. This resulted in the set-up of an important new data base which gave rise to new questions. In 1991, a new project was initiated: the ARCHIMEDE-argile project. The main objective of this project was to better understand the pore water chemistry of Boom Clay (Griffault et al., 1996; Beaucaire et al., 2000). The project included field work, field sampling and in situ Eh-pH measurements in the HADES URF, laboratory investigations and analyses, and modelling. In 1997, Dierckx published a report on 'Boom Clay in situ pore water chemistry'. In this report, a mean value for the ionic composition of the EG/BS piezometer was given and compared with other piezometers. A major conclusion of this study was that the EG/BS piezometer is not an optimal reference for studying Boom Clay pore water, and that more measurements are necessary to better understand the Boom Clay pore water chemistry. The Boom Clay pore water composition is considered in many studies, such as the study on the variability of the clay characteristics and its pore water, the study of dissolved organic matter, Eh and pH studies, migration experiments, geochemical modelling, ... In each of these studies, only a particular part of the pore water characteristics is considered. Information was fragmentary. The objective of this report is to evaluate the most reliable technique(s) to obtain representative pore water samples, to determine the variation of the pore water composition in the Boom Clay, to present a coherent geochemical model for explaining the origin of the Boom Clay pore water composition, and to propose a reference pore water composition to be used in the laboratory experiments and for speciation calculations and assessments of perturbation of the Boom Clay. Therefore, the first step was to put together the information on the Boom Clay pore water geochemistry from the various studies. This report is thus not the result of an integrated study on the Boom Clay pore water composition. Consequently, some important information may still lack if it was not considered in one of the above mentioned studies. This report describes the pore water sampling and analytical techniques, the results and interpretation of a series of studies carried out in situ in the HADES URF and in the laboratories. Pore water sampling is done in situ from various piezometers, or by the mechanical squeezing or leaching of clay cores in the laboratory. These three pore water sampling techniques are compared and evaluated. A large data-set on pore water composition is available, however, because of the different sampling techniques, the different design of the piezometers, and the different filter materials used, it is not always unambiguous to interpret these data. The reliability and significance of these data have therefore been considered by statistical analyses and geochemical modelling. This enabled to define a reference Boom Clay pore water composition at the Mol site. Finally, some conclusions and recommendations for further research on the pore water geochemistry are given. 13 1 Physical and mineralogical characteristics of Boom Clay 1.1 Physical characteristics The petrophysical and hydraulic parameters of the Boom Clay are summarised in the table below (Table 1-1). Table 1-1: Petrophysical and hydraulic parameters of Boom Clay (compiled from Baeyens et al., 1985; Henrion et al., 1985; Volckaert et al., 1997; SAFIR 2, 2001) Parameter Unity Value Bulk density (sat.) [t/m³] 1.9 - 2.1 Average grain density [t/m³] 2.65 Water content [% dry wt] 19 – 24 Total porosity [vol. %] 36 – 40 Hg injection porosity Macro-porosity Micro-porosity [vol. %] No reliable data: due to swelling and water content, outgassing causes serious change in the Boom Clay fabric Specific surface [m²/g] 44 In situ temperature [°C] 16 Thermal conductivity [W/mK] 1.68 Specific Heat [J/kgK] 1400 Heat Capacity [MJ/m²K] 2.8 Seismic Velocity Vp [m/s] 1300 1852 1700 2000 Hydraulic conductivity Lab. experiments In situ field testing [m/s] (from migration experiments) (laboratory samples) (DSI sonic-logging) (Vs = 526) (uphole sonic logging) (seismic velocity analysis) Vert. 1.3 - 3.4x10-12; Horiz. 3.5 - 7.9x10-12 Vert. 2.1x10-12; Horiz. 4.5x10-12 14 1.2 Mineralogical characteristics The Boom Clay is a sedimentary deposit, mainly composed of siliciclastic minerals, fossils and organic matter. Identification and semi-quantitative analyses of the minerals is generally performed by X-ray diffraction, also in the case of the Boom Clay (see references below). As an alternative, Wouters et al. (1999) used the dual range Fourier Transform Infrared Spectroscopy method (FT-IR) and demonstrated the accurateness of this technique for the measurement of most minerals, including clay minerals. 1.2.1 General mineralogical composition of Boom Clay The mineralogical composition of Boom Clay consists of clay minerals (up to 60 wt%), quartz (~20 wt%), feldspars (~10 wt%), and minor amounts of muscovite, biotite, and some heavy minerals. The clay mineralogy is dominated by illite, smectite, illite/smectite interstratifications, and kaolinite. Chlorite, degraded chlorite and illite/chlorite interstratifications are also present. The authigenic mineral assemblage in the Boom Clay includes apatite, glauconite, authigenic quartz, carbonates (calcite and siderite, 1-5 wt%) and pyrite (1-5 wt%). Gypsum is present as a weathering product. A summary of the Boom Clay mineralogical composition is given in the NEA 'Clay Club' Catalogue of the Characteristics of Argillaceous Rocks (Volckaert et al., 1997). More recent mineralogical analyses (see references in Van Keer and De Craen, 2001; and De Craen et al., 2004b) enabled to update the summary table mentioned below (Table 1-2). Table 1-2: Mineralogical composition of Boom Clay. Values in % total dry wt. (SAFIR 2, 2001; updated with data from various mineralogical studies, see references in Van Keer and De Craen, 2001 and De Craen et al., 2004b) 30-60 % Clay minerals 10-45 % Illite 10-30 % Smectite + illite/smectite ML 5-20 % Kaolinite 0-5 % Chlorite 0-5 % Chlorite/smectite ML Quartz 15-60 % K-Feldspars 1-10 % Albite 1-10 % 1-5 % Carbonates 1-5 % Calcite present Siderite present Dolomite present Ankerite Pyrite 1-5 % Organic Carbon 1-5 % Others Glauconite, apatite, rutile, anatase, present ilmenite, zircon, monazite, xenotime present 15 1.2.2 Clay-mineralogical composition of Boom Clay A lot of effort is done to characterise the clay-mineralogical composition of the Boom Clay. Already in the early seventies, an extensive regional sedimentological study of Boom Clay was performed by Vandenberghe (Vandenberghe, 1974, 1978). In 1976, the clay mineralogical composition of samples taken from the exploratory borehole at the Mol site was studied in detail by Thorez (Thorez, 1976). However, some discrepancies exist between these studies. On the one hand, Thorez (1976) mentioned the presence of vermiculite and the absence of kaolinite. On the other hand, Vandenberghe (1974; 1978) illustrated the presence of kaolinite, while the occurrence of vermiculite was not mentioned. To clarify this contradiction, Vandenberghe and Thorez (1985) used different techniques to identify this mineral. The results obtained do not indicate the presence of vermiculite. In the eighties and nineties, additional clay mineralogical studies were carried out on a limited series of samples (Table 1-3) taken at the Mol site. Samples were taken from clay cores from exploration boreholes, at the underground research laboratory (Ouvry, 1986; Push et al., 1987; Rousset, 1988; Baldi et al., 1990; Goemaere, 1991; Merceron et al., 1993; Griffault et al., 1996), and in the second shaft. The corresponding data set is enlarged by the work of Vandenberghe (1978), Decleer et al. (1983), Laenen (1997) and De Craen et al. (2000) (Table 1-4), who analysed the mineralogical and geochemical variations of the Boom Clay in detail. Table 1-3: Clay mineralogical composition of the Boom Clay near the underground research facility. Clay mineral amounts are relative percentages of the total clay content in the <2 µm fraction; n: number of samples. (from Van Keer and De Craen, 2001) Ouvry 1986 n=2 35 Push et al. 1987 n=? 15 Rousset Vandenberghe Merceron et al. Griffault et al. 1988 1990* 1993 1996 n=? n=1 n=3 n=3 54 16 Smectite Mixed layers 10 4 50 38 Illite/smectite traces Chlorite/smectite Illite 25 46 23 41 25 34 Kaolinite 30 39 18 35 20 19 Chlorite + degraded traces 4 4 traces 9 chlorite * Since the sample was grinded, only the qualitative characterisation of the <2 µm fraction is meaningful. 16 Table 1-4: Generalised clay mineralogical composition of the Boom Clay in %. Clay mineral amounts are relative percentages in the <2 µm fraction; n: number of samples; min: minimum; max: maximum. (from Van Keer and De Craen, 2001) Illite Smectite Illite/Smectite Smectite + Illite/Smectite Chlorite/Smectite Kaolinite Chlorite + degraded chlorite Vandenberghe 1978 n = 30 outcrop min max mean 37 59 48 9 28 16 0 27 12 Decleer et al. 1983 n = 21 outcrop mean 24 67 min 6 25 4 Laenen De Craen et al. 1997 2000 n = 243 n = 40 outcrop Mol-1 borehole max mean min max mean 28 18 20 60 40 70 40 18 9 20 60 40 Traces 10 2 31 14 19 6 10 14 1 45 6 30 3 2 2 30 10 It is generally accepted that the clay fraction is dominated by illite, smectite, illitesmectite interstratifications and kaolinite. Chlorite, degraded chlorite and illitechlorite interlayers were only found in small amounts. The reported semi-quantitative clay percentages differ from one author to the other (see Table 1-3 and Table 1-4). These differences are mainly due to the application of different quantification methodologies. 15 5 17 2 Boom Clay pore water sampling and analytical techniques 2.1 Pore water sampling techniques Pore water extraction from argillaceous rocks is done by either in situ or laboratory techniques. For Boom Clay, being a relatively soft clay and having a high water content, almost all standard pore water extraction techniques are feasible (Sacchi and Michelot, 2000). In situ pore water extraction from Boom Clay is realised by using piezometers. Various types of piezometers are placed in different directions and with filters at different depths (levels) into the clay. Piezometry requires the drilling of boreholes in which porous filterscreens are placed, that are mounted on supporting tubes. In general, sealing off of a piezometer in a borehole is necessary to avoid geochemical alterations of the rock due to the intrusion of atmospheric reactive gasses. However, the natural convergence of Boom Clay automatically seals off the porous filterscreens of the piezometers. This property avoids the use of engineered sealing materials, such as packers or backfill materials. Laboratory pore water extraction techniques used for Boom Clay are mechanical squeezing and leaching. Both techniques require the sampling and preservation of clay cores, in a way that all possible geochemical perturbations are minimised. Since Boom Clay is sensitive to air-oxidation, the clay cores have been immediately vacuum-packed in sample-bags made out of aluminium-coated poly-ethylene sheets. This protects the clay core as much as possible from oxidation and also from drying out. Samples for pore water characterisation were then stored in a nitrogen-filled glove box (oxygen level < 10 mg/l), or in PVC tubes filled and flushed with argon to create the best feasible anaerobic conditions. Whenever possible, long-term storage is done in dark and cooled facilities where the temperature is around 4 °C. To prevent geochemical perturbations (in particular oxidation) during sample preparation prior to analyses, the sample preparation should always be performed in a glove-box (oxygen level < 10 mg/l). 2.1.1 Piezometers Besides the initial use of piezometers to determine hydraulic and mechanical properties of the Boom Clay (pore water pressure, effective stress, hydraulic conductivity, ...), they are also used to obtain samples of Boom Clay pore water. These samples are used to determine specific pore water characteristics or to serve as feed water in all kinds of laboratory experiments. Piezometers have been designed in different shapes and dimensions containing singleor multiple screens. The piezometer design which is mostly used is the cylindrical design. Here, a cylindrical porous filter screen is mounted on a supporting tube equipped with (a) filter chamber(s). Each individual filter chamber (filter screen) is normally equipped with one or two small diameter water pipes, closed at the gallery side with valves (see Figure 2-1). The valves allow the sampling of Boom Clay pore water out of the piezometer whenever needed. Because of the relative high pressures (lithostatic as well as hydraulic) and the mechanical constructability, the first piezometers were all made of durable stainless steel alloys. In November 2000, a first 18 non-metallic piezometer, as part from the ORPHEUS-set up, has been constructed and installed in the HADES-underground laboratory. An overview of the different piezometers and the filter material used is given in Annex 1 and Annex 2. Multi-piezometer composed of stainless steel. Also the filters are composed of stainless steel. Reference piezometer R55I, Connecting Gallery Close up of the filter (5 cm length). Reference piezometer R55I, Connecting Gallery Piezometer composed of PVC. At the inside, a filter chamber is present next to the filter in which the pore water is collected. Two small-diameter water pipes enable to sample the pore water in the URF. MORPHEUS piezometer, Test Drift Pore water sampling and pressure measurement. MORPHEUS piezometer, Test Drift Figure 2-1: Design of a multi-piezometer for in situ pore water sampling. 19 In situ piezometric water extraction is realised by connecting a recipient (usually isolated from air to prevent oxidation) to one of the water pipes of the piezometric filter screen. After opening the water valve, the hydraulic pressure drop will generate a water flow into the recipient. The flow rate is governed by the hydraulic conductivity of the Boom Clay in the vicinity of the filter screen. It is important to note that by applying this in situ pore water extraction technique only the free or unbound pore water can be obtained and collected. Solutes and colloids (such as mobile organic material) will simultaneously flow out under the condition that they are small enough to pass through the hydrodynamic tortuous network of water conducting pores. Therefore, this in situ pore water extraction technique has to be considered as a kind of ultra-filtration technique in which the filtration pressure is equal to the in situ pore water pressure, and, in which the effective pore size of the Boom Clay, in the vicinity of the piezometer, governs the filtration efficiency. Such ultra-filtration efficiency is expressed by the "Molecular Weight Cut Off" number (MWCO). It is questionable whether a fixed piezometric-MWCO number can be given. The reason for this is that the effective pore size can not always be considered as constant. Mean pore size around each piezometer can differ from location to location and is likely to be altered by the Excavation Disturbed Zone (EDZ) of HADES (caused during the excavation of the underground laboratory), and, the excavation disturbed zone around each individual piezometer (caused by the borehole drilling). The piezometer EDZ arises from the necessity to drill an oversized borehole and the consecutive convergence of the Boom Clay during piezometer installation. A supplementary pore size reduction might occur during the piezometric pore water sampling itself: if a piezometer is opened, the water pressure drops to atmospheric pressure, leaving only the lithostatic pressure to act on the clay skeleton. In other words, the drop of pore water pressure leads to an increase of the effective stress if one assumes that the total stress will remain equal. This imposes a squeezing effect onto the clay fabric that yields to lower pore sizes. Besides these engineered perturbations on piezometric pore water extraction, the natural spatial variability of Boom Clay mineralogy might also influence the effective pore size linked to each specific piezometric filter screen. 2.1.2 Squeezing The technique applied in this study is mechanical squeezing. Pore fluids are pressed out of saturated Boom Clay cores by mechanical pressure (Figure 2-2). Squeezing is analogous to the natural process of consolidation, caused by the deposition of material during geological times, but at a greatly accelerated rate. Mechanical squeezing is a widely used technique for the extraction of pore water from low-permeability clays (Sacchi and Michelot, 2000). The squeezing technique and its application to clayey sediments were studied amongst others at the British Geological Survey (Reeder et al., 1998, 1999). Part of the study was performed on the Boom Clay (Reeder et al., 1992, 1994, see also Sacchi and Michelot, 2000). The squeezing technique was further studied in the "Natural analogue study on Boom Clay" (De Craen et al., 2000), and in the study "Natural evidence on the long-term behaviour of trace elements and radionuclides in the Boom Clay" (De Craen et al., 2001). To prevent oxidation of Boom Clay cores by oxygen in air, sample preparation is always performed in anaerobic conditions, in a nitrogen-filled glove box (oxygen 20 level < 10 mg/l). In the glove-box, the samples are first taken out of the aluminiumcoated poly-ethylene sheets, in which they were packed immediately after drilling. The outer rim of the clay core, which has been inevitably in contact with air during the drilling, is then removed to eliminate possible effects of oxidation. Subsequently, the clay core is transferred to the 'squeezing cell', made of stainless steel type 316 (resistant to corrosion and high tensile strength). The sample chamber has a diameter of 8 cm and a height of 10 cm. The squeezing cell is then removed from the glove box and is put under a hydraulic press (COMPAC EMAC HP100). Important changes in the pore water chemistry occur with increasing pressures (references in Sacchi and Michelot, 2000). Indeed, squeezing experiments on Boom Clay have indicated that the pore water chemistry remains more or less the same when a pressure lower than 30-35 MPa is applied, but important changes in the pore water chemistry were observed when higher pressures were applied (De Craen et al., 2000). Therefore, it was decided to squeeze all Boom Clay samples with a relatively low and constant pressure of 30 MPa during one week (for details and arguments see De Craen et al., 2000). After the squeezing, the water samples were stored at 4 °C before chemical or radiochemical analyses. With this technique and the procedure applied, 40 to 50 millilitres of water is generally collected out of about 700 g wet clay. This is about 30 to 35 % of the total water content. Note: As mentioned above, samples for pore water characterisation were vacuumpacked in aluminium-coated poly-ethylene sheets and stored in a nitrogen-filled glove box (oxygen level < 10 mg/l), or in PVC tubes filled with argon (to prevent oxidation as much as possible) at 4 °C. Clay cores which were vacuum-packed in aluminiumcoated poly-ethylene sheets but stored in air and room temperatures, are often oxidised, resulting in an unrepresentative pore water composition (De Craen, 2001; De Craen et al., 2002a, De Craen et al., 2003). Figure 2-2: Set-up for the extraction of pore water by mechanical squeezing of clay cores. 21 2.1.3 Leaching A leaching method involves batch experiments and modelling of equilibrium chemistry of the resulting extracts. Different from a piezometric or a mechanical squeezing technique in which pore water samples are collected directly, a leaching method add a leachant to the clay sample, hence leads to a dilution and processes associated with it. Bradbury and Baeyens (1998) developed a method by which they determined the soluble salt concentrations of Oplinus clay by the dilution factor using different solid to liquid ratios (S/L ratio) and the cation concentrations through cation exchange and mineral solubility equilibriums. In this report, we follow the general idea of the method but also study the effect of colloids, which is a distinguished characteristic of Boom Clay. Moreover, the interpretation of our results opens some new aspects and demonstrates potential differences between Oplinus Clay and Boom Clay. The experimental procedure is illustrated in Figure 2-3. Boom Clay samples from the HADES 2001/4 borehole were grinded, suspended, and agitated in NaHCO3 solution of 0.01 M. The bicarbonate solution was used as the leachant because Boom Clay pore water is basically a dilute NaHCO3 solution. Some samples were suspended in distilled water for comparison. Experiments were conducted in glove boxes to protect the clay samples from oxidation. The oxygen content of the glove boxes is about 2 ppm but generally below 10 ppm. For practical reasons, some experiments were carried out in an Ar glove box and some others in an Ar/CO2 (g) glove box. The CO2 content in the glove box was 0.4 percent to mimic the supposed in situ partial pressure of CO2 (10-2.4 atm). Four different S/L ratio were used: 25, 50, 200, and 800 gram wet clay per litre of solution. The leaching duration was 2 to 3 months in which a steady aqueous concentration of the major ions were reached. After the leaching, samples of the suspension were centrifuged at 21,255 g for 2 hours before the chemical analysis for major cations and anions. Some samples were further filtered by 0.45 µm filters and YM3 (3000 MWCO) centriplus ultrafilters to study the possible effect of clay particulates or colloids on the concentration of clay water components. solution: NaHCO3 (0.01 M) or water chemical analysis for cations and anions clay centrifugation supernatant filtration (0.45 µm) ultrafiltration (3000 MWCO) Figure 2-3: Shematic presentation of the experimental procedure of pore water extraction by leaching. 22 2.2 Analytical techniques 2.2.1 pH, Eh, pCO2, electrolytic conductivity and dissolved gases 2.2.1.1 pH Three analytical principles exist to determine pH-values: colorimetric, electrochemical and electronic (Omega Engineering, Inc. 2001, Meier et al., 1989; Poghossian et al., 2002). The colorimetric principle relies on a detectable colour change of a dye as function of the varying hydrogen concentration. The electrochemical principle makes use of a measurable electrical potential change of a pH sensitive electrode, as function of pH variations. The electronic principle relies on a signal change in an Ion Sensitive Field-Effect Transistor (ISFET) if the hydrogen concentration of a sample changes. As the latter principle is a fairly new one that still needs a lot of scientific development, no analyses based on this principle are considered in this report. The simplicity of the colorimetric principle is its main advantage, but colorimetric techniques suffers from a lack of accuracy: ± 0.1 pH units on the measurement is the best accuracy that can be achieved (Characteristics of a Fiber Optical pH meter, DBE Technology GmbH). A second important drawback of colorimetric methods is that they are often difficult to interpret and this especially in coloured samples (e.g. colour changes in coloured samples, as in Boom Clay pore water). Therefore, the use of colorimetric principles can only give a rough indication of the pH-value (Omega Engineering, Inc. 2001 pH Technical reference guide). The electrochemical principle, however, is scientifically recognised as being an accurate, robust and reliable principle, that is applicable under almost every physicochemical measurement condition. To illustrate this, it is worthwhile to mention that the U.S. Geological Survey only approves pH determinations that are based on this measurement principle (Radtke et al., 2003). Therefore, the pH of Boom Clay pore water is generally measured with electrochemical based pH electrodes. A drawback of the electrochemical principle is the leaching of salt from the pH electrode, more specifically the reference electrode which is normally build in a pH electrode. For environmental reasons, the commonly used salt to stabilise the reference signal is potassium chloride. In aqueous samples this salt is inert and does not influence the pH of the solution. However, when measuring soil samples, the leached potassium chloride can disturb the pH measurement through secondary effects like: ionexchange, suspension effect and varying solid–liquid ratios. As these three effects counteract each other, it is sometimes difficult to quantify the overall effect. An electrochemical pH measurement system consists of three parts: a pH indicator electrode, a reference electrode and a mV/pH meter. For each of these three components, numerous models and types exist. The choice of the complete pH measuring system depends on the specific measurement requirements: nature of the samples (aqueous, organic, semi solid, ...), sample conditions (temperature, pressure, electrolytic conductivity, ...) and system demands (laboratory or in situ measurement, batch or on line measurement, desired response time, electrode stability, data). From the measurement requirements point of view, laboratory pH measurements of Boom Clay pore water samples are not very demanding. Typically the samples are aqueous, not pressurised, at room temperature and possess sufficient electrolytic conductivity to be measurable (see Section 2.2.1.4). However, to determine the true in situ pH value of Boom Clay pore water, laboratory measurements are questionable for two reasons: 23 the low pH buffering capacity and the unavoidable physico-chemical perturbations of Boom Clay pore water (e.g. loss of carbon dioxide through degassing, perturbation through oxidation of dissolved organic matter, ...). The physico-chemical perturbations appear at three stages: during the sample collection, when the samples are manipulated and during the time of pH-measurements in the laboratory. Because of the perturbations associated with laboratory pH measurements of Boom Clay pore water, the pH values reported in most earlier publications or reports were not always representative of the real in situ pH. Because laboratory pH measurements fail to render the true in situ pH value of Boom Clay pore water, pH measurement under undisturbed physico-chemical conditions are imperative. However, the requirements for such an in situ pH measurement system are very demanding. The main idea is that the integrity of Boom Clay pore water has to be maintained during the in situ pH measurements. To realise this, the following measurement requirements are needed: measuring under in situ hydraulic pressure to maintain pressure depending dissolution chemistry (e.g. calcite solubility, dissolved gasses, ...), avoiding contact of the Boom Clay pore water with the atmosphere to prevent gas exchanges (e.g. CO2 loss, O2 intrusion, ...), and, monitoring of the pH evolution to determine the moment of geochemical equilibrium and to quantify reaction kinetics. The first meaningful attempts to measure in situ pH values were made during the CERBERUS project (Beaufays et al., 1994). In this project, the pH measurements were done with electrochemical pH-electrodes screwed in a non-pressurised heated flow-through cell. Also during the ARCHIMEDE-argile project (Beaucaire et al., 2000; Griffault et al., 1996) in situ pH measurements have been done using a Fiber Optical pH measurement system, based on colorimetric principles. As mentioned above the inaccuracy of such systems is larger then ± 0.1 pH units, and this, even without taking into account the errors linked to the disturbed geochemistry (temporarily and locally) of the Boom Clay pore water inside the piezometer, due to the opening of the piezofilter waterline to introduce the optical pH-fibre. Since 1996, sound efforts have been made to implement glass electrode technology for in situ measurements of pH (De Cannière et al., 1997). The installation of the ORPHEUS experiment in 2000 (Moors et al., 2002) was the result of the best available technology and gained expertise to fulfil the requirements for representative in situ pH measurements. The ORPHEUS set up (see Figure 2-9) is composed of a flow-through cell equipped with robust and rugged solid-polymer filled pH electrodes (Xerolyt®, Mettler-Toledo). The flow-through cell is placed in a closed circuit configuration between the inlet and outlet water pipe of a piezometric filterscreen. The closed circuit configuration maintains dissolved gas-equilibria. The polymer filled Xerolyt-electrodes ensure long term stability and combine this quality with the accuracy and reliability of common electrochemical glass electrodes. A circulation pump (Milton Roy Solenoid Diaphragm Metering Pump) provides water circulation inside the closed circuit. This water circulation ensures the contact of the Boom Clay pore water with the Boom Clay solid phase and the pH electrode. As the system is closed, geochemical equilibrium of the circulating Boom Clay pore water with the surrounding solid Boom Clay will be reached after a certain time. A data acquisition system monitors the evolution of pH measurements and helps to indicate when geochemical equilibrium is reached. A controlled-atmosphere cabinet (argon flushed) hosts the pump and flow through cell of the pH measurement system. This controlled-atmosphere cabinet provides additional protection of the experimental set 24 up against unwanted gas interactions. At the end of a measurement campaign, the collected measurements are corrected for any electrode drift (see Annex 3 for details on the electrode drift test). As mentioned above, the inevitable leaching of potassium chloride when using glass electrodes can cause secondary effects that can cause an overall effect on the measured pH value. Chemical analyses and geochemical modelling might aid to correct the measurements, not only for electrode drift but also for geochemical drift. 2.2.1.2 Eh To some extent, a similar reasoning as for pH measurements goes for redox measurements. However, the correct interpretation of measured redox potential values is far more complicated. Basically any redox reaction can be represented by the following equilibrium reaction: i red Ox + ne − ↔ Red i ox Every oxidation and reduction reaction generates a current flowing in opposite directions (ired and iox). These currents (sum of both is called exchange current) have to be strong enough so that they are measurable (detectable) with a redox measurement system (Schüring et al., 2000). Colorimetric redox indicators are not sensitive enough to determine the redox state of natural waters. The only accepted measuring principle to measure Eh in natural waters is the electrochemical principle. And, even with measuring systems based on this principle, it is questionable whether or not the measured values represent the true redox state (Christensen et al., 2000). A classical electrochemical redox measurement consists of three parts: a redox indicator electrode, a reference electrode and a sensitive millivolt meter. The ideal requirements for each of these components are the following: a reference electrode has preferentially following characteristics: its potential does not depend on changing redox conditions, it has no internal electrical resistance, and, it is in perfect electrical contact with the media in which it is submersed. The indicator electrode should have following characteristics: it behaves absolutely inert (i.e. it does not participate to any of the on going redox reactions inside the medium), on the other hand, it catalyses every redox reactions at its surface, and, has no double layer or coating that might limit the passing of the exchange current. The redox meter which is electrically connected to the electrodes must have an extremely high input resistance, and no voltage offset. As for pH measurement systems, from each of these three components, numerous models and types exist. Unlike for pH measurement systems the choice of a complete Eh measurement system is not solely based on objective criteria. Often, a compromise between habits and measurement requirements has to be found and is used. Bearing these facts in mind, it is obvious that analytical redox measurements will seldom reflect the theoretically calculated values. However, the empirical use of measured redox values, under the best possible conditions, will always yield valuable information about the redox-tendency of the measured medium (Christensen et al., 2000; Nordstrom et al., 2003), in our case the Boom Clay pore water. Combined with other analytical results and chemical calculations a clear insight of the Boom Clay pore water redox condition can be obtained. 25 As discussed in Section 2.2.1.1., the reliability of laboratory measurements, in view of rendering a representative in situ Eh value of Boom Clay pore water, is doubtful. Again, as for pH measurements, a very low buffering capacity of redox active species is present and physico-chemical redox perturbations of any sample of Boom Clay pore water, under laboratory conditions, are unavoidable (possible irreversible redox reactions, ...). On top of these sample handling problems, the limitations of each electrochemical redox measurement system complicate the interpretation of laboratory redox measurements to obtain a representative in situ Eh value for Boom Clay pore water. The implementation of redox electrode technology for in situ measurements is, as for pH-electrodes, far from evident. But the advantage of in situ measurements is to be able to have measurement conditions which are the closest to the real conditions. This advantage makes it worthwhile to invest time and work in in situ measurements. Again, high water pressure and long term electrode stability demand appropriate precautions and measures to obtain accurate and reliable in situ values of the redox tendency of Boom Clay pore water. The best solution is the simultaneous use of the flow-through cell set-up for pH electrodes and redox electrodes. Specifically for redox measurements, the following extra precautions are foreseen: the anaerobic atmosphere prevents oxygen perturbation, the exclusive use of polymer or ceramic materials allows to circulate Boom Clay pore water that never has come in contact with redox active metal surfaces, and the use of different indicator electrode materials allows qualitative comparison of the redox measurements. In contrast with pH and as explained above, just monitoring and mathematically correcting the measured in situ redox values will seldom yield to representative Eh values. Nevertheless, in situ redox measurements obtained with an experimental set up in which all precautions are taken, will render the most representative redox values of Boom Clay pore water. To eliminate as much as possible errors linked to the redox measurement system, an electrode functionality and an electrode drift test (see Annex 3 for details of the tests) is performed. These tests assure the good functioning of the used redox electrode(s) during the whole duration of the in situ measurement and, therefore, help with the correct interpretation of the redox results. 2.2.1.3 pCO2 and other dissolved gasses The measurement of the partial pressure of carbon dioxide and other dissolved gasses in argillaceous rocks is a technical challenge. Different methods have been considered (Henrion et al., 1985). The partial pressure is either calculated based on the relation of pCO2 with the measured pH and the inorganic carbonate chemistry or the partial pressure is measured based on the physico-chemical principle on which Henry's law relies. In its simplified form the law of Henry can be written as follows: pA = xA K A With pA the partial pressure of solute A in the gas phase, xA the mole fraction of solute A in the liquid phase and KA a constant called Henry's law constant with the dimension of pressure. The law of Henry thus states that there exists a linear relationship between the concentration of a gaseous solute dissolved in a solution and 26 the concentration of the same gaseous solute in the gas phase, in contact with the solution (assuming that both phases are equilibrated). Thus, it is sufficient to determine the concentration of a dissolved gas either in the gas phase or in the solution to know its concentration in the remaining other phase. The first attempt to measure the partial pressure of CO2 in Boom Clay was done by Henrion (Henrion et al., 1985). He assumed, in analogy with common CO2 measurements on frozen pieces of pole ice, that a large piece of frozen Boom Clay will keep its initial content of CO2. The opportunity to sample a piece of frozen Boom Clay was given during the construction of the underground laboratory. To be able to excavate the first access shaft of the HADES-laboratory, the surrounding soil (including a huge Section of the Boom Clay) had to be frozen. From this frozen Boom Clay, a large piece was sampled. This piece of frozen Boom Clay was put in a aluminium-lined sample bag. The sample bag was immediately sealed off to prevent escape of gasses. After unfreezing, the CO2 content of the gas atmosphere inside the gastight sample bag was measured. From this measurement the partial pressure of CO2 was determined assuming the relationship with the dissolved carbonate system (CO2, CO2(aqua), H2CO3*, HCO3-, CO32-). Because of the significance of the dissolved carbonate system (e.g. pH controlling mechanism, ...) in relation to radionuclide migration and speciation, the knowledge of representative in situ pCO2 values for Boom Clay pore water has become a priority. This knowledge is also essential for geochemical modelling and to evaluate the response of Boom Clay towards changing environmental conditions (e.g. alkaline plume, sodium nitrate perturbation, ...). The best solution to obtain representative in situ values for pCO2, is to use a set up in which the experimental boundaries are imposed and controlled by real in situ conditions. Because, under these conditions no gas phase is present in Boom Clay pore water and because it is easier to measure CO2 in a gas phase, an artificial gas phase has to be introduced which can equilibrate with the Boom Clay water. A schematic presentation of an experimental set up is given in Figure 2-4. In this set up Henry's law is valid: a gas phase is present by creating a static inert (argon) gas bubble in the barrel, and equilibrium can be obtained by circulating the Boom Clay pore water from the barrel towards a piezometric filterscreen and vice versa. In the pores of the piezometric filterscreen the circulating Boom Clay pore water will continuously re-equilibrate with the Boom Clay formation and its minerals. Inside the barrel the circulating Boom Clay pore water will equilibrate with the static gas phase by Henry's law principle. Periodically, the gas phase will be analysed for CO2 measurements. After a certain time, it is expected to measure stable carbon dioxide concentrations. From these CO2 concentrations and the total gas pressure, the pCO2 can be determined. Stable isotope composition analysis (δ 13C and/or δ 18O) to try to determine the origin of the CO2 will be considered after pCO2 quantification. 27 Gas-phase sampling Pinlet Poutlet level Water + dissolved gasses Figure 2-4: Schematic representation of the experimental setup for pCO2 measurement. The same set up allows the simultaneous determination of all other dissolved gasses, in Boom Clay pore water, under the condition that they are present in measurable quantity. It is expected to find gasses such as nitrogen (N2), Hydrogen Sulfide (H2S), Helium (He), alkanes (Methane (CH4), Ethane (C2H6), ...) and maybe some other volatile organic components. At first the gas samples will be analysed with a massspectrometer as most of the molecule masses of the expected gasses differ significantly. 2.2.1.4 Electrolytic Conductivity (EC) The electrolytic conductivity (EC) of a solution is proportional to the quantity of dissolved ions (charge carriers) present in the Boom Clay pore water. The obtained value provides also a very good idea of the Total Dissolved Solids present in Boom Clay pore water. A conductivity cell connected with a conductivity meter is the standard analytical technique which is used to measure Boom Clay pore water conductivity. The conductivity cell consists of a glass support on which two or more platinum bands are fixed. These platinum bands are coated with platinum black to limit polarization effects. Any conductivity cell is characterized by a cell constant to allow normalization and standardization of different measurement systems. The conductivity meter is in fact a power supply which generates an alternating current (AC) potential onto two platinum bands of the conductivity cell. The measured current is proportional to the conductivity or EC-value of the liquid in which the cell is submersed. An EC measurement serves more as a quality indicator rather then as an analytical parameter. The reason for this is that it is a bulk parameter from which only limited analytical information can be gathered. 28 2.2.2 Other techniques 2.2.2.1 The total organic carbon content (TOC) The total organic carbon content in Boom Clay (TOC) is measured with a hightemperature TOC analyser. The total carbon content (TC) is determined by injection of an aliquot of the sample, without any pre-treatment, in a combustion tube at 680 °C. Catalytic oxidation transforms the TC of the sample completely to CO2 and H2O. After drying, the CO2 concentration is measured by a non-dispersive infrared detector. The inorganic carbon content (TIC) is determined by injection of an aliquot of the sample, without any pre-treatment, in a TIC reactor, which contains acidified water (20% H3PO4) at room temperature. In this acidic environment, all forms of IC are purged out of the solution as CO2. After drying, the CO2 concentration is measured by a non-dispersive infrared detector. The total organic carbon content (TOC) is made by the difference of TC and TIC. TOC measurements were performed with a Dohrman DC-190 High Temperature TOC analyser at the Laboratories of Geological Disposal, Waste and Disposal Department, and Analyses and Applied Radiochemistry, Nuclear Chemistry and Services, SCK•CEN, Belgium. 2.2.2.2 UV/VIS spectrometry A UV/VIS spectrometer is used for the characterisation of Dissolved Organic Carbon (DOC). The instrument used is a PERKIN ELMER, Lambda 40 at the Laboratory of Geological Disposal, Waste and Disposal Department, SCK•CEN, Belgium. Two radiation sources, a deuterium lamp (UV) and a halogen lamp (VIS), cover the working wavelength range of the spectrometer. The monochromator used is based on holographic grating, while the exit slit restricts the spectrum segment to a nearmonochromatic radiation beam. The slits provide a spectral band pass of 0.5, 1, 2 or 4 nm. This monochromatic beam is reflected on a beam splitter, allowing 50 % of the radiation to pass through the sample cell and 50 % of the radiation to pass through the reference cell (dual beam). The passed beams continue their way onto a photodiode detector. Absorbance is normally measured at 280 nm for the determination of DOC content. 2.2.2.3 The cation concentration (Ca, Fe, K, Mg, Na, Si) The cation concentration (Ca, Fe, K, Mg, Na, Si) in the pore water is measured by Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES). Samples are diluted ten times and acidified with 1% HNO3 / 5% HCl; higher dilutions may be necessary when the concentrations are too high. The diluted samples are filtered through a 0.45 µm filter. Analysis is performed on a simultaneous ICP-AES with radial plasma view and SCD (Segmented-array Charged-coupled device Detector). The results obtained with the principal spectral line of each element are confirmed by measuring an alternative spectral line. Furthermore, spectral interferences are accounted for by the use of interelement corrections. Cation concentrations were measured at the Laboratory of Analyses and Applied Radiochemistry, Nuclear Chemistry and Services, SCK•CEN, Belgium. 29 2.2.2.4 The anion concentration (F-, Cl -, Br -, HPO42-, NO3-, SO4 2-) Ion chromatography (IC) is used for the analyses of the anions Cl -, Br -, HPO42-, NO3and SO4 2- in the pore water. For the extraction of the anions, a Dionex AG4A-SC guard column and a Dionex AS4A-SC analytical column are used. To avoid overload of the separation column, samples are usually diluted ten times. Higher dilutions may be necessary when the concentrations are too high. The diluted samples are analysed by the use of a classical ion chromatograph with a 1.8 mM Na2CO3 / 1.7 mM NaHCO3 solution as eluent and suppressed conductivity detection. For the measurement of the concentration of F- in the pore water, the ion selective electrode (ISE) is used. Samples (and standards) are diluted with TISAB (Total Ionic Strength Adjustment Buffer), which provides a nearly uniform ionic strength background, adjusts pH and breaks up complexes. The fluoride concentration is determined by direct potentiometry using a combined fluoride / reference electrode. Anion concentrations were measured at the Laboratory of Analyses and Applied Radiochemistry, Nuclear Chemistry and Services, SCK•CEN, Belgium. 2.2.2.5 The trace element concentration Trace elements are measured by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS). The pore water samples are first diluted in 2 % nitric acid short before analysis. An internal standard is added to improve the precision and to correct for matrixeffects (mainly signal suppression). During analysis, the sample solution is nebulised into flowing argon gas and passed into an inductively coupled plasma where the elements are separated according to mass, detected, multiplied and counted. Trace element concentrations were measured with an Elan 5000 Perkin Elmer ICPMS at the Laboratory of Analyses and Applied Radiochemistry, Nuclear Chemistry and Services, SCK•CEN, Belgium. 2.2.2.6 The stable isotope composition The stable isotope composition is measured by a gas source mass spectrometer. The methodology first involves the conversion of the element of interest into a gas. In general, hydrogen is analysed as H2, oxygen and carbon are both analysed as CO2, and sulphur is usually analysed as SO2. The gas is then purified and introduced into the mass spectrometer for analyses. Stable oxygen and hydrogen isotope analyses were determined at the Stable Isotope Laboratory at BGS, UK, using a VG-isogas dual-inlet twin analyser gas source mass spectrometer. Determination is carried out on carbon dioxide and hydrogen gases, for O and H respectively, prepared from the original sample according to the procedures described by Darling et al. (1992). The mass spectrometer has a dual collector arrangement for each of the two nuclides and a dual inlet system to permit the rapid switching between the sample gas and a standard gas with calibrated 18O/16O or 2 H/1H. Data are expressed in ‰ relative to the Standard Mean Ocean Water (SMOW). The overall method precision is estimated to be better than ± 4 ‰ for δ2H and ± 0.2 ‰ for δ18O, based on the repeated determination of independent quality control standards. 30 Oxygen isotope analyses of sulphate were performed at the Isotope Geoscience Unit of SURRC, Glasgow, UK, following the procedure reported in Hall et al. (1991). Combustion of an intimate mixture of the sample plus spectrographically pure graphite took place in a platinum crucible at a temperature around 1200 °C. Evolved SO2 gas was then analysed on a VG SIRA II mass spectrometer and standard corrections were applied to the raw data. Reproducibility is about ± 0.2 ‰. Sulphate isotope compositions were determined at the Isotope Geoscience Unit of SURRC, Glasgow, UK. Sulphur isotope analyses of pure BaSO4 precipitates were carried out according to the standard procedures of Coleman and Moore (1978), involving combustion at 1120 °C of an intimate mixture of the sample, excess Cu2O and a pure SiO2 catalyst. Product SO2 gas was analysed on a VG SIRA 10 mass spectrometer and standard corrections were applied to the data. Data are expressed in ‰ relative to the Canyon Diabolo Troilite (CDT). Reproducibility is about ± 0.2 ‰ for δ34S, based on the repeated analyses of internal and international standards. 2.2.2.7 The radiochemical composition 238 U, 234U, 232Th, 230Th and 226Ra isotope concentrations in Boom Clay pore water were measured at the Laboratory of the Section Mineralogy and Petrography, Royal Museum for Central Africa, Tervuren, Belgium. Samples were acidified with HNO3 and an internal standard was added. For the accuracy, U is measured in an internal standard of water (SLRS 4). Analyses were performed with a Finnigan Element 2 high resolution-inductive coupled plasma-mass spectrometer (HR-ICP-MS). HR-ICPMS is similar to the ICP-MS technique previously described with a few modifications. The mass spectrometer used for detection is a quadripole followed by a magnetic sector instead of just a quadripole. The main advantages are that most interferences can be resolved providing essentially interference-free analysis. The HR-ICP-MS provides detection limits in the ng/l (ppt) to pg/l (ppq) range. Many isotope ratios can be determined to better than ±0.1%. The 14C was studied by UPS – GdR Tandétron, Gif-sur-Yvette, France. 14C measurements were performed on total dissolved inorganic carbon and total dissolved organic carbon. 14C measurements on total dissolved inorganic carbon were performed by acidification of the water sample, followed by reduction of the produced CO2 to graphite by hydrogen, and measurement of the graphite isotope ratios by accelerator mass spectrometry. 14C measurements on total dissolved organic carbon were performed by isolation and purification of the fulvic acids by CuO/Cu2O, reduction of the produced CO2 to graphite by hydrogen, and measurement of the graphite isotope ratios by accelerator mass spectrometry. 31 2.3 Sample locations In order to study the Boom Clay pore water composition and the in situ geochemical conditions, several specific piezometers are considered: EG/BS Extension Gallery Bottom Shaft ARCHIMEDE Acquisition et Régulation de la Chimie des Eaux en milieu argileux. SPRING 116 Source Piezonest at RING 116 of the Test Drift gallery ORPHEUS Oxidation Reduction Potential and pH Experimental Underground Station MORPHEUS Mobile ORganic matter and Pore water extraction in the Hades Experimental Underground Site These piezometers are all installed in the HADES URF (Figure 2-5). They were typically designed either for the sampling of Boom Clay pore water, or for the measurement of geochemical parameters such as the pH and the Eh. A detailed description of the various piezometers is given below and summarised in Annex 1. The many other piezometers in the HADES URF are not described here, since they are not used for geochemical purposes. Each pore water sample remains a code in which the name of the piezometer, filter material, sample number and date of sampling is included (see Annex 1). Figure 2-5: Location of the piezometers considered in this report, in the HADES underground facility (Dimensions are not to scale). In Belgium, the reference level is: "Tweede Algemene Waterpassing" (TAW). It is important to note that for historical and practical reasons TAW is not always used. Some depths-values use the earths surface (Below Surface Level, BSL) or drilling table (Below Drilling Table, BDT) as reference level. For the HADES URL Shaft 1, the conversion between TAW and BSL is: TAW = BSL + 25.60 [m]. Also for 32 practical reasons, distances relative from gallery lining are used. The indication given in this case is: distance "Extra DOS", referring to distance relative to the outside surface of the lining. 2.3.1 EG/BS piezometer The EG/BS (or EGBS/2; Neerdael, 1984) piezometer was installed in September 1983. It is one of the oldest piezometers still in operation. The initial goal of this piezometer was to collect large amounts of pore water. The EG/BS piezometer is a vertically orientated piezometer located at the bottom of the First Shaft. Figure 2-6 shows a schematic view of the complete piezometer-construction at its location. Coarse sand (0.71 – 1.25 mm) was used to enhance the water-draining capabilities of this piezometer. The stainless steel cylindrical filter screen of this piezometric set up, has a length of 60 mm and a diameter of 56 mm. It is made from high porosity seamless filter tube made by "Krebsöge", quality: SIKA R5, material: 1.4404 (AISI 316 L/B), pore size distribution: 7 to 16 µm. The coarse sand column, in which the stainless steel filter screen is placed and centralised, is about 13 m long (260 m to 247 m BSL) with a diameter of only 85 mm. This coarse sand column hydraulically interconnects the pore water from the Boom Clay between septaria-levels S40 and S60. This large range encompasses also the silty "double band" of which the hydraulic conductivity is two to three times higher then for the rest of the Boom Clay (Wemaere et al., 2002). Physically and chemically, the water collected with this EG/BS piezometer will be proportionally influenced by the characteristics of the "double band". The reason of including this piezometer into the current report is that it is the piezometer of which the most diverse analyses are available and this over a period of more than 20 years. Major disadvantage for geochemistry are the influences of the coarse sand backfill material, the bentonite top-cover seal and the presence of the "double band". COARSE SAND: 0.71 – 1.25 mm Figure 2-6: Schematic drawing of the complete EG/BS (EGBS/2) piezometerconstruction, shown at its location on the bottom of the First Access Shaft. Depth in BSL. (from Neerdael, 1984). 33 2.3.2 ARCHIMEDE piezometers In the frame of the ARCHIMEDE-argile project, in which the better understanding of the Boom Clay pore water chemistry was the main objective, several piezometers were installed in the Boom Clay. The ARCHIMEDE #1 piezometer was installed in March 1992 in the ANDRA gallery of the HADES URF, between sliding ribs 24 and 25. It is a semi-horizontal piezometer (3% inclined upwards) oriented towards the east. It is 15 m long with a diameter of 60 mm. This piezometer is used for the sampling and chemical analyses of Boom Clay pore water. The ARCHIMEDE #2 piezometer was installed in April 1992 in the ANDRA gallery of the HADES URF, between sliding ribs 4 and 5. It is also a 15 m long horizontal piezometer oriented towards the east, but with a diameter of 140 mm. This piezometer is used for in situ pH measurements. Both piezometers are entirely composed of stainless steel and contain five filter screens, also composed of stainless steel. A schematic view of the location of the ARCHIMEDE piezometers is shown in Figure 2-7. Figure 2-7: Schematic presentation of the location of the various boreholes and piezometers in the frame of the ARCHIMEDE-argile project (from Griffault et al., 1996) 34 2.3.3 Spring 116 Because of the presence of coarse sand around the EG/BS piezometer, the EG/BS piezometer probably provides Boom Clay pore water which is geochemically disturbed. Therefore, a new piezometer equipped with large filter screens has been installed in the HADES laboratory in October 1999. This piezometer is called SPRING 116. The purpose of this piezometer is to provide sufficient quantities of representative Boom Clay pore water as feed and reference material for laboratory experiments. The piezometer is placed horizontally in the Boom Clay, and is located in the Test Drift part of the HADES URF, at ring 116, pointing towards the east. The SPRING 116 piezometer is entirely made of stainless steel and contains in total four large surface filter screens. These are made from high porosity seamless filter tube made by "Krebsöge", quality: SIKA R5, material: 1.4404 (AISI 316 L/B), pore size distribution: 7 to 16 µm. A schematic presentation is given in Figure 2-8. The spring 116 piezometer consists of four filter screens with the following dimensions: an outside diameter of 149 mm, an inside diameter of 142 mm, and a length of 1500 mm. The first screen starts at about 5 m (Extra Dos) and the last deepest screen ends at 12 m (Extra Dos). Although the four different filters can be sampled separately, in total, about six meters of filter screen is placed horizontally in the Boom Clay (see Figure 2-8. The hydraulic interconnections of this substantial length of Boom Clay results in a high water flow (approximately 350 ml per day) and, consequently, the capability of collecting large amounts of Boom Clay pore water. Figure 2-8: Schematic presentation of the SPRING-116 piezometer, with four large filter screens of 1500 mm close to each other. 2.3.4 ORPHEUS piezometer The ORPHEUS piezometer was installed in November 2000 at ring 116 of the Test Drift gallery. This piezometer is horizontally oriented towards the west (installed just in front of the SPRING 116 piezometer). Figure 2-9 shows a schematic overview of the ORPHEUS setup. This figure visualises the goal of the ORPHEUS experiment, which is to study in situ the geochemistry of undisturbed Boom Clay pore water. For this purpose, the piezometer of ORPHEUS is the first that is entirely constructed out of polymer based and metal free materials. With this construction, Boom Clay pore water has never been in contact with metal surfaces, and, at the same time it is not oxidised since it can be sampled under protective atmosphere conditions. To evaluate the influence and constructability of different metal free porous materials, the 35 ORPHEUS piezometer contains four filter screens composed of four different materials: Sintered glass Polyethylene Carbo Schumatherm Each of these filter screens is 250 mm long with a fixed outside diameter of 120 mm. The inside diameters vary little from each other due to the four different fabrication processes. The mean pore sizes of the materials are: for glass between 10 and 16 µm (porosity-class P4), for Polyethylene 40 µm (Filtroplast 40), for carbon 90 µm (Carbo 40), and, for Schumatherm 60 µm (Schumatherm 30). A data-sheet of the used filter materials can be found in Annex 2. Every filter screen is equipped with two PEEK (Poly-Ether-Ether-Keton) water pipes, 1/8" diameter, to conduct the pore water into the controlled atmosphere cabinet (anaerobic environment). PEEK is chosen as material for water pipes because it has an extreme low oxygen permeability (oxygen permeability coefficient = 8.3 x 10-19 [m.s-1.Pa-1] which is at least four orders of magnitude lower then any other polymer tubing material), which guarantees the best possible protection of the Boom Clay pore water against oxidation. As extra precaution, the free space inside the piezometer supporting tube and all voids around the water pipes have been (back)filled with a hard type protective polymer (Stycast W19). pH Eh DAQ P Cabinet with controlled atmosphere + flow-through cell with electrodes Shumatherm filter Carbo filter Poly Ethylene filter Sintered Glass filter Figure 2-9: Schematic presentation of the ORPHEUS set up and its piezometer. The four different filter materials are clearly visualised by their specific colour. 2.3.5 MORPHEUS piezometer The MORPHEUS piezometer, installed in May 2001 in the HADES laboratory (Test Drift between ring 11 and 12), is a vertically oriented piezometer designed to study the variability of the Boom Clay pore water composition underneath the HADES laboratory. In contrast to the other vertically oriented piezometer EG/BS, the MORPHEUS piezometer allows pore water sampling at 12 distinct stratigraphic levels of the Boom Clay, including the level of the "double band". Each filter of this 36 multipiezometer is only 10 cm long. In view of studying the geochemistry of Boom Clay pore water, MORPHEUS is an unique piezometer, that allows the sampling of pore water from separate Boom Clay layers such as: organic rich layers, carbonate rich layers, more silty layers, ... . Figure 2-10 gives a schematic view of this piezometer positioned next to the Boom Clay layering. All the porous filter screens of this piezometer are made out of "Schumatherm" filters (see Annex 2 for material specification). Schumatherm is used for its chemically inert characteristics. The mean pore size of the used Schumatherm is 60 µm. The filter screens are mounted onto a PVC supporting tube and are all equipped with two water pipes. For economic reasons, the water pipes are made of nylon and not of PEEK. The nylon water pipes lead the water into teflon coated stainless steel sample cylinders. Although the pore water collected from MORPHEUS is assumed to be less protected against oxidation, the setup still guarantees the sampling of Boom Clay pore water with only limited geochemical disturbances. m Extra DOS 15 Boom Clay lithostratigraphy m TAW MORPHEUS piezometer -212.27 S70 F23 20 -217.27 S61 F20 S60 25 F18 -222.27 F15 S50 30 -227.27 F13 F12 F10 Putte Member DB 35 White bands: clayey layers F6 Grey bands: silty layers Black bands: septaria layers, indicated by Sxx -232.27 S40 DB: double band (two very silty layers) F4 F2 Terhagen Member 40 F9 F8 Filters in the MORPHEUS piezometer indicated by Fxx -237.27 1m Figure 2-10: View of the MORPHEUS piezometer and its relative positioning towards the Boom Clay layering. 37 3 Boom Clay pore water composition and characteristics 3.1 Chemical composition According to Henrion et al. (1985), the Boom Clay pore water in Mol is equivalent to a 1.25 g/l NaHCO3 solution, rich in humic acids with a large molecular size spectrum, and a CO2 pressure of 10-2.5 atm. Because of the ionic mobilities in compacted Boom Clay larger than 10-10 m² sec-1 (Henrion et al., 1985), and the low hydraulic conductivity in the order of magnitude of 10-12 m sec-1 (De Cannière et al., 1996), it is assumed that the pore water is in equilibrium with the solid phase and that the pore water composition is constant throughout the thickness of the formation. The last few years, several new piezometers were installed in the Boom Clay, providing pore water at different locations in the clay. A lot of pore water samples were taken and analysed, resulting in a large dataset. This enabled us to study the variability of the pore water chemistry, and to define a reference Boom Clay pore water composition at the Mol site. 3.1.1 Major elements The ionic composition of Boom Clay pore water, sampled from various piezometers in the HADES URF, Test Drift, is given in Table 3-1. For each piezometer, the minimum, maximum, mean, and median values of the ionic concentrations in Boom Clay pore water are given. The same data is visualised in Figure 3-1. Note that the various piezometers are designed differently, and that they are not all composed of the same filter materials (see Section 2.3 Sample location). Moreover, the filters are often positioned in different layers of the Boom Clay. • For the EG/BS piezometer, 74 analyses were considered. (1 filter) • For the ARCHIMEDE piezometer, 14 analyses were considered (4 filters, 2 to 5 analyses per filter) • For the SPRING 116 piezometer, 8 analyses were considered. (4 filters, 2 analyses per filter) • For the ORPHEUS piezometer, 14 analyses were considered. (4 filters, generally 4 analyses per filter). • For the MORPHEUS piezometer, 44 analyses were considered. (12 filters, generally 4 analyses per filter). For most of the elements, the variation of concentration within each piezometer is limited to a few mg/l (Table 3-1). Also, when comparing the five piezometers, some small variations can often be observed from one piezometer to another. Up to now, we can conclude from Figure 3-1 that the general Boom Clay pore water composition can be considered to be comparable in the various piezometers. However, a detailed discussion on the variability of the Boom Clay pore water composition will be given further in this report (Section 3.4 statistical analysis). 38 Table 3-1: Chemical composition of Boom Clay pore water sampled from various piezometers. (Min.=Minimum, Max.=Maximum, Med.=Median, n/a= not analysed) EG/BS mg/l ARCHIMEDE #1 SPRING 116 Min. Max. Mean Med. Min. Max. Mean Med. Min. ORPHEUS MORPHEUS Max. Mean Med. Min. Max. Mean Med. Min. Max. Mean Med. Ca 3.1 5.4 3.9 3.9 1.6 2.7 1.8 1.8 2.2 4.7 2.8 2.3 2.4 4.6 2.9 2.7 1.3 3.0 1.9 1.8 Fe 0.5 4.6 1.1 1.0 0.1 0.2 0.2 0.2 0.3 1.2 0.6 0.5 0.1 0.3 0.2 0.2 0.1 1.6 0.3 0.2 Mg 1.7 4.0 3.0 3.0 1.0 1.7 1.5 1.5 1.9 4.0 2.5 2.1 1.9 3.7 2.6 2.5 1.1 2.8 1.7 1.6 K 5.2 13.1 9.3 9.4 7.0 10.2 8.1 7.8 8.7 12.0 10.3 10.1 8.7 10.2 9.4 9.0 6.2 9.7 7.5 7.5 Si 1.2 21.2 3.0 2.3 3.4 4.8 4.1 4.2 2.6 4.5 3.5 3.5 5.0 10.3 6.7 5.6 4.1 20.8 5.5 4.6 Na 390 467 410 410 269 290 280 279 298 390 332 320 290 330 309 300 340 440 364 360 F - 2.7 4.3 3.2 3.1 0.2 3.0 1.6 1.7 2.6 2.9 2.7 2.7 2.8 3.0 2.9 2.9 2.4 3.5 2.9 2.8 - 23.5 34.8 26.0 25.9 17.0 18.4 17.6 17.7 18.5 24.8 20.4 19.3 20.1 25.9 22.6 22.0 22.8 30.1 25.4 25.0 - Br 0.5 0.8 0.5 0.5 n/a n/a n/a n/a 0.4 0.7 0.5 0.5 0.4 2.0 1.2 1.2 0.4 0.7 0.6 0.6 SO42- 0.3 1.5 0.6 0.4 2.1 4.8 3.4 3.3 <0.25 4.2 1.3 0.6 1.1 7.7 3.7 3.7 0.4 5.6 1.3 0.8 HCO3- 728 4425 1048 871 702 763 731 729 n/a n/a n/a n/a n/a n/a n/a n/a 835 1092 909 887 TIC 143 871 206 171 n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a 164 215 179 175 DOC 78 160 96 89 n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a n/a 97 263 141 124 Cl 39 Figure 3-1: Chemical composition of Boom Clay pore water sampled from various piezometers, visualised in box plots. The box has lines at the lower quartile, median and upper quartile values. The whiskers are lines extending from each end of the box to show the extent of the rest of the data, with a maximum length of 1.5 times the interquartile range. Outliers are data with values beyond the end of the whiskers and plotted with a +. 40 The chemical composition of Boom Clay pore water sampled from the MORPHEUS piezometer has been compared to pore water obtained by the squeezing and by the leaching of clay cores. Clay cores from the HADES 2001/4 drilling (in which the MORPHEUS piezometer was installed) were selected at the same stratigraphical depths as the piezofilters to allow comparison of the pore water composition. As an example the results from the MORPHEUS filter 18 and the corresponding clay core are given in Table 3-2 and Figure 3-2. Note that pore water sampled from the MORPHEUS piezometer is not filtered prior to analyses. This is because earlier analyses have indicated that filtration of the pore water with a 0.45 µm filter does not influence the analytical results. In contrast, pore water obtained by the squeezing of clay cores is always filtered over a 0.45 µm filter before analyses. For F 18, the pore water obtained by the leaching of clay cores is filtered by both 0.45 µm and YM3 (3000 molecular weight cut off) filters. These results are included in Table 3-2. For the cations Ca, Fe and Mg, large differences in concentrations were observed after filtration at different pore size of the filters, suggesting the presence of colloids. For K and Si, no colloids prevail so species are present as truly soluble forms. For the anion species, filtration with YM3 did not influence the measured concentrations. The procedure to derive the leaching concentrations is given in Annex 4 at the end of this report. Table 3-2: Comparison of the Boom Clay pore water composition sampled from the MORPHEUS piezometer filter 18 and from the squeezing and leaching of clay cores at the corresponding stratigraphical depth (sample H 18, -222.625 m TAW). HADES 2001/4 Squeezed clay cores H 18 HADES 2001/4 Leaching experiments H 18 mg/l MORPHEUS Piezometer Filter 18 0.45 µm filtration 0.45 µm filtrat. YM3 filtrat. Ca Fe Mg K Si Na 1.3 0.2 1.4 7.2 4.4 340 2.6 0.4 1.5 5.2 7.2 254 1.5 – 7.6 0.35 – 2.8 1.3 – 4.3 7.4 3.4 - 1.3 < 0.05 1.6 7.6 3.8 - FClBrSO42TIC DOC 2.8 24.8 0.6 3.6 2.4 26.6 0.7 7.0 23.6 16.8 < 1.9 16.8 170 110 112 74 120 (fixed by 0.01 M NaHCO3) 2708 41 Figure 3-2: Comparison of the Boom Clay pore water composition sampled from the MORPHEUS piezometer filter 18 and from the squeezing and leaching of clay cores at the corresponding stratigraphical depth (-222.625 m TAW): MORPHEUS water (•), squeezed water (), leached water filtrated at 0.45 µm ( | or ), and leached water filtrated at YM3 (). 42 From the above table and figure, it can be concluded that the Boom Clay pore water composition, at a depth of -222.625 m TAW, is comparable for the major cations, provided that the leached samples are filtered by a YM3 filter. For the anions, piezometer waters have a similar composition as the squeezed waters. The leached waters have a pronounced different composition due to the double layer properties as discussed in Annex 4. The DOC content is extremely high in the leached waters. This is probably because both the mobile and (part of the) immobile organic matter are put in suspension, in contrast to pore water obtained from piezometers or squeezing where mostly only the mobile fraction of the organic matter is measured. Similar conclusions can be drawn for all the other pore water samples from which the three different extraction techniques were compared. These observations indicate that the measured pore water composition partly depends on the extraction technique. A few squeezed pore water samples have substantially higher contents of Ca, Mg, Na, K and SO42-. The pore water composition of these samples is severely modified because oxidation affected the clay cores (De Craen et al., 2002a, 2004a, 2004b). This indicates once more that methodology of sampling, sample preservation and sample treatment is very important. Only well-preserved clay cores are suitable for the characterisation of the pore water! Note that these data (modified pore water composition as a result of oxidation) are not included in this report, since they are not representative of in situ Boom Clay pore water. 3.1.2 Trace elements The trace element composition of Boom Clay pore water, sampled from piezometers, is given in Table 3-3. Unfortunately, trace elements were not often analysed. Therefore, it is not possible to compare the data. In anaerobic conditions, the presence of nitrate (NO3–) and nitrite (NO2–) is not expected in the sediment. Nitrate and nitrite are easily detected by ion chromatography in the mg/l range. For normal pore waters, their concentrations are usually < 1-2 mg/l, and often below the limit of detection of the instrumental techniques used. The only time NO3– was measured the last decade in the Boom Clay, was in squeezed water from a Boom Clay lump sampled in 1999 at the interface with the concrete blocks of the lining of the small shaft used to host the Reseal-II in situ experiment. The concentration was anomalously high (90 mg/l) for an unknown reason. Ammonium (NH4+) can also indicate chemical or microbial perturbations and was evidenced for the first time by CEA in the water samples from the piezometer # 2 of the ARCHIMEDE-argile project (Merceron et al., 1993b). This borehole probably encountered an oxidative perturbation during the drilling operations, since it remained open to air 1 week before installation of the piezometer. 43 Table 3-3: Trace element composition of Boom Clay pore water sampled from various piezometers. (Min.=Minimum, Max.=Maximum, n/a = not analysed) MORPHEUS Min. Max. EG/BS Min. ORPHEUS Max. Min. Max. SPRING 116 Min. Max. Li mg/l < 0.2 < 0.2 < 0.2 < 0.2 n/a n/a < 0.2 < 0.2 B mg/l 6.7 7.6 6.7 9.7 6.7 11.3 7.4 8.6 Al mg/l < 0.2 0.4 < 0.2 0.89 < 0.2 0.25 < 0.2 0.25 Mn mg/l 0.015 0.036 < 0.05 < 0.05 n/a n/a < 0.05 0.14 Ni mg/l n/a n/a < 0.2 < 0.2 n/a n/a < 0.2 < 0.2 Zn mg/l n/a n/a < 0.2 < 0.2 n/a n/a < 0.05 < 0.2 Se µg/l 0.05 * 5* n/a n/a n/a n/a n/a n/a Rb µg/l 1.9 9.2 n/a n/a n/a n/a n/a n/a Sr µg/l 40 100 90 130 n/a n/a 60 130 Y µg/l < 0.5 0.9 n/a n/a n/a n/a n/a n/a Zr µg/l 3.1 58.0 n/a n/a n/a n/a n/a n/a I µg/l 590 840 660 740 n/a n/a n/a n/a Cs µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Ba µg/l 10 60 n/a n/a n/a n/a 13 55 La µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Ce µg/l < 0.5 1.2 n/a n/a n/a n/a n/a n/a Pr µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Gd µg/l < 0.5 0.6 n/a n/a n/a n/a n/a n/a Tb µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Dy µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Ho µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Hf µg/l < 0.5 < 0.5 n/a n/a n/a n/a n/a n/a Th µg/l 0.005 * < 0.5 n/a n/a n/a n/a n/a n/a U µg/l 0.200 * 3.5 < 0.5 1.5 n/a n/a < 0.25 0.43 * measured with a HR- ICP-MS (better resolution than ICP-MS) The trace element composition of Boom Clay pore water is slightly different in pore water sampled from piezometers and pore water obtained by the squeezing of clay cores (no data are available on pore water from leaching experiments). This can be illustrated by comparing pore water samples from the MORPHEUS piezometer with pore water obtained by the squeezing of the HADES 2001/4 clay cores at corresponding stratigraphical depths (Table 3-4). In a lot of squeezed pore water samples, the trace elements are more abundant compared to piezometer-derived pore water. The higher amount of trace elements in squeezed pore water samples is not fully understood. Possibly it is an artefact of the squeezing technique. 44 Table 3-4: Comparison of the trace element composition of Boom Clay pore water sampled from the MORPHEUS piezometer and pore water obtained by the squeezing and leaching of clay cores at corresponding stratigraphical depths (Min.=Minimum, Max.=Maximum, n/a = not analysed) MORPHEUS HADES 2001/4 piezometer Squeezed clay cores Min. Max. Min. Max. Al mg/l < 0.2 0.4 < 0.2 0.4 Mn µg/l 15 36 60 296 Rb µg/l 1.9 9.2 2.4 50 Sr µg/l 40 100 61 425 Y µg/l < 0.5 0.9 <1 32 Zr µg/l 3.1 58.0 <5 71 I µg/l 590 840 430 610 Cs µg/l < 0.5 < 0.5 < 0.5 3.8 Ba µg/l 10 60 5 51 La µg/l < 0.5 < 0.5 <1 63 Ce µg/l < 0.5 1.2 <1 180 Pr µg/l < 0.5 < 0.5 <1 21 Gd µg/l < 0.5 0.6 <1 13 Tb µg/l < 0.5 < 0.5 <1 2.2 Dy µg/l < 0.5 < 0.5 <1 9.4 Ho µg/l < 0.5 < 0.5 <1 1.6 Hf µg/l < 0.5 < 0.5 <1 2.8 Th µg/l 0.005 * < 0.5 <1 48 U µg/l 0.200 * 3.5 < 0.5 18 * measured with a HR- ICP-MS (better resolution than ICP-MS) 3.1.3 pH, pCO2, and alkalinity of Boom Clay The concentration of hydrogen ions (hydrated) is very low for waters that are not strongly acidic. To quantify the concentration (activity) of hydrogen ions, the more convenient expression is pH, the negative base-10 logarithm of the hydrogen-ion activity in moles per litre. The pH of most natural waters is related to reactions between carbon dioxide (partial pressure, pCO2) and its water soluble species following the reactions: CO2(g) + H2O ⇔ H+ + HCO3- (3.1) CO2(g) + H2O ⇔ 2 H+ + CO32- (3.2) 45 In the case of the Boom Clay, since the sediment is water saturated, no free gas phase is normally present. The pCO2 of the Boom Clay is therefore related only to the dissolved CO2 as carbonic acid: CO2 (g) + H2O ⇔ H2CO3 (aq) (3.3) Another quantity relating the solution pH and the water soluble carbonate species is alkalinity. Alkalinity is defined as the capacity of a water to neutralise acid and is measured by titration with strong acid (HCl or H2SO4) to an end point around pH 4.5. Different from most quantities determined by chemical analysis, e.g., pH and pCO2 that are intensity functions, alkalinity (also for acidity) is a capacity function so that in principle all basic species including hydroxide (OH-), silicate, borate, phosphate, and natural organic ligands should also contribute to the alkalinity. In practice however, only the dissolved carbonic acid is of quantitative importance as shown in equation (3.4). carbonate alkalinity = mHCO3- + 2 mCO32- ≈ total alkalinity where mHCO3- and mCO32- are the molarity of bicarbonate and carbonate, respectively. By definition, the carbonate alkalinity is only part of the total alkalinity, but for the Boom Clay pore water the difference of the two quantities is negligible. Reactions among the species related to pH, pCO2, and alkalinity are in general fast in natural waters and can therefore be treated by chemical equilibrium principles. This should be particularly the case in the Boom Clay because of the extremely long resident time of the pore water. 3.1.3.1 pH of Boom Clay pore water The techniques to determine the Boom Clay pH was discussed already in the Section 2.2.1.1 and will not be repeated here. Until now, the most acknowledged Boom Clay pH value is 8.2 determined in the project of ARCHIMEDE-argile (Beaucaire et al., 2000; Griffault et al., 1996). This value was measured in situ by an optode under the Boom Clay hydraulic pressure. The same value was also determined in the same project by a flow cell with a combined glass electrode. The recent in situ pH measurement (Moors et al., 2002) using a closed-circuit technique in the same ARCHIMEDE-argile piezometer revealed a value of 8.0. The latter measurement was to improve the pH sensing technique, for example, by replacing the optode sensor with a pressure resisting polymer filled pH electrode under a 1.8 MPa hydraulic pressure (see section 2.2.1.1). Two problems were however encountered in this closed-circuit measurement: 1. after 463 days of circulation of clay water, the pH electrode was found having a drift of 120 mV (about 2 pH unit). Because of the electrode drift, other pH values before the final reading (pH = 8 after correcting the drift) were not reliable so we did not know if the pH 8 was the stable value; 2. due to the continuous circulation of the clay water between the measuring cell (with pH electrode in it) and the filter of the piezometer, a leakage of KCl from the electrode to the clay water was observed (Table 3-5). The water with (3.4) 46 a high concentration of K ion was pumped back to the clay and induced cation exchange reactions as evidenced by the elevated concentration of Na, Ca, and Mg ions in the pore water. Table 3-5: Comparison of water compositions collected at the ARCHIMEDE-argile piezometer (No. 1) for the two pH measurement campaigns. The reference water composition derived from the current work is also given. mg/kg water K Na Ca Moors et al., 2002 50 760 14.3 12.8 0.5 421 Beaucaire et al., 20002 8.2 282.8 1.9 1 This work, table 5-1 1 2 3 7.2 359 2.0 Mg 1.7 1.6 Fe Cl SO4 Br Si 7.8 0.8 5.2 0.2 17.7 3.8 0.6 4.2 0.2 26 2.2 3 0.6 3.4 the water compositions were given in the certificates of chemical analysis MC/PT/C072021/01-032/pt (cations) and -033/pt (anions) it is not clear if the water was the same from which the pH was measured taken from the average of MORPHEUS water compositions Although it is clear from Table 3-5 that the water from the closed-circuit pH measurement (Moors et al., 2002) has an unusual high concentration of K, Na, Ca, Mg, and Cl due to the leak of KCl, it is not clear to what extent the pH value was affected. One mechanism that might have impacted on the system pH is the cation exchange, inducing calcite precipitation: >X2:Ca + 2 K+ + HCO3- ⇔ 2>X:K + Calcite (CaCO3) + H+ The reaction suggests that the process releases acid (H+) and will decrease the pH. Besides, the high concentration of K was circulated around the filter for more than one year, so K ions have been exchanged onto the surrounded clay surfaces. The pH value measured this way is therefore considered as being perturbed to a certain extent. Apart from the in situ measurements, all other reported pH values for the Boom Clay were measured in surface laboratories in pore water samples that were unavoidably contacted with air. The pH measurement in a surface laboratory is normally performed in the open air by immersing a pH electrode into water samples collected from the underground gallery. Depending on the in situ pCO2 of the water samples, i.e., higher or lower than that of the atmosphere, the pH reading may increase (degassing) or decrease (ingassing) from the initial pH reading, which is mostly close to the in situ value. To minimise the extent of de- or ingassing of CO2 (g) while measuring a pH in a surface laboratory, a pH reading should be recorded as soon as possible to avoid prolonging the contacting time between water samples and the air. In practice however, a quick pH measurement has not been routinely performed. A pH reading is normally recorded when the reading stops drifting based on a given criterion for the pH meter. Because of the problem of de- or ingassing of CO2 (g), a pH measurement in a surface laboratory is normally considered as unreliable. Dierckx (1997) reported the pH of EG/BS water as 9.5, which is higher than the available in situ pH values of 8.0 and 8.2, so it is considered as being caused by artefacts and not representative for real Boom Clay. The exact cause of the high pH in EG/BS water is not clear but might relate to the leaching of the filling material between the filter and the clay formation. Earlier study performed by batch experiments upon leaching clay 47 samples in distilled water resulted in a pH of about 9.3 (Baeyens et al., 1985b). This value is one pH unit higher than the in situ value of 8.0 and 8.2 because of the low pCO2 (g) in the N2 (g) or Ar (g) filled glove boxes where pH was measured. The average pH of 12 MORPHEUS water samples collected in April, 2003 is 8.03 (Figure 3-3). However, the same samples revealed a pH lower than 7.5 after two months of storage at room temperature instead of 4 °C (routinely applied) suggesting possible effects of bacterial activity (Figure 3-3). Related to the problem of de- or ingassing of CO2 (g), Figure 3-3 demonstrates that a laboratory measured pH is influenced by factors such as the sample storage time and temperature, sample stirring during the measurement, and the time that the sample was in contact with air. The high pH approaching to the value of 9.3 is due to the complete degassing of CO2 (g) originally present in the water samples. 9 at the time of sampling 1 month storage pH 8.5 2 month storage 8 45 min stir in air 48 hrs stir in air 7.5 7 -240 -235 -230 -225 -220 -215 depth, m Figure 3-3: Laboratory pH measurement on MORPHEUS water samples in air and at room temperature (~23°C). The in situ temperature of pore water is about 16 °C. Water samples were stored at room temperature in closed vessels under slightly overpressure before they were measured at open air. Because of the difficulties encountered in both in situ and laboratory pH measurements, an alternative way to acquire the pH of the Boom Clay is through speciation calculation using representative pore water compositions. The basic model is to assume a chemical equilibrium between calcite and the pore water. The pore water composition is therefore constrained by the calcite dissolution/precipitation reaction: Calcite + H+ ⇔ Ca2+ + HCO3(3.5) At a chemical equilibrium state, i.e., calcite has a saturation index (SI) of 0, the pH can then be calculated if Ca2+ and HCO3- are known. However, water analyses in 48 general only provide total calcium (not free Ca2+) and total dissolved inorganic carbon (not bicarbonate) concentrations and therefore pH cannot be readily estimated using only the equation (3.5). A speciation calculation, normally performed with geochemical computer codes, takes into account all constraints given in equations (3.1) to (3.5), and the pH can be fitted to the measured calcium and TIC concentrations. Figure 3-4 shows the modelling results by allowing calcite to be in equilibrium with MORPHEUS waters with measured Ca and TIC concentrations. Within a varied pH range (imposed), the saturation index of calcite changes and the pH at which the SI is 0 corresponds to the pH of the in situ pore water. The results of Figure 3-4 suggest that the pH of MORPHEUS water is in the range of 8.3 to 8.6. Calcite saturation index (log Q/K) 0.8 0.6 mor-1 0.4 pH 8.3 0.2 mor-2 mor-3 0 mor-4 -0.2 pH 8.6 mor-5 -0.4 Archimede -0.6 -0.8 7.75 8 8.25 8.5 8.75 9 pH Figure 3-4: Saturation indices (SI) calculated at 16 °C using MORPHEUS water compositions as function of pH. Mor-1 to mor-5 are data from 5 statistic groups of water samples (see section 3.4.1 latter). Data from the ARCHIMEDE-argile project is also plotted for comparison. pH at SI = 0 corresponds to in situ pH value. Data from ARCHIMEDE-argile were fitted in the same way: the pH is about 8.6 which is higher than 8.2, the in situ value measured. In other words, ARCHIMEDEargile water is undersaturated with respect to calcite at pH 8.2. Two possibilities may have caused the observed undersaturation: (1) the pH of 8.2 measured was too low; (2) the assumption of calcite equilibrium is not valid. Considering the difficulties encountered in an in situ pH measurement, the modelling approach for pH estimation is particularly interesting for the purpose of a long term prediction, for which a general equilibrium condition should be established. Table 3-6 summarizes the pH values measured at the underground laboratory, surface laboratories, and estimated by speciation modelling using measured pore water compositions. Note that laboratory measurements show the largest extent of variation suggesting an unfavourable condition for pH determination. 49 Table 3-6: Summary of pH values measured at the underground laboratory, surface laboratories, and estimated by speciation modelling In situ (underground laboratory) batch (surface laboratory) model fit (speciation modelling) 8.21(optode, ARCHIMEDE) 8.02 (polymer filled electrode) 8.23 (MORPHEUS) 7.54 (MORPHEUS) 9.55 (EG/BS) 9.2-9.36 8.6 (ARCHIMEDE) 8.3 ~ 8.67 (MORPHEUS) 1: ARCHIMEDE piezometer (Beaucaire et al., 2000); 2: ARCHIMEDE piezometer (Moors et al., 2002), disturbed by the leakage of KCl; 3: measured shortly after the sampling; 4: measured after several weeks storage at room temperature (unpublished results); 5: water from the silty layer and the borehole was filled with gravel (Dierckx, 1997); 6: measured in a glove box under inert atmosphere but no CO2 (g). The solid/liquid ratio is 1:1 (Baeyens et al., 1985b); 7: see figure 3-4 for results fitted to measured [Ca] and TIC concentrations for MORPHEUS waters. 3.1.3.2 Partial pressure of CO2 (g) in Boom Clay The only pCO2(g) measurement on the Boom Clay was performed by Henrion et al., (1985) and a value of 10-2.5 atm (25 °C) was reported. The measurement was not in situ but carried out on preserved clay samples in surface laboratory by an out-gassing technique. Assuming that the measured pCO2 is equal to its fugacity, Henrion et al., (1985) also calculated the Boom Clay pH, using equations (3.1) to (3.3), as being in a range of 8.5 to 8.8 at varied total inorganic carbon content (TIC, the sum of bicarbonate and carbonate concentrations). Different from the approach of Henrion et al., (1985), that is, to estimate pH from the measured pCO2, Griffault et al., (1996) calculated pCO2 from the measured pH. Using the in situ measured pH value of 8.2 (see section 3.1.3.1), a pCO2 of 10-2.4 atm was calculated at TIC of 12 mM based on equation (3.1). It is well known that pCO2 and pH measurements are technically challenging. This is because the Boom Clay is susceptible to the loss of CO2 (g) if in contact with air. Measurement in an absolutely closed system is technically difficult. The so far measured pCO2 of the Boom Clay is about 10 times higher than the atmospheric pCO2 of 10-3.5 atm. Any contact with air tends to lower the pCO2 of the water sample due to the loss of dissolved CO2 and many attribute it to a so called degassing or out-gassing process. The loss of dissolved CO2 can be caused by two different processes: • • degassing due to the difference in CO2 solubility in water; loss of dissolved CO2 due to the re-equilibrium with the atmospheric CO2 partial pressure. The first process might occur when a clay or pore water sample was oversaturated in respect of CO2 (g) under the in situ high pressure. When the sample is brought to low atmospheric pressure, the CO2 solubility decreases and CO2 (g) will escape from the 50 sample. Such degassing happens when one opens a bottle of sparkling water and CO2 gas bubbles escape from the bottle. Whether or not such a degassing might happen to Boom Clay or its pore water depends on the in situ saturated of CO2. Whether the partial pressure of CO2 is equal to the atmospheric pressure can be estimated by comparing CO2 solubility and the TIC measured in the Boom Clay pore water. The Henry’s law constant for CO2 (g) is 1.6 × 103 atm at 298 K (Atkins, 1994). The solubility of CO2 (g) in 1 litre of water at 25 °C (i.e., 55.5 mol) can be approximated as: n (CO2) ≈ 1atm × 55.5 = 34.7 mmol 1.6 × 10 3 atm where n is the number of mols CO2 dissolved when its partial pressure equals to the atmospheric pressure of 1 atm. The solubility of CO2 at the in situ temperature of 16 °C is even higher and equals to 45 mmol/l. The TIC of a Boom Clay pore water is about 15 mmol/l which is less than half of the calculated solubility of CO2 . This suggests that Boom Clay pore water is not oversaturated with respect to CO2 (g) under the in situ pressure and therefore will unlikely experience degassing due to the difference in CO2 solubility. The second process causing the loss of CO2 from samples is due to the re-equilibrium with the atmospheric CO2 partial pressure. The loss of CO2 will increase pH as shown in equation (3.1) and (3.2). A Boom Clay pore water containing 15 mmol/l of inorganic carbon should have a pH of about 9.3 if in equilibrium with air (see Figure 3-5 below for measured value at room temperature). The re-equilibrium process is however affected by kinetics. Stumm and Morgan (1996) stated that an establishment of water-atmosphere equilibrium is primarily limited by slow gas transfer reactions. To a lesser extent, the re-equilibrium can also be slowed down by unfavourable mixing. Figure 3-5 shows the pH change of a MORPHEUS water when measured in air under a strong mechanical stirring. The pH approaches 9.3, the value in equilibrium with air, only after 48 hours. This suggests that a complete degassing of CO2 even in a surface laboratory needs more than 2 days of time to accomplish. The initial step of degassing was however quite fast, within one hour, the pH increased about half a pH unit. From foregoing discussions plus the results shown in section 3.1.3.1, it can be concluded that the pH measurements performed in the surface laboratory do not provide representative values due to possible bacterial effects and the loss of CO2. Degassing of CO2 in situ due to the solubility difference is unlikely in the Boom Clay. Since gas transfer reactions are in general slow in kinetics, 'on site' pH measurement in piezometer water using a carefully designed flow cell might provide relevant pH values. The difference between an 'on site' measurement and the currently ongoing in situ closed-circuit measurement is, that an 'on site' measurement does not have to be performed at the high in situ hydraulic pressure. This will relax most of the constraints on instrumentation, e.g., to resist to the high in situ hydraulic pressure. A conventional glass electrode may be used in place of a high pressure resistant electrode, for which a routine calibration for pressure effects is not straightforward. Another advantage of a piezometer flow-through cell measurement is that the operation is simple and therefore can be performed for all piezometers from which pore waters are sampled. 51 9.25 9 calculated pH 9.3 (25 °C) pH 8.75 8.5 8.25 8 0 10 20 30 40 50 time, hr Figure 3-5: pH evolution for a MORPHEUS water as function of time with a mechanical stir. The calculated pH is 9.3 if the water is in equilibrium with air at 25 °C. The same as in the case of the pH, the partial pressure of CO2 (g) can also be calculated by assuming calcite equilibrium with water in terms of measured compositions. The calculation can be done in the same way as shown in Figure 3-4 but the principle variable is pCO2 instead of pH. Since in both cases the TIC concentration is a measured value (not imposed by the model), the pCO2 (g) can be derived directly from the calculation shown in Figure 3-4 but plot pCO2 as the x axis instead of pH (figure not shown). The corresponding results are: pCO2 is in a range of 10-2.4 to 10-2.8 atm while pH changes from 8.3 to 8.6. Until now the pCO2 (g) has been either measured experimentally or calculated using a measured pH value without knowing the exact mechanism controlling the parameter. Coudrain-Ribstein et al., (1998) reviewed literature data of pCO2 in confined aquifers without a gas phase and concluded that pCO2 is principally constrained only by mineral assemblage. For sedimentary formations, one example for such CO2 control mechanism is as: 5 calcite + chlorite + 5 CO2 ⇔ kaolinite + silica + dolomite + 2 H2O (3.6) At an equilibrium state, all minerals involved in the reaction have a unity activity so that the pCO2 can be easily calculated if all stability constants of the involved minerals needed for balancing the equation (3.6) are known. In addition, the equilibrium is in principle only dependent on temperature since the system is free of a gas phase and the equilibrium won’t be affected by the absolute pressure to a big extent. The problem with this approach is that the uncertainty on the values of the thermodynamic database is usually large. This is because the stoichiometry and thermodynamic 52 properties of some of the involved minerals are poorly known. This is also the main drawback of the thermodynamic modelling approach. The pCO2 controlling mechanism like equation (3.6) should also be applicable to Boom Clay since all minerals appearing in the equation are present in Boom Clay (see section 3.1.3.2). The only question is whether or not dolomite exist in Boom Clay. Van Keer and De Craen (2001) reviewed five representative studies on Boom Clay mineralogy and only Griffault et al., (1996) reported dolomite in the Boom Clay based on the XRD analysis performed at ERM (Etudes Recherches Materiaux). The same institute again identified dolomite in the Boom Clay samples in the recent EC Eco-clay –II project (Bouchet, 2003). Wouters et al., (1999) determined 1.1 % magnesite in Boom Clay samples. At the present stage and for the purpose of geochemical modelling, we consider dolomite as a proxy of magnesium rich carbonate mineral which likely exists in Boom Clay. Based on the current version of MOLDATA (Wang, 2003), the Boom Clay pCO2 is calculated to be 10-3.1 atm at 16 °C following the reaction: clinochlore-14A + 5 CO2(g) + 5 calcite ⇔ kaolinite + chalcedony + 5 dolomite-dis + 2 H2O where: clinochlore-14A: chlorite (Mg5Al2Si3O10(OH)8) calcite: CaCO3 kaolinite: Al2Si2O5(OH)4 chalcedony: SiO2 dolomite-dis: CaMg(CO3)2 dis-ordered form of dolomite According to Coudrain-Ribstein et al. (1998), at 20°C, the pCO2 controlled by the same mineral assemblage is 10-2.8 bar which is about 1.5 times higher than the value 10-3.0 atm calculated by MOLDATA at the same temperature. The difference is surely due to the difference between the two thermodynamic databases. Another well known database Wateq4f (Ball and Nordstrom, 1991) calculates pCO2 at 20 °C in a range of 10-2.8 to 10-3.1 atm using respectively amorphous silica and quartz to represent the silica mineral. Note that different databases may use different crystallinities to represent the same mineral identified by, e.g., X-ray diffraction measurement. Modellers may also make decisions on what minerals to use based on observed pore water composition, e.g., we use chalcedony in equation (3.7) to replace the silica mineral, since the silicon concentration measured in the pore water of Boom Clay is very close to the solubility of chalcedony but much higher than the solubility of quartz. The same goes for dolomite where a disordered form of dolomite is often used instead of a well crystalline phase. This is because dolomite has large variations in solubility depending on its crystallinity. The aqueous concentration of magnesium has been found mostly governed by the solubility of a disordered form of dolomite rather than by the well crystallized form (Coudrain-Ribstein et al., 1998). Although the magnesium concentration in Boom Clay is explained by ion exchange in this report (see section modelling), the solubility of disordered dolomite calculated on the basis of MOLDATA is about 2 mg/kg water, which is in good agreement with the magnesium concentration found in Boom Clay pore water. (3.7) 53 Besides the reaction (3.7), other possible mineral assemblages were proposed by Coudrain-Ribstein, et al., (1998). Table 3-7 summarizes some reactions relevant for Boom Clay, together with the calculated pCO2 values using MOLDATA. From Table 3-7, it is seen that except the assemblage of illite which resulted in a much too low pCO2, other groups result in a pCO2 equal to or slightly lower than the value of 10-2.5 atm (25 °C) measured by Henrion et al. (1985). Note that the current version of MOLDATA (04-01) contains some questionable data concerning aluminium species and minerals as pointed out by Nordstrom et al. (1990). Current review, data selection, and modification of MOLDATA (04-01) is in progress and the pCO2 values given in Table 3-7 should be recalculated after the MOLDATA is updated. Another point observed from Table 3-7 is the effect of temperature. The temperature increase from 16 to 25 °C increases the pCO2 with a factor of 2, as predicted. The temperature effect has been studied in the Cerberus experiment (Noynaert et al., 1998) in which Boom Clay was heated up to 80 °C. Although no direct pCO2 was measured in the experiment, the on site pH measurement evidenced a pH drop to 6~7 from the original value of 8.7. This pH drop has been previously explained as being due to the pyrite oxidation or bacterial activities. The effect of temperature increase, which causes an increase in PCO2 so a decrease in pH, should not be overlooked in the future modelling on the effect of heating on Boom Clay. Table 3-7: Possible mineral assemblages for controlling pCO2 in sediments and the calculated pCO2 values at 16 and 25 °C mechanisms * pCO2 (atm), 16/25 °C clinochlore-14A + 5 CO2(g) + 5 calcite ⇔ kaolinite + chalcedony + 5 dolomite-dis + 2 H2O 10-3.1/10-2.8 kaolinite + 3 CO2 + 3 calcite + phlogopite ⇔ muscovite + 2 chalcedony + 2 H2O + 3 dolomite-dis 10-3.0/10-2.7 phlogopite + 3 CO2 + 3 calcite ⇔ K-feldspar + 3 dolomite-dis + H2O 10-2.9/10-2.5 20 illite + 5 CO2 + 2 H2O + 5 calcite ⇔ 5 dolomite-dis + 12 muscovite + 5 kaolinite + 24 chalcedony 10-4.9/10-4.6 * calculated using the MOLDATA (04-01) database clinochlore-14A: Mg5Al2Si3O10(OH)8, chlorite kaolinite: Al2Si2O5(OH)4 calcite: CaCO3 phlogopite: KAlMg3Si3O10(OH)2, mica group muscovite: KAl3Si3O10(OH)2, mica group chalcedony: SiO2 dolomite-dis: CaMg(CO3)2 K-feldspar: KAlSi3O8 illite: K0.6Mg0.25Al1.8Al0.5Si3.5O10(OH)2 54 The change of pCO2 due to the temperature variation has not been realised in a context of sample storage and experimental protocols. The so far applied procedure is to store clay samples at 4 °C before use. Depending on the time of storage, a possible decrease of pCO2 in clay samples may cause a change in pore water chemistry. This is particularly important for laboratory experiments where squeezing and leaching techniques are used to extract pore waters from clay samples. Finally, Table 3-8 summarizes pCO2 values of Boom Clay either measured experimentally or estimated based on pore water analysis and chemical speciation calculations. Table 3-8: Values of pCO2 of Boom Clay obtained by laboratory measurement and estimations based on model simulation measurement 10-2.5 (25°C), outgassing clay core1 estimations 10-2.4 (25°C), ARCHIMEDE data2 10-2.8~10-2.4 (16°C), MORPHEUS waters3 10-3.1~10-2.5 (25 to 16 °C), mineral assemblages4 1: outgassing clay cores (Henrion et al., 1985); 2: calculated from the measured pH and alkalinity (Beaucaire et al., 2000; Griffault et al., 1996); 3: calculated based on the assumption of calcite equilibrium and the Ca, TIC concentrations measured in MORPHEUS waters; 4: calculated using the MOLDATA database (see Table 3-7). It must be stressed that although attempts have been made to measure or calculate the pCO2 of Boom Clay, the real mechanism governing the parameters is not understood. On the one hand, current activities for a more accurate and representative pCO2 measurement should be continued, on the other hand future studies to understand the mechanism of the CO2 evolution in Boom Clay should be pursued. Last but not least, the role of natural organic matter and bacterial activity in regulating the pCO2 in Boom Clay has not been evaluated so far. 3.1.3.3 Alkalinity of Boom Clay pore water As defined in section 3.1.3, alkalinity is the capacity of a water to neutralise acid. Rounds and Wilde (2001) also use acid neutralising capacity (ANC) which is essentially the same but measured in water samples without filtration (0.45 µm). For Boom Clay pore water collected through piezometers, our titration data (not shown) illustrated no difference between the alkalinity and the ANC so we do not differentiate alkalinity from ANC in this report. The alkalinity of Boom Clay pore water was reported as 12.1 mmol/l in the ARCHIMEDE-argile project (Griffault et al., 1996). Much lower values, e.g., ~7 55 mmol/l were reported for the pore waters collected by mechanical squeezing, probably due to the pyrite oxidation (Reeder et al., 1994). Recent measurement on MORPHEUS waters from different depth resulted in a mean value of 14.9 meq/l HCO3- or 909 mg/l (see Figure 3-6 below). It has been unclear as to what extent other solutes than bicarbonate, in particular natural organic matter, contribute to the total alkalinity of Boom Clay pore water. As a rough estimate, the contribution of other solutes can be derived by comparing the total alkalinity to the total inorganic carbon content expressed as bicarbonate. Figure 3-6 shows the comparisons in 12 MORPHEUS water samples. It is clear from Figure 3-6 that the total alkalinity agrees very well with the TIC suggesting that other solutes, including natural organic matter, do not contribute significantly to the total alkalinity. It is notable though that the alkalinity is systematically higher than TIC for about 2 %, which might be due either to the real contribution of other solutes or to the underestimation of TIC by assuming only bicarbonate existing in water samples. 1200 Alkalinity, mg HCO3-/l 1000 800 Titration TIC 600 400 200 0 -217.1 -220.8 -222.6 -226.0 -227.2 -227.8 -229.1 -229.8 -230.3 -231.8 -233.8 -235.2 depth, m Figure 3-6: Alkalinity and total inorganic carbon (TIC) content (as HCO3-) measurements performed in 12 MORPHEUS water samples collected at different depth. Water samples were collected in April, 2003. Different from pH and pCO2, alkalinity is a conservative quantity and independent of pCO2 (Drever, 1997). The alkalinity is therefore not susceptible to the loss of CO2 in the process of a water sampling. This is because neither the pCO2 nor the dissolved H2CO3 (aq) will contribute to the charge balance of the system. Although the increase of pCO2 will increase the TIC through either: 56 or CO2 + H2O + CO32– ⇔ 2 HCO3- (3.8) CO2 + H2O ⇔ H+ + HCO3- (3.9) In both reactions, the increased alkalinity due to the increase of [HCO3-] will be counterbalanced by the disappearance of CO32– and the production of H+, respectively. The net change of alkalinity will thus be zero. The alkalinity is no longer conservative if redox reactions between carbon species start to play a role, e.g., through biological processes. One important concern has been to what extent the oxidation of the water sample will affect the alkalinity. The most probable alkali consuming reaction in Boom Clay pore water might be the oxidation of Fe(II) and the precipitation of iron hydroxide following the reaction: 2 Fe2+ + 4 HCO3- + 0.5 O2 + 5 H2O ⇔ 2 Fe(OH)3 + 4 H2CO3 Boom Clay pore water in general contains less than 1 mg/l of Fe, which is 0.018 mmol/l. If such a concentration of Fe2+ precipitates, the water will lose, according to equation (3.10), twice this molarity of HCO3-, i.e., 0.036 meq/l as alkalinity. This is about 0.3% of the total 15 meq/l alkalinity measured in MORPHEUS waters, which is negligible. 3.1.3.4 Conclusions of pH/ pCO2 As the most important parameter, the Boom Clay pH and its controlling mechanisms are still not conclusive. Apart from the well known in situ ARCHIMEDE-argile measurement (pH 8.2), the new in situ measurement (pH 8.0) suffered from the leak of KCl from the electrode and demonstrated the technical difficulties in pursuing a good quality pH measurement. The model simulation, using water compositions from a new piezometer (MORPHEUS) suggests a pH range of 8.3 to 8.6. The observed variation in water compositions suggests that variations in pH or related pCO2 should exist in the collected water samples. As an absolutely non-disturbed geochemical condition is not achievable, the variation in pH or pCO2 may well be caused by the disturbances introduced while installing piezometers and/or collecting waters. The extent of such disturbances is of course hard to quantify. It is therefore more objective to use a modelled reference pH instead of measured pH values that are, to certain degrees, functions of the system disturbances. 3.1.4 Redox processes and redox potential in Boom Clay Redox is the acronym of 'reduction and oxidation', that is, processes occurring in a system involving multi-valent elements. These elements are therefore called redox sensitive elements. A detailed description on redox chemistry is out of the scope of contamination this report and can be found in standard text books such as Appelo and Postma (1999), Langmuir (1997), and Hem (1985). We hereafter only give a brief definition of redox potential and precautions one should bear in mind when applying the concept to the interpretation of Boom Clay geochemistry. The redox potential is a numerical index of the intensity of oxidising or reducing conditions within a system. It is the potential, expressed as Eh in volt or millivolt, (3.10) 57 developed by a redox reaction involving transfer of electrons. The subscript “h” implies that the potential is relative to the standard potential of hydrogen (H2/H+) electrode. Positive values of Eh indicate that the system is relatively oxidising, and negative values suggest that it is relatively reducing. Another widely used concept equivalent to the redox potential is pE, defined as the negative logarithm of the electron concentration: (3.11) pE = -log(e-) The value of pE can be related to Eh at 25 °C and 1 atm pressure as: pE = FE h = 16.9 E h 2.303RT where F is the Faraday’s constant (96.42 kJ/volt gram equivalent), R the gas constant (8.314 × 10-3 kJ/deg.mol), and T the absolute temperature. In the definition, pE is analogous to pH, in reality, there are practical difficulties in application of pE (or Eh) comparing to pH. Protons exist in water as its hydrated form (e.g., H3O+), whereas electrons do not exist as free species. Also, most reactions involving protons are reversible but those involving electrons are not. Most importantly, electron transfers are often very slow suggesting that redox reactions in natural system are often kinetically controlled. In general, redox reactions only proceed at high rate if microbial catalysis is involved. Inorganic reactions involving electron transfers occur only at an immeasurable rate. Application of the equilibrium theory to redox processes has been proven invalid for many natural systems (Lindberg and Runnells, 1984). Common laboratory measurements of Eh may provide a qualitative indication of the system redox conditions but cannot be used quantitatively unless the equilibrium state of the system is thoroughly evaluated. Comparing to normal surface and sub-surface waters where the redox potential is difficult to define, the Boom Clay redox potential should be thermodynamic indicative for two specific reasons: (1) water movement in Boom Clay is extremely slow so that the water can be considered approaching to an equilibrium with the surrounding rocks; (2) in compact Boom Clay microbial activities are likely very low because of the small pores. The redox potential is most likely imposed only by chemical interactions involving inorganic components. 3.1.4.1 Redox potential in Boom Clay Due to the presence of pyrite and natural organic matter, the Boom Clay is reducing and therefore has negative redox potentials. The first reported value of the Boom Clay redox potential was -280 mV measured in a surface laboratory by Baeyens et al. (1985a) in a Boom Clay slurry. Many subsequent studies have focused on in situ measurement of Eh and a range of values at -250 to -400 mV has been reported (De Cannière et al., 1996). In the study of ARCHIMEDE-argile (Beaucaire et al., 2000; Griffault et al., 1996), although no direct Eh values were reported, the interpretations on the water composition suggested a similar range of Eh at -240 to -400 mV. Recent efforts pursuing Eh-pH measurement at the in situ hydraulic pressure (see section 3.1.4.1) report a redox potential Eh of -310 ± 30 mV (Moors et al., 2002). (3.12) 58 Although the redox potential of Boom Clay has been proven difficult to measure and interpret, plausible processes controlling the measured Eh can be anticipated using EhpH diagrams in combination with the measured mineralogical and pore water compositions. Figure 3-7 plots the Eh-pH diagram of the Fe-S-C system at concentrations representative for Boom Clay pore water. The lines of equilibrium shown in the figure are drawn from the reaction between the two species which are separated by the line. For example, the equilibrium between pyrite and siderite is as follows: Pyrite + 9H2O + CO2(g) = 18H+ + 2SO42- + Siderite + 14e- logK = -91.2 (3.13) Eh = 0.38 - 0.076pH + 0.0084log a[SO42-] - 0.0042log f[CO2(g)] (3.14) Following the equation (3.14), under the reference Boom Clay condition, e.g., at pH of 8.2, pCO2 of 10-2.4 atm, and taking a sulphate concentration as 0.1 mg/l (~1 µM), the redox potential is -283 mV, which is the upper limit of the pyrite stability field. The sulphate concentration of 0.1 mg/l is about the lowest measured in all piezometer waters. Except some severely oxidised waters, e.g., as studied by De Craen (2001), the sulphate concentration in the Boom Clay pore water varies from a few to hundred mg/l. Since Boom Clay is a marine sediment, the threshold of the present-day sulphate concentration can be calculated from the seawater ratio between sulphate and chloride. According to the seawater composition given by Drever (1997), the ratio of chloride and sulphate in seawater is about 19.3. Taking the chloride concentration in Boom Clay pore water as 26 mg/l, the sulphate concentration is 3.6 mg/l and the redox potential calculated by equation (3.14) is -270 mV. This value might be taken as the maximum redox potential for a non-disturbed Boom Clay. From the mineralogical composition of the Boom Clay given in Table 1-2, it can be seen that both pyrite and siderite are present in Boom Clay. So, the calculated Eh based on equation (3.14) should be a reasonable estimate assuming a chemical equilibrium. Without further information on the redox equilibrium state of the nondisturbed Boom Clay, Figure 3-7 provides the current-state conceptual model for describing redox processes in Boom Clay. This model suggests that the measured reducing Eh is controlled by pyrite-siderite equilibrium under the in situ partial pressure of CO2 (g). If the Boom Clay pH is well buffered, the only factor influencing the system redox condition is the concentration of sulphate. This suggests that it might be practically interesting to use the sulphate concentration as an indicator to evaluate the degree of perturbation on water samples caused by oxidation1. Again, the seawater ratio of sulphate and chloride can be used to define the sulphate concentration. As discussed in the previous paragraph, 3.6 mg/l sulphate fixed by seawater SO4/Cl ratio is a reasonable threshold value. A significant higher concentration of sulphate than 3.6 mg/l should indicate severe oxidation of Boom Clay. A lower sulphate concentration is the result of biochemical sulphate reduction processes. In argillaceous sediments, such as the Boom Clay, bacterial sulphate reduction processes occur soon after deposition, in the shallow burial realm. However, it can also relate to 'recent' renewed activity of, or contamination with sulphate reducing bacteria. 1 provided that the bacterial activity, e.g., sulphate reducing bacteria, is low. 59 1.2 1 +++ Fe .8 Eh (volts) .6 .4 Fe(OH)3 ++ Fe .2 0 › Pyrite› › –.2 › Troilite –.4 œ Siderite –.6 –.8 16°C 0 2 4 6 8 10 12 14 pH Figure 3-7: Eh-pH diagram of Fe-S-C system at pCO2 of 10-2.4 atm and sulphate concentration of 1 µM. The boundary between the siderite and pyrite fields represents the anticipated Boom Clay conditions. Squares are values measured in the Cerberus experiment at 80°C (Noynaert et al., 1998); The triangle is measured in the Cerberus experiment at 23°C; and circle is measured by Moors et al., 2002. 3.1.4.2 Redox capacity of Boom Clay pore water The redox buffering capacity of Boom Clay pore water isolated from the solid phase is considered to be very low (De Cannière et al., 1996). Pirlet (2003) reported a measured value of 0.14 meq/l and interpreted it as being dominated by dissolved natural organic matter. 3.1.4.3 Conclusion of Boom Clay redox conditions Both measurements and model interpretation support the conclusion that the reference redox potential (Eh) of the non-disturbed Boom Clay should be lower than -270 mV. This reference value is only indicative that Boom Clay is relatively reducing and implies that laboratory experiments should be performed in an anaerobic environment. However, because of the slow rate of redox reactions and the extremely low concentrations of redox species in Boom Clay pore waters, it is not recommended to use the reference Eh value to constrain the redox condition for laboratory experiments. Future work should focus on mechanisms controlling the redox potential and especially the redox equilibrium state of Boom Clay. 60 3.1.5 Electrolytic Conductivity (EC) The average Electrolytic Conductivity (EC) value for Boom Clay pore water is about 1700 [µS.cm-1]. This value corresponds to a Total Dissolved Salt content (TDS) of around ≈ 935 [mg.l-1] (for bicarbonate waters TDS, in milligrams per liter, is calculated by multiplying the EC value, in microSiemens per centimetre, by 0.55). 3.1.6 Dissolved organic carbon (DOC) and its effect on pore water composition Dissolved Organic Carbon (DOC) is, by definition, the most mobile organic fraction. Mobile organic matter is commonly divided in three groups (Stevenson, 1982). The fraction that is not soluble in alkali is the humin fraction, whereas the humic and fulvic fractions are soluble in alkali. The humic acids precipitate below pH 2, while the fulvic fraction remains in solution. The most important property of natural organic matter with respect to environmental chemistry is its polyfunctional structure. The various functional groups are responsible for the mixed complexation of cations. Humic and fulvic acids are mostly classified as colloidal matter, with a size variation arbitrarily defined between 1 nm and 1 µm. Because of their solubility at alkaline pH, and because of their relatively small molecular size, fulvic and humic acids are mobile in porous and fractured media. The DOC content is actually a Total Organic Carbon (TOC) measurement of pore water after a filtration at 0.45 µm. For piezometer water there is no significant difference between TOC and DOC (Van Geet, 2004). For squeezing and leaching only TOC values are available. 3.1.6.1 Presence of TOC in Boom Clay pore water The sampling of mobile organic matter from Boom Clay is performed in three ways (see Section 2.1). Firstly, pore water is extracted in situ from the clay by means of piezometers. Secondly, Boom Clay samples are squeezed to extract the water content. Finally, organic matter is leached from Boom Clay samples at different solid/liquid ratios. Subsequently, TOC is measured with a high temperature TOC analyser. Pore waters sampled from piezometers contain variable amounts of TOC in function of time. This is shown for the MORPHEUS piezometer in Figure 3-8. Two important observations are made. Firstly, it can be noticed that a very large fluctuation of TOC is present. The reason for this is not known. Secondly, all filters, except F8 (the filter within the double band), show a decreasing TOC content during about 2 years of continuous water sampling. After these two years, a steady state seems to be installed for the TOC. For F8, in which a much higher flow rate (about 4 times higher) is present, a steady state was probably reached already when sampling started. Decreasing TOC contents with time have also been observed in other piezometers (personal communication, H. Moors). Furthermore, a similar trend has been observed by sequential leaching of Boom Clay solid phase with synthetic Boom Clay water (Figure 3-9; Maes et al., 2003). The latter is interpreted by Maes et al. (2003) as a characteristic dilution pattern showing the presence of an easily soluble organic matter pool, and an organic matter fraction for which the release is dictated by an adsorption/distribution mechanism. 61 450 400 350 Filter 2 Filter 4 300 Filter 6 TOC [mgC/l] Filter 8 Filter 9 250 Filter 10 Filter 12 200 Filter 13 Filter 15 Filter 18 150 Filter 20 Filter 23 100 50 0 0 100 200 300 400 500 600 700 800 900 1000 Time [days] OM conc. (measured as Abs. at 280 nm) Figure 3-8: Evolution of the TOC content in the 12 filters of MORPHEUS, since its installation. 2.0 1.8 1.6 1.4 1.2 S/L 0.02 1.0 S/L 0.06 0.8 S/L 0.15 0.6 S/L 0.25 0.4 0.2 0.0 1 2 3 4 5 extraction step Figure 3-9: Leaching tests with Synthetic Boom Clay water (absence of OM) performed at different solid/liquid ratios (1 week intermittent equilibration) (Maes et al., 2003). 62 The TOC measurements of the MORPHEUS piezometer (mean TOC values at steady state conditions) were compared with the TOC measurements of pore waters obtained by the squeezing and the leaching of clay cores at the same stratigraphic depths. Different amounts of total organic carbon are measured for the three techniques (De Craen et al., 2002b). The difference in TOC measurements related to the extracting technique is clearly evidenced for borehole HADES 2001/4, where a core was retrieved for squeezing and leaching and where the MORPHEUS piezometer was installed. Table 3-9 summarises the TOC measurements of the pore water for the 3 techniques at all 12 different levels, together with the TOC of the sediment. Table 3-9: TOC measurements of the solid and the pore water at 12 different levels within the Boom Clay at the corresponding depths of the MORPHEUS piezometer. TOC of the pore water is analysed by extracting water in 3 different ways, namely squeezing of a clay core, leaching of a clay sample, or in situ sampling from the MORPHEUS piezometer. Pore water samples were not filtered. F2 F4 F6 F8 F9 F10 F12 F13 F15 F18 F20 F23 Solid (wt%) 1.37 2.7 2.93 2.67 1.82 1.89 2.25 2.79 2.51 3.27 1.26 1.14 Squeezing (mg C / l) 42.8 50 43.4 81.5 69.9 78.7 105.1 73 41 73.9 54 89 Leaching (mg C / l) 1356.7 1168.3 3250.4 2883.3 - 3335.0 2686.9 2119.5 1822.2 2806.8 2295.5 2569.8 MORPHEUS (mg C / l) 101.1 136 98.7 215.7 120.5 118.9 122.8 119.2 196.6 109.1 127.7 264.9 For none of the pore water TOC values, a correlation was found with the TOC present within the sediment. Moreover, no correlation was found between the pore water TOC measurements of any of the extracting techniques. It is clear that squeezing results in very low TOC values compared to the other techniques, while leaching results in very high measurements. It is believed that the piezometer gives the most reliable results. For squeezing it is assumed that due to porosity and pore size decrease the larger molecules cannot be evacuated from the sample. During leaching, quite the opposite is expected. A slurry is prepared, so that a filtration through pores is no longer active. A piezometer is assumed to keep the in situ conditions as close as possible. Moreover, migration experiments of 14C labelled organic matter through Boom Clay samples illustrated that low molecular size organic matter is more easily transported through the clay than higher molecular weight organic matter (Dierckx et al., 2000). Boom Clay, thus, seems to act as a non-perfect ultra filter. From the preceding it is clear that it is quite difficult to define the real TOC content of Boom Clay pore water. If it is agreed that a steady state TOC production of a piezometer filter is the most representative TOC value, than a mean TOC content of about 115±15 mg C / l (range between 96 and 146 mg C / l) seems most appropriate. However, it should be noted that much higher TOC values are recorded within the double band, namely a mean value of about 220 mg C / l. 63 3.1.6.2 TOC versus UV measurements Two techniques are frequently used for TOC measurements, namely UV absorbance and a high temperature TOC analysis. For UV absorbance, two frequencies are mostly reported in literature, namely 256 and 280 nm. For Boom Clay pore water samples, the absorption at 280 nm is mostly used. One should be careful, however, in interpreting results from both techniques. For a long time it was believed that there was a linear correlation between TOC measurements and UV absorbance measurements for mobile organic matter from Boom Clay. Henrion et al. (1985) described a linear correlation (Figure 3-10), i.e. TOC=23.2xAbs+2.5, for a series of diluted extracts of Boom Clay samples. A similar trend, namely TOC=22.7xAbs+0.011 was obtained by Maes et al. (2002), once again on diluted extracts. With the installation of the MORPHEUS piezometer, the correlation between both techniques could not be established (Figure 3-10). Through time a constant UV signal was measured (Figure 3-11), although a fluctuating TOC value was measured. Henrion et al. (1985) also mentioned some measurements on piezometer water that did not fit the obtained linear regression on diluted extracts. The pore water organic matter contains a higher TOC content for the same UV280 absorbance as compared to the linear relationship found by Henrion et al. (1985). This indicates that a part of the organic carbon in the piezometer waters is UV280 insensitive. Figure 3-10: UV-VIS absorbance measurements at 280 nm versus Total Organic Carbon content for the different filters in the MORPHEUS piezometer, measured 800 days after installation. The trend line and data from Henrion et al. (1985) for piezometer water and organic matter extracts are given for comparison. 64 U.V.-Vis.-measurements at 280 nm y 7 6 Filter 2 5 Filter 4 U.V.-Vis.-absorbance Filter 6 Filter 8 4 Filter 9 Filter 10 Filter 12 Filter 13 3 Filter 15 Filter 18 Filter 20 2 Filter 23 1 0 0 100 200 300 400 500 600 700 800 900 1000 Time [days] Figure 3-11: Evolution of the UV absorbance at 280 nm in the 12 filters of MORPHEUS, since its installation. 3.1.6.3 Characteristics of the mobile organic matter in Boom Clay An overview of the current knowledge on Boom Clay organic matter characteristics is given in Van Geet et al. (2003). A short overview will be given here. A first important characteristic of organic matter is its molecular size distribution. Two techniques have been used to determine this ditribution, namely ultrafiltration and Flow Field Flow Fractionation (FFFF). For ultrafiltration, two experimental set-ups were used. The first uses Amicon filters at 1, 10, 50 and 100 kDa and a gas pressure was used to force the Boom Clay pore water through the filters. The second set-up uses centrifugal ultrafiltration units (Pall Life Sciences: Omega membranes) of 1, 10, 30 and 100 kDa. It should be noted that the Amicon filters consist of regenerated cellulose, while the Omega membranes consist of Poly Ether Sulfones. Both types of filters are pre-conditioned by keeping them in bidistillated water for one week, refreshing the water every day. Blanco experiments were performed for these pre-conditioning technique and did not show any measurable TOC content. The first experiment was performed on piezometer waters at 9 different stratigraphical levels (F23, F20 and F18 were not used) and sampled 352 days after installation of the MORPHEUS piezometer. The results are shown in Figure 3-12, illustrating that about 45 % of the mobile organic matter is smaller than 1000 Da and 45 % is larger than 50,000 Da. This might correspond with the bimodal structure that is observed in GPC measurements during the TRANCOMClay project, although no calibration has been performed (Dierckx et al., 2000). The second experiment was performed on piezometer water at 6 different stratigraphical levels and sampled 853 days after installation of the MORPHEUS piezometer. Results of the distribution are given in Figure 3-13. Again a bimodal distribution is obtained. However, most of the mobile organic matter is now smaller than 10 kDa, except for filter F8 (positioned in the double band, a more silty layer) that contains mostly mobile organic matter between 30 and 100kDa. 65 DOC molecular size distribution (Amicon UF), incorporating 100k filtration 55 Relative contribution (%) 45 35 F2 F4 25 F6 F8 F9 15 F10 F12 F13 5 F15 50k to 100k -5 10k to 50k 1k to 10k <1 000 Molecular weight (Da) Figure 3-12: Molecular weight distribution of the pore water organic matter found in the different filters in 9 stratigraphical levels in the MORPHEUS piezometer, sampled 352 days after installation, by means of Amicon ultrafilters. 80 70 relative contribution (%) 60 50 F6 F8 F12 F18 F20 F23 40 30 20 10 0 -10 100k-.45 30k-100k 10k-30k 1k-10k <1k -20 molecular weight (Da) Figure 3-13: Molecular weight distribution of the pore water organic matter found in the different filters found at 6 different stratigraphical levels in the MORPHEUS piezometer, sampled at 853 days after piezometer installation, by means of centrifugal ultrafiltration units (Omega membranes) 66 The pore water analysed originates from the same filters, but only differs in the timing of sampling, which also results in different DOC concentrations. A comparison of both data sets (Figure 3-14) shows that there exists a good correlation between both experimental set-ups, except for the ultrafitration at 100kDa. The Amicon ultrafiltration gives an overestimation of the fraction 50 to 100 kDa, compared to the centrifugal ultrafiltration fraction 30 to 100 kDa. It is unclear whether this difference is related to an experimental error (poor pre-conditioning of ultrafilter), different forces with which the organic matter is forced through the filter, or to the difference observed in DOC between the two moments of sampling. Thang et al. (2001) performed a flow field flow fractionation (FFFF) on EG/BS piezometer pore water. A calibration of the technique with polystyrene sulphonate was used to determine quantitatively the size fraction of the Boom Clay mobile organic matter. Their study illustrates that the most important fraction of mobile organic matter has a molecular weight below 4000 Da. It should be noted, however, that the authors mention possible deviations of the exact values when using other calibration material. F6 TOC content after Pall Life Sciences UF (mg C/l) 135 125 115 105 y = 1.3511x - 33.799 95 2 R = 0.9553 85 75 75 95 115 135 155 175 TOC content after Amicon UF (mg C/l) 195 Figure 3-14: Example of the TOC contents measured after two types of ultrafiltration (Amicon and centrifugal ultrafiltration units) for filter 6 of the MORPHEUS piezometer, showing a good correlation between both techniques for most ultrafilters(◊), except for the 100 kDa UF (■). A second characteristic of mobile OM is its functional group capacity. The latter has been determined during the TRANCOM-Clay project (Dierckx et al., 2000). This functional group capacity was measured on different sources of Boom Clay organic matter and by means of different techniques. The functional group capacity in the pHrange 7-8 was taken to be representative for the Boom Clay environment and used for 67 comparison (Table 3-10). It was concluded that three types of functional groups are needed to describe the titration curves within the studied pH-range. Table 3-10: Comparison of the functional group capacity in the pH-range 7-8 for different organic matter samples (Dierckx et al., 2000). Name of sample Extraction technique Measuring technique Capacity [Eq/mg] EG/BS (V103) Piezometer pore water Titration 5.95 EG/BS (V3A) Piezometer pore water Titration 6.19 EG/BS (3) Piezometer pore water Titration 5.10 Total extracted Boom Clay Leaching BC with SCW with an L/S ratio of 1/2 Titration 1.80 BCPHA Concentration of EG/BS and then isolation, purification and transformation to the proton form of the HA Titration 2.85 BCEHA Successive extraction of HA from Boom Clay with NaHCO3 0.015 M in air under close to in situ carbonate concentration conditions Cobalt hexamine method 2.1 BCEHA (2) Similar as for BCEHA with an additional extraction step consisting in bringing the Boom Clay to pH 3 to break salt bridges Titration 1.65 A last characteristic of Boom Clay organic matter is its molecular organic chemistry. The latter has not yet been established due to the experimental difficulty of such a characterisation. However, a recent technique of electrospray ionisation quadrupole time-of-flight mass spectrometry has been proposed and tested on pore water from the Mol aquifer below the Boom Clay. Plancque et al. (2001) used this technique for the characterisation of the fulvic acids occurring in the aquifer below the Boom Clay. The fulvic acid solution seems to be a complex mixture of several hundred molecular structures. Tandem mass spectrometry experiments demonstrated losses of 18 Da (water molecule) and of 44 Da (CO2 molecule), which indicates the presence of carboxylic functional groups. A representative molecular structure of the molecules, which are most likely to be self-assembled in the fulvic acid samples, is given in Figure 3-15. Consequently, the fulvic acids of the Mol aquifer contain two families having homologous substructures with alkyl chains terminated by CH3 or NH2. Both families contain a mixture of species with a range of numbers of carboxyl functions, alkyl chain lengths and of numbers of substituents on the aromatic ring. 68 Figure 3-15: Representative molecular structure proposed for fulvic acid from the Mol aquifer (Plancque et al., 2001). 3.1.7 Evaluation of extraction techniques and recommendations for water sampling and storage At the present time, pore water samples from piezometers are considered to be the most representative for the determination of the in situ pore water composition. This is because piezometer waters experience minimum laboratory manipulations and therefore suffer minimum artefacts. However, pore waters obtained in the laboratory from well-preserved clay cores also provide valuable data, which are, in a lot of cases, not attainable by the piezometer technique. Consequently, the applied pore water sampling technique should be chosen as a function of the kind of study/analyses/experiments one wants to perform. Squeezed pore water samples can be considered as representative for the in situ conditions, up to a certain degree. These waters generally have a comparable major element composition, but higher amounts of trace elements and lower amounts of dissolved organic matter are generally observed compared to piezometer waters (see Section 3.1). Furthermore, the slightest oxygen perturbation of the clay core strongly affects the geochemical composition of the pore water Leached pore water samples have a composition which is strongly different from piezometer water and squeezed water (see Section 3.1). The leaching technique needs a lot of laboratory manipulations and is therefore applied under conditions different from that of in situ. Among many factors, system pH (pCO2), ionic strength, solid to liquid ratio, and microbial activity are the most probable factors influencing these water compositions. Physical changes during pore water extraction (decreasing porosity during squeezing; increasing porosity during leaching) also influence the composition of the obtained pore water, in particular the content of natural organic matter. This is because the natural organic matter in Boom Clay has a wide distribution of molecular weight and a large fraction is present in colloidal form. The most obvious evidence is that the squeezed water is colourless comparing to the yellowish to brown piezometer water. This indicates the removal of the coloured dissolved organic matter by squeezing. In 69 contrast, the leaching technique resulted in deep brownish water containing the highest amount of natural organic matter (De Craen et al., 2002b). It is clear that great care should be taken during the drilling of the borehole, the anaerobic preservation and the handling of the clay cores, to prevent oxygen perturbation. The drilling of the borehole should be carefully planned. In the HADES URF, boreholes are generally air drilled. The main disturbance of air drilling consists of a desaturation and oxidation of the clay. These processes can be minimalised when the drilling is performed in a non-oxidising environment, for example when nitrogen or argon is used. Pore water sampling from piezometers at a low flow rate probably provides the most representative pore water, because the system is the least disturbed. Dehydration and oxidation of clay cores can be prevented with some simple precautions. The clay cores should be isolated from atmospheric air as soon as possible. This means that, immediately after drilling (and at the drilling site), the clay cores should be vacuum-packed in Al-coated poly-ethylene sheets, flushed with nitrogen or argon, then evacuated and sealed. The clay cores should then be stored at 4°C in anaerobic conditions. In the laboratory, oxidation of the clay cores can be prevented by working in anaerobic glove boxes. The outer rim of the clay core, which has been inevitably in contact with air during the drilling, should be removed to eliminate possible effects of oxidation. For the sampling of the pore water, sampling containers, or dark bottles should be used to avoid organic matter degradation as a result of UV radiation. Finally, the samples should be stored at low temperatures (at 4 °C) to avoid bacterial activity in the pore water. 70 3.2 Isotope geochemistry 3.2.1 Stable isotopes The stable isotope composition of Boom Clay pore water was studied in the frame of the ARCHIMEDE-argile project (Reeder et al., 1994; Griffault et al., 1996; Beaucaire et al., 2000). In this study, a profound analysis of the Lower Rupelian aquifer, at a regional scale, was also performed. This allowed for comparison between the Boom Clay and its underlying aquifer. Other studies also focussed on the characterisation of the groundwater in the aquifers above and below the Boom Clay at a regional scale (Beaufays et al., 1994; Marivoet et al., 2000) with no or few analyses on the Boom Clay pore water. During the PHYMOL project, stable isotope analyses were performed on 4 pore water samples obtained by the distillation and the squeezing of clay cores from the Zoersel borehole, and on 20 pore water samples obtained by the squeezing of clay cores from the Weelde borehole (Marivoet et al., 2000; Philoppot et al., 2000). No samples were taken at the Mol site. δ18O values range from -5.3 to -8.4 ‰. δ2H values range from 43.1 to -58.5 ‰. Pore water samples obtained by distillation always have slightly more depleted δ18O and δ2H values. No interpretation was however proposed on these values. In the ARCHIMEDE-argile project, one pore water sample from the ARCHIMEDEpiezometer #1, and 5 pore water samples obtained by the squeezing of clay cores were analysed for their stable isotope composition. The results are given in Table 3-11. Table 3-11: Stable isotope data of Boom Clay pore water, data from the ARCHIMEDE-argile project (Reeder et al., 1994; Griffault et al., 1996). SMOW = Standard Mean Ocean Water, CDT = Canyon Diabolo Troilite. Boom Clay pore water Sample reference δ18O water δ2H water δ18O sulph. δ34S sulph. ‰ vs SMOW ‰ vs SMOW ‰ vs SMOW ‰ vs CDT -12.5 -10.0 -37.1 -39.1 -6.6 -25.9 ARCHIMEDE piezo #1 P1,8m -7.28 -54.9 Squeezed clay cores TD R97/98 WH 1,15-1,35 m TD R97/98 WH 2,35-2,55 m A07478 7,00-7,10 m A07479 8,15-8,25 m A07480 14,20-14,40 m -7.07 -7.05 -7.10 -7.25 -6.97 -52.3 -54.5 -54.0 -52.3 -53.7 71 The present day isotope composition of ocean water is more or less constant with δvalues very near to zero (Hoefs, 1997). In contrast, the isotope composition of meteoric waters vary linearly (linear relationship is described as the "meteoric water line"; IAEA, 1992) on a global scale and are dependent on geographic location. The isotope composition of the mean worldwide precipitation is estimated to be δ18O = -4 ‰ and δ2H = -22 ‰ (Craig and Gordon, 1965). The isotope composition of Boom Clay pore water is δ18O = -7 ‰ and δ2H = -53 ‰. These values suggest that the pore water is not of marine origin. A meteoric origin can explain the δ18O values, but not the δ2H values. In Figure 3-16, the isotope composition of Boom Clay pore water is compared to the meteoric water line and groundwater of various Rupelian aquifers (for details see Griffault et al., 1996). 2 H (‰ vs SMOW) 0 M WL -10 Boom Clay pore water (ARCHIMEDE piezo #1) -20 -30 Boom Clay pore water (squeezed clay cores) -40 Rupelian aquifers -50 -60 -10 -8 -6 -4 -2 0 18 δ O (‰ vs SMOW) Figure 3-16: δ18O versus δ2H of Boom Clay pore water compared to the Meteoric Water Line (MWL) and groundwater of Lower Rupelian aquifers from various locations (from Griffault et al., 1996). Groundwaters from the Lower Rupelian aquifer are in the range of values close to meteoric water line (Figure 3-16). This indicates that the recharged conditions are either those of present day or climatically little different from these (Griffault et al., 1996). The oxygen and hydrogen isotope composition of Boom Clay pore water fall under the Meteoric Water Line. This is the case for pore water collected from the piezometer as well as for pore water derived from the squeezing of clay cores (Figure 3-16). 2Hdepletion is a common observation in water extracted from clayey formations. It is therefore considered as an artefact of the extraction technique, causing stable isotope fractionation (Griffault et al., 1996). According to the chemical analyses performed by Reeder et al. (1994), sulphate is the dominant anion in most squeezed pore waters (values between 1130 and 11100 mg/l) and substantial quantities of thiosulphate are often present (200 – 500 mg/l). In order to characterise the origin of the dissolved sulphate, the δ18O and δ34S isotope 72 composition of the dissolved sulphate is measured. The low δ18O and δ34S values for dissolved sulphate (see Table 3-11) suggest that sulphide oxidation indeed occurred in these Boom Clay pore water samples. Also De Craen (2001) and De Craen et al. (2002a and 2004a) explained these high concentrations of sulphate and thiosulphate as the result of oxidation. 3.2.2 Radioisotopes 3.2.2.1 U-Th isotopes In the frame of a regional programme of hydrochemical and isotopic measurements, uranium series disequilibrium studies were performed on groundwater samples collected from sand layers above and below the Boom Clay in Mol (Ivanovich and Wilkins, 1988). Unfortunately, Boom Clay pore water itself was not sampled. Recently, uranium series disequilibrium studies were performed on pore water derived from the MORPHEUS piezometer. The uranium content (238U) in the pore water is generally less than 1 µg/l. Higher uranium contents are present in pore water sampled from septaria level S50 (1.3 µg/l), and in pore water sampled from an organic-rich layer at the base of the Putte Member (2.2 µg/l). The 234U/238U activity ratios are indicative of radioactive disequilibrium, with 234U/238U activity ratios between 1 and 5. An excess of 234U relative to 238U, however, is a common observation in natural waters (Fleisher, 1988). It is explained as the result of α recoil, either as the direct recoil ejection of a recoil atom into the pore water, or indirectly by recoil leaching (the preferential etching of recoil damage and associated preferential solubility of the products of α decay). Squeezed pore water samples always contain higher amounts of uranium (generally about 5 µg/l, but values up to 18 µg/l are also measured) compared to pore water derived from piezometers. The higher uranium content can be explained by the presence of colloids in the squeezed pore waters. The 234U/238U activity ratios of squeezed pore water samples (De Craen et al., 2000; De Craen et al., 2004b) show radioactive disequilibrium, with ratios between 2 and 6. These results fall in the same range as the results of the pore water sampled from the MORPHEUS piezometer. 3.2.2.2 14C In the frame of the PHYMOL project (Marivoet et al., 2000), 14C measurements were performed on Boom Clay pore water. Unfortunately, no 14C was detected (with a detection limit of 60 000 years). 3.2.2.3 36Cl Until now, 36Cl measurements have not been performed on Boom Clay pore water. 73 3.3 Spatial variability Although the mineralogical and geochemical properties of the Boom Clay are considered to be rather constant throughout the deposit (Vandenberghe, 1978; De Craen et al., 2000), Boom Clay is characterised by a banded nature or layering. This layering is the result of variations in grain-size, organic matter and carbonate content (Vandenberghe, 1978). Thus a vertical variability exists in the Boom Clay deposit. Several of these layers were found to have enough specific characteristics to be recognised whenever they were found in outcrops, clay cores or even in geophysical logs, the so-called 'marker horizons' (see references in Van Keer and De Craen, 2001). Examples of marker horizons are: a pinkish layer, calcareous-rich layers containing septarian carbonate concretions, the double band, the boundary between organic-poor and organic-rich layers (corresponding to the boundary between the Terhagen Member and the Putte Member). These layers can be followed on a regional scale. In general, the composition of Boom Clay doesn't change much laterally, although some slight variations in the clay mineralogical composition can be recognised (Laenen, 1997; De Craen et al., 2000; Van Keer and De Craen, 2001). Whether these variations in Boom Clay compositions are also reflected in the pore water composition is considered in the following paragraphs. 3.3.1 Vertical variability To study the vertical variability of the Boom Clay pore water composition, the MORPHEUS piezometer was designed. Pore water is sampled at 12 different stratigraphic levels. The piezofilters are positioned in silty clay / clayey silt, in organic-poor / organic-rich clay, in septaria layers, and in the double band. In Figure 3-17, the concentration of some major elements is plotted against depth. The chemical composition of the pore water is comparable in most of the filters, except at a depth of 230.28 m TAW. At this level, a concentration peak can be recognised for most of the elements. At the same level, much more dissolved organic matter is measured in the pore water (see Section 3.1.5). This sample corresponds to the double band. Thus, apart from the double band, the chemical composition of the pore water seems to be comparable for most of the filters, suggesting that the variability of the Boom Clay composition is not reflected in its pore water chemistry. A more detailed study on the vertical variability of the pore water composition by statistical analyses (Section 3.4), however, will disprove this conclusion. 74 HADES borehole 2001/4 -216 -218 -220 dep th (m TA W ) Putte Member -222 -224 -226 -228 -230 Terhagen Member -232 -234 -236 0 2 4 6 8 10 Ca Fe Mg K 0 10 20 30 concentration (mg/l) concentration (mg/l) Si F- Cl- 0 200 400 600 800 1000 concentration (mg/l) Br- Na HCO3- Figure 3-17: Chemical composition of Boom Clay pore water sampled in the MORPHEUS piezometer. 3.3.2 Lateral variability The lateral variability of the Boom Clay pore water composition at the Mol site can be studied by comparing the pore waters sampled from the various piezometers. Variations in pore water chemical composition at the Mol site are small. Moreover, these variations might be related to other factors, such as different types of filter materials, as explained in more detail in Section 3.4.1. To study the lateral variability of the Boom Clay pore water composition on a regional scale, squeezed pore water samples from various boreholes were analysed (Figure 3-18): the Doel-2b borehole, the Zoersel borehole, the Mol-1 borehole, and the HADES borehole 2001/4 (also at Mol). In these four boreholes, the zone between S40 and S50 was considered (De Craen et al., 2000; De Craen et al., 2004b). 75 URF Underground Research Facility Figure 3-18:Location of the boreholes used in the study of the lateral variability of the Boom Clay: the Doel-2b borehole (in Doel), the Zoersel borehole (in Zoersel), the Mol-1 borehole (in Mol), and the HADES borehole 2001/4 (also in Mol). The chemical composition of squeezed Boom Clay pore waters are given in Table 3-12. As mentioned above, the pore water of the double band always has a different chemical composition, and therefore, the double band is not taken into account for the calculation of the mean values. It should be mentioned that the clay cores from the HADES 2001/4 borehole were well-preserved and squeezing was performed soon after the drilling of the clay cores. In contrast, clay cores from the Doel-2b, Zoersel and Mol-1 boreholes have been preserved for a few years in comparable (but not ideal) conditions. These clay cores might have been oxidised. Oxidation of the clay cores affects the chemical composition of the pore water (De Craen, 2001; De Craen et al. 2002a; De Craen et al., 2003). However, some elements, such as chloride, are not influenced by oxidation. Furthermore, the squeezed pore water samples from the HADES 2001/4 clay cores were filtered at 0.45 µm before analyses of the cations. Filtration was not done on pore water samples from the other boreholes. This explains why higher amounts of Mg and Si are present in pore water samples of the Mol-1 borehole compared to the HADES 2001/4 borehole. Small clay particles, colloids, were probably measured as well, resulting in higher amounts of some elements, such as Mg and Si. 76 Table 3-12: Chemical composition of squeezed Boom Clay pore water in various boreholes. Doel-2b borehole Zoersel borehole HADES 2001/4 borehole Mol-1 borehole mg/l Min.-max. mean Min.-max. mean Min.-max. mean Min.-max. mean Ca 8.4 – 339 40.7 3.5 – 10.3 5.6 3.6 – 9.3 5.1 1.2 – 8.4 3.5 Fe 0.12 – 10.1 1.08 0.16 – 0.62 0.37 0.4 – 8.2 2.4 <0.05 – 11.1 0.9 Mg 7.7 – 425 62.8 6.0 – 20.0 11.7 3.4 – 34.2 18.8 0.9 – 10.4 2.8 K 10.1 – 115 27.5 4.9 – 14.4 7.6 4.8 – 29.5 8.5 3.9 – 15.0 6.8 Si 5.1 – 11.5 7.2 5.9 – 10.5 8.3 9.1 – 33.5 15.0 6.4 – 11.3 8.0 Na 381 – 1780 953 365 – 777 502 238 – 1240 378 225 – 730 356 - F Cl 0.9 – 1.4 1.1 1.1 – 1.9 1.7 2.2 – 3.0 2.4 1.4 – 3.0 2.2 - 317 – 4000 1230 248 – 347 323 20.4 – 52.2 28.5 20.6 – 39.9 27.3 - 2.1 – 11.2 4.0 1.26 – 1.75 1.45 0.5 – 5.6 1.2 0.43 – 1.43 0.80 53 – 507 215 6 – 618 172 32 – 999 188 3.6 - 700 155 247 - 742 447 448 - 1209 616 422 - 1122 561 422 - 707 575 Br SO42HCO3 - The most important lateral variability of the Boom Clay pore water chemistry on a regional scale is probably the variation of the NaHCO3 -type water in Mol to a NaCl/NaHCO3 -type water in Doel (Figure 3-19). The mixing of the NaHCO3-type water with NaCl is clearly increasing from the east (Mol) to the west (Doel). A gradient from the east to the west is also present for some other elements. These variations might be linked with the general change of the water-type. However, this interpretation should be taken with some care, because possible oxidation of the samples might also have influenced the pore water chemistry. Doel-2b borehole DoTP - samples 83 Zoersel borehole ZTP - samples mg / l Depth (m) 0 1000 2000 3000 4000 vv vvv vvv 150 84 151 85 86 vv vvv vvv 87 vv vvv vvv 153 vvvvv vvv 256 vv vvvvvv 257 vv vvvvvv 258 vv vvv vvv 156 90 91 vv vvv vvv 259 vvvvv vvv vv vvv vvv vvvvv vvv 158 vv vvvvvv 260 261 vv vvv vvv vv vvvvvv 263 160 94 161 95 Na Cl HCO3- 162 Na Cl HCO3- 258 259 vvvvvvvv vvvvvvvv vvvvvvvv 260 261 262 262 159 93 vvvvvvvv vvvvvvvv 92 257 vvvvvvvv vvvvvvvv 157 255 256 vvvvvvvv vv vvvvvv vv vvv vvv 1000 2000 3000 4000 255 vvvvv vvv 155 0 254 Depth (m) vvvvv vvv vvvvvvvv vv vvv vvv mg / l Mol-1 borehole MTP - samples vvvvv vvv 152 154 89 1000 2000 3000 4000 vv vvv vvv vv vvv vvv 88 mg / l 0 Depth (m) vvvvvvvv 263 264 264 265 265 266 Na Cl HCO3- Figure 3-19: Lateral variability on a regional scale of the NaHCO3 and NaCl concentration in the Boom Clay. 77 3.4 Data quality 3.4.1 Statistical Analysis Several piezometers starting from the URF are available for the characterisation of the Boom Clay pore water in Mol. However, the question raises whether we can provide one mean composition from the waters sampled and analysed from those different piezometers. It is clear that several factors may influence the measured composition, so that a statistical analysis is appropriate. In this Section we will discuss all factors that might have an influence on the measured pore water composition, the factors that will be analysed statistically, the data used for the statistical treatment, and the results. It should be noted that this is only a statistical treatment of data. When statistically significant differences are found, one still needs to look for the possible geochemical causes for these differences. 3.4.1.1 Factors that might influence pore water composition Pore water sampling from piezometers is a quite simple procedure. However, many processes are involved so that many factors might influence the final pore water composition. The following factors are assumed to be the most important. • Spatial variability Boom Clay is sometimes treated as a homogeneous material. However, it is known that natural layering does occur and variances in mineralogy, grain size, etc. are present (see Section 1 and Section 3.3). Consequently, it should be considered that the 3D position of the filter within the Boom Clay might have an effect on the pore water composition. • Evolution in time Placing a piezometer causes a disturbance of the host rock (see Section 2). It is known that this disturbance will be restored. However, it should be tested if an evolution in time of the pore water composition is noticed. • Effect of filter material As described in Section 2, different filter materials can be used in the design of a piezometer. The effect of this filter material should be tested as well. • Effect of sampling conditions The final pore water sampling can be performed in aerobic or anaerobic conditions. This also might influence the measured composition. It is thus clear that many factors might influence the pore water composition. In this study, the effect of only some of the factors is studied. The factor of spatial variation is only partly studied. It is known that the natural layering within the Boom Clay is very continuous parallel to the bedding. Moreover, only the Boom Clay pore water from the Mol site is used. The used piezometers are laterally only separated by a maximum of 67 m of clay. Consequently, the effect of spatial variation is studied in one dimension, namely perpendicular to the bedding. The effect of time evolution is also only partly considered. Actually, samples that might have an influence of the disturbance due to the emplacement of the piezometer are excluded. Since it is known that SO42- will be produced during installation due to the oxidation of the surrounding clay, a criterion based on the sulphate content is defined. It is stated that the sulphate 78 content should be constant and low (<6 mg/l). The effect of filter material is taken into consideration in this study. However, it should be noted that only a very limited amount of data is available to check this influence. The effect of sampling in anaerobic or aerobic conditions is not incorporated in this statistical evaluation. It is assumed that the latter has no important influence on the results. Furthermore, it should be noted that different analysis techniques might influence the measured composition. However, this influence is not considered because of the lack of sufficient data. Moreover, the oldest results, date from 1996 and we have checked that since then, no new techniques became in operation at the Nuclear Chemistry and Services, SCK•CEN. 3.4.1.2 Data and statistical techniques To evaluate the effect of the above described factors, different piezometers are available. Within this statistical evaluation, 5 piezometers are considered, namely EG/BS, MORPHEUS, ORPHEUS, SPRING 116 and ARCHIMEDE #1 (see Annex 5). EG/BS is used already a long time for many laboratory experiments and consequently provides a large amount of data (70 analyses of major elements are available). MORPHEUS contains 12 filters at different layers. For 11 of these filters 4 analyses are available. ORPHEUS is a horizontal piezometer with 4 filters of different materials. For each filter material, a different amount of data is available, with a maximum of 5 analyses (n ≤ 5). For SPRING 116, a horizontal piezometer with 4 stainless steel filters, only two analyses for each filter are available (n=2). ARCHIMEDE #1 is a horizontal piezometer with 5 filters; 2 to 5 analyses are available for the 4 deepest filters. Although pore water sampling is taking place already a long time, the amount of useful data is limited. Especially the amount of data to test the effects of filter material and the effects of spatial variability is low. This results from the fact that the piezometers that might give an answer to these questions were recently installed in the URF. Most other piezometers are in the horizontal plane, or were used for pore water pressure measurements. For the statistical evaluation, only the major elements occurring in the pore water are included: B, Br, Ca, Cl, F, Fe, K, Mg, Na, Si. TIC is not available for all the measurements and is only used if available. For easy recognition, each pore water sample got a code in which the name of the piezometer, filter material, sample number and date of sampling is included (see Annex 1). For the statistical evaluation it is first tested whether the pore water sampled from different filters of one single piezometer can be grouped to one single population. This is done element per element using an analysis of variance (ANOVA1). This analysis provides a probability value indicating whether all filters can be grouped to one single population (H0: µ1=µ2=...=µm). This is the case when the probability p>0.05 (95% confidence interval). The basic assumptions when using the ANOVA test are: • • All data are normally distributed. The variance within each of the populations is equal. 79 A limited amount of data is available, but the assumptions can be supposed to be true. A description of the ANOVA technique and the check of the assumptions is given in Annex 6. Next, several elements will be considered together to answer the following questions: • • Is there an effect of the filter material on the measured pore water composition? Is there an effect of the spatial variability on the measured pore water composition? To this end, a multivariate analysis of variance (MANOVA) is used. This MANOVA allows to determine whether the entire set of means of all variables (elements) is different from one group to the next. In MANOVA several linear combinations of the original variables (elements) are defined, so that the largest separations between groups occur. From these linear combinations an estimate of dimension (d) of the groups means is defined. If the means were all the same, the dimension would be 0. If the means differed, but fell along a line, the dimension would be 1, and so on. For each dimension a probability (p) can be calculated. A scatter plot of the linear combinations that result in the largest separation between groups will thus show more separation between groups than a scatter plot of any pair of original variables. If the estimated dimension is 2, than this scatter plot most probably shows all variation between the groups. The basic assumptions when using the MANOVA test are • • • • Data are independent. Data are normally distributed. No big difference of the variances between groups of the same variables. MANOVA is sensitive to outliers. A description of the MANOVA technique and an evaluation of the assumptions is given in Annex 6. 3.4.1.3 Results of the statistical analyses First of all the ANOVA analysis of each piezometer will be discussed. Later on, the MANOVA analysis related to the questions whether filter material and spatial variability influence the pore water composition will be given. EG/BS EG/BS is a vertical piezometer surrounded with coarse sand, so that a large vertical segment of the Boom Clay is draining into the filter. EG/BS is in use already for a long time, so that measurements over a longer period are available. The first results date from 1996. For all measurements available, the sulphate value is very low and can be assumed as being constant. The first 3 measurements have an unknown starting date of sampling and were excluded from the statistical evaluation. For all elements a visual inspection of the analytical results did not define any systematic time trend. However, it was noticed that two groups can be distinguished in time. The first group consists of the measurements from 1996-03-13 till 1999-04-07. The second group contains data from 2000-08-08 till 2003-02-17. In between the two groups, a serious time lack of data is present, and based on the visual inspection a shift in the mean value of both groups is possible. It is unknown whether anything happened during this 80 time gap that might have affected the composition of the pore water. For all major elements listed above, 69 or 70 data points are available, except for Br for which only 26 data points are available. For TIC, 52 data points are available. For most of the elements, group 1 contains 45 (or 44) data points and group 2 contains 25 data points. Table 3-13 lists the p values obtained with the ANOVA test. For the elements Na, K, TIC, Cl and F, there is significant evidence that these elements belong to one single group. However, for the other half of elements considered (Ca, Fe, Mg, B and Si) this is not the case. Consequently, the most conservative option would be to take into account that both groups may be different. However, it should be noted that the most abundant elements (Na, TIC) can be considered to belong to one single group. Table 3-13: ANOVA probability values for each element testing the hypothesis that the two groups distinguished in time in the EG/BS piezometer belong to the same group. P-values above 0.05 (in bold) are accepted as significant. K Mg Na Si B Ca Cl F Fe HCO3 8.8·10-8 0.017 0.090 0.2353 0.010 0.826 0.810 0.002 0.250 0.016 ARCHIMEDE #1 The ARCHIMEDE #1 piezometer is a semi-horizontal piezometer (3% inclined upwards) with 5 filters at respectively 15, 14, 8, 7 and 3 m from the gallery. Pore water analyses are only performed for the 4 deepest filters. From the deepest to the closest filter, respectively, 2, 5, 5 and 2 analyses are available. The sulphate criteria are met in all the analyses. However, not all the major elements were always measured. Therefore, the ANOVA 1 test is limited to Cl, F, Na, K, Ca and Mg. The results of the ANOVA 1 test suggest that all the analyses can be considered as belonging to one single group (see Table 3-15). Table 3-14: ANOVA probability values for several element testing the hypothesis that the filters of the ARCHIMEDE #1 piezometer belong to the same group. P-values above 0.05 (in bold) are accepted as significant. Cl F Na K Ca Mg 0.59 0.78 0.21 0.85 0.21 0.26 SPRING 116 The SPRING 116 piezometer is a horizontal piezometer containing 4 large stainless steel filters. Filter 1 is the deepest filter, while filter 4 is the nearest one. For each of these filters only 2 measurements are available. This is too few for statistical analyses. However, if the groups do not seem to differ, all measurements can be combined to increase the number of data points in a further statistical evaluation. In all these measurements the sulphate content is below 6 mg/l. For filters 1, 2 and 3 the sulphate content seems to be more or less constant, but for filter 4 the sulphate content still 81 shows a decreasing trend (more than 2 sulphate measurements are available for deduction of these conclusions). However, all data samples are used. It is assumed that for each filter the data of each element follows a normal distribution (see Annex 6). Table 3-15 shows the probability values that all filters belong to 1 group. For most of the elements (Na, Ca, Mg, Fe, Cl and Si) this probability is very low and thus it can be concluded that not all filters belong to the same group. From a multicomparison of the ANOVA results it can be concluded that filter 1 has the most deviating values compared to the other filters. Therefore, an ANOVA test was also performed on filters 2, 3 and 4, only. Table 3-15 gives the p-values, showing that, apart from Mg, all elements of these 3 filters seem to belong to one group. Consequently, filters 2, 3 and 4 are considered as one group with 6 data points, while filter 1 is considered as a different group with only 2 data points. Up to now, it is unknown what caused the difference between the filters of this piezometer. Table 3-15: ANOVA probability values for each element testing the hypothesis that the filters of the SPRING 116 piezometer belong to the same group. P-values above 0.05 (in bold) are accepted as significant. B Br Ca Cl F Fe K Mg Na Si All filters 0.074 0.661 0.003 0.002 0.232 0.011 0.062 0.003 Filters 2, 3 and 4 0.303 0.977 0.268 0.282 0.211 0.258 0.555 0.0149 0.175 0.173 0.001 0.043 ORPHEUS The ORPHEUS piezometer is a horizontal piezometer containing 4 different filters, each of another material, namely: PolyEthylene (PE), Glass, Carbo, and a sintered aluminium silicate (Schuma). For the PE, glass and Schuma filters respectively 5, 4 and 3 measurements are available for which a constant and low (<6 mg/l) sulphate concentration can be deduced. For the Carbo filter only 1 measurement is available that fits the sulphate criteria stated above. However, for an ANOVA test, at least two measurements are needed. Therefore the measurement with the lowest but one sulphate content, i.e. 7.7 mg/l, is included as well. Consequently, one should be very cautious in making conclusions on this filter material. For all the major elements listed above data are available. For TIC, however, no data are available. Table 3-16 lists the probability values that all filters belong to one group for each element. It is clear that only for the elements Ca and Fe no distinction can be made on the filter material. For all other elements there is a significant difference. Table 3-16: ANOVA probability values for each element ANOVA testing the hypothesis that the 4 different filter materials in the ORPHEUS piezometer belong to the same group. P-values above 0.05 (in bold) are accepted as significant. B Br Ca Cl F Fe K Mg Na Si 9.5·10-7 0.035 0.364 0.001 0.029 0.253 0.001 0 0 1.43·10-11 82 MORPHEUS The MORPHEUS piezometer is a vertical piezometer with 12 different filters (each 10 cm high) at several distinguished layers. Due to some technical errors the top filter did not provide water during a long time and consequently only one analysis of the pore water composition of this filter is available. Therefore, this filter was not included in the statistical evaluation. For each of the other filters, 4 measurements are available. In all 4 of them the sulphate content is very low and can be assumed to be constant. In all measurements, data for all the major elements listed above and for the TIC are available, except B for which only 2 data points are available. As only 4 measurements are available, a histogram cannot be provided, and thus it is assumed that the measurements of each filter are normally distributed (see Annex 6). Table 3-17 gives the p-values that are obtained for each element, considering all 11 filters. It can be concluded that, except for B and Br, none of the elemental analysis of all filters belongs to 1 group. Box plots of each element and a multicomparison of the ANOVA analysis illustrates that filter F8, located within the double band, seems to differ extremely from all other filters for most of the elements. Consequently, it was tested if all filters except F8 of the double band, belong to one group considering each element apart. From Table 3-17, it can be concluded that excluding F8 allows to consider the elements B, Br, Fe, TIC and K as originating from one group. However, for the elements Na, Ca, Mg, Cl, F and Si there is no significant evidence that they belong to the same group, even when excluding F8. Table 3-17: ANOVA probability values for each element testing the hypothesis that the measured element concentrations in the MORPHEUS filters belong to one single group. P-values above 0.05 (in bold) are accepted as significant. B Br Ca Cl F Fe HCO3 K Mg Na Si All filters 0.970 0.078 2.11·10-7 0 4.3·10-8 0.033 3.6·10-5 0.006 4.9·10-9 9.9·10-16 0.028 All filters except F8 0.964 0.380 0.005 1.9·10-14 0.003 0.307 0.211 0.087 0.021 1.1·10-6 0.035 3.4.1.4 Effect of the filter material on the pore water composition Is there any effect of the filter material on the pore water composition? Two piezometers are used to test the effect of the filter material. The ORPHEUS piezometer is a horizontal piezometer with 4 different filter materials. SPRING 116 is also a horizontal piezometer with 4 stainless steel filters. For the SPRING 116 piezometer, only the filters 2, 3 and 4 are used, as filter 1 seems to be significantly different and only contains 2 data points (see ANOVA above). From the assumption that the spatial variability can be reduced to a one-dimensional vertical problem, it can be assumed that possible differences between the pore water composition of ORPHEUS and SPRING 116 are only due to the filter material. A MANOVA test is now used to compare these different filter materials. However, it should be noted once more that only a limited amount of data is available and results should be treated with care. The obtained results might indicate whether the raised question should be re- 83 analysed in future when more data points are available. All major elements listed above are taken into consideration together. From a grouped scatter plot of all elements and for all filter materials, it is clear that B and Si have extremely high values in the sintered glass filter of ORPHEUS compared to the other filter materials (Figure 3-20). This clearly indicates a leaching of both elements from the sintered glass filter material. Figure 3-20: Cross plot of the concentration of all elements (see diagonal) in function of the filter material. It is noticed that B and Si (outer columns and rows) of the sintered glass filter (open circles) are extremely different from the values for the other filter materials. Therefore these two elements were removed from the MANOVA analysis, as to be able to compare all filter materials. The MANOVA test shows a significant probability to result in a 2D space (p=0.070), so that a 2D scatter plot illustrates the maximum difference between all groups (Figure 3-21). From this graph it can be concluded that the water provided by the poly-ethylene (PE) and sintered glass (SG) filter materials is very similar. The first 3 stainless steel (SS) filters of SPRING 116 (SP-SS group 2) show a small difference with the PE and SG group. A larger difference is noticed for the Schumatherm (ST) filter and the Carbo (CA) filter. However, it should be noted that for the latter two filters only a very limited amount of data is available. The elements mostly influencing the separation are F, Cl and Na. 84 Figure 3-21: MANOVA result for all filter materials, illustrating the maximum separation between groups. Pore water collected with a PE or SG filter seems to have a very comparable composition. SS gives a slight deviation, while ST and CA create the largest deviations. With the limited amount of data available, it can be concluded that the filter material has an effect on the measured pore water composition. Table 3-18 gives an overview of the filter materials tested and the elements contributing to differences. From this table it can be concluded that the sintered glass (SG) filter causes an increase in B and Si content. Apart from these elements there is no difference between the SG and PE filter materials. The CA and ST filter materials only differ in their Mg content. As this is a rather minor element, it can be concluded that CA and ST hardly differ from each other. A small difference (except for B and Si) between the PE–SG and CA–ST pore water composition is observed. However, additional data points are certainly necessary to confirm this observation. The stainless steel (SS) filters cause an increase in Fe content and depleted concentrations for several other elements. Finally, it can be concluded that Na, Cl, Mg and F are the most important elements influencing the separation between all groups. Once again it should be noted that additional data points in the future are needed to confirm or disapprove the observed trends. 85 Table 3-18: Overview of the elements influencing the difference in pore water composition for each filter material studied. For each comparison the elements are ordered from left to right with decreasing contribution to difference. The elements marked in grey are more abundant in the material mentioned at the top of the column for that comparison. PE PE SG CA SG CA ST SS2 Si, B Mg, Cl, Na Cl Fe, Si, Cl, F Si, Mg, B, Cl, Na Si, B, Mg, Cl Si, Fe, B, Cl, F Mg Br, Fe, Mg, Si, Cl ST Br, Si, Mg, Cl, F SS1 Fe, Ca, Mg, Cl, Na, K SS2 3.4.1.5 Effect of the spatial variability on the pore water composition Is there any effect of the spatial variability on the pore water composition? To evaluate this question, it is necessary to compare pore water of piezometers with the same filter material. As the question of spatial variability around the URF is reduced to a vertical 1D problem, we only need a vertical piezometer with different filters of the same material, like MORPHEUS (all Schumatherm filters). However, as one filter in ORPHEUS also contains a Schumatherm filter in a layer that is not sampled in MORPHEUS, it can be included in the MANOVA test. Consequently, 12 different locations are taken into account. For this test all major elements are taken into account, except B, as only a limited amount of data are available for this element. A first MANOVA test results in a 4D separation of several groups. A scatter plot of the two most important separations illustrates that the pore water sampled in the ORPHEUS piezometer and the pore water provided by F8 of MORPHEUS is clearly different from all other filters (Figure 3-22). The elements causing this separation are Na and Cl (and to a lesser extent F, Ca and Mg). A boxplot for Na and Cl for all filters at different depths is given in Figure 3-23. Filter F8 of MORPHEUS is providing water from the "double band", a more silty layer within the sampled Boom Clay interval. The presence of a geologically particular layer close to the gallery, that might influence the pore water composition of the ORPHEUS filter is not known up to now. To decrease the dimensionality of the separation between several groups (and to optimise the visualisation), those filters (F8 and the ORPHEUS filter) are excluded from a new MANOVA test. This new test results in a 3D separation (p=0.177). From this MANOVA test, it can be concluded that filters F18 and F20 are very similar (results not shown). Distinguishing other groupings, however, is very difficult. The elements mostly influencing this 3D separation are once again Na and Cl. 86 Figure 3-22: MANOVA result for all Schumatherm filters at different depths, illustrating the two largest linear combinations of elements to optimise the separation between groups. Pore water sampled at the gallery (gal) and at the double band (F8) seem to have a different composition from all other sampling filters. a b Figure 3-23: Boxplot for Na (a) and Cl (b) for all depths studied, showing the major difference of the pore water sampled at the double band and in the ORPHEUS filter. 87 It is clear that only a limited amount of data are available to answer the proposed question. However, it might be justified to try to group the filters of MORPHEUS into geologically justified sets. Therefore, a new MANOVA was performed on the remaining filters by excluding not only B, but also Na and Cl. The new separation is 2D (p=0.433), so that a maximum separation is visualised in Figure 3-24. Now, several groupings can be made. A first group encompasses F20, F18 and F15, which are all located above S50, a septarian horizon within the Putte member. A second group contains filters F6, F9, F12 and F13, which are all located between the base of the Putte member and S50. One filter located in the same area seems to have a different composition of pore water, namely F10. For F10 there is no geological peculiarity known. A last group is formed by filters F2 and F4 that are located within the Terhagen member. It should be stressed once more that the grouping made here is probably very conservative as the amount of data is very limited and there is not really a reason to reject the Na and Cl elements from the analysis. The most important elements contributing to these groupings (causing the largest "difference") are Mg, Ca and F, which are present in only relatively low amounts in the Boom Clay pore water. Hence, the fact that Na and Cl play a major role in the Boom Clay pore water composition is probably a more important conclusion from this analysis. Figure 3-24: MANOVA result for filters of MORPHEUS, except the one in the double band (F8), illustrating the maximum separation between groups, based on all major elements except B, Na and Cl. Pore water sampled at Terhagen (F2 and F4) or between base Putte and S50 (F6, F9, F12, F13) or above S50 (F15, F18, F20) seem to be distinguishable. 88 3.4.1.6 Conclusions of the statistical analyses The amount of data presently available has certainly revealed some statistical differences of the measured pore water composition. First of all it is clear that there is an effect of the filter material on the measured pore water composition. The use of a glass filter will increase the amount of B and Si within the pore water. Stainless steel filters increases the Fe content. Apart from B and Si, no difference is observed between the SG filter and a PE filter. The CA and ST filter only differ in Mg content. However, additional data points are needed to increase our confidence in these results. Apart from the filter material, there also seems to be an influence of the vertical position of the filter. Without any doubt the pore water composition of the double band is different from the rest of the Boom Clay. With the limited amount of data and the exclusion of Na and Cl from the analysis, a distinction can be made between the pore water composition of the Terhagen member, the base of the Putte member up to S50, and the Putte member above S50. However, it should be noted that the latter needs confirmation or rejection when more data points are available. Within the SPRING 116 piezometer having 4 different stainless steel filters and positioned in the same layer, a distinction between the deepest filter and the three filters closer to the gallery was observed. Up to now it is not known what causes this difference. Some suggestions might be a relict disturbance caused by the excavation of the gallery, the effect of oxidation caused by coring not completely removed yet in the first three filters, or a lateral variation even on short distance in the same layer. Continued follow-up of this piezometer might clarify this. The EG/BS filter provided many data through time. There is no continuous or systematic evolution found in time. However, two groups of results separated by a time gap can be distinguished. It is unknown what has caused this difference. Finally, it can also be concluded that Na and Cl play a major role in discriminating several groups of pore water composition. 3.4.2 Charge balance and equilibrium state of the pore water To evaluate the quality of a chemical analysis, one may look at the accuracy of the analysis and the equilibrium state of the measured water composition. The precision of the analysis is determined by the sensitivity of the analytical technique itself and will not be discussed here. The accuracy of a water analysis is normally assessed by the degree of charge imbalance calculated from the given water composition. The principle of electroneutrality requires that the ionic species in any water sample must remain charge balanced. The degree of charge imbalance is thus a measure for the error of a water analysis: Charge Imbalance (%) = ( sum cations − sum of anions ) × 100 ( sum cations + sum anions ) where cations and anions are expressed in meq/l. It is obvious that a charge imbalance is inevitable in a chemical analysis but an acceptable charge imbalance should be less than 5 % for a good water analysis (Appelo and Postma, 1999). (3.1) 89 For the Boom Clay pore water, the major contributions of the cationic charge are from Na+, K+, Ca2+, and Mg2+. Negative charges are from the total alkalinity and other conservative anions Cl-, F-, and SO42-. The total alkalinity is the total titratable bases and is equal to the sum of equivalents of HCO3-, CO32–, hydroxide (OH–), silicate, borate, phosphate, and natural organic ligands. The charge balance equation for Boom Clay pore water can therefore be simplified to: mNa+ + mK+ + 2 mCa2+ + 2 mMg2+ = alkalinity + mCl- + mF- + 2 mSO42where m refers to the molarity of the different species. Figure 3-25 plots the charge imbalance for the MORPHEUS water collected in April, 2003. It is seen that the charge imbalance is well below 2 % suggesting that the analysis is quite accurate. Also shown in Figure 3-25 is the charge imbalance calculated considering the TIC as the total alkalinity. In the latter case, the charge imbalance is higher than using the alkalinity but still below 5 %. The use of TIC to replace the alkalinity in equation (3.2) has been practiced for calculating the charge imbalance of some previously studied Boom Clay water samples (Noynaert et al., 1998; De Cannière et al., 1994a; De Cannière et al., 1994b) and some high values of charge imbalance were found specifically for waters of high pH (9 and above). That was probably because the use of TIC to represent the alkalinity underestimated the total negative charges of the water by neglecting the contribution of CO32–. The approximation that the equivalent of TIC is equal to the total alkalinity is only valid at a pH below 8. At a higher pH range, the use of the TIC to calculate the charge imbalance needs a representative pH measurement which generally has not been available (see section pH and pCO2). A precise charge imbalance should be calculated using the total alkalinity and not the TIC. The charge imbalance was more pronounced in a lot of squeezed water samples as evidenced by Reeder, et al. (1992) and De Craen (2001). These waters were mostly severely oxidised so the possible reason for the observed charge imbalance is the lack of data for thiosulphate. Anionic deficit of 10 to 20 % has been observed in those waters. Another useful indication for evaluating the quality of the water sampling and analysis is the saturation state of the water in terms of minerals that are in contact with the water. As will be discussed in section 4 on modelling, the calcite saturation index is used widely to evaluate the equilibrium state of a given water composition. Nordstrom and Munoz (1994) pointed out that the calcite saturation index should approach to zero in almost all types of water but not surpassing it. A water analysis showing significant oversaturation of calcite normally suggests that either the sampling procedure is inadequate or the chemical analysis is not accurate. For the Boom Clay pore water compositions discussed in this report, a direct calculation of the saturation state is not possible because of the lack of relevant pH measurements. A reverse calculation has been performed therefore assuming a priori that calcite is in equilibrium with the MORPHEUS water compositions and that the resulted pH values are in a range of 8.3 to 8.6 (see section 3.1.3). This pH range is higher than the in situ pH measurements of 8 to 8.2, suggesting that the MORPHEUS waters are slightly undersaturated with calcite at the in situ measured pH values. No indication of oversaturation of calcite is observed, so the quality of water analysis should be acceptable in terms of the calcite saturation state. (3.2) 90 4.5 3.5 charge imbalance, % 2.5 1.5 0.5 -0.5 -217.1 -220.8 -222.6 -226.0 -227.2 -227.8 -229.1 -229.8 -230.3 -231.8 -233.8 -235.2 alkalinity TIC -1.5 -2.5 -3.5 -4.5 depth, m Figure 3-25: Charge imbalance calculated for MORPHEUS water compositions (collected in April, 2003). 91 4 Model simulation of pore water chemistry The present-day chemical composition of the Boom Clay pore water is the result of the early diagenesis and the subsequent water-rock interactions, mass transfers, and the mixing of groundwaters from the surroundings with the Boom Clay pore fluid. Broad interrelationships among these processes can be discerned by application of chemical thermodynamics. In addition, because of the low hydraulic conductivity, the Boom Clay pore water is practically immobile so that a chemical equilibrium is likely established between the pore water and the minerals. The system can therefore be evaluated by principles of chemical equilibrium. We present in this section equilibrium model simulations, calibrated with the detailed analysis of Boom Clay pore water as discussed in previous Sections, to produce a probable interpretation of the measured water compositions. 4.1 Equilibrium model and water-rock interaction An equilibrium model describes the distribution of chemical components in a system containing fluid, minerals, and gases at a chemical equilibrium state. The system is constrained at the initial state by known temperature, pressure, and composition. At the equilibrium, the redistribution of the involved chemical components among species in fluid, minerals, and gases is so that the value for the total Gibbs free energy of the system is minimised, that is, the lowest potential for chemical reactions. The chemical equilibrium model consists of two parts: the constraint of chemical equilibrium, and the constraint of conservation of mass. The constraint of chemical equilibrium is a set of mass action expressions in terms of equilibrium constants. The constraint of conservation of mass is a set of mass balance equations, one for each component. The combination of the mass action and the mass balance equations results in a set of nonlinear equations to which the mathematical solutions describe the final distribution of the chemical components. For the simulation on water-rock interactions of the Boom Clay, the pore water composition is calculated at the equilibrium state as the result of interactions between the pore fluid and minerals. 4.2 Computer code and thermodynamic database Most of calculations were performed using the computer code The Geochemist’s Workbench 3.2.2 (Bethke, 2001). Some calculations were done with the new GUI (graphic user interface) version of the code 4.0.2 (Bethke, 2002). In calculations where temperature corrections were needed, the latest working version of 4.0.3 was used (only available to SCK•CEN at the present time). The version 4.0.3 allows to fix the values for the ion exchange selectivity coefficients at 25°C while the solubilities of the minerals are calculated at 16°C, the temperature of in situ Boom Clay. The thermodynamic database used is the MOLDATA (04-01) (Wang, 2004), a database maintained at SCK•CEN for the study of geochemistry and migration of radionuclides in Boom Clay. MOLDATA is a database derived from the EQ3/6 database version 8 release 6 which has its roots in the SUPCRT database (Johnson et al., 1991) developed at Lawrence Livermore National Laboratories (LLNL). The database supports activity coefficients calculated by an extended form of the Debye- 92 Hückel equation (the B-dot equation). Concerning aqueous species and mineral stability constants, MOLDATA is basically identical to the LLNL database except some corrections of known errors. The only difference, relevant to this report, is that MOLDATA contains a sub-dataset for ion exchange selectivity coefficients. This ion exchange dataset is calibrated with the pore water composition of the Boom Clay and literature data on ion exchange so that the data are considered to be specific for the Boom Clay conditions. An example of input and output files from geochemical modelling simulations is given in Annex 7. 4.3 Mineral solubility and ion exchange Solubility is an important mechanism controlling the compositions of natural waters since many common minerals dissolve and precipitate readily and reach solubility equilibrium. However, it is also known that many other minerals do not readily reach solubility equilibrium in the temperature range of 0-100 °C. In a clay rich system such as the Boom Clay, which contains 60 wt% of clay minerals and which is practically free of metal oxides, ion exchange is as important as solubility in regulating the pore water composition. In the framework of the EC ARCHIMEDE-argile project (Griffault et al., 1996), two distinguished approaches have been applied for the interpretation of measured Boom Clay pore water composition. The first approach (Beaucaire et al., 2000) stressed the difficulties in defining the ion exchange complexes and used only solubility constraints to explain measured concentrations of major ions present in the Boom Clay pore water. The second approach (Sanjuan et al., 1994) applied a combined solubility and ion exchange model and claimed a better fit of the simulation to the measured data. In comparing the two basic approaches, Pearson (2001) noticed that the addition of ion exchange to the model at least reproduced equally well the observed water composition in Mont Terri project. Our scoping calculations revealed that neither pure solubility, nor ion exchange models explain the observed Boom Clay pore water composition satisfactorily. We therefore in this report explore a combined solubility and ion exchange model. Concerning solubility mechanisms, a common difficulty is to decide priorily what are the reactive minerals and if they are in chemical equilibrium with the pore water. Nordstrom and Munoz (1994) summarised a guide to which minerals are likely to reach solubility equilibrium (given sufficient time) and which ones are not likely based on mineral reactivity, kinetics, and degree of complexity of mineral stoichiometry. Following this guide we can classify the Boom Clay minerals (Table 1-2) according to their reactivity and their plausibility to reach an equilibrium with the pore fluid: • • • calcite, siderite, and pyrite are likely in chemical equilibrium with the pore water. These secondary minerals are reactive and have a simple stoichiometry. The simple stoichiometry facilitates geochemical modelling since the mineral formulas and the mass action laws are easily defined; illite, smectite, feldspars, and chlorites are reactive but unlikely reach chemical equilibrium with the Boom Clay pore water due to either the slow kinetics at low temperatures, incongruent dissolutions, or complex stoichiometries. These minerals are generally not considered suitable to be included in equilibriumbased model simulations; kaolinite might reach an equilibrium but has a complex stoichiometry; 93 • quartz is not reactive. A common mineral that is considered as being in equilibrium for almost all types of water-rock interactions is calcite (Langmuir, 1997; Nordstrom and Munoz, 1994; Pearson et al., 1978). This has also been recognised and applied in previous studies of Boom Clay (Beaucaire et al., 2000; Griffault et al., 1996). Calcite is therefore taken as the control mineral for the calcium concentration in the Boom Clay pore water through the following dissolution reaction: Calcite + H+ ⇔ Ca2+ + HCO3- (4.1) Iron has never been taken into account in previous studies of the Boom Clay, mainly because its concentration and behaviour are not supposed to influence the geochemistry of the Boom Clay to a noticeable extent. Another reason might have been that iron is a redox sensitive element and the water sampling and preservation procedure will influence the analytical results. In this report, siderite is used to constrain the iron concentration under an undisturbed in situ condition. We are aware of, however, the sensitivity of the iron concentration to the redox condition, evidenced in a batch experiment (Wang et al., 2002). Siderite dissolution likely follows two reactions depending on pH: Siderite + H+ ⇔ FeHCO3+ (4.2) Siderite ⇔ FeCO3 (aq) (4.3) where FeHCO3+ species dominate at pH less than 8.4 and FeCO3 (aq) is important at higher pH values. If only Fe(II) prevails, the concentration of the free Fe2+ in the Boom Clay pore water is about an order of magnitude lower than the two ion pair species. Pyrite is an important mineral since it is sensitive to oxidation and contributes largely to the redox buffering capacity of the Boom Clay. It is not uncommon in literature that pyrite is used to control the aqueous concentration of sulphate in a pore water. However, an accurate quantification of pyrite dissolution through reaction modelling needs an accurate redox potential measurement. As discussed in the Section of redox (Section 3.1.4), Boom Clay redox measurements have been proven elusive and technically challenging. On the other hand, due to the marine origin of Boom Clay, there is no evidence that the sulphate content of the present-day pore water is exclusively constrained by the dissolution of pyrite. We therefore use pyrite to buffer the system redox but use a sulphate concentration measured from piezometer waters as the initial condition. The pyrite-sulphate couple, i.e., FeS2/SO42- will fix the redox potential. This is more workable and sound since the sulphate analysis is more reliable than the redox measurement. The redox potential of the Boom Clay can be calculated as demonstrated in Section 3.1.4 if the system pH, alkalinity, sulphate and total iron concentrations are known: FeS2 + 8 H2O + HCO3- ⇔ 16 H+ + 2 SO42- + FeHCO3+ + 14 e- (4.4) In the Boom Clay, illite, smectites, feldspars, and chlorites are important minerals in terms of weight percentage (see Table 1-2). Although they belong to groups of reactive minerals, they unlikely reach chemical equilibrium with the pore water at low 94 temperatures so that they are not taken in this report as controlling minerals for simulating equilibrium solubilities. However, it is important to note that although the pore water of the Boom Clay may not achieve stoichiometrical solubility equilibrium with these minerals, they do exist in the Boom Clay and they do react with the pore water and affect the overall water-rock mass balances. Beaucaire, et al., (2000) considered albite and microcline (K-feldspar) as the minerals controlling Na and K concentrations and found comparable results to the measured water compositions (ARCHIMEDE-argile project). Typical dissolution reactions of these minerals as written in the LLNL database are as follows: Illite ⇔ 1.2 H+ + 0.25 Mg2+ + 0.6 K+ + 2.3 AlO2- + 3.5 SiO2(aq) + 0.4 H2O (4.5) Montmor-Na (Smectite) + 6 H+ ⇔ 0.33 Mg2+ + 0.33 Na+ + 1.67 Al3+ + 4 H2O + 4 SiO2(aq) (4.6) Albite ⇔ AlO2- + Na+ + 3 SiO2(aq) (4.7) K-Feldspar ⇔ AlO2- + K+ + 3 SiO2(aq) (4.8) Clinochlore-14A (chlorite) + 8 H+ ⇔ 2 AlO2- + 3 SiO2(aq) + 5 Mg2+ + 8 H2O (4.9) Kaolinite is also reactive but complex in stoichiometry. The relatively faster kinetics comparing to illite and feldspars makes kaolinite-pore water equilibrium possible: Kaolinite ⇔ 2 H+ + 2 AlO2- + H2O + 2 SiO2(aq) Kaolinite is commonly used as the controlling mineral for the Al concentration in natural waters. The thermodynamic stability constant of kaolinite has been a subject of lively discussions (Nordstrom and Munoz, 1994). Based on a self-consistent solubility dataset (Nordstrom et al., 1990), at 16 °C and pH of 8 to 8.5, the Al concentration controlled by the dissolution of kaolinite at the Si concentration of Boom Clay pore water (~5 mg/l) is in a range of 2 to 8 µg/kg water which is about an order of magnitude lower than the Al concentration normally measured in unfiltered Boom Clay piezometer waters. This may be explained either by the possible presence of Al colloids/particulates in water samples or the choice of thermodynamic data for kaolinite in terms of crystallinity. Aluminium is known to form colloids in natural waters. To remove colloids, a careful ultrafiltration of water samples is necessary using filters of pore size in a nanometer range. Beaucaire et al., (2000) found that the measured Al concentration in the Boom Clay pore water was generally overestimated if the water sample was not carefully filtered. They found that the ultrafiltration of a water sample at 10 nanometer (about 200,000 MWCO) resulted in a representative and truly dissolved Al concentration which was in a solubility equilibrium with kaolinite. The second possible explanation might be that the kaolinite presence in Boom Clay is less stable than the assumed crystalline kaolinite. Coudrain-Ribstein and Gouze (1993) studied the Dogger aquifer (Paris Basin) and concluded that kaolinite in equilibrium is not a well-crystallised mineral but a kind of disordered form, that is less stable and has a higher solubility. Ultrafiltration has not been generally practiced in the procedure of sampling and analysis of Boom Clay waters. Pre-filtration on water samples should be performed in future analyses to better define the Al concentration. Aluminium is not a routinely measured element in the Boom Clay water characterisation campaign. As aluminosilicates are not considered as being (4.10) 95 in equilibrium with pore water for the current simulation, the Al concentration will not influence the other major ions concentration. For the purpose of modelling when Al is to be treated, we currently use kaolinite as the solubility controlling mineral. We recommend to carry out more measurements on the Al concentration in Boom Clay pore waters in the future. Quartz consists of 40 wt% of the Boom Clay. As the second most abundant element in the Earth’s crust, Si in quartz is however rarely found in equilibrium with natural waters. This is because most of the dissolved silica observed in natural waters results originally from the chemical breakdown of silicate minerals in processes of weathering. These processes are irreversible, and the silica concentration in water is likely controlled either by kinetics of dissolution processes, surface processes like sorption, or by precipitation of secondary minerals of less organised structures. Boom Clay pore waters are generally oversaturated in terms of quartz. The Si concentration in Boom Clay pore waters is close to the solubility of chalcedony, a more soluble phase but having the same stoichiometry as quartz. We therefore use chalcedony to model the concentration of Si. Although the use of chalcedony in place of quartz is a widely applied approach in water rock interaction modelling, e.g., Beaucaire et al., (2000) and Bradbury and Baeyens (1998), the existence of chalcedony in the Boom Clay is still to be demonstrated. Besides the solubility approach, ion exchange is a well known process regulating the water composition in clays. Ion exchange is an adsorption process which removes one solute from the aqueous phase and releases another from the clay surface. Exchange reactions occur at the surfaces of clay particles where the isomorphic substitution results in a permanent surface charge. Ion exchange reactions follow a mass action law such as, for example, for a potassium-sodium exchange: >X:Na + K+ ⇔ >X:K + Na+ (4.11) with a mass action coefficient expressed as: KC (K-Na)= [> X : K ]{Na + } = N K {Na + } [> X : Na ]{K + } N Na {K + } In this equation, KC is the selectivity coefficient, the “>” sign means exchange complexes on the clay surface, that is, >X:Na and >X:K express the sodium and potassium exchange complexes on the surface of the clay. {Na+} and {K+} are the activities in the aqueous phase. Depending on molal or equivalent fractions being adopted for expressing the quantity of the exchange complexes within [ ], KC takes different forms, namely, the Gaines-Thomas, Vanselow, or Gapon convention. In this report, we use the Gaines-Thomas convention, i.e. the surface exchange complex is expressed as an equivalent fraction of the total ion exchange capacity of the clay. The activity of the exchange complex is therefore written as equivalent fractions NNa and NK, respectively, for Na and K occupancies. Limited only to metal cations, the capacity of the Boom Clay to exchange cations is the cation exchange capacity (CEC), which is an important characteristic of Boom Clay and represents the major buffering sink for the cation composition in the pore water. According to the well established ion exchange selectivity behaviour in common soils and sediments, for example, the data given by Appelo and Postma (1999), sodium (Na), potassium (K), calcium (Ca), and magnesium (Mg) are dominant cations 96 contributing to the major part of exchange complex on clays. In addition to the K-Na exchange reaction given in reaction (4.11), Ca-Na and Mg-Na exchange reactions are as follows: 2 >X:Na + Ca2+ ⇔ >X2:Ca + 2 Na+ 2+ KC (Ca-Na) = + 2 >X:Na + Mg ⇔ >X2:Mg + 2 Na KC(Mg-Na) = { } { } N Ca Na + 2 N Na Ca 2+ { } {Mg } N Mg Na + 2 N Na 2 (4.12) 2 2+ (4.13) If other cations occupancies are negligible, Na, K, Ca, and Mg should take all exchange sites so that: NNa + NK + NCa + NMg = 1 (4.14) Ion exchange is an important mechanism regulating the water composition of the present-day marine sediments, analogues to the Boom Clay. Sea water is Na+ and Cldominant and sediment in contact with sea water will have a large Na+ occupation on clay surfaces. Fresh water, commonly rich in Ca2+ and HCO3-, infiltrates into the sediment resulting in an exchange of Ca2+ onto clay surfaces and Na+ in return to the water. The water therefore changes from the Ca-HCO3 type to the present-day NaHCO3 type. Ion exchange in the Boom Clay has been studied by Baeyens (1982), Baeyens et al. (1985), and Griffault et al. (1996). The first two papers were actually based on the same experiment, so we consider them as one study. Ion exchange properties of the Boom Clay derived from these studies are summarised in Table 4-1. Parameters needed for modelling the ion exchange in the Boom Clay will be taken from Table 4-1 with some re-evaluation as given below. Table 4-1: Summary of ion exchange data determined from previous studiesa Ion exchange properties Baeyens (1982) and Griffault et al. (1996) Baeyens et al. (1985) CEC, meq/100 g clay 24 24 NNa 0.365 0.36 NK 0.16 0.096 NCa excluded 0.159 NMg 0.475 0.154 NH Not considered 0.229b KC (K-Na) 10 15 KC (Mg-Na) 3.8 Not consideredc KC (Ca-Na) Not applicable 2.1 a KC were expressed following the Gaines-Thomas convention in Baeyens (1982) and the Vanselow convention in Griffault et al. (1996) b the proton occupancy was reported by Sanjuan et al. (1994) c magnesium concentration was considered being controlled by the dissolution of dolomite, not by ion exchange 97 Both studies resulted in exactly the same CEC value of Boom Clay, i.e., 24 meq/100 g clay. Griffault et al. (1996) tried to divide experimentally the total CEC into two fractions, namely the exchange capacity from clay and from natural organic matter. Baeyens (1982) and Baeyens et al. (1985) estimated that the contribution of natural organic matter to the total CEC is about 5-10 meq/100 g clay. They did not separate the two fractions and the CEC was determined as a total value. We in this study treat the CEC as the total without making difference between inorganic and organic fractions. Separation between clay and the natural organic matter makes sense only if the two phases can be described by two distinct mechanisms attributed to two sets of model parameters for the clay and the natural organic matter. As seen from Table 4-1, both studies defined only one set of parameters for ion exchange in terms of selectivity coefficients and exchange occupancies, therefore it makes no sense to split the total CEC into two fractions. The studies of Baeyens (1982) and Baeyens et al. (1985) were characterised by the exclusion of calcium exchange, that is, no exchangeable calcium occupancy on Boom Clay surface. It was considered that calcium in the Boom Clay exists exclusively as calcite. This was explained by the fact that the carbonate contents measured in the Boom Clay matched stoichiometrically the total amount of calcium in the clay samples. To our opinion, this way of reasoning depends on the extent of errors associated with the quantification of calcite and the total calcium concentration on clay. Considering the CEC of 24 meq/100 g clay and a 0.2 equivalent fraction occupancy of calcium on the clay, the amount of exchangeable calcium expressed by Baeyens et al. (1985) as calcite is about 8 % of the total amount of the calcite mineral. This percentage is within the error range normally found in the X-ray diffraction method used to determine the quantity of calcite. Since no error range on the calcite and the total calcium determinations was given in the work of Baeyens et al. (1985), it is difficult to judge if the calcium exchange occupancy exists or not. In any case, whether or not calcium is part of the exchange complex is very important for choosing an ion exchange model for Boom Clay. Some further discussion will follow in the Section 4.4.2 of modelling results. The pH value of the water derived from the batch leaching experiment of Baeyens and co-workers is 9.2 to 9.3 (Baeyens et al., 1985, Table II), which is higher than the pH of the Boom Clay as estimated in Section 3.1.3. This is probably because the experiments were performed in a glove box under inert atmosphere to prevent the clay from oxidation. Cautions were apparently taken to maintain the in situ redox condition of the Boom Clay but the importance of keeping a correct CO2 (g) partial pressure was not realised. Glove boxes filled with (an) inert gas(es) contain practically no CO2(g), and the clay suspension in equilibrium with (or partly in equilibrium with) the gas phase likely experienced severe degassing in terms of CO2(g). The pH will increase when the system was degassed, i.e., decrease in partial pressure of CO2(g), following the reaction: CO2 (g) + H2O ⇔ H+ + HCO3- (4.15) An increase in pH will change the system chemistry in terms of calcite dissolution, carbonate to bicarbonate ratio, and ion exchange reactions. A higher pH due to the degassing of CO2(g) leads to a decrease in calcite solubility, i.e., lower calcium concentration than it would be under the in situ partial pressure of CO2(g). If cation exchange between calcium and sodium occurred, i.e., not excluding Ca from the 98 exchange complex as concluded by Baeyens et al. (1985), the concentrations of other exchangeable cations in the aqueous phase, i.e., Na, K, and Mg, will also decrease accordingly. Under the conditions of the Baeyens experiment, i.e., pH of 9.2 to 9.3, the calcium concentration controlled by the calcite dissolution is 3 × 10-5 molal, which is identical to the value measured by Baeyens et al. (1985). This is an indication that the batch water composition reported by Baeyens et al. (1985) was likely extracted at a relative higher pH than the expected value for in situ Boom Clay. It must be noted however that the effect of dilution must also play a role since the water was extracted at a solid to liquid ratio of 1:1 g/ml. However, as demonstrated by Henrion et al. (1985) in the same project, the influence of CO2(g) degassing dominated over the dilution effect. In the ARCHIMEDE-argile project (Griffault et al., 1996; Sanjuan et al., 1994) the ion exchange composition was interpreted differently comparing to that of Baeyens (Table 4-1). The fact that about 0.2 (NH) equivalent fraction of cation occupancy was attributed to protons was remarkable. Since no explanations were given in neither of the two papers regarding why such high proton occupancy was derived, we assume that the NH value was probably resulting from the difference between the total CEC and the sum of individual cation occupancies measured by selective extraction experiments. It has been noticed from the Table 18 in Griffault et al. (1996) that: (1) the sum of the cation exchange occupancies of Na, K, Ca, and Mg is about 0.8, i.e., 0.2 in deficit; and (2) no proton occupancy was reported. It is not uncommon that the sum of extracted cations turns out being less than the total CEC in ion exchange experiments. Bradbury and Baeyens (1998) found the deficit of about 25% in Opalinus clay, which is comparable to the 23 % being noticed in the ARCHIMEDE-argile project for Boom Clay. However, Bradbury and Baeyens (1998) explained that the difference is most likely due to the uptake of the index cation by the amphoteric functional groups existing at the edge sites of the clay, in other words, the total CEC measurements tend to overestimate the true exchange capacity. Amphoteric functional groups on clay behave similarly to the hydroxyl groups on oxi- hydroxides and react with solutes in aqueous phase through surface complexation reactions (Dzombak and Morel, 1990). These reactions occur on the variable charge sites and are different from ion exchange reactions taking place on the permanent charge sites and must be therefore treated differently. We follow the notion of Bradbury and Baeyens (1998) that the sum of the extracted cations is a better measure for the total CEC than the global CEC measurement using an index cation. The CEC measured in the ARCHIMEDE-argile project should therefore be 18.5 meq/100 g clay instead of 24 meq/100 g clay. In addition to the above given argument, a high proton exchange occupancy is unlikely due to the low selectivity of proton to clay surfaces. Bradbury and Baeyens (1998) pointed out that the Na-H selectivity coefficient is about unity on clays. At a pH value of 8-9 for the Boom Clay, the proton occupancy is not expected to be significant. In this report, we do not consider proton exchange. We do not, at this stage, consider amphoteric groups on Boom Clay since no data available for characterising the edge sites. As a result, we consider the CEC derived from the ARCHIMEDE-argile project as being 18.5 meq/100 g clay, and comprising of Na, K, Ca, and Mg as exchangeable cations. Another concern about the model used in the ARCHIMEDE-argile project is the use of dolomite to control the magnesium concentration in the pore water. First of all, the 99 question is still open if dolomite exist or not in Boom Clay (see Section 3.1.3). Secondly, since the measured Ca/Mg ratio in Boom Clay pore waters is relatively constant, ion exchange approach will also explain the measured Ca and Mg concentrations. In this report, we use ion exchange to estimate the magnesium concentration, assuming that dolomite does not exist. It is important to remember that mineral dissolution mechanisms, including solubility of dolomite (either as pure dolomite phase or as a proxy of magnesium rich calcite), illite, and chlorite, may also contribute to the total concentration of magnesium measured in the pore water especially under severely perturbed conditions. 4.4 Equilibrium model for the simulation of the pore water composition of Boom Clay The equilibrium model is based on a system containing 1 liter of water in contact with 5 kg dry clay, that is, a water content of about 17 wt%, as found in clay samples collected in the MORPHEUS drilling (calculated as weight of the water / weight of the wet clay; De Craen et al., 2004b). The amount of minerals are roughly taken as the weight percentage of minerals given in Table 1-2. Except in the cases of severe perturbations, for example, pyrite oxidation or alkaline plume - in which large amount of minerals will dissolve - the amount of minerals present in an equilibrium model simulation is not important since the concentration of dissolved species is much smaller than the total quantity of minerals present. The individual equivalent fraction of adsorbed cations will be discussed and derived from the following Section. According to the arguments given in the previous Section, we can summarise a general model to be used for simulations (Table 4-2). The model is a set of constraints, each for one element of interest in Boom Clay pore water, that regulate the water composition. Table 4-2: Models constraints applied in the simulation of the Boom Clay pore water composition Element or variables Constraints Na ion exchange K ion exchange Ca Calcite Mg ion exchange Al Kaolinite Fe Siderite Si Chalcedony SO42Fixed initial con. (imposed)1 2 C pCO2 (imposed) pH balancing charge Eh Pyrite 1 2 initial value 2.31 mg/l, the average value found in one of the statistic groups from MORPHEUS water compositions (Section 3.4.1); in the range of 10-2.8 to 10-2.2. 100 4.4.1 Mineral stability constants and ion exchange parameters The stability constants used in solubility calculations are listed in Table 4-3. All values are given for a temperature of 16°C, which is the in situ temperature of the Boom Clay. Stability constants at a specific temperature are interpolated using a polynomial fit at a temperature span of 0-300°C. Table 4-3: Minerals considered in the model simulation and their dissolution constants minerals Chalcedony Kaolinite Calcite reactions Chalcedony = SiO2 (aq) -3.9397 + 3+ Kaolinite + 6 H = 2 SiO2(aq) + 2 Al + 5 H2O + 2+ - 8.2205 - 1.9825 2+ -0.0224 Calcite + H = Ca + HCO3 + logK (16°C) Siderite Siderite + H = HCO3 + Fe Pyrite Pyrite + H2O + 3.5 O2(aq) = 2 H+ + Fe2+ + 2 SO42- 225.0954 As discussed in Section 3 and shown in Table 4-1, both existing ion exchange models have some limitations. Because we have not performed ion exchange experiments for the purpose of this report, to apply ion exchange mechanisms in modelling, we need to derive ion exchange parameters based on the two existing studies with some modifications. The model of Baeyens et al. (1985) does not involve calcium exchange. Our scoping calculations revealed that, without the calcium exchange mechanism, the generally observed variation of major cations cannot be explained. This can be elucidated as follows: • The calcium concentration is controlled by the solubility of calcite; • The pH/pCO2 variation is the only system variable imposed by still unknown mechanisms (see discussion in the Section 3.1.3); • Variations in concentration of sodium, potassium, and magnesium are regulated by ion exchange and directly linked with the change in calcium concentration, which in turn, is controlled by the pH/pCO2 dependent calcite dissolution. Based on the above reasoning, the dissolved calcium ion must be exchanged with other cations present on the Boom Clay surface to have any impact on variations of major cation concentrations (unless ion exchange does not play any role, which is highly unlikely). We therefore in this report do not use ion exchange data from Baeyens et al. (1985) and do consider the calcium ion exchange reaction. As one of the most important model assumptions, the existence or the non-existence of calcium exchange complex on Boom Clay should be demonstrated by future experiments. 101 The ion exchange data collected in the ARCHIMEDE-argile project (Griffault et al., 1996) demonstrated the presence of calcium exchange complex on the Boom Clay (Table 4-1). Except the questionable high proton occupancy claimed in the model interpretation (Sanjuan et al., 1994), we will use the ion exchange data derived from the ARCHIMEDE-argile project as follows: • We consider the CEC value is equal to the sum of the extracted cation concentrations, i.e., CEC = [Na] + [K] + [Ca] + [Mg], where [] is the concentration in equivalents used for expressing cation occupancies; • According to the data given in Table 18 in Griffault et al. (1996), CEC = 8.7 + 2.3 + 3.8 + 3.7 = 18.5 meq/100 g clay; The individual cation occupancies are: NNa = 8.7/18.5 = 0.47; NK = 2.3/18.5 = 0.12 NCa = 3.8/18.5 = 0.2 NMg = 3.7/18.5 = 0.2 With known cation occupancies and using the mass action equations (4.11) to (4.13), ion exchange selectivity coefficients can be calculated if the cation activity ratios in the aqueous phase are known. To calibrate the ion exchange model, we will use the real pore water composition derived from the collected piezometers to calculate the selectivity coefficients. Boom Clay pore water is basically a low ionic strength NaHCO3 water of 15 mmolal. The total pool of cations in the aqueous phase represents therefore only 1 % of the total CEC. This means that the ion exchange complex on the Boom Clay is large enough so that the composition of the exchanging surface remains invariant. In other words, the equivalent fractions (N terms) in equations (4.11) to (4.13) are constant values. Taking KC, i.e., the selectivity coefficient as a constant under certain chemical conditions, the activity ratio of exchangeable cations in the aqueous phase should be invariant. This implies that, if ion exchange is the dominant mechanism, the activity ratio of exchangeable cations in the aqueous phase should be a constant value. Figure 4-1 plots the measured concentration ratios and the calculated activity ratios of major cations from 5 statistic groups of 44 MORPHEUS water samples. Although the measured concentration ratio is different from the free cation activity ratio expressed in the ion exchange reactions (equation 4.11 to 4.13), it gives a good indication as if ion exchange mechanism is important or not. Figure 4-1 indicates that the K/Na activity ratio is constant suggesting that it is fixed by ion exchange between the two cations. The Mg/Na2 activity ratios are more scattered but have a mean value around 0.19. The Ca/Na2 activity ratio varies in a range of 0.1 to 0.14. This variation, although small, cannot be explained at the present time. One aspect can be considered in the future is to carry out more careful sample preservation and handling. Calcium is susceptible to precipitation when water samples were brought to a surface laboratory because of CO2 degassing and the consequent pH rise. On site filtration and immediate acidification of water samples to pH < 2 can overcome the precipitation of calcite. 102 cation concentration/activity ratio 0.35 0.3 0.25 con. Mg/Na^2 act. Mg/Na^2 con. Ca/Na^2 act. Ca/Na^2 con. K/Na act. K/Na 0.2 0.15 0.1 0.05 0 1 2 3 4 5 statistic groups Figure 4-1: Measured concentration ratios (closed symbols) and calculated activity ratios (open symbols) of major cations in 5 statistic groups of 44 MORPHEUS water samples. For a modelling purpose at the present stage, the activity ratio of Ca/Na2 is taken as the average value of the 5 statistic groups, i.e., around 0.13. Activity ratios derived from the MORPHEUS water compositions for the purpose of ion exchange model calibration are: K+/Na+ = 0.012; Mg2+/(Na+)2 = 0.19; Ca2+/(Na+)2 = 0.13. With the known cation occupancies and free cation ratios, selectivity coefficients for the ion exchange on Boom Clay can be readily calculated using equations (4.11) to (4.13). The results are as follows: KC (K-Na) = 21.28; KC (Mg-Na) = 4.76; KC (Ca-Na) = 6.96 The value of selectivity coefficient for potassium KC (K-Na) is higher than literature value generally reported for common soils and sediments, e.g., KC (K-Na) = 4-7 (Appelo and Postma, 1999). The higher KC (K-Na) calibrated by MORPHEUS water compositions suggests that Boom Clay has a higher selectivity for potassium. This may be due to the high content of illite clay which is known for its high potassium selectivity. Thellier and Sposito (1988) reported a KC (K-Na) value of 15 for Silver Hill illite, the value was taken by the ARCHIMEDE-argile project to explain the composition of Boom Clay pore water. Selectivity coefficients for magnesium and calcium calibrated for MORPHEUS water compositions are in good agreement with literature values compiled by Appelo and Postma (1999), e.g., KC (Mg-Na) = 4 and KC (Ca-Na) = 6. 103 It is important to note that when the ion exchange pool in the pore water is small compared to the ion exchange complex on the clay surface, one can readily calculate major cation concentrations by fixing the cation ratios without knowing the exact composition of the ion exchange complex on the clay surface. This means that we do not really need the values of cation occupancies and selectivity coefficients to calculate the major cation concentrations. Those open questions concerning the cation exchange compositions as discussed in previous Sections therefore do not influence our present calculations as far as non-disturbed pore water composition is to be calculated. To make the model intrinsic and especially to apply the model to conditions of severe perturbations where the dissolved cation concentration is not small comparing to the total CEC, we do need the knowledge about the exchange complex and selectivity coefficients of Boom Clay. The cation exchange composition and the selectivity coefficients derived in this Section are reference model parameters calibrated on the most extensive set of water compositions, and are therefore considered as plausible interpretations for Boom Clay pore water chemistry. 4.4.2 Results of model simulations and discussions With the model parameters derived from the previous Section, the Boom Clay pore water compositions can be calculated using the constraints given in Table 4-2. Figure 4-2 shows the variations in concentration of the major cations that are controlled by calcite dissolution (Ca) and ion exchange reactions (Mg, K, Na). Major cation concentration, mM 0.25 0.2 0.15 K Mg Ca 0.1 0.05 0 320 340 360 380 400 420 440 [Na], mg/kg H2O Figure 4-2: Variations in concentration of the major cations in 44 water samples sampled from the MORPHEUS piezometer. Dots are measured data and lines are calculated values. The system pH varies in the range of 8.3 to 8.6 in equilibrium with calcite (16°C). 104 From the modelling result of Figure 4-2, it is seen that the combined calcite dissolution and ion exchange model represents the measured data well. Although plotted as the concentration of major cations versus sodium concentration, the system variable in the calculation was pCO2. As shown in Figure 3-4, the equilibrium pH of MORPHEUS waters was estimated in the range of 8.3 to 8.6, which corresponds to pCO2 of 10-2.3 to 10-2.8 atm. This variation in pCO2 resulted in a variation in calcium concentration controlled by calcite dissolution. The calcium in turn exchanges with Boom Clay to release Na, K, and Mg into aqueous phase, hence regulating the major cation concentrations in the Boom Clay pore water. Other cation concentrations, i.e., Fe, Si, and Al, that are governed by the solubility of minerals, are plotted in Figure 4-3. The iron concentration calculated based on the siderite solubility agrees generally with the measured data, except that few data points at the high end of the sodium concentration. These points are data collected at the double band from where higher concentrations of ions are in general observed (F8, MORPHEUS piezometer). Some batch experiments have suggested the presence of Fe-containing colloids. Although colloids may or may not be present in the piezometer waters, future experiments with the determination of the Fe concentration using careful filtration is essential. Silicon concentrations are invariant and the measured concentrations are in agreement with the calculated solubility of chalcedony. 1.0E+00 cation concentration, mM 1.0E-01 1.0E-02 Si Fe Al Al model 1.0E-03 1.0E-04 1.0E-05 1.0E-06 320 340 360 380 400 420 440 460 [Na], mg/kg H2O Figure 4-3: Variations in concentration of Fe, Si, Al in 44 water samples collected from the MORPHEUS piezometer. Dots are measured data and lines are calculated values. The system pH varies in the range of 8.3 to 8.6 in equilibrium with calcite (16°C). 105 4.5 Concluding remarks • Although statistically very different, the observed variations found in the MORPHEUS waters can be generally explained by principles of chemical equilibrium. • pH and pCO2 values are critical in choosing the model. More field measurements of pH and pCO2 are needed to better constrain the model. • A combined ion exchange and solubility model explains well the observed water compositions. • The calcium concentration is controlled by the calcite solubility, provided that the system pH or pCO2 is imposed. • The concentrations of Na, K, and Mg can be satisfactorily simulated by ion exchange reactions. • The measured iron concentrations agree, for most of data points, with the calculated values based on the solubility of siderite. However, the iron concentration determined piezometer waters without filtration may involve colloids as suggested by the batch leaching experiments (see Annex 4). Future iron determination with careful filtration is needed to test the siderite control mechanism. • The silicon concentration can be explained by the solubility of chalcedony. • A slight variation in composition of the Boom Clay pore water is generally observed and attributed to the change of pCO2 (g). However, the cause of the pCO2 (g) variation is not clear at the moment. It is therefore difficult to assign a single composition representing the Boom Clay pore water. At present, the pore water composition is therefore best represented by a reference model which can be used to calculate the specific water composition under the given chemical condition. 4.6 Future work needed to improve the model The model presented in this report is based on pH and pCO2 values in equilibrium with calcite. As the most important model assumption, the calcite equilibrium state should be experimentally demonstrated. Although a lot of high quality water samples became available recent years, the lack of field pH and/or pCO2 measurements hampered the evaluation of the equilibrium state of these waters. It is desirable to have more accurate and an increased amount of field pH/ pCO2 measurements. It is also recommended that pH, pCO2, and the water composition should be determined at the same time for the same sample to better define the system and to evaluate the system equilibrium state unambiguously. The need of well-thought procedures for the sampling, handling and preservation of the water samples should be emphasised. On site acidification of water is necessary before analysis of metal ion concentrations. The aluminium concentration in the pore water should be re-evaluated with the support of careful pre-filtration or ultrafiltration procedures. 106 The ion exchange complex should be re-measured to confront with the existing data. The most important issue is whether or not the calcium exchange complex exists in the Boom Clay. Finally, the current model uses an imposed pH and pCO2 as the system variable without knowing the mechanisms controlling the observed variation of pH or pCO2. First of all, more efforts should be made to identify possible artefacts associated with the piezometer water sampling. It is desirable to be able to quantify the extent and the rate of degassing, the degree of bacterial activities in water samples, and the extent of Boom Clay oxidation due directly to the installation of a piezometer. Secondly, the intrinsic cause of the pCO2 variation can be studied through the planned heater test, i.e., the effect of a temperature increase on the variation of the pCO2 and the water composition. The effect of temperature on the Boom Clay pCO2 can also be studied by a surface laboratory experiment. A pCO2 measurement can be performed on a well preserved clay core with the ‘out-gassing’ technique but under different temperatures. 107 5 Reference Boom Clay pore water composition at the Mol site As described in the statistical analysis of available data (Section 3.4.1), many factors can influence the measured Boom Clay pore water composition. A major factor is the spatial variability. In that sense, it is not possible to define one single mean of the Boom Clay pore water composition at the Mol site, as: • • For some filter materials a straightforward chemical perturbation is noticed (e.g. B and Si increase with glass filter), but other elementary differences between filter materials are unexplained. A natural spatial variation is present, and including all available data does not provide a Gaussian distribution of measured concentrations for all elements (for some elements a bimodal distribution is noticed, while for others a normal distribution is present). However, as a basic input for geochemical modelling and performance assessment, a reference Boom Clay pore water composition is needed. As a mean composition cannot be derived statistically from the measurements, we propose to use a reference Boom Clay pore water composition as given in Table 5-1. This reference composition is modelled taking into account the current knowledge of Boom Clay mineralogy and calibrated towards the MORPHEUS water samples available. From the statistical analysis performed, it can be concluded that the MORPHEUS piezometer is a good choice to perform this calibration. First of all, a quite extensive and complete dataset is available. Secondly, no major disturbance of the borehole or surrounding host rock is observed (in contrast with EG/BS). Thirdly, the Schumatherm filters do not have a major effect on the analysed pore water composition. Only the measured Mg content can differ somewhat from the PE, glass and Carbo filters. A comparison with stainless steel filters is less straightforward due to the statistically differentiated groups in the SPRING 116 piezometer. Fourthly, the extent of the influenced zone due to pore water sampling is smallest for the MORPHEUS piezometer, compared to the EG/BS and SPRING 116 piezometers. The ORPHEUS piezometer has an even smaller influenced zone, but due to in-situ pH-Eh measurements, the system might be more disturbed (leak of KCl, and possibly also glycerine and acrylate from electrodes, pers. comm. from N. Bogaerts, Elscolab). Finally, the MORPHEUS piezometer comprises at least some of the spatial variation noticed around the HADES URF as indicated by the variation of measured concentrations (also shown in Table 5-1). For laboratory experiments, a synthetic Boom Clay pore water composition is also needed to mimic the real Boom Clay water. Within the framework of Belgian R&D for radioactive waste disposal in Boom Clay, two names of synthetic water have appeared in literatures, namely SIC and SCW, standing for synthetic interstitial clay water and synthetic clay water, respectively. A subdivision of SIC was also made between the waters with and without humic acid (Lemmens, 2001), i.e., SIC (with humic acid) and SICZH (without humic acid). Maes (2000) also reported a modified SCW in comparison to the water used by Dierckx et al., (2000). Although rather confusing in terminology, these synthetic waters have basically two compositions as given in Table 5-2. 108 Table 5-1: The reference Boom Clay pore water composition and the measured MORPHEUS water composition. The major ion concentrations of the reference water are calculated by cation exchange and mineral dissolution reactions that are calibrated against the measured MORPHEUS water compositions. composition reference MORPHEUS water* water -217~ -235 m TAW mg/l mmol/l mg/l mmol/l Na K Ca Mg Fe Si Al 359 7.2 2.0 1.6 0.2 3.4 0.6E-3 15.6 0.2 0.05 0.06 0.003 0.1 2.4E-5 348-431 6.7-8.3 1.5-2.9 1.3-2.6 0.10-0.68 4.2-5.5 0.03-0.06 15.1-18.7 0.17-0.21 0.04-0.07 0.05-0.11 0.002-0.012 0.1-0.2 1.1-2.2E-3 HCO3TIC (mg C/l) alkalinity (meq/l) Cl total S SO42HPO42NO3F Br B 878.9 181.3 15.12 26 0.77 2.2 14.4 15.1 173-206 14.9 24-30 na 0.63-2.31 na§ ~0.4 2.6-3.3 ~0.6 ~7 14.4-17.2 DOC (mg C/l) Cs Sr U pH pCO2 (atm) Eh (mV) temperature (°C) conductivity (µS.cm-1) ionic strength * 0.7 0.02 0.02 0.7-0.8 6.5E-3-0.02 6.4E-3 0.13-0.17 7.5E-3 0.6 120-200 8.5 10-2.62 -274 16 0.016# <0.5 (µg/l) 46-90 (µg/l) 0.3-1.2 (µg/l) na na na ~16 1700 <4E-9 (M) 5-10 E-7 (M) 1-5 E-9 (M) the range observed in 5 statistic groups of 44 measurements. na: not analysed. # calculated by the B-dot method (Helgeson and Kirkham, 1974; Helgeson, 1969). § not measured in MORPHEUS waters but in general below the detection limit of 0.5 mg/l. 109 It is apparent from Table 5-2 that SIC and SCW are similar in major components and the difference between them will unlikely have any impact on experimental results. From Table 5-1 and the detailed interpretation given in this report, an undisturbed Boom Clay pore water is basically a solution of 15 mM NaHCO3, rich in organic matter. It is therefore our opinion that a synthetic Boom Clay pore water made of 15 mM NaHCO3 will suffice to perform most of the laboratory experiments. It will be the decision of experimentalists if additional components should be added according to the objective of the experiment. In case a more detailed water composition is needed, Table 5-1 provides the reference. Table 5-2: Compositions of synthetic interstitial clay water (SIC) and synthetic clay water (SCW) used in the laboratory experiments*. composition elemental reference composition composition mg/l SIC SCW mg/l NaHCO3 NaCl MgSO4 Na2SO4 KCl NaF MgCl2•6H20 H3BO3 FeCl2 1250 44 12 1.5 20 8 1170 10 Na K Ca§ Mg Fe B humic acid 150 * 0.3 25 11 22 43 3 TIC (mg HCO3-) Cl SO42F SIC SCW 363 330 10.5 13 saturated saturated 2.4 2.6 1.3 7.5 909 33.7 10.6 3.6 848 27.3 0.2 4.9 359 7.2 2.0 1.6 0.2 878.9 26 2.2 - detailed procedures for preparation of SIC and SCW are given in Lemmens (2001) and Maes (2000) respectively. See also Annex 8. § saturated with calcite by equilibrating synthetic water with calcite. 110 111 6 Conclusions In the frame of the methodological studies on the Boom Clay as the reference host rock for the geological disposal of radioactive waste in Belgium, a good understanding of the Boom Clay pore water composition is necessary. Indeed, in the laboratory, representative Boom Clay pore water is used in the experiments. Furthermore, the Boom Clay pore water composition is a basic input for geochemical modelling and performance assessment calculations. It is therefore important to carefully know the Boom Clay pore water composition. In this report, different technique(s) to obtain representative pore water samples were evaluated. The Boom Clay pore water was sampled, chemical analyses were performed and geochemical parameters such as the pH, Eh, pCO2, and the alkalinity were studied. This was done in order to determine the variation of the pore water composition in the Boom Clay, to present a coherent geochemical model for explaining the origin of the Boom Clay pore water composition, and to propose a reference pore water composition to be used in the laboratory experiments and for speciation calculations and assessments of perturbation of the Boom Clay. Evaluation of the different pore water extraction techniques Pore water extraction from the Boom Clay is done by either in situ or laboratory techniques, all carried out at SCK•CEN. In situ pore water extraction is realised by using various types of piezometers, which are placed in different directions and at different depths into the clay. In the laboratory, the pore water is extracted from wellpreserved clay cores either by mechanical squeezing or leaching. Great care should be taken during the drilling of the borehole, and the anaerobic preservation and handling of the clay cores, to prevent oxygen perturbation. At the present time, piezometer water is considered to be the most representative for the in situ pore water. This is because piezometer waters experience minimum laboratory manipulations and therefore suffer minimum artefacts. Squeezed pore water is comparable to piezometer-derived water when considering the major ionic composition, but not for trace elements and organic matter. Squeezed pore water samples can thus be considered as representative for the in situ conditions, up to a certain degree. Comparing to the piezometer and squeezing techniques, batch leaching experiments provide comparable results for the major cation composition if the samples are carefully filtered. Due to the electrostatic properties of the Boom Clay, i.e., double layer phenomena, the leaching waters reveal a very different anion composition compared to the waters extracted from compacted clay using piezometers and squeezing techniques. This aspect deserves a more detailed study in the future. Chemical composition of the Boom Clay pore water The Boom Clay pore water is basically a NaHCO3 solution of 15 mM, containing an important amount of dissolved organic matter (about 115±15 mg C / l). The observed major cation concentrations can be explained by cation exchange and mineral dissolution/precipitation mechanisms. The current geochemical model assumes the equilibrium of calcite, siderite, pyrite, and chalcedony, and the cation exchange between Ca, Na, K, and Mg. 112 pH, pCO2, and alkalinity of the Boom Clay As the most important parameter, the Boom Clay pH and its controlling mechanisms are still not conclusive. Apart from the well known in situ ARCHIMEDE-argile measurement (pH 8.2; Beaucaire et al., 2000; Griffault et al., 1996), the new in situ measurement (pH 8.0; Moors et al., 2002) suffered from the leak of KCl from the electrode and demonstrated the technical difficulties in pursuing a good quality pH measurement. An alternative way to acquire the pH of the Boom Clay is through speciation calculations. This model simulation, using water compositions from the MORPHEUS piezometer, suggests a pH range of 8.3 to 8.6. Both pH and pCO2 measurements are technically challenging. This is because the Boom Clay is susceptible to the loss of CO2 (g) if in contact with air. Although attempts have been made to measure or calculate the pCO2 of Boom Clay, the real mechanism governing the parameters is not understood. On the one hand, current activities for a more accurate and representative pCO2 measurement should be continued, on the other hand future studies to understand the mechanism of the CO2 evolution in Boom Clay should be pursued. Until now, the calculated pCO2 value in the range of 10-2.4 to 10-2.8 atm is used. The total alkalinity of Boom Clay pore water is 12.1 mmol/l and agrees very well with the total inorganic carbon content suggesting that other solutes, including natural organic matter, do not contribute significantly to the total alkalinity. Redox processes and redox potential of the Boom Clay The maximum redox potential Eh is about -270 mV; probably controlled by the equilibrium of pyrite and siderite under the in situ geochemical conditions. A lower redox potential is possible as the result of interactions involving natural organic matter mediated by biochemical processes. Spatial variability The statistical analysis of the available data at the Mol site (to about 40 m around the HADES URF) has shown that a vertical spatial variability (perpendicular to the bedding) is present within the Boom Clay pore water composition. This vertical variability shows no gradient, and is mostly influenced by the elements Na, Mg, Ca and Cl. The mechanism behind these variations in major cations is explained by cation exchange and calcite dissolution/precipitation. The ultimate cause of these chemical reactions is assumed to be due to the spatial variability in pCO2 and pH, although the reason of this is not yet understood. Nevertheless, if the assumed pCO2 variation exists, the pore water seemed to respond rapidly to reach a chemical equilibrium with the clay. Because transport in the Boom Clay is diffusion-controlled, the spatial variability in the pore water composition can still be present, even on small scales. An important horizontal variability in the pore water composition is present at a regional scale, where a seawater contribution is obvious towards the west (NaHCO3 dominated versus NaHCO3-NaCl mixed waters). 113 Model Simulation of the Boom Clay pore water chemistry Although statistically very different, the observed spatial variations in pore water composition can be explained by principles of chemical equilibrium and cation exchange. The current geochemical model assumes the equilibrium of calcite, siderite, pyrite, and chalcedony, and the cation exchange between Ca, Na, K, and Mg. The model presented in this report is based on pH and pCO2 values in equilibrium with calcite. The lack of accurate field pH and/or pCO2 measurements hampered the evaluation of the equilibrium state of these waters. Furthermore, as the most important model assumption, the calcite equilibrium state should be experimentally demonstrated. In order to test the calibrated geochemical model in a wider range of conditions, some recommendations for further study are formulated (see Section 7). Reference Boom Clay pore water composition at the Mol site Due to the spatial variability, one single mean Boom Clay pore water composition at the Mol site cannot be given. However, a modelled reference composition, taking into account the current knowledge of Boom Clay mineralogy and calibrated towards a dataset including spatial variability, is provided. 114 115 7 Recommendations A systematic follow-up of the pore water composition, with a systematic set of analyses and a centralised data-base is necessary in order to: • • • • study the variability of the pore water composition (spatial variability); follow the evolution of the perturbations (variability with time); improve the statistical data set; calibrate the geochemical model in a wider range of conditions. Well-though procedures for the sampling, handling and preservation of the pore water samples should be emphasised. Regarding to the chemical analyses, appropriate on site filtration should be practiced to remove possible particulates and colloids. Pre-acidification of sample to a pH < 2 is required to preserve the concentrations of the major cations. This is especially important for cations that precipitate easily such as Ca and Fe. Sample preservation at low temperature and/or in the presence of bactericide like thymol is needed to restrict bacterial activity. Aluminium and total sulphide (HS- and H2S) should be analysed. More accurate in situ pH measurements are needed. Hence, the technique for in situ pH measurement should be evaluated. The currently used closed-circuit technique encountered several important problems, such as the electrode drift and the leakage of KCl of the electrode into the pore water. In the future, the electrode drift should be calibrated. Also the influence of the KCl leakage on the pore water chemistry and the possibly associated pH drop should be studied in more detail. In order to test the calibrated geochemical model in a wider range of conditions, the following aspects should be further studied: • Although a lot of high quality water samples became available recent years, the lack of field pH and/or pCO2 measurements hampered the evaluation of the equilibrium state of these waters. It is necessary to have more accurate (see above) and an increased amount of field pH/ pCO2 measurements. It is also recommended that pH, pCO2, and the water composition should be determined at the same time for the same sample to better define the system and to evaluate the system equilibrium state unambiguously; • In addition to the pCO2 measurements, the CO2 sources and controlling mechanism should be studied; • Further mineralogical analysis is needed to reveal if dolomite exists or not. This is directly related to the way how magnesium containing minerals should be modelled; • Cation exchange experiments should be performed to indicate if a calcium exchange complex exists or not on the surface of the Boom Clay; • The nature and the extent of biochemical processes (as an artefact in the surroundings of the piezometer) should be evaluated to better scope the effect of sulphate reduction, variation on pCO2 and pH, organic matter oxidation, methane formation… 116 117 8 Acknowledgements This work would not have been possible without the technical support of Marc Van Gompel, Louis Van Ravestyn, Frank Vandervoort and Tom Maes. We are very greatful to them. We also thank Norbert Maes, Isabelle Wemaere and Christelle Cachoir for reviewing particular sections of this report. Geert Volckaert, Ann Dierckx, Elie Valcke and Pierre De Cannière are thanked for the review of the entire document and the many fruitful discussions concerning the Boom Clay pore water chemistry. Finally, NIRAS/ONDRAF is thanked for the financial support. 118 119 9 References Appelo, C. A. J. (1977) Chemistry of water extracted from compacting clay layers: A model based on Donnan equilibrium, Chemical Geology, 19, 91-98. Appelo, C. A. J. and Postma, D. (1999) Geochemistry, groundwater and pollution, A. A. Balkema, Rotterdam. Atkins, P. W. (1994) Physical chemistry, Oxford University Press, Oxford, Melbourne, Tokyo. Baeyens, B. (1982) Strontium, cesium and europium retention in Boom Clay: A potential repository site for nuclear waste, PhD thesis, Faculteit Der Landbouwwetenschappen, K. U. Leuven, September 1982, Belgium. Baeyens, B., Maes, A., Cremers, A., and Henrion, P. N. (1985a) Aging effects in Boom Clay, Radioactive Waste Management and the Nuclear Fuel Cycle, 6, 409423. Baeyens, B., Maes, A., Cremers, A., and Henrion, P. N. (1985b) In situ physicochemical characterization of Boom Clay, Radioactive Waste Management and the Nuclear Fuel Cycle, 6, 391-408. Baldi, G., Hueckel, T., Peano, A. and Pellegrini (1990) Further check and development of the thermo-hydro-geomechanical model for Boom Clay and clay based buffer material with an emphasis on thermo-mechanics of water solid system. Final report. Bergamo: ISMES, WAS 380.83.7.I(S). Ball, J. W. and Nordstrom, D. K. (1991) User's manual for WATEQ4F, with revised thermodynamic data base and test cases fro calculating speciation of major, trace, and redox elements in natural waters, U.S. Geol. Surv. Open-file Rep. 91-183: 189 pp. Beaucaire, C., Pitsch, H., Toulhoat, P., Motellier, S. and Louvat, D. (2000) Regional fluid characterisation and modelling of water-rock equilibria in the Boom clay Formation and in the Rupelian aquifer at Mol, Belgium. Applied Geochemistry, 15, 667-686. Beaufays, R., Bloomaert, W., Bronders, P., De Cannière, P., Del Marmol, P., Henrion, P., Monsecour, M., Patyn, J. and Put, M. (1994) Characterisation of the Boom Clay and its multilayered hydrogeological environment. Final report, Project EUR 14961 EN. Beaufays, R., De Cannière, P., Fonteyne, A., Labat, S., Meynendonckx, L., Noynaert, L., Volckaert, G., Bruggeman, A., Lambrechts, M., Vandervoort, F., (1994) Cerberus – A demonstration test to study the near-field effects of an HLW canister in an argillaceous formation. Activity report 1990-92, Contract No FI2W/0003, Contractor Ondraf/NIRAS, EUR 15718. Bethke, C. M. (2001) The Geochemist's Workbench, release 3.2, A User's Guide to Rxn, Act2, Tact, React, and Gtplot, Hydrogeology Program University of Illinois. Bouchet, A. (2003) Mineralogical study of three cores of Boom Clay subjected to percolation and through-diffusion experiment (Preliminary report), ERM 03 132 AB 220. 120 Bradbury, M. H. and Baeyens, B. (1998) A physicochemical characterisation and geochemical modelling approach for determining pore water chemistries in argillaceous rocks, Geochimica et Cosmochimica Acta, 62/5, 783-795. Characteristics of a Fiber Optical pH meter., DBE Technology GmbH subsidiary from Deutsche Gesellschaft zum Bau und Betrieb von Abfallstoffe mbH, accessed on 5th December 2003 at http://www.dbetec.de/e_tec_geotechnik_1.7.html Coleman, M.L. and Moore, M.P. (1978) Direct reduction of sulphate to sulphur dioxide for isotopic analysis. Analytical chemistry, 50, 1594-1595. Coudrain-Ribstein, A. and Gouze, P. (1993) Quantitative study of geochemical processes in the Dogger aquifer, Paris Basin, France, Applied Geochemistry, 8, 495506. Coudrain-Ribstein, A., Gouze, P., and de Marsily, G. (1998) Temperature-carbon dioxide partial pressure trends in confined aquifers, Chemical Geology, 145, 73-89. Craig, H. and Gordon, L. (1965) Deuterium and oxygen-18 variations in the ocean and the marine atmosphere. In: Symposium on marine geochemistry. Graduate school of Oceanography, Univ. Rhode Island, Occ. Publ. No 3:277. Cremers, A. and Maes, A. (1986) Radionuclide partitioning in environmental systems: A critical analysis, In: Application of distribution coefficients to radiological assessment models, T. H. Sibley and C. Myttenaere, Eds., 4-14, Elsevier Applied Science Publishers: London. Darling, W.G., Talbot, J.C. and Warrington A.W. (1992) Updated procedures for the measurementt of 18O/16O and 2H/1H in natural water samples. Rep. Br. Geol. Surv. Hydrogeology GP. WD/92/11. De Cannière, P., Moors, H., Lolivier P., De Preter P., and Put, M. (1996) Laboratory and in situ migration experiments in the Boom Clay. CEC, Nuclear Science and Technology Series. Contract N° F12W-CT90-0039. Final Report EUR 16927 EN. De Cannière, P., Dierckx, A., Moors, H., Wang, L., Lolivier, F., and Put, M. (1997) Migration studies in the Boom Clay (laboratory and in situ). In: Geological Disposal of conditioned high level waste. Tasks: 1.2 to 5.2 & Operation and maintenance of the URF. Progress report to NIRAS/ONDRAF for the first semester of 1997. Contract CCHO-95/268, KNT 9094610. Report R-3240, Volume 2 (of 3). De Cannière, P., Fonteyne, A., Moors, H., Put, M., and Van Gompel, M (1994a) Migration studies, Task 2.1, 2.3 and 2.4, In: Geological disposal of conditioned high-level and long lived radioactive waste, progress report to NIRAS/ONDRAF for the second semester of 1993, SCK•CEN Report, R-2992, Mol, Belgium. De Cannière, P., Moors, H., Lolivier, P., De Preter, P., and Put, M. (1996) Laboratory and in situ migration experiments in the Boom Clay. Luxembourg, European Commission. EUR 16927EN. De Cannière, P., Moors, H., Put, M., and Wang, L. (1994b) Migration studies, Task 2.1, 2.3 and 2.4, In: Geological disposal of conditioned high-level and long lived radioactive waste, progress report to NIRAS/ONDRAF for the second semester of 1994, SCK•CEN Report, R-3053, Mol, Belgium. De Craen, M. (2001) High sulphate concentrations in squeezed Boom Clay pore water. Topical report in the frame of "Geological Disposal of Conditioned High- 121 level and Long-lived Radioactive Waste. Task 2.9: Natural evidence on the longterm behaviour of trace elements and radionuclides in the Boom Clay". NIRAS/ONDRAF contract CCHO 2000-773/00/00, KNT 90 01 1467, November 2001, R-3564. De Craen, M., Delleuze, D., Volckaert, G., Sneyers, A., Put, M. (2000) The Boom Clay as natural analogue. SCK•CEN Final report to NIRAS/ONDRAF for the period 1997-1999, Contract nr. CCHO-98/332, KNT 90 98 1042, , R-3444. De Craen, M., Van Geet, concentrations in squeezed cores. Poster presentation Engineered Barriers for December 9-12, 2002. M., Wang, L. and Put, M. (2002a) High sulphate Boom Clay pore water: evidence of oxidation of clay at the International Meeting "Clays in Natural and Radioactive Waste Confinement", Reims, France, De Craen, M., Van Geet, M., Wang, L. and Put, M. (2002b) Natural organic matter in Boom Clay (Mol, Belgium) and its pore water obtained by different extraction techniques. Poster presentation at the International Meeting "Clays in Natural and Engineered Barriers for Radioactive Waste Confinement", Reims, France, December 9-12, 2002. De Craen M., Van Geet M., Wang L. and Put M. (2004a) High sulphate concentrations in squeezed Boom Clay pore water: evidence of oxidation of clay cores. Physics and Chemistry of the Earth, 29, 91-103. De Craen M., Wang L. and Weetjens E. (2004b) Natural evidence on the long-term behaviour of trace elements and radionuclides in the Boom Clay, SCK•CEN Final report to NIRAS/ONDRAF for the period 2000-2003, Contract nr. CCHO 2000773/00/00, KNT 90 01 1467. R-3926, Mol, Belgium. Decleer, J., Viaene, W. and Vandenberghe, N. (1983) Relationships between chemical, physical and mineralogical characteristics of the Rupelian Boom Clay, Belgium. Clay Minerals 18, 1-10. Dierckx, A., Put, M., De Cannière, P., Wang, L., Maes, N., Aertsens, M., Maes, A., Vancluysen, J., Verdickt, W., Gielen, R., Christiaens, M., Warwick, P., Hall, A. and Van Der Lee, J., 2000. Transport of Radionuclides due to Complexation with Organic Matter in Clay formations (TRANCOM-clay). European Commission Nuclear Science and Technology. Final Report EUR 19135. Dierckx, A. (1997) Boom Clay in situ porewater chemistry, SCK•CEN Report, BLG734, Mol, Belgium. Drever, J. I. (1997) The geochemistry of natural waters: Surface and groundwater environments, Prentice-Hall, Upper Saddle River, New Jersey. Dzombak, D. A. and Morel, F. F. M. (1990) Surface Complexation Modelling, John Wiley & Sons, New York. Fleischer, R.L. (1988) Alpha-recoil damage: Relation to isotopic disequilibrium and leaching of radionuclides. Geochim. Cosmochim. Acta, 52, 1459-1466. Goemaere, E. (1991) Révision critique de l' analyse par diffraction des rayons X de matériaux et minéraux argileux: applications à quelque problèmes géologiques et pédologique à intérèt géotechniques. Université de Liège: unpublished PhD-thesis. Griffault, L., Merceron, T., Mossmann, J.R., Neerdael, B., De Cannière, P., Beaucaire, C., Daumas, S., Bianchi, A. and Christen, R. (1996) Participation au 122 projet "ARCHIMEDE - ARGILE": Acquisition et Régulation de la Chimie des Eaux en Milieu Argileux pour le projet de Stockage de Déchets Radioactifs en Formation Géologique. Rapport Final. Contrat CCE N° F12W-CT92-0117. Griffault, L, Merceron, T., Mossmann, J. R., Neerdael, B., De Cannière, P., Beaucaire, C., Daumas, S., Bianchi, A., and Christen, R. (1996) Acquisition et régulation de la chimie des eaux en milieu argileux pour le project de stockage de déchets radioactifs en formation géologique, Project <Archimède argile>, Rapport final, EUR 17454 FR. Hall, A.J., Boyce, A.J., Fallick, A.E. and Hamilton, P.J. (1991) Isotopic evidence of the depositional environment of Late Proterozoic stratiform barite mineralisation, Aberfeldy, Scotland. Chemical Geology (Isotope Geoscience Section), 87, 99-114. Helgeson, H. C (1969) Thermodynamics of hydrothermal systems at elevated temperatures and pressures, American Journal of Science, 267, 729-804. Helgeson, H. C and Kirkham, D. H (1974) Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures, II debye-Huckel parameters for activity coefficients and relative partial molal properties, American Journal of Science, 274, 1199-1261. Hem, J. D (1985) Study and interpretation of the chemical characteristics of natural water, Third Edition, U.S Geological Survey Water-supply Paper 2254. Henrion, P.N., Monsecour, M., Fonteyne, A., Put M. and De Regge, P. (1985) Migration of radionuclides in Boom Clay. Radioactive Waste Management and the Nuclear Fuel Cycle, 6 (3-4) 313-359. Hoefs, J. (1997) Stable Isotope Geochemistry. Springer, Berlin, 201 pp. IAEA (1992) Statistical treatment of data and environmental isotopes in precipitations. Technical Report Series 331. Ivanovich, M. and Wilkins, M.A. (1988) Uranium series disequilibrium measurements at Mol (Belgium) and Whiteshell (Canada), CEC, Nuclear Science and Technology, Final Report, EUR 11629 EN. Johnson, J. W., Oelkers, E. H., and Helgeson, H. C. (1991) SUPCRT92: A software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bars and 0° to 1000°C., Earth Sciences Department, Lawrence Livermore Laboratory. Laenen, B. (1997) The geochemical signature of relative sea-level cycles recognized in the Boom Clay. K.U.Leuven: Unpublished PhD-thesis. Langmuir, D. (1997) Aqueous Environmental Geochemistry, Prentice Hall, New Jersey. Lemmens, K. (2001) Bereiding, bewaring en gebruik van synthetisch kleiwater, werkinstructie IW. WD. 062, versie 1.0. Lindberg, R. and Runnells, D (1984) Groundwater redox reactions: an analysis of equilibrium state applied to Eh measurements and geochemical modeling, Science, 225, 925-927. Maes, N. (2000) Composition of synthetic clay water and preparation procedure for TRANCOM-II, in memo 00/C072028/NMa/R-152. 123 Maes, N., Wang, L., Delécaut, G., Beauwens, T., Van Geet, M., Put, M., Van der Lee, J., Warwick, P., Hall, A., Walker, G., Maes, A., Bruggeman, C., Bennett, D., Higgo, J. and Galson, D., 2002. Migration Case Study: Transport of radionuclides in a reducing Clay Sediment (TRANCOM II) 2nd Progress Report. Maes N., Wang L., Delécaut G., Beauwens T., Van Geet M., Put M., Weetjens E., Marivoet J., Van der Lee J., Warwick P., Hall A., Walker G., Maes A., Bruggeman C., Bennett D., Hicks T., Higgo J., Galson D. (2004) Migration Case Study: Transport of radionuclides in a reducing clay sediment (TRANCOM-II) - Final Scientific&Technical Report, BLG-988, SCK-CEN, Mol, Belgium. Marivoet, J., Van Keer, I., Wemaere, I., Hardy, L., Pitch, H., Beaucaire, C., Michelot, J.-L., Marlin, C., Philippot, A.C., Hassanizadeh, M. and van Weert, F. (2000) A PalaeoHYdrogeological study of the MOL site (PHYMOL project). European Commission Nuclear Science and Technology. Final Report, EUR 19146 EN. Meier, P., Lohrum, A., Ammann, J., (1989) Practice and Theory of pH Measurement, An outline of pH measurement. Information and practical hints., Copyright © 1989 by Mettler-Toledo AG, CH-8902 Urdorf/Switzerland. Merceron, T., Mossmann, J.R., Neerdael, B., De Cannière, P., Beaucaire, C., Daumas, S., Bianchi, A. and Christen, R. (1993a), Projet Archimede-Argile, Acquisition et régulation de la chimie des eaux en milieu argileux. Rapport d'avancement semestriel n°3 - ANDRA, 696 RP BRG 93-001. Merceron, T., Mossmann, J.R., Neerdael, B., De Cannière, P., Beaucaire, C., Daumas, S., Bianchi, A. and Christen, R. (1993b), Projet Archimede-Argile, Acquisition et régulation de la chimie des eaux en milieu argileux. Rapport d'avancement semestriel n°4 - ANDRA, 696 RP BRG 93-004. Moors, H., Wang, L., Vandervoort, F., De Cannière, P., and Dierckx, A. (2002) Assessment of the in situ pH and Eh of Boom Clay, Poster presentation, Clays in Natural and Engineered Barriers for Radioactive Waste Confinement, Reims, France, December 9-12, 2002. Neerdael, B. (1984) Lentille "sableuse" au sein de l'argile de Boom – Forage de reconnaissance. Inner report SCK•CEN, Geotechnology: C84/46/D6350/BN/lsb/R/123. Nordstrom, D. K. and Munoz, J. L. (1994) Geochemical Thermodynamics, Blackwell Scientific Publication, Oxford. Nordstrom, D. K., Plummer, L. N., Langmuir, D., Busenberg, E., May, H. M., Jones, B. F., and Parkhurst, D. L. (1990) Revised chemical equilibrium data for major water-mineral reactions and their limitations, In: Chemical modeling of aqueous systems II, D. C. Melchior and R. L. Bassett, Eds., American Chemical Society: Wachington, DC. Noynaert, L., Volckaert, G., De Cannière, P., Meynendoncks, P., Labat, S., Beaufays, R., Put, M., Aertsens, M, Fonteyne, A., and Vandervoort, F. (1998) The Cerberus project: Demonstration test to study the near-field effects of an HLW canister in argillaceous formation, Final report, EUR 18151 EN. Omega Engineering, Inc. (2001) pH Technical reference guide. Accessed on 9th December 2003 at http://www.omega.com/techref/ph.html 124 Ouvry, J.F. (1986) Etude physique et rhéologique des argiles congelées. Aplication à l'argile profonde de Boom (Belgique). PhD-thesis. Bureau de Recherches Géologiques et Minières report 103, 247p Planque, G. Amekraz, B., Moulin, V., Toulhoat, P. and Moulin, C., 2001. Molecular structure of fulvic acids by electrospray with quadrupole time-of-flight mass spectrometry. Rapid Communications in mass spectrometry, 15, pp. 827-835. Pearson, F. J. (2001) Modelling Clay Pore-Water: Principles and Input Data Selection, Chapter 6 in Technical Note 2000-36, Mont Terri Project. Pearson, F. J., Fisher, D. W., and Plummer, L. N. (1978) Correction of ground-water chemistry and carbon isotopic composition for effects of CO2 outgassing, Geochimica et Cosmochimica Acta, 42, 1799-1807. Pirlet, V. (2003) The investigation of the neptunium complexes formed upon interaction of high-level waste glass and Boom Clay medium, PhD thesis, faculty of sciences, University of Liege. Planque, G. Amekraz, B., Moulin, V., Toulhoat, P. and Moulin, C., 2001. Molecular structure of fulvic acids by electrospray with quadrupole time-of-flight mass spectrometry. Rapid Communications in mass spectrometry, 15, pp. 827-835. Poghossian, A.; Berndsen, L.; Lüth, H.; Schultze, J.W.; Schöning, M.J., MultiParameter-Detektions-System unter dem Einsatz eines einzigen Transducerprinzips sowohl für (bio-)chemische als auch physikalische Sensoren, in: VDI/VDEGesellschaft (ed.) Sensoren und Meßsysteme 2002, VDI-Verlag (2002) 137-140. Pusch R., Börgesson, L. and Elström, M. (1987) Alteration of isolating properties of dense smectite clay in repository environment as exemplified by seven prequarternary clays. SKB Technical report 87-29, 110 p. Radtke, D.B., Busenberg, E., Wilde, F.D., and Kurklin, J.K. (2003), pH Field Measurements, National field manual for the collection of water-quality data: U.S. Geological Survey Techniques of Water-Resources Investigations, book 9, chapter 6.4 Reeder, S. and Cave, M.R. (1999) Evaluation of the containment properties of geological and engineered barriers by pore-water extraction and characterization. In: Chemical Containment of Waste in the Geosphere. (Editors Metcalfe R. and Rochelle C.A.) Geological Society, London, Special Publications, 157, 265-273. Reeder, S., Cave, M.R., Bath, A.H., Entwisle, D.C., Inglethorpe, S.J., Pearce, J.M., Trick, J.K., Blackwell, P.A. and Green, K.A. (1994) A study of Boom Clay Drillcore from Mol in Belgium: Chemical and Isotopic Characterisation of PoreWaters and Clay-Mineralogy. Brit. Geol. Surv. Tec. Rep. WI/93/12C. Reeder, S., Cave, M.R., Entwisle, D.C., Trick, J.K. (1998) Extraction of water and solutes from clayey material: a review and critical discussion of available techniques. Brit. Geol. Surv. Tech. Rep., WI/98/4C, 60. Reeder, S., Cave, M.R., Entwisle, D.C., Trick, J.K., Harmon, K.A., Blackwell, P.A., Mitchell, N. and Cook, J.M. (1992) The extraction and analysis of pore fluids from the Boom Clay Drillcore, Mol, Belgium. Brit. Geol. Surv. Tec. Rep. WI/92/7C. Rounds, S. A. and Wilde, F. D. (2001) Alkalinity and acid neutralizing capacity, chapter 6.6, In: U. S. Geological Survey TWRI Book 9. 125 Rousset, G. (1988) Comportement mécanique des argiles profondes. Application au stockage des déchets radioactifs. Unpublished PhD-thesis, Ec. Net. Ponts & chaussees. Sacchi, E. and Michelot, J.-L. (2000) Porewater Extraction from Argillaceous Rocks for Geochemical Characterisation. Methods and Interpretations. Radioactive Waste Management. NEA. OECD Publications, pp. 185. Safety Assessment and Feasibility Interim Report 2 (SAFIR 2) (2001) NIROND 2001-05. Sanjuan, B., Mossmann, J. R., and Merceron, T. (1994) Modelling boom clay formation porewater chemistry: ion exchange versus dissolution precipitation mechanisms, Conference abstract, Goldschmidt Conference Edinburgh. Stumm, W. and Morgan, J. J. (1996) Aquatic Chemistry, John Wiley & Sons, Inc., New York, Chichester, Brisbane, Toronto, Singapore. Thang, N.M., Geckeis, H., Kim, J.I. and Beck, H.P., 2001. Application of the flow field flow fractionation (FFFF) to the characterisation of aquatic humic colloids: evaluation and optimization of the method. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 181, pp. 289-301. Thellier, C and Sposito, G. (1988) Quaternary cation exchange on Silver-Hill illite, Soil Sci.Sco.Am.J., 52, 979-985. Thorez, J. (1976) Rapport d'analyse mineralogique: contenu qualitatif et semiquantitatif en mineraux argileux dans l'argile de Boom, au site de Mol (CEN). Intern report, 325/07/030 MiUL. Van Geet, M., Maes, N. and Dierckx, A., 2003. Characteristics of the Boom Clay organic matter, a review. Geological Survey of Belgium, Professional Paper 2003/1 Nr. 298. Van Geet, M. (2004) Characterisation of Boom Clay organic matter: mobile and immobile fraction. SCK•CEN interim report to NIRAS/ONDRAF for the period 2001-2003, contract nr. CCHO 2000-773/00/00, KNT 90 01 146, R-3884. Van Keer, I. and De Craen, M. (2001) Sedimentology and diagenetic evolution of the Boom Clay: State of the art. Long-Term Performance Studies of the Geological Disposal of Conditioned High-Level and Long-Lived Radioactive Waste. Report to NIRAS/ONDRAF contract CCHO-98/332 / KNT 90.98.1042, R-3483. Vandenberghe, N. and Thorez, J. (1985) XRD-onderzoek van de 14 Å componenten in de kleimineralenfractie van de Boomse klei in de boring te Mol 31W-237. Brussel: Belgian Geological Survey: internal document, 7 p. Vandenberghe, N. (1974) Een sedimentologische studie van de Boomse Klei. K.U.Leuven: Unpublished PhD-thesis. Vandenberghe, N. (1978) Sedimentology of the Boom Clay (Rupelian) in Belgium. Verhandeling Koninklijke Academie voor Wetenschappen, Letteren en Schone Kunsten van België, Klasse Wetenschappen XL, 147 p. Volckaert, G., Neerdael, B., Manfroy, P., Lalieux, Ph., De Cannière, P. and Labiouse, V. (1997) Characteristics of Argillaceous Rocks: A Catalogue of the Characteristics of Argillaceous Rocks Studied with Respect to Radioactive Waste Disposal Issues: 126 Belgium, Canada, France, Germany, Italy, Japan, Spain, Switzerland, United Kingdom, and Unite States. - Boom Clay. Revision number 2 - 05/5/1997. Wang, L. (2004) MOLDATA: A Thermodynamic Database maintained at SCK•CEN for study migration of radionuclides in Boom Clay, version 04-01, SCK•CEN Report, in preparation. Wang, L., Dierckx, A., De Cannière, P., and Maes, A (2002) Uranium release from boom clay in bicarbonate media, Radiochim.Acta, 90, 515-520. Wemaere I., Marivoet J., Labat S., Beaufays R. & Maes T. (2002) Mol-1 borehole (April-May 1997) Core manipulations and determination of hydraulic conductivities in the laboratory. Geological Disposal of Conditioned High-Level and Long-Lived Radioactive Waste. Report to NIRAS/ONDRAF, R-3590. Wouters L., Herron, M., Abeels V., Hagood M. and Strobet J. (1999) Inovative applications of dual range Fourier transform infrared spectroscopy to analysis of Boom Clay mineralogy. Aardk. Mededel., 9, 159-168. 127 10 Annexes Annex 1: Overview of the general characteristics of the various piezometres considered in this report (EG/BS, ARCHIMEDE, SPRING 116, ORPHEUS, MORPHEUS). Annex 2: Materials used as piezometric filterscreen Annex 3: Assessing the performance of pH and Eh electrodes used for in situ measurement campaigns Annex 4: The procedure, results, and interpretations for batch leaching experiments to determine the concentration of Boom Clay pore water components Annex 5: Boom Clay pore water geochemistry: analytical data used in the statistical analyses Annex 6: Statistics: methodology Annex 7: The input and output files from geochemical modelling simulations Annex 8: Prescriptions for the preparation of synthetic Boom Clay water 129 Annex 1: Overview of the general characteristics of the various piezometres considered in this report (EG/BS, ARCHIMEDE, SPRING 116, ORPHEUS, MORPHEUS). piezometer name piezometer emplacement piezometer location piezometer material piezometer length piezometer diameter filter amount filter material filter size vertical depth (m TAW) EGBS/2 September 1983 vertical piezometer, downward from HADES, located at the bottom of the first schaft stainless steel ~13 m stainl. steel 6 cm, coarse sand 8.5 cm 1 stainless steel stainl. steel 9 cm, coarse sand 120 cm -227.6 ARCHIMEDE #1 March 1992 semi-horizontal piezometer (3 % inclined upwards) towards the east, HADES URL, ANDRA Gallery, between sliding ribbs 24 and 25 stainless steel 15 m 60 mm 5 ? -196.5 ARCHIMEDE #2 April 1992 horizontal piezometer towards the east, HADES URL, ANDRA Gallery, between sliding ribbs 4 and 5 stainless steal 15 m 140 mm 5 ? -196.5 SPRING 116 October 1999 horizontal piezometer towards the east, Test Drift, ring 116 stainless steel 12 m 15 cm 4 November 2000 horizontal piezometer towards the west, Test Drift, ring 116 PVC 8m 12 cm 4 MORPHEUS May 2001 vertical piezometer, downward from HADES, Test Drift, between ring 11 and 12 PVC 40 m 15 cm 12 152 cm 152 cm 152 cm 152 cm 25 cm 25 cm 25 cm 25 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm 10 cm -196.5 ORPHEUS stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel stainless steel sintered glass poly ethylene carbo schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm schumatherm -196.5 -217.07 to -217.17 -220.77 to -220.87 -222.57 to -222.67 -225.98 to -226.08 -227.18 to -227.28 -227.78 to -227.88 -229.08 to -229.18 -229.73 to -229.83 -230.23 to -230.33 -231.78 to -231.88 -233.79 to -233.89 -235.19 to -235.29 distance from the code piezometer- code stalen gallery (in m, filter extra dos Test Drift) stainl.steel 30 m, EGBS-SS coarse sand 22-34 m 3m 7m 8m 14 m 15 m 3m 7m 8m 14 m 15 m 5.44 - 6.94 7.16 - 8.66 8.88 - 10.38 10.60 - 12.10 5.25 - 5.50 5.90 - 6.15 6.45 - 6.70 7.10 - 7.35 18.12 - 18.22 21.92 - 22.02 23.72 - 23.82 27.13 - 27.23 28.33 - 28.43 28.93 - 39.03 30.23 - 30.33 30.88 -30.98 31.38 - 31.48 32.93 - 33. 03 34.94 - 35.04 36.34 - 36.44 AR1-SS-03 AR1-SS-07 AR1-SS-08 AR1-SS-14 AR1-SS-15 AR2-SS-03 AR2-SS-07 AR2-SS-08 AR2-SS-14 AR2-SS-15 SP-SS-4 SP-SS-3 SP-SS-2 SP-SS-1 OR-ST OR-CA OR-PE OR-SG MO-ST-23 MO-ST-20 MO-ST-18 MO-ST-15 MO-ST-13 MO-ST-12 MO-ST-10 MO-ST-09 MO-ST-08 MO-ST-06 MO-ST-04 MO-ST-02 EGBS-SS/yyyymmdd AR1-SS-03/yyymmdd AR1-SS-07/yyymmdd AR1-SS-08/yyymmdd AR1-SS-14/yyymmdd AR1-SS-15/yyymmdd AR2-SS-03/yyymmdd AR2-SS-07/yyymmdd AR2-SS-08/yyymmdd AR2-SS-14/yyymmdd AR2-SS-15/yyymmdd SP-SS-4/yyyymmdd SP-SS-3/yyyymmdd SP-SS-2/yyyymmdd SP-SS-1/yyyymmdd OR-ST/yyyymmdd OR-CA/yyyymmdd OR-PE/yyyymmdd OR-SG/yyyymmdd MO-ST23/yyyymmdd MO-ST-20/yyyymmdd MO-ST-18/yyyymmdd MO-ST-15/yyyymmdd MO-ST-13/yyyymmdd MO-ST-12/yyyymmdd MO-ST-10/yyyymmdd MO-ST-09/yyyymmdd MO-ST-08/yyyymmdd MO-ST-06/yyyymmdd MO-ST-04/yyyymmdd MO-ST-02/yyyymmdd 130 Annex 2: Materials used as piezometric filterscreen For more then 20 years multipiezometers were all made of stainless steel (sometimes in combination with classic sand-filter systems). The main raisons for this choice were the machinability, and the pressure and corrosion resistance of stainless steel. As new materials became available, the applicability of these materials as piezometric filterscreen was investigated. Nowadays, the choice of piezometric filterscreen is a strategic item in the construction phase of a multi-piezometer, that can be adapted in function of the intended uses of multi-piezometers. Coarse sand The filterscreen of the EG/BS piezometer consists of a column of purified coarse sand with a granulometry of 0.71 to 1.25 mm. Stainless steel (SS) The stainless steel filterscreens of SPRING 116 and the collecting chamber of the EG/BS piezometer are manufactured by GKN Sinter Metals (Germany). The filter elements are produced by cold isostatic pressing of metal powder followed by a sintering process that bonds and fuses the powder particles to each other. The filter elements have the following characteristics: • • • • material: stainless steel, 1.4404 (AISI 316 L/B); filter type: SIKA-R5 IS which stands for self-supporting structural element porosity: 30 % (DIN ISO 30911-3) average CCE pore diameter: 9 µm (ASTM F 902) permeability coefficients: α = 0.8 x 10-12 m2; β = 0.9 x 10-7 m (DIN ISO 4022) Glass (SG) The glass filterscreen which is part of the ORPHEUS multi-piezometer is manufactured by ROBU GLASFILTER-GERÄTE GMBH (Germany). It has the trade-name VitraPOR, which stands for a complete series of glass filter-products. The filter element has the following characteristics: • • • material: Borosilicate glass 3.3 (DIN-ISO 3585) porosity: porosity grade 4, ISO 4793 designation P16 nominal pore size: 10-16 µm Filtroplast (PE) The plastic filterscreen which is part of the ORPHEUS multi-piezometer is manufactured by Schumacher Umwelt- und Trenntechnik GmbH (Germany). The filterelements are know as FILTROPLAST. FILTROPLAST is a porous sintered plastic product of pure Polyethylene (PE-HD). Rigid elements of different dimensions and shapes are manufactured from special PE granules of defined size and particle distribution in a controlled sintering process. The filtration and aeration properties of the product depend on the pore size, which is a function of both the raw material and the production parameters. 132 The filter element has the following characteristics: • • • material: High Density Polyethylene, grade 40 porosity: 45 % mean pore size: 40 µm Carbo (CA) The carbon filterscreen which is part of the ORPHEUS multi-piezometer is manufactured by Schumacher Umwelt- und Trenntechnik GmbH (Germany). The filterelements are known as CARBO. CARBO consists of pure carbon (98 % C). The various types of CARBO are based on different grain size fractions that are linked by carbon bridges which were built from tar during a sintering process. The material is very resistant against chemical reactions due to its binder-free structure. The filter element has the following characteristics: • • • material: Carbon (98 % C), grade 40 porosity: 35 % mean pore size: 90 µm Shumatherm (ST) The Schumatherm filterscreens are used as filterscreens for the MORPHEUS multi piezometer and are part of the ORPHEUS multi-piezometer. Schumatherm is manufactured by Schumacher Umwelt- und Trenntechnik GmbH (Germany). Schumatherm is a high-quality fireclay rich in mulite(a), obtained by sintering of refractory clay and subsequent crushing. This raw material is sintered with an alumo-siliceous bond. The binding phase imparts good mechanical properties and is mostly of amorphous structure. Schumatherm filter elements have the following properties: • • • material: fireclay rich in mulite, grade 30. Mullite is a nonstoichiometric compound of approximate composition Al6Si3O15. It is rare as mineral but commonly used in artificial Al2O3–SiO2 systems at high temperature. porosity: 37 % mean pore size: 60 µm Annex 3: Assessing the performance of pH and Eh electrodes used for in situ measurement campaigns. Introduction Electrodes used for in situ measurements are generally submitted to harsh conditions (e.g. high pressure, long contact time between electrode and the measuring medium, influence of the ligand and complex-forming foreign species, ageing, ...). To demonstrate the performance of such electrodes over the whole period of in situ measurement an adapted testing procedure is required. This adapted testing procedure is composed out of two successive tests: • a first test that allows to interpret the offset, the deviation with respect to the zero point and the slope or linearity of the electrode at moment of testing, and, • a second test that provides evidence on the origin of the deviation and the quantification of the electrode drift. Background Ideal pH-electrodes display 0 mV when immersed in a solution of pH = 7 and respond linear, with a slope of 59.16 mV per pH unit (@ 25 °C) when immersed in solutions having other pH values. Similarly, ideal Eh-electrodes display 0 mV when immersed in a pH buffer solution of pH = 8.5 saturated with quinhydrone and respond linear with an identical slope of 59.16 mV per pH unit (@ 25 °C) when immersed in quinhydrone saturated solutions buffered at other pH-values. The differences between the theoretical values and the measured ones, determines the offset values of the tested pH or Eh-electrode. Linearity is evaluated by comparing the calculated electrode slopes, based on the electrode measurements in different pH buffered solutions, with the theoretical slope. Offset values within ±10 mV and slopes deviating ±5 % from the theoretical values are allowed for most laboratory applications. This kind of testing, especially for pH electrodes, is used in many standard calibration protocols and electrode functionality checks. However, for electrodes used in long term in situ measurements, the deviation of the offset can become quite large. The logic source for this deviation is a change of the chloride concentration inside the in situ reference electrode, and, consequently, a change of the reference potential. To check and quantify this change of reference potential, an additional test is used. For this test, a separate reference electrode, of the same type and filling as the in situ reference electrode, is required and connected to a pH-meter together with the reference electrode of the in situ electrode. If the offset of the in situ electrode is solely due to a drift of the electrodes reference potential than, immersing the in situ reference electrode in conjunction with a separate reference electrode in an aqueous solutions, either pH, Eh or salt buffered, should always display the same value. A value equal to the previously determined offset potential. In this case, the data set collected during the in situ measurement campaign can be appropriately corrected. The correction is done assuming a linear drift of the reference potential over the measurement period. All data points are pro rata corrected with the calculated drift-slope. Procedure The first test: Determination of the electrode offset and linearity pH-electrodes Three certified buffer solutions are used: 4.00, 7.00 and 9.00. The pH-electrode, connected to its pH-meter, is successively immersed in each of the three buffer solutions. When the meter 134 reading has stabilised the displayed value (preferably in mV) is recorded and compared with the theoretical value. This test is repeated at the beginning and at the end of an in situ measuring campaign. An electrode is considered as functioning properly if the absolute– and relative differences of the measured values are smaller than ± 0.15 pH units. If the absolute differences are larger than 0.15 pH-units, but the relative differences are smaller than 0.15 pH-units, then only the linearity of the pH-electrode is acceptable. In this case a supplementary test is needed to determine the origin and to verify the offset of the electrode. Eh-electrodes As for pH-electrodes, three certified pH buffer solutions are used. To these solutions and just before the test, an excess of Quinhydrone is added. The Eh-electrode, connected to its Ehmilli-voltmeter, is successively immersed in each of the three solutions. When the meter reading has stabilised the displayed value is recorded and compared with the theoretical value. This test is repeated at the beginning and at the end of an in situ measuring campaign. An electrode is considered as functioning properly if the absolute– and relative differences of the measured values are smaller than respectively ± 20 and ± 10 mV. If the absolute differences are larger than 20 mV, but the relative differences are smaller than 10 mV, then only the linearity of the Eh-electrode is acceptable. In this case a supplementary test is needed to determine the origin and to verify the offset of the electrode. The second test: determination of the value and the origin the electrode drift This test is similar for pH and Eh-electrodes. It is used to check the signal deviation of the reference electrode, that is "build in" in pH and Eh-electrodes. For this test a separate properly functioning reference electrode is required, preferably of the same type as the in situ reference electrode. The one lead of this separate reference electrode is connected to one of the two signal inputs of a pH-milli-voltmeter. The lead of the reference electrode of the in situ pHelectrode is connected to the remaining electrical signal input of the milli-voltmeter. The indicator electrode lead, or glass membrane signal lead, is left unconnected. Both reference electrodes are successively immersed in the three certified buffer solutions of the first test, and, if possible in a non buffered electrolyte solution. Typically, in a solution containing 3 moles per litre of potassium chloride. When the meter reading, which indicates during these measurements the deviation between two reference electrodes, has stabilised the value is recorded and evaluated. Evaluation From the results of the first test we learn whether or not the electrode functions properly (absolute differences between measured values and theoretical values), and if not, whether the response behaviour is linear or not (relative differences = comparison of the differences between the measurements). If linearity is considered to be good, the second test allows to interpret the origin and to verify the electrode drift. To conclude that the in situ electrode offset is solely due to a drift of the reference electrode, the relative differences between the measured values of the second test, and in all buffer solutions, may not exceed ± 10 mV. Also, the recorded values of this second test have to be equal (within a range of ± 10 mV) to the offset values determined in first test. An example of an evaluation report is given. The change of the potassium chloride concentration inside the reference electrode is controlled by diffusion. The larger the concentration difference between reference electrode electrolyte and measuring solution, the stronger the rate of out-diffusion. For standard laboratory application this out-diffusion can easily be neglected. However, for long lasting in situ measurement campaigns this out-diffusion cannot be neglected. Due to this potassium chloride leaching problem and the non uniform construction of electrochemical electrodes, the true evolution of the reference electrode drift is unknown. The easiest approach is to consider a linear process, and, proportionally correct all measured data with the appropriate slope or drift speed of the in situ electrode. This approach is used until experimental evidence will come up with the correct interpretation of the evolution of the drift speed. 136 Example of an "evaluation report" of in situ used pH and Eh electrodes: Archiméde 2 - assessment of the electrode performance First measurement period: 1-March-2000 to 3-July-2001 pH-electrode Electrode data Electrode type: Serial number: Laboratory tested on: Method: Test result: Ingold, Xerolyt, glass-pH electrode 8238228 29-February-2000 Standard "Metrohm" electrode test procedure good electrode Field data Electrode placed in flow-through cell on 1-March-2000 Chronological event history (see logbook ARCHIM2.xls) Electrode taken out of flow-through cell on 3-July-2001 Covering a period of 463 days Test methods 1) Potential measurement in three different pH-buffer solutions Table 1: Results of the potential measurement in pH-buffer solutions Time/Evaluation [dd-mm-yyyy] Theoretical 29-February-2000 Difference 3-July-2001 Difference Buffer 4 [mV] 177 181 +4 56 -125 Buffer 7 [mV] 0 4 +4 -124 -124 Buffer 9 [mV] -118 -110 +8 -239 -121 2) Reference-Reference measurement Table 2: Results of the Reference-Reference measurement Time/Evaluation [dd-mm-yyyy] Theoretical 29-February-2000 Difference 3-July-2001 Difference Buffer 4 [mV] 0 +3 +3 -119 -119 Buffer 7 [mV] 0 +3 +3 -120 -120 Buffer 9 [mV] 0 +4 +4 -121 -121 3M KCl [mV] 0 n.a. n.a. - Conclusions for the pH-electrode performance From the potential measurements in pH-buffer solutions we can calculate an average difference of 123.3 (± 2.1) mV over the pH-region 4 to 9. From the Reference-Reference measurement we calculate an average difference of 120.0 (± 1.0) mV over the pH-region 4 to 9. We therefore conclude that: • the measured difference is solely due to a drift of the reference electrode • the drift is in a logical direction (depletion of KCl results in higher pH values) • the end value of the drift can be taken as: 120 mV If one considers a linear drift, measurement correction should be done with a drift-speed or slope = 0.25918 mV per day or -0.00438 pH units per day 138 Example of an "evaluation report" of in situ used pH and Eh electrodes: Archiméde 2 - assessment of the electrode performance First measurement period: 1-March-2000 to 3-July-2001 Pt-Eh-electrode Electrode data electrode type: serial number: Tested on: Method: Test result: Ingold, Xerolyt, Platinum redox-electrode 8484707 8-June-1999, 29-February-2000 SCK•CEN redox-electrode test procedure Good Electrode Field data Electrode placed in flow-through cell on 1-March-2000 Chronological event history (see logbook ARCHIM2.xls) Electrode taken out of flow-through cell on 3-July-2001 Covering a period of 463 days Test methods 1) Potential measurement in three different Eh-buffer solutions Table 1: Results of the potential measurement in pH-buffer solutions Time/Evaluation [dd-mm-yyyy] Theoretical 8-June-1999 Difference 29-February-2000 Difference 3-July-2001 Difference Buffer4 + Quinhydrone [mV] 266 264 -2 263 -3 142 -124 Buffer 7 + Quinhydrone [mV] 89 88 -1 89 0 -36 -125 Buffer 9 + Quinhydrone [mV] -30 -29 +1 -29 +1 -143 -113 2) Reference-Reference measurement Table 2: Results of the Reference-Reference measurement Time/Evaluation [dd-mm-yyyy] Theoretical 29-February-2000 Difference Buffer 4 + Quinhydrone [mV] 0 n.a. - Buffer 7 + Quinhydrone [mV] 0 n.a - Buffer 9 + Quinhydrone [mV] 0 n.a - 3M KCl [mV] 0 n.a - Conclusions for the Pt-Eh-electrode performance From the potential measurements in Eh-buffer solutions we can calculate an average difference of 121 (± 7) mV over the Eh-region +266 to -30 mV. The Reference-Reference measurements have not been performed. Therefore, we can only base the conclusions on the Quinhydrone measurements and evaluate the observed differences at the start and the end of the measurement period. We can assume that: • the measured difference is probably due to a drift of the reference electrode • the observed drift is logical (depletion of KCl results in apparently lower Eh values) • the final value of the reference drift can be taken as: 121 mV If one considers a linear drift, measurement correction should be done with a drift-speed or slope = -0.26 mV per day 140 Example of an "evaluation report" of in situ used pH and Eh electrodes: Archiméde 2 - assessment of the electrode performance First measurement period: 1-March-2000 to 3-July-2001 Au-Eh-electrode Electrode data electrode type: serial number: Tested on: Method: Test result: Ingold, Xerolyt, gold redox-electrode 9084656 8-June-1999, 29-February-2000 SCK•CEN redox-electrode test procedure Good Electrode, slightly over the linearity limit Field data Electrode placed in flow-through cell on 1-March-2000 Chronological event history (see logbook ARCHIM2.xls) Electrode taken out of flow-through cell on 3-July-2001 Covering a period of 463 days Test methods 1) Potential measurement in three different Eh-buffer solutions Table 1: Results of the potential measurement in pH-buffer solutions Time/Evaluation [dd-mm-yyyy] Theoretical 8-June-1999 Difference 29-February-2000 Difference 3-July-2001 Difference Buffer4 + Quinhydrone [mV] 266 269 +3 254 -12 144 -122 Buffer 7 + Quinhydrone [mV] 89 87 -2 89 0 -31 -120 Buffer 9 + Quinhydrone [mV] -30 -28 +2 -28 +2 -134 -104 2) Reference-Reference measurement Table 2: Results of the Reference-Reference measurement Time/Evaluation [dd-mm-yyyy] Theoretical 29-February-2000 Difference Buffer 4 + Quinhydrone [mV] 0 n.a. - Buffer 7 + Quinhydrone [mV] 0 n.a - Buffer 9 + Quinhydrone [mV] 0 n.a - 3M KCl [mV] 0 n.a - Conclusions of the Au-Eh-electrode performance From the potential measurements in Eh-buffer solutions we can calculate an average difference of 115 (± 10) mV over the Eh-region +266 to -30 mV. The Reference-Reference measurements have not been performed. Therefore, we can only base the conclusions on the Quinhydrone measurements and evaluate the observed differences at the start and the end of the measurement period. We can assume that: • the measured difference is probably due to a drift of the reference electrode • the observed drift is logical (depletion of KCl results in apparently lower Eh values) • the final value of the reference drift can be taken as: 115 mV If one considers a linear drift, measurement correction should be done with a drift-speed or slope = -0.25 mV per day 142 Annex 4: The procedure, results, and interpretations for batch leaching experiments to determine the concentration of Boom Clay pore water components Background In parallel with using the piezometric and mechanical squeezing techniques, the batch leaching experiment was performed for a purpose of comparison. Summaries of the techniques and the results are given in Section 2.1 and 3.1 of this report. Different from the piezometer and squeezing techniques, the batch leaching method does not sample pore water directly without diluting the system, i.e., the addition of water to clay samples is necessary in batch experiments. Due to the addition of water to the system, the resulting solution composition cannot directly be used as the real pore water composition of the Boom Clay without interpretations or modelling. This annex documents the details of the procedure, results, and interpretations for the batch leaching technique. The experiment was designed in the way that the approach of Bradbury and Baeyens (1998), referred as the B&B approach hereafter, can be used for the data interpretations. The essential feature of the B&B experiment is to measure the water composition change as a result of leaching as a function of varied clay to the solution ratio (solid to liquid ratio, S/L). The chemical principles of the B&B approach are based on the dilution effect for soluble salts (NaCl, NaHCO3 or Na2SO4), the cation exchange equilibrium for major cations, and the solubility equilibrium for solubility controlled phases like calcite and dolomite. Because of the general similarities between the Opalinus Clay and the Boom Clay, we expected that the B&B approach, at the stage of experiment, should work for the Boom Clay as well. The major difference between our experiments and that of B&B is that we did look at the effect of colloids by filtration techniques and B&B did only centrifugation. In addition, we mostly used a NaHCO3 solution as the leachant while B&B used only distilled water. We didn’t perform the exhaustive cation exchange extraction, e.g., using nickelethylenediamine as B&B did. Different from the Opalinus Clay for which the data of ion exchange complex was not available (to our knowledge), Boom Clay ion exchange has been well studied (Baeyens et al., 1985). The experimental procedure The experimental procedure is illustrated in Figure A4-1. Boom Clay samples from the HADES 2001/4 borehole were grinded, suspended, and agitated in a NaHCO3 solution of 0.01 M. Bicarbonate solution was used as the leachant because Boom Clay pore water is basically a dilute NaHCO3 solution. Some samples were suspended in distilled water for comparison. Experiments were conducted in glove boxes to protect the clay samples from oxidation. The oxygen content of the glove boxes is about 2 ppm but generally below 10 ppm. For practical reasons, some experiments were carried out in an Ar glove box and some others in an Ar/CO2 (g) glove box. The CO2 content in the glove box was fixed at 0.4 percent to mimic the supposed in situ partial pressure of CO2 (10-2.4 atm). Four different S/L ratio were used: 25, 50, 200, and 800 gram wet clay per litre of solution. The leaching duration was 2 to 3 months in which a steady aqueous concentration of the major ions were reached. After the leaching, samples of suspension were centrifuged at 21,255 g for 2 hours before the chemical analysis for major cations and anions. Some samples were further filtered by 0.45 µm filters and YM3 (3000 MWCO) centriplus ultrafilters to study the possible effect of clay particulates or colloids on the concentration of clay water components. 144 solution: NaHCO3 (0.01 M) or water chemical analysis for cations and anions clay centrifugation supernatant filtration (0.45 µm) ultrafiltration (3000 MWCO) Figure A4-1: Schematic illustration of the batch leaching experiment to determine the concentration of Boom Clay pore water components Results and discussion General The detailed experimental results are given in the end of this annex from Table A4-1 to A4-5. The general trend of the observation is similar to that of the B&B experiments, i.e., the concentration of cations, except Si and K, and all anions increases linearly with the S/L. Our data show however important differences comparing to the data of B&B and are explained in detail hereafter. The pH value of the leaching suspension The pH was measured at the end of the leaching period inside the glove boxes. The pH electrode was immersed in suspension before centrifugation, so the pH was recorded in suspension, not in solution. Figure A4-2 shows the pH values of all samples as a function of the S/L ratio, the type of infill gas in the glove box, and the type of leachant used. The pH in the Ar box is higher than the N2/CO2 box as expected because of the lower pCO2 in the Ar box. Also the pH in the Ar box is around 9 and is higher than the anticipated pH of Boom Clay at pH around 8.5 due to degassing of CO2(g). In the N2/CO2 box, the pH is around 8.2 at low S/L and increases as a function of the S/L. This contradicts the findings of B&B in which they observed that the lower pH are associated with higher S/L. B&B explained that the pH decrease as a function of S/L was the result of calcite/dolomite solubility equilibrium using the Ca/Mg activity ratio of 1.35 according to their solubility data. As will be seen from the later Sections on Ca and Mg concentrations, the Ca/Mg concentration ratio in our batch extracts is about 0.5 (after ultrafiltration) which has been explained as being controlled by cation exchange reactions (see modelling Section 4). The increase of pH found in our batch experiments as a function of S/L is likely due to the increase of the concentration of bicarbonate released into the solution following the reaction: CO2(g) + H2O = H+ + HCO3- (A4-1) HCO3- As the S/L ratio increases, more will be released from the clay to the liquid phase and so the concentration of HCO3 will rise. Following the reaction A4-1, at a constant pCO2, the H+ activity should decrease, i.e., increasing the pH. 9.2 9 8.8 pH 8.6 8.4 8.2 Ar box 8 N2/CO2 box 7.8 distilled water N2/CO2 box 7.6 0 100 200 300 400 500 600 700 800 900 S/L, g/L (wet clay/NaHCO3 solution) Figure A4-2: pH values in suspensions of leaching experiments. The role of S/L on the apparent leached concentrations Following the approach of B&B, the S/L ratio is the key system variable to derive concentrations of leached components from clay. For non-filtered samples, our data agrees with the data of B&B in the sense that the concentration generally increases as S/L. Figure A4-3 and -4 show the cases of Ca and Cl from one experiment as an example for cations and anions. Other data show similar behaviours and are therefore not plotted here. All data are listed in Table A4-1 and A4-2 at the end of this annex. 8 7 [Ca], mg/L 6 5 centrifuge 0.45 µm YM3 4 3 2 1 0 0 100 200 300 400 500 600 700 800 900 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-3: Calcium concentrations in the aqueous extracts as function of S/L. 146 3 2.5 [Cl], mg/L 2 centrifuge 0.45 µm YM3 1.5 1 0.5 0 0 100 200 300 400 500 600 700 800 900 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-4: Chlorine concentrations in the aqueous extracts as function of S/L. In the case of Ca, concentrations in the centrifuged extracts and in the 0.45 µm filtered extracts are identical suggesting that no coarse clay particulates containing Ca are present in the extracts. The important finding is that the Ca concentration is independent of S/L after the extracts were ultrafiltered by YM3 filters. This evidences the possible existence of colloids in the extracts. B&B didn’t use ultrafiltration to remove colloids, so we can’t compare our data with theirs. The detailed discussion about the filtration will be given in the next Section. In the case of Cl, concentration increases in all three samples indicating no colloids involving Cl are present in the extracts, as expected. The effect of filtration From the example of Ca as shown in Figure A4-3, the YM3 ultrafiltration effectively removed colloids, so the ‘real’ dissolved Ca concentration is independent of S/L. From this result, we conclude that the extracts obtained only by the centrifugation and the 0.45 µm filtration contain colloids and cannot be used for the determination of soluble concentration of Boom Clay components. The concentration of extracts should be obtained from the samples ultrafiltered by filters of nanometre scale. Because of the colloids, the observed S/L effect as given in the above Section is actually an effect of colloids. When the extracts were not ultrafiltered, more clay was added to the system, more colloids would release into the suspension as the result of agitation. This behaviour has been observed in our earlier study (Wang et al., 2002) and also found by Cremers and Maes, (1986) but interpreted as being due to the effects of natural organic matter. The real nature of colloids, i.e., if they are real clay colloids or natural organic matter colloids, is still not clear. As discussed in the Section of background, the S/L is the key system variable in the work of B&B and the effects of S/L are the central information for data interpretation. Our data however suggest no S/L effect for cations if ultrafiltration is applied, so our data cannot be interpreted in the same way as that of B&B. Also for the anion extraction data, our results indicate strong effects of clay compaction, so the anion concentration cannot be determined only by dilution, as done by B&B. The detailed procedure to derive the extracted cation and anion concentrations will be further elaborated when discussing the result of each element in later Sections in this annex. It is not clear from the work of B&B if colloids were also present in their systems since their samples were not ultrafiltered (or not reported). It is important to note however that the system of Opalinus Clay can be quite different from Boom Clay in terms of colloids stability. Opalinus Clay has a much higher ionic strength, up to 0.5 molal comparing to the low ionic strength of Boom Clay of 0.02 molal. It is possible that colloids are much less stable in Opalinus Clay because of the high ionic strength induced peptisation. Also, the Opalinus Clay may contain less natural organic matter especially the immobile fraction at the surfaces of clay. The presence of natural organic matter may enhance the stability of colloids. The effect of glove box atmosphere Because of the restricted availability of glove boxes, some experiments were performed in the Ar filled glove box and others were done in the N2/CO2 box. Figure A4-5 and -6 show the Ca and Cl concentrations measured under the two atmospheres. 12 10 [Ca], mg/L 8 Ar box N2/CO2 box 6 4 2 0 0 100 200 300 400 500 600 700 800 900 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-5: Calcium concentrations in the aqueous extracts as function of S/L under the Ar and the N2/CO2 atmospheres. 8 Ar box N2/CO2 box N2/CO2 box sample 2 7 [Cl], mg/L 6 5 4 3 2 1 0 0 100 200 300 400 500 600 700 800 900 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-6: Chlorine concentrations in the aqueous extracts as function of S/L under the Ar and the N2/CO2 atmospheres. 148 Figure A4-5 indicates that the evolutions of the Ca concentration are similar under the two atmospheres. The Ca concentration is slightly higher in the N2/CO2 box than in the Ar box. This is due to slightly higher solubility of calcite when CO2 is present, i.e., lower pH as indicated by Figure A4-2. The Cl concentrations are scattered. One sample (cross) shows a higher Cl concentration and is less independent on S/L in the N2/CO2 box. The other sample (triangle, sample 2) shows a similar behaviour to the case of the Ar box except one point at the S/L of 50 g/L. Both data points at 50 g/L in the N2/CO2 box are considerably higher than other data points suggesting experimental artefacts at this specific S/L. In general, there is no distinguished effects of the glove box atmosphere on the measured Cl concentration. The effect of leachant type Different from the work of B&B, we use in most of the experiments a NaHCO3 (0.01 M) solution as the leachant instead of distilled water. The use of NaHCO3 is to minimise the disturbances to the original Boom Clay geochemistry because Boom Clay pore water is a 15 mM NaHCO3 solution. The approach of B&B is indirect, i.e., the measurements must be interpreted by a kind of model before being able to calculate the real pore water compositions. Therefore, the type of leachant is not important, to our opinion, as long as the composition of the leachant is well defined. According to experiences, even a slight disturbance on Boom Clay geochemistry may cause severe changes of the pore water composition. We therefore decided to use NaHCO3 as the leachant. We also used distilled water for one sample in parallel to check the influence of the type of leachant (Figure A4-7) on the concentration of the extracts. 9 8 concentration, mg/L 7 6 Ca, NaHCO3 Ca, distilled water Cl, NaHCO3 Cl, distilled water 5 4 3 2 1 0 0 100 200 300 400 500 600 700 800 900 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-7: The effect of leachant type on the extracted concentrations of Ca and Cl as a function of S/L. For both the Ca and Cl cases, distilled water seems to extract more Ca and Cl into the aqueous phase comparing to the NaHCO3. The measurements in the distilled water samples show also less dependency on S/L. However, it is difficult to observe a general difference between the case of distilled water and the case of NaHCO3. Chlorine concentration in the extracts For easily soluble Cl, B&B assume that the source of Cl in the extracts can only be the original pore water in the Opalinus Clay. They found exactly the 1:1 correspondence between the Cl concentration and the dilution at the different S/L. To their data, the original Cl concentration in the pore water can be easily calculated by: CCl , pore = CCl ,ext S L where CCl,pore is the Cl concentration in the original pore water, CCl,ext is the Cl concentration in the extracts of batch leaching experiment, and S/L is the solid to liquid ratio used. B&B derived from 15 extracts a mean value of 12.3 mmol/kg Opalinus Clay. Apparently, the calculated CCl,pore in the case of Opalinus Clay is independent of S/L although the CCl,ext is dependent on S/L due to the dilution effect. Following the same approach, we also calculated the CCl,pore based on our leaching data but found out that the CCl,pore for Boom Clay is dependent on the S/L as shown in Figure A4-8. 100 converged at high S/L [Cl], mg/L 10 C(Cl, ext) C(Cl, pore) 1 0.1 0 200 400 600 800 1000 Solid/liquid, g/L (wet clay/NaHCO3 solution) Figure A4-8: Chlorine concentrations in the aqueous extracts as function of S/L. See text for the definitions of CCl,pore and CCl,ext. Although the CCl,ext (triangles in Figure A4-8) increases linearly as a function of S/L, the same as the case of B&B for the Opalinus Clay, the CCl,pore (cross symbols in Figure A4-8) calculated for the Boom Clay is not a constant value but decreases as a function of S/L. Also interesting is that the CCl,pore and CCl,ext are converging at the high end of the S/L. The observation of Figure A4-8 is not fully understood at the present time. A likely explanation is related to the colloids feature of the Boom Clay in terms of the double layer structure, hyper-filtration phenomena, and anion exclusion mechanism. The Boom Clay is known to have very small pores and the double layer is overlapped. Under such condition, anions are not free to move so under the confined conditions, i.e., when collecting pore water via piezometers or mechanical squeezing techniques, only part of the total Cl is extractable, so the Cl concentration is smaller than the inventory Cl concentration. In the batch experiment where the confined clay core is suspended, Cl ions are freed from the pores and the apparent concentration increases. This is probably why the Cl concentration is higher in a more diluted system. As the S/L increases, a negative potential increases (so as the stability of colloids) so 150 the Cl ions are trapped between pores in a similar way as under the in situ or squeezing conditions. This is at the moment a statement without being tested by rigorous calculations. Theories exist, for example, Donnan equilibrium in suspensions (Appelo, 1977), and should be applied to our experimental systems in the near future. Following this reasoning, the Cl concentration determined at the highest S/L should be close to the concentration measured in piezometer and squeezing waters. The Cl concentration measured at the lowest S/L should approach the Cl inventory in Boom Clay. With the known Cl inventory CCl,pore, i.e., the number of moles of Cl per kilogram of wet Boom Clay, we can calculate the Cl concentration in Boom Clay ‘total water’, the amount of water measured by the method of evaporation, if the water content in the sample is known: CCl = CCl , pore WH 2 O where CCl is the Cl concentration in Boom Clay total water, WH2O is the total water content in the sample from which the extraction was performed. Using all the data presented in Figure A4-8 and the average water content of 16.5 wt% (determined separately), CCl is calculated as being in the range of 15 to 425 mg Cl/kg water. As the highest S/L is favourable for determination of in situ Boom Clay pore water concentration, we calculated the average value from 11 samples of the HADES 2001/4 borehole and the value is 18 mg/kg water, which is lower than 26 mg/kg water as the reference Cl concentration. It is important to note that the value of 26 mg/kg water is the Cl concentration under the confined condition and can be higher than the real pore water concentration of Cl, due to the anion exclusion effect. The batch experiments show that the Cl inventory concentration can be as high as 425 mg/kg water. If the hyper-filtration and anion exclusion effects exist as expected, the Cl inventory can only be determined with batch experiments. Fluorine concentrations in the extracts If the Cl concentration in the Boom Clay is controlled by the double layer properties, the same is expected for F. A similar behaviour is indeed observed in the extracts for F, i.e., the extracted concentration increases linearly as a function of S/L but the inventory concentration decreases with S/L. The range of F concentration in the batch accessible water is: 21 to 104 mg/kg water. The average of 11 measurements on the highest S/L is 26 mg/kg water which is about 10 times higher than the value found in MORPHEUS waters as 2.6 to 3.3 mg/kg water. Sulphate and thiosulphate concentrations in the extracts The evolution of SO42- concentration in the extracts are similar comparing to that of Cl and F. The inventory concentration measured is in the range of 16 to 2542 mg/kg of batch accessible water. The high concentration is due to the oxidation of some samples. The average of 11 samples is 31 mg/kg water which is about 10 times higher than the reference water composition of 2 mg/kg. Thiosulphate ion was only measured in some samples suggesting the oxidation of these samples. Figure A4-9 shows the concentration of SO42- vs that of S2O32- and the linear relationship indicates the oxidation of the samples. 40 [S2O32-], ppm 30 20 10 0 0 40 80 120 160 200 2- [SO4 ], ppm Figure A4-9: Concentration of S2O32- as a function of SO42- in the extracts. Calcium concentration in the extracts As shown in the Figure A4-3, the Ca concentration is only independent to S/L if the extracted water was filtered by YM3 filters. The centrifuged and the 0.45 µm filtered samples contain colloids and hence cannot be used for the determination of the dissolved Ca concentration. We therefore only use the data from 8 determinations performed on two clay samples (Table A4-1). The average Ca concentration found is 1.5 mg/kg water which is close to the reference value of 2 mg/kg water. Iron concentration in the extracts Similar to Ca, iron was also found in colloidal fraction. Ultrafiltration data show all 8 determinations (Table A4-1) are under the detection limit of 0.05 mg/kg. This is an important finding suggesting that the Fe concentration measured in piezometer waters may contain colloids. Further tests are needed to clarify: (1) if the low Fe concentration found in batch system is due to the oxidation of the samples, i.e., a precipitation of Fe(III) oxi- and hydroxides; or (2) the high concentration of Fe, as we always assumed controlled by siderite solubility, is the result of the involvement of colloids. Magnesium concentration in the extracts Colloids containing Mg are also observed in the extracts. The average value of 8 measurements (Table A4-1) from the ultrafiltered samples is 1.8 mg/kg which is in good agreement with the 1.6 mg/kg derived from the reference water composition. Potassium concentration in the extracts There is no evidence that colloids involving K ions are present in the extracts. All data are quite consistent and the average value from the 8 ultrafiltration determinations is 8.9 mg/kg water which agrees with the value of reference water composition. Different from the case of Ca, Fe, and Mg, where non-filtered samples give higher concentrations due to colloids, the average of all the non-filtered K determinations is identical to the measurements in the ultrafiltered samples and is also 8.9 mg/kg. 152 Silicon concentration in the extracts There are no Si colloids observed in the extracts. Ultrafiltered samples result in a value of 3.8 mg/kg while the average of all the non-filtered samples is 2.8 mg/kg. The ultrafiltered value agrees better with the value 3.4 mg/kg derived from the reference water composition but we take the latter value since it is an average from many more determinations. Aluminium concentrations in the extracts The contents of Al from all samples were under the detection limit given as 0.2 mg/kg. The Al concentration of Boom Clay pore water should be much lower than that value. Water analysis in the future should determine Al with an improved detection limit. Natural organic matter concentration in the extracts Natural organic matter is present in Boom Clay pore water as colloids. The centrifugation and the 0.45 µm filtration do not result in different concentration of TOC (total organic carbon) suggesting no coarse particulate materials containing natural organic matter are present in the extracts. Our experiments with YM3 ultrafiltration failed due to the leaching of organic matter from the filter material. The concentration of TOC is therefore derived from non-filtered samples. From 11 measurements, the TOC ranges from 1284 to 5857 mg C/kg water. Following the reasoning given in the case of chlorine, if the value at the highest S/L represents most closely the value of the pore water, the average TOC from 11 samples is 2289 mg C/kg. This value is somewhat 10 times higher than the value determined in MORPHEUS water. If the electro-double layer properties such as hyper-filtration and anion exclusion are effective in the Boom Clay, they will affect the migration of natural organic matter as well. There has been a commonly observed feature in Boom Clay when collecting pore water from piezometers: the TOC at the beginning stage of water collection is always high and gradually decrease till a certain plateau value (Van Geet, 2004). This is perhaps related to the anion exclusion phenomena as explained by Appelo (1999) so that the natural organic matter molecules are hesitant to enter the pores with negative potential and will show an earlier breakthrough than water. Concluding remarks • the B&B approach for batch experiments cannot be directly applied to Boom Clay suspensions mostly because of the involvements of colloids (for cations) and the electro double layer related behaviours (for anions) of the Boom Clay. The colloids and the double layer properties may well be the reasons for the observed differences between Opalinus Clay and Boom Clay; • for the major cations, the batch leaching method provides comparative results to those determined by piezometric and squeezing techniques if ultrafiltration is applied to remove the colloids; • for the anions, the batch leaching method in general results in higher concentrations comparing to piezometric and squeezing techniques suggesting that an interpretation using double layer theory is needed to correct the in situ compaction of the Boom Clay; • colloids containing Ca, Mg, and Fe are evidenced by ultrafiltration experiments. This finding may suggest that colloids are of the secondary mineral nature. No colloids of K and Si are found indicating that no clay colloids are present. Future experiments should focus on the determination of Al concentration and the possible involvement of Al colloids to test the above statements; • finally, the batch leaching experiment provides a way to determine the inventory concentration of Cl and other anions in Boom Clay. The inventory concentration of, e.g., Cl is important for the corrosion study of overpack materials. Due to the in situ compaction of the Boom Clay and the related double layer properties, piezometric and squeezing techniques tend to underestimate the total inventory concentration of anions. 154 Table A4-1: Cation concentrations measured in batch leaching extracts. Glove box atmosphere Ar. S/L 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 No. (1)-0.5 (1)-1 (1)-4 (1)-16 (2)-0.5 (2)-1 (2)-4 (2)-16 (3)-0.5 (3)-1 (3)-4 (3)-16 (4)-0.5 (4)-1 (4)-4 (4)-16 (5)-0.5 (5)-1 (5)-4 (5)-16 (6)-0.5 (6)-1 (6)-4 (6)-16 (7)-0.5 (7)-1 Al <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 Ca 1.26 1.47 2.5 6.9 1.51 1.79 3.0 7.4 1.49 1.61 2.9 7.4 1.67 1.85 2.6 6.1 1.56 1.60 2.6 6.4 1.63 1.60 2.7 6.8 1.68 1.93 0.45µm YM3 Fe 0.12 0.10 0.20 0.83 0.06 0.13 0.29 0.95 1.50 1.48 0.08 1.64 1.30 0.11 2.8 1.2 0.37 7.6 1.3 1.15 1.64 1.40 0.08 1.86 1.57 0.08 2.5 1.6 0.20 6.0 2.2 0.55 0.05 0.09 0.22 0.86 0.06 0.08 0.24 1.03 0.06 0.13 0.45µm YM3 0.35 0.39 1.06 2.80 0.29 0.26 0.67 1.60 <0.05 <0.05 <0.05 <0.05 <0.05 <0.05 <0.05 <0.05 Mg 1.17 1.22 1.82 4.2 1.28 1.40 2.05 4.4 1.17 1.36 2.03 4.6 1.24 1.53 1.84 4.1 1.19 1.16 1.68 3.7 1.12 1.22 1.74 3.9 1.29 1.44 0.45µm YM3 K 6.9 8.1 9.3 10.6 7.1 8.0 9.6 11.2 1.30 1.77 6.7 1.50 1.72 8.1 2.09 1.57 9.0 4.3 1.4 9.8 1.32 1.76 8.4 1.58 1.92 10.8 1.89 1.88 11.0 3.8 2.6 14.3 7.5 8.0 9.2 10.0 7.4 7.9 9.0 9.7 7.5 8.4 0.45µm YM3 Si 2.5 2.4 3.2 3.3 2.5 2.4 2.7 3.3 6.4 6.3 2.4 7.4 7.3 2.4 8.5 8.4 2.9 10.0 8.6 3.8 7.8 7.6 2.7 10.2 10.2 2.6 10.0 9.8 2.8 13.0 13.0 3.0 2.3 2.7 3.0 3.8 2.2 2.3 3.3 3.5 2.7 2.7 0.45µm YM3 Na 242 241 250 283 239 244 254 289 3.5 3.3 241 3.1 3.2 245 3.7 3.8 257 10.0 4.8 278 3.4 3.6 249 3.5 3.4 254 3.9 4.1 272 6.0 4.0 354 243 246 259 285 245 242 256 278 249 251 200 800 25 50 200 200 200 200 200 200 800 25 50 200 800 25 50 200 800 25 50 200 800 (7)-4 (7)-16 (8)-0.5 (8)-1 (8)-4 (8)-4-1 (8)-4-2 (8)-4-3 (8)-4-4 (8)-4-5 (8)-16 (9)-0.5 (9)-1 (9)-4 (9)-16 (10)0.5 (10)-1 (10)-4 (10)16 (11)0.5 (11)-1 (11)-4 (11)16 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 <0.2 3.5 9.9 1.37 1.54 2.7 2.3 2.22 2.7 2.4 2.6 7.2 1.63 1.93 3.1 8.7 0.34 1.27 0.09 0.11 0.29 0.18 0.16 0.25 0.21 0.24 0.99 <0.05 0.12 0.32 1.20 2.27 5.7 1.23 1.22 1.86 2.01 2.15 2.28 1.93 1.83 4.4 1.18 1.31 1.96 5.2 9.1 9.7 6.7 8.2 9.6 9.1 9.2 9.2 9.3 9.0 11.2 6.7 7.6 8.6 11.0 3.1 3.3 2.0 2.4 2.8 3.4 3.3 3.5 3.2 2.9 3.1 2.6 2.8 3.0 3.5 259 289 244 257 290 260 268 269 266 269 313 253 259 271 337 <0.2 1.29 <0.2 1.63 <0.2 2.5 <0.05 0.10 0.27 1.28 1.18 1.69 7.8 8.6 10.0 2.4 2.4 2.5 249 253 297 <0.2 3.9 0.35 2.55 11.6 2.5 318 <0.2 1.12 <0.2 1.41 <0.2 2.15 <0.05 0.09 0.30 0.93 1.22 1.50 6.5 7.6 8.6 2.1 2.2 2.6 244 247 258 <0.2 4.5 0.43 2.65 9.6 2.9 294 156 Table A4-2: Anion concentrations measured in batch leaching extracts. Glove box atmosphere Ar. S/L 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 800 25 50 200 200 200 200 200 200 800 25 50 200 800 No. (1)-0.5 (1)-1 (1)-4 (1)-16 (2)-0.5 (2)-1 (2)-4 (2)-16 (3)-0.5 (3)-1 (3)-4 (3)-16 (4)-0.5 (4)-1 (4)-4 (4)-16 (5)-0.5 (5)-1 (5)-4 (5)-16 (6)-0.5 (6)-1 (6)-4 (6)-16 (7)-0.5 (7)-1 (7)-4 (7)-16 (8)-0.5 (8)-1 (8)-4 (8)-4-1 (8)-4-2 (8)-4-3 (8)-4-4 (8)-4-5 (8)-16 (9)-0.5 (9)-1 (9)-4 (9)-16 F0.26 0.42 1.07 3.27 0.35 0.53 1.46 3.7 0.30 0.44 1.27 3.12 0.43 0.52 1.26 2.82 0.32 0.50 1.37 3.48 0.31 0.44 1.35 3.26 0.32 0.53 1.50 3.8 0.32 0.46 1.40 1.20 1.21 1.24 1.22 1.27 3.10 0.37 0.56 1.58 3.7 Cl0.55 0.61 0.92 2.30 0.66 0.64 0.81 2.35 0.56 0.74 0.86 2.22 1.00 8.1 1.31 2.15 0.47 0.75 0.83 2.18 0.67 0.61 0.77 2.69 1.44 0.66 0.77 2.36 1.09 1.20 2.05 0.83 1.01 0.92 0.93 0.86 2.82 0.91 0.48 1.12 2.50 0.45 µm 0.85 1.02 1.22 2.77 1.77 8.2 1.62 2.87 YM3 Br<0.25 <0.25 <0.25 <0.25 0.27 <0.25 <0.25 <0.25 1.12 <0.25 1.11 0.27 1.22 <0.25 2.49 <0.25 1.38 <0.25 8.3 0.37 1.79 <0.25 2.89 <0.25 <0.25 <0.25 <0.25 0.34 <0.25 0.28 <0.25 0.28 0.43 0.32 0.34 0.30 0.30 <0.25 <0.25 1.06 0.66 0.52 0.54 0.34 0.25 0.32 0.26 0.35 0.28 HPO42<0.5 <0.5 0.73 1.60 <0.5 <0.5 0.71 1.47 <0.5 <0.5 0.66 1.58 <0.5 <0.5 <0.5 0.50 <0.5 <0.5 0.80 1.70 <0.5 <0.5 0.75 1.52 <0.5 <0.5 0.77 1.58 <0.5 <0.5 17.1 0.70 0.65 1.53 0.55 0.61 1.05 <0.5 <0.5 0.74 1.30 SO420.79 1.03 1.19 4.40 1.03 1.40 2.20 6.4 0.96 0.62 1.01 2.22 10.5 16.7 35.0 166 0.97 0.62 1.28 3.78 1.18 0.74 1.10 2.91 0.64 0.69 1.07 2.96 9.6 6.6 18.1 24.5 27.8 18.7 19.6 22.5 55.1 4.35 5.55 15.4 58.5 0.45 µm 1.18 1.17 1.60 3.85 10.4 16.7 35.8 157 YM3 2.08 1.35 1.63 2.85 10.7 17.2 34.8 165 S2O32(1) <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 2.82 32.6 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 <1 4.06 <1 <1 5.23 3.89 4.31 10.1 <1 <1 2.73 11.2 25 50 200 800 25 50 200 800 (10)-0.5 (10)-1 (10)-4 (10)-16 (11)-0.5 (11)-1 (11)-4 (11)-16 0.38 0.62 1.49 3.9 0.33 0.57 1.41 3.9 1.75 0.55 2.23 2.04 0.73 2.03 1.18 1.95 0.35 <0.25 0.26 0.29 <0.25 0.46 0.25 0.34 <0.5 <0.5 15.6 0.66 <0.5 1.15 0.60 1.09 3.98 7.4 19.3 71 1.74 3.23 7.1 23.2 <1 <1 1.76 5.05 <1 <1 <1 2.96 158 Table A4-3: Major cation and anion concentrations measured in the N2/CO2 box and in distilled water leachant for the purpose of comparison. S/L 25 50 200 800 (3) - II - 0,5g (3) - II - 1g (3) - II - 4g (3) - II - 16g Ca [ppm] 2.07 2.08 3.1 9.6 Fe [ppm] 0.05 0.09 0.32 1.39 Mg [ppm] 1.89 1.88 2.24 5.7 K [ppm] 9.2 13.1 10.7 11.7 Si [ppm] 2 2.1 2.8 2.8 Cl [ppm] 2.55 7.2 2.83 2.96 SO4 [ppm] 1.29 1.2 1.76 4.76 25 50 200 800 (4) - II - 0,5g (4) - II - 1g (4) - II - 4g (4) - II - 16g 3.5 2.79 2.9 6.7 <0,05 <0,05 0.22 0.51 2.24 2.07 2.2 4.4 9.3 11.8 11.9 14.1 2.1 2.2 3 2.4 1.12 3.07 1.19 2.77 8.1 11.9 37.1 168 25 50 200 800 (4) - H2O - 0,5g (4) - H2O - 1g (4) - H2O - 4g (4) - H2O - 16g 4.6 3.1 3.1 7.5 0.17 0.09 0.35 0.78 3.1 1.82 1.55 3.8 9.6 9.8 8.2 10.7 2.4 1.8 1.9 1.8 2.16 2.46 6.7 2.97 7.2 13.5 38.1 166 Table A4-4: Concentrations of natural organic matter in batch leaching extracts. Glove box atmosphere Ar. Samples centrifuged at 21,255 g for 2 hrs. S/L 25 50 200 800 1 TOC ext [ppm] 19 28 80 319 pore 2 TOC ext 4688 3407 2438 2417 [ppm] 21 38 118 370 pore 3 TOC ext 4972 4621 3573 2799 [ppm] 19 32 118 395 pore 4 TOC ext 4640 3899 3588 2994 [ppm] 21 35 73 245 pore 5 TOC ext 5152 4296 2217 1852 [ppm] 16 25 75 290 pore 6 TOC ext 3884 2972 2276 2199 [ppm] 20 26 84 319 pore 7 TOC ext 4812 3155 2551 2418 [ppm] 24 42 126 488 pore 8 TOC ext 5857 5075 3824 3696 [ppm] 24 32 91 366 pore 9 TOC ext 5833 3853 2748 2773 [ppm] 19 36 123 450 pore 10 TOC ext 4507 4417 3718 3407 [ppm] 16 32 73 170 pore 11 TOC ext pore 3976 3856 2198 1285 [ppm] 15 28 63 190 3583 3347 1894 1441 Table A4-5: Comparisons of the TOC content as the result of centrifugation, filtration, and ultrafiltration of the leaching extracts. Glove box atmosphere N2/CO2 TC IC TOC St. Dev. TC IC TOC St. Dev. (3) - 0,5 not filtered 125.6 108.5 17.11 0,45µm 127.7 110.5 17.18 0.77 0.836 YM3 5,103 217.7 4885 (3) - 1 not filtered 137.5 110.6 26.85 0,45µm 157.7 111.6 46.12 47.40 1.778 4.963 (4) - 0,5 not filtered 127.5 109.8 17.73 0,45µm 127.8 110.3 17.48 YM3 3,873 163.1 3710 (4) - 1 not filtered 148.7 113.8 34.93 0.672 1.436 36.53 2.807 YM3 3,974 205.8 3769 (3) - 4 not filtered 215.2 113.1 102 YM3 4,157 185.5 3971 (3) - 16 not filtered 476.5 119 357.5 0,45µm 217.3 114.1 103.2 0,45µm 479.7 119 360.7 YM3 4,312 185.6 4127 29.42 1.244 0.706 25.47 1.798 1.677 21.25 0,45µm 144.2 113.7 30.5 YM3 4,562 170.9 4392 (4) - 4 not filtered 180.5 113.7 66.77 0,45µm 178.7 113.6 65.1 YM3 4,221 126.9 4094 (4) - 16 not filtered 333.7 115 218.7 0,45µm 332 113.8 218.2 YM3 4,938 153.5 4784 1.595 0.00 0.825 0.841 21.39 3.106 1.452 42.33 160 Annex 5: Boom Clay pore water geochemistry: analytical data used in the statistical analyses EG/BS (statistical group 1) 162 EG/BS (statistical group 2) ARCHIMEDE #1 reference Mol(8) Mol(8) Mol(8) Mol(8) Mol(8) Mol(7) Mol(7) Mol(14) Mol(14) Mol(14) Mol(14) Mol(14) Mol(15) Mol(15) N°piezo A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 A50001 N°Echant A07502 A07505 A07509 A07510 A07513 A07501 A07514 A07503 A07506 A07507 A07508 A07512 A07504 A07511 Prof 8 8 8 8 8 7 7 14 14 14 14 14 15 15 date perlvt pH 09.07.1992 8,5 28.10.1992 8,86 06.11.1992 17.11.1992 24.03.1993 09.07.1992 8,5 24.03.1993 09.07.1992 8,7 28.10.1992 8,8 17.11.1992 8,7 30.11.1992 8,8 24.03.1993 09.07.1992 8,7 24.03.1993 only 1 group based on ANOVA1 analysis of Cl, F, Na, K, Ca, Mg mean 8,695 SPRING 116 ppm alcalini 732,24 762,75 762,75 713,934 732,24 713,934 701,73 726,138 ppm SO4 4,803 4,51482 3,3621 3,65028 3,16998 4,3227 2,30544 4,3227 3,74634 2,97786 3,07392 2,78574 2,8818 2,11332 ppm NO3 31 0,806 0,62 1,116 0,31 0 0,248 0,62 0,434 <0.372 1,24 0,248 0 0,31 ppm Cl 17,725 18,434 17,016 17,016 17,725 17,725 17,725 17,725 17,725 18,0795 18,0795 17,3705 17,725 17,016 ppm Br 7,8302 730,7145 3,430714 2,842462 17,64904 7,8302 ppm F 0,152 1,9 1,672 1,71 3,04 0,152 2,85 0,152 1,729 2,09 1,691 2,85 0,152 2,85 ppm S 2O 3 n.d. n.d. n.d. n.d. n.d. 0,67278 n.d. n.d. n.d. n.d. 1,00917 n.d. ppm PO4 0,66479 0,66479 0,66479 0,66479 0,56982 0,28491 0,66479 0,66479 0,56982 0,47485 0,75976 0,56982 0,37988 0,66479 ppm SIO2 9,012 10,2136 9,6128 10,2136 ppm Na 285,076 275,88 268,983 273,581 280,478 ppm K 7,82 7,82 7,429 7,429 10,166 ppm Ca 2,004 2,004 1,6032 1,6032 1,92384 ppm Mg 1,2155 1,7017 1,4586 1,4586 1,7017 287,375 10,166 2,72544 1,67739 275,88 278,179 278,179 280,478 289,674 280,478 7,429 7,038 7,038 9,384 7,82 8,211 1,76352 1,6032 1,6032 1,92384 1,6032 1,8036 1,55584 1,4586 1,33705 1,7017 0,9724 1,4586 ppm Li ppm NH4 0,0902 0,029842 0,5412 0,029842 0,43296 0,029842 0,46904 3,7884 ppm Fe ppm Mn ppm Al 0,072605 0,170314 0,17872 0,488966 0,161965 0,071422 0,035883 n.d. 7,2096 8,4112 9,012 8,4112 7,2096 1,642143 0,840975 0,590171 8,811733 279,5201 8,145833 1,84702 5,0512 0,029842 0,39688 0,029842 <0.054 0,006038 <0.054 3,4276 1,9844 4,1492 0,17872 0,032964 0,028059 0,23457 0,065928 0,028059 0,161965 0,076916 0,028059 1,474807 0,025875 2,033108 0,164758 0,151085 0,030015 164 ORPHEUS MORPHEUS (major elements) 166 MORPHEUS (trace elements) Annex 6: Statistics: methodology ANOVA1 First it is checked whether data provided by the different filters of one single piezometer all belong to the same population. In case of the EG/BS, consisting of 1 major filter, two different groups in time are compared. This is done element per element using an analysis of variance (ANOVA1). For this ANOVA test, the mean ( xi ), standard deviation (si) and amount of samples (ni) of each group (k groups) is calculated. The grand mean ( x ) is calculated as the mean of all groups together. Next the between group variability (SSB) is calculated as : k ( SS B = ∑ ni ⋅ xi − x i =1 ) 2 and the degrees of freedom for the between group variability is dfB=k-1. The variance between groups is then defined as s B2 = SS B . df B The within groups variability is defined as SSW = ∑ (ni − 1) ⋅ si2 . The degrees of freedom are defined as dfW=N-k, with N the number of samples of all groups together SS and the variance within groups is defined as sW2 = W . The ratio of the variance df W s B2 between groups and the variance within groups is the F statistic: Fstat = 2 . Under the sW null hypothesis, namely that the mean value of each group is not differing, this test statistic has an F sampling distribution with dfB and dfW degrees of freedom. The probability (p) value can then be calculated. The null hypothesis is accepted if p>0.05. Based on this information it will be possible to consider all filters of one piezometer as one single population or to assume several groups within one piezometer. The basic assumptions when using the ANOVA 1 test are All data are normally distributed The variance within each of the populations is equal. As only a limited amount of data are available, both assumptions cannot be tested on all data sets. The EG/BS data are the largest and thus these are used to test the first assumption of normallity of the data. Figure A6-1 illustrates the normal-quantile-quantile plots for all elements of both data sets discriminated in the EG/BS data. From these plots it is concluded that all elements are normally distributed, although some tailings are sometimes observed. For the other filters, not enough data points are available to check the normallity. However, it is assumed that the assumption of normally distributed data can be transposed to all filters of all piezometers. 168 170 Figure A6-1: Normal – quantile-quantile plots of all elements of the two groups of data discriminated in the EG/BS data set (left: EG/BS1, referring to the data from 1996-03-13 till 1999-04-07, and right EG/BS2, refering to the data after from 200008-08 till 2003-02-17). The second assumption of homogeneity of variance is especially important when the number of measurments in each group are not equal. Therefore, this should be checked for the EG/BS and ORPHEUS data sets. The Brown-Forsythe test can be used to check for homogeneity of variance in the data considered. This test is simple to execute and is robust to nonnormality. The steps are: • Calculate the median score, Mdi, for each of the i groups; • Replace each observed score, Yij, with a new variable, Zij, which is equal to the absolute difference of the observed score and group median. That is: Zij=|Yij-Mdi| • Run an ANOVA on the new scores, Zij. The overall F is a test of the hypothesis that all groups are drawn form populations having the same variance. The results of this Brown-Forsythe test is given in Table A6-1. It can be concluded that for EG/BS and ORPHEUS data sets, there is no need to worry about a violation of the homogeneity of variance assumption. Table A6-1: P-values of the Brown-Forsythe test to check the hypothesis that there is a homogeneity of variance. P-values above 0.05 (in bold) are accepted as significant. Ca Cl F Fe HCO3 K Mg Na Si EG/BS 0.92 0.38 0.15 0.07 ORPHEUS 0.16 0.19 0.19 0.72 0.90 0.61 0.18 0.40 0.32 0.52 0.30 0.38 0.66 MANOVA Multivariate analysis of variance (MANOVA) is simply an ANOVA with several dependent variables. Instead of a univariate F value, we would obtain a multivariate F value (Wilks' lambda) based on a comparison of the error variance/covariance matrix and the effect variance/covariance matrix. Although Wilks' lambda has been used here, there are other statistics that may be used, including Hotelling's trace and Pillai's criterion. Testing the multiple dependent variables is accomplished by creating new dependent variables that maximise group differences. These artificial dependent variables are linear combinations of the measured dependent variables. The basic assumptions when using the MANOVA test are: • Data are independent • Data are normally distributed • No big difference between the variances between the groups of the same variables • MANOVA is sensitive to outliers First of all it is assumed that the data are independent. Moreover, the normality of data is assumed as mentioned before in the ANOVA test. The assumption of homogeneity of variance is especially important when the group samples are not equal. Therefore, it should only be considered for the question on the effect of filter material on pore water composition. A possible test of the homogeneity of variance is Box's M test. This test could, however, not be applied on the question of the effect of filter material on pore water composition, since the matrix is singular. It is clear that additional data are needed to confirm the obtained results so far. To avoid any problems with outliers, these have been removed before performing the MANOVA test. Data are considered outliers when they fall outside the range of 1.5 times the interquartile range away from the 25th or 75th percentile of the sample. 172 Annex 7: The input and output files from geochemical modelling simulations Input file This input calculates the variation of the Boom Clay pore water composition while the system pCO2 is varying between 10-2.8 to 10-2.2 atm at 16°C in equilibrium with the minerals calcite, pyrite, siderite, chalcedony, and kaolinite. An ion exchange complex of 0.925 eq per 5 kg of clay, i.e., 18.5 meq/100 g clay is also present. # React script, saved Tue Dec 09 2003 by lwang data = "C:\Program Files\Gwb\Gtdata\MOLDATA.dat" verify surface_data = "C:\Program Files\Gwb\Gtdata\MOLDATA ION EX.dat" exchange_capacity IonEx = 0.925 eq work_dir = "N:\USERS\Lwang\Projects\Topical reports\Pore water chemistry\GWB" temperature = 16 swap Calcite for Ca++ swap Pyrite for O2(aq) swap Siderite for Fe++ swap CO2(g) for HCO3swap Chalcedony for SiO2(aq) swap Kaolinite for Al+++ 1 kg free H2O free kg Calcite = .15 free gram Pyrite = 100 free gram Siderite = 5 fugacity CO2(g) = .00158489319 free gram Chalcedony = 2000 free gram Kaolinite = 1000 total mol Na+ = .01417 total mol Mg++ = 5.41e-5 total mol K+ = .000168 total mg/kg SO4-- = 2.31 balance on H+ total mg/kg Cl- = 26 slide log fugacity of CO2(g) to -2.2 suppress >X2:Fe >X3:Al alter >X:K -1.328 -1.328 -1.328 -1.328 -1.328 -1.328 -1.328 -1.328 alter >X2:Ca -.843 -.843 -.843 -.843 -.843 -.843 -.843 -.843 alter >X2:Mg -.678 -.678 -.678 -.678 -.678 -.678 -.678 -.678 extrapolate precip = off 174 Output file This output only lists the results at the step 30 of the simulation which has been used as the reference Boom Clay pore water composition as given in Table 5-1 of this report. The full output file is too lengthy to be included in this annex and can be obtained if desirable. Step # 30 Xi = 0.3000 Temperature = 16.0 C pH = 8.487 Eh = -0.2744 volts Pressure = log fO2 = pe = 1.013 bars -71.013 -4.7833 Ionic strength = 0.016057 Activity of water = 0.999974 Solvent mass = 0.999986 kg Solution mass = 1.001316 kg Solution density = 1.019 Chlorinity = 0.000733 molal Dissolved solids = Rock mass = Carbonate alkalinity= g/cm3 1329 mg/kg sol'n 3.254925 kg 756.71 mg/kg as CaCO3 IonEx sorbing surface: Exchange capacity = Reactants 0.925 eq moles moles grams cm3 remaining reacted reacted reacted ---------------------------------------------------------------------------CO2(g) -- sliding fugacity buffer -- Minerals in system moles log moles grams volume (cm3) ---------------------------------------------------------------------------Calcite 1.498 0.175 149.9 55.32 Chalcedony 33.29 1.522 2000. 755.2 Kaolinite 3.874 0.588 1000. 385.5 0.8335 -0.079 100.0 19.95 0.04316 -1.365 Pyrite Siderite 5.000 _____________ (total) Aqueous species 3255. molality mg/kg sol'n act. coef. 1.268 _____________ 1217. log act. --------------------------------------------------------------------------Na+ HCO3- 0.01531 351.6 0.8829 -1.8690 0.01442 878.9 0.8829 -1.8950 Cl- 0.0007320 25.92 0.8789 -3.1916 NaHCO3(aq) 0.0002941 24.67 1.0000 -3.5315 CO3-- 0.0002409 14.43 0.6104 -3.8326 K+ 0.0001855 7.244 0.8789 -3.7877 SiO2(aq) 0.0001149 6.894 1.0000 -3.9397 CO2(aq) 0.0001084 4.766 1.0000 -3.9648 Mg++ 5.535e-005 1.344 0.6443 -4.4478 Ca++ 3.928e-005 1.572 0.6257 -4.6094 SO4-- 2.251e-005 2.159 0.6051 -4.8659 NaCO3- 9.503e-006 0.7877 0.8829 -5.0762 CaCO3(aq) 6.437e-006 0.6434 1.0000 -5.1913 MgHCO3+ 5.709e-006 0.4865 0.8829 -5.2975 MgCO3(aq) 4.585e-006 0.3861 1.0000 -5.3387 CaHCO3+ 4.042e-006 0.4081 0.8829 -5.4475 HSiO3- 3.394e-006 0.2613 0.8829 -5.5234 NaHSiO3(aq) 2.207e-006 0.2206 1.0000 -5.6563 OH- 1.686e-006 0.02864 0.8809 -5.8281 FeCO3(aq) 1.619e-006 0.1873 1.0000 -5.7908 NaCl(aq) 1.385e-006 0.08086 1.0000 -5.8584 NaSO4- 1.378e-006 0.1638 0.8829 -5.9149 FeHCO3+ 1.245e-006 0.1453 0.8829 -5.9589 Fe++ 3.884e-007 0.02166 0.6257 -6.6143 MgSO4(aq) 9.967e-008 0.01198 1.0000 -7.0014 CaSO4(aq) 4.140e-008 0.005629 1.0000 -7.3830 AlO2- 2.401e-008 0.001414 0.8829 -7.6736 FeOH+ 2.036e-008 0.001481 0.8829 -7.7453 MgCl+ 2.007e-008 0.001198 0.8829 -7.7515 KSO4- 1.897e-008 0.002561 0.8829 -7.7760 (only species > 1e-8 molal listed) Exchanging species molality moles act. coef. activity log activity ------------------------------------------------------------------------------------>X:Na 0.4339 0.4339 1.0811 0.4690 -0.3288 >X:K 0.1114 0.1113 1.0811 0.1204 -0.9195 >X2:Ca 0.09532 0.09531 2.1621 0.2061 -0.6860 >X2:Mg 0.09458 0.09458 2.1621 0.2045 -0.6893 Mineral saturation states log Q/K log Q/K ---------------------------------------------------------------Nontronite-Na 3.6185s/sat Cristobalite(alp -0.2915 Nontronite-Mg 3.5426s/sat Greenalite -0.3026 Nontronite-Ca 3.4947s/sat Dolomite-dis -0.3125 Nontronite-K 3.3517s/sat Magnesite -0.3858 Nontronite-H 2.4546s/sat Sanidine_high -0.3942 Stilbite 2.2431s/sat Albite -0.4100 Mesolite 1.6165s/sat Boehmite -0.4626 Celadonite 1.4773s/sat Coesite -0.5542 Dolomite-ord 1.2994s/sat Gibbsite -0.5955 Dolomite 1.2994s/sat Saponite-H -0.6214 Muscovite 1.2476s/sat Beidellite-Na -0.6399 Cronstedtite-7A 1.0652s/sat Beidellite-Mg -0.7159 Hematite 1.0306s/sat Analcime -0.7471 Minnesotaite 0.9181s/sat Cristobalite(bet -0.7609 Annite 0.9048s/sat Beidellite-Ca -0.7637 Maximum_Microcli 0.8662s/sat Dawsonite -0.8067 K-Feldspar 0.8647s/sat Monohydrocalcite -0.8106 Montmor-Na 0.6158s/sat Smectite-high-Fe -0.8356 176 Magnetite 0.5481s/sat Phlogopite -0.8453 Montmor-Mg 0.5439s/sat Scolecite -0.8973 Saponite-Na 0.5424s/sat Beidellite-K -0.9066 Saponite-Mg 0.4675s/sat Paragonite -0.9483 Montmor-Ca 0.4232s/sat SiO2(am) -1.0811 Saponite-Ca 0.4186s/sat Pyrophyllite -1.1561 Daphnite-14A 0.4157s/sat Mordenite -1.2239 Montmor-K 0.3533s/sat Ferrosilite -1.3049 Quartz 0.2797s/sat Albite_high -1.7893 Saponite-K 0.2756s/sat Beidellite-H -1.8036 Tridymite 0.0982s/sat Clinoptilolite-h -1.9538 Illite 0.0934s/sat Clinoptilolite-C -2.0052 Clinoptilolite-N 0.0540s/sat Ripidolite-14A -2.0697 Clinoptilolite-h 0.0537s/sat Jadeite -2.3127 Goethite 0.0489s/sat Clinoptilolite-K -2.4232 Talc 0.0206s/sat Laumontite -2.4750 Calcite 0.0000 sat Chrysotile -2.5484 Chalcedony 0.0000 sat Natrolite -2.5708 Kaolinite 0.0000 sat Chamosite-7A -2.5782 Siderite 0.0000 sat Clinoptilolite-h -2.6250 0.0000 sat Pyrite Lansfordite -2.7484 Diaspore -0.0432 Huntite -2.7944 Ice -0.1042 C -2.8648 Aragonite -0.1448 Kalsilite -2.9715 Smectite-low-Fe- -0.2040 (only minerals with log Q/K > -3 listed) Gases fugacity log fug. ----------------------------------------------H2O(g) CO2(g) 0.01481 -1.829 0.002399 -2.620 H2(g) 2.383e-008 -7.623 CH4(g) 1.536e-009 -8.814 H2S(g) 5.785e-010 -9.238 CO(g) 1.859e-014 -13.731 HCl(g) 4.093e-019 -18.388 SO2(g) 4.046e-025 -24.393 S2(g) 1.529e-030 -29.815 C2H4(g) 6.335e-034 -33.198 Na(g) 3.505e-059 -58.455 K(g) 8.817e-062 -61.055 Cl2(g) 2.766e-064 -63.558 O2(g) 9.705e-072 -71.013 Mg(g) 2.052e-098 -97.688 Ca(g) 5.390e-122 -121.268 C(g) 4.188e-125 -124.378 Al(g) 2.671e-145 -144.573 Si(g) 6.140e-158 -157.212 In fluid Original basis total moles moles Sorbed mg/kg moles Kd mg/kg L/kg ------------------------------------------------------------------------------>X:Na 0.925 Al+++ 7.75 2.44e-008 0.000657 Ca++ 1.59 4.98e-005 1.99 Cl- 0.000733 0.000733 26.0 Fe++ 0.877 3.27e-006 0.183 H+ -23.1 -0.000162 -0.163 0.0953 3.81e+003 H2O 74.0 HCO3- 1.56 0.0151 920. 0.112 0.000186 7.24 0.111 4.35e+003 Mg++ 0.0946 6.58e-005 1.60 0.0946 2.30e+003 Na+ 0.449 0.0156 359. 0.434 9.96e+003 O2(aq) -2.92 -4.49e-009 -0.000143 K+ 55.5 9.99e+005 SO4-- 1.67 2.40e-005 2.31 SiO2(aq) 41.0 0.000120 7.23 Sorbed fraction log fraction -----------------------------------------------Ca++ 0.9995 -0.000 K+ 0.9983 -0.001 Mg++ 0.9993 -0.000 Na+ 0.9652 -0.015 Elemental composition In fluid total moles moles Sorbed mg/kg moles mg/kg ------------------------------------------------------------------------------Aluminum 7.747 2.438e-008 0.0006569 Calcium 1.593 4.981e-005 1.994 181.1 Carbon 1.556 0.01510 Chlorine 0.0007334 0.0007334 25.97 Hydrogen 126.5 111.0 1.118e+005 0.1825 Iron 0.8766 3.273e-006 0.09465 6.577e-005 1.596 161.6 55.55 8.877e+005 0.1115 0.0001855 7.245 Silicon 41.03 0.0001205 3.379 Sodium 0.4495 0.01562 358.6 Sulfur 1.667 2.405e-005 0.7701 Magnesium Oxygen Potassium 0.09531 3815. 0.09458 2296. 0.1113 4348. 0.4339 9961. 178 Annex 8: Prescriptions for the preparation of synthetic Boom Clay water (SBCW) General Boom Clay pore water is basically a NaHCO3 solution of 15 mM. A 15mM NaHCO3 solution can thus be used in the experiments. SBCW as used in the section 'Geological Disposal' The directives for the preparation of Synthetic Boom Clay Water which is used in the section 'Geological Disposal' is recently published by Maes et al. (2000 and 2004). The directives are the following: If you are planning to work in anaerobic conditions, make sure your water is degassed! Always leave the bottle open in the glovebox, to allow the last ppm O2 to diffuse out. Be aware of the oxygen level while you do that. Then, close the bottle. To prepare 1 L of synthetic clay water, add the following products to 1 L of high quality water (demineralised, bidistilled, milli-Q,...) : Table 1: Weight of salts needed for the preparation of 1 L of SBCW Salt Quantity (mg) sorted by descending order NaHCO3 1 170 mg H3BO3 43 mg KCl 25 mg MgCl2 · 6 H2O 22 mg (hygroscopic salt !) NaF 11 mg NaCl 10 mg FeCl2 3 mg (very sensitive to oxidation !) Na2SO4 0.3 mg (10 ml of 3 mg dissolved in 100 ml) TDS 1 284.3 mg (-12 mg H2O) = 1 272.3 mg (anhydr.) TDS: Total Dissolved Salts (control of mass balance in Table 2). • • • • MW 83.996 61.834 74.555 203.218 41.988 58.443 126.751 142.041 (Mole·L-1) 1.39 E-02 6.95 E-04 3.35 E-04 1.08 E-04 2.62 E-04 1.71 E-04 2.37 E-05 2.11 E-06 — 1.55 E-02 Supersaturation with 1000 mg CaCO3 to adjust Ca2+, followed by one week of stirring. Bubbling with 0.4 CO2 % gas mixture until constant pH (carrier gas Argon or N2) Always filter through a 0.22 µm filter before use. In case of solubility experiments, perform an ultrafiltration through a 30 000 MWCO filter. Bubbling with 0.4 CO2 % gas mixture until constant pH. If this SBCW is to be used in the presence of organic matter, organic matter is added AFTER filtration. We advise to analyse the solution before using it, the minimum analysis required is the Fe, Ca and inorganic carbon (IC) content and pH. Table 2:Reference to check the analyses of the SBCW Cations Na+ K+ Mg2+ Ca2+ Fe2+ Sum cations (eq.g.) (mole · L-1) 1.44 E-02 3.35 E-04 1.08 E-04 — 2.37 E-05 1.50 E-02 MW 22.990 39.098 24.305 40.078 55.845 (mg · L-1) 330.3 13.1 2.6 — 1.3 347.3 Anions HCO3ClFSO42Sum anions (eq.g.) (mole · L-1) 1.39 E-02 7.70 E-04 2.62 E-04 2.11 E-06 1.50 E-02 MW 61.017 35.453 18.998 96.064 (mg · L-1) 849.9 27.3 5.0 0.2 882.4 Neutral Species B(OH)3 (mole · L-1) 6.95 E-04 MW 61.834 (mg · L-1) 43.0 TDS (mg · L-1) 1 272.8 SBCW as used in the section 'Waste Characterisation' For the composition of Synthetic Boom Clay Water as used in the section 'Waste Characterisation', we refer to the SCK•CEN document WI.WD.062 (Lemmens, 2001).