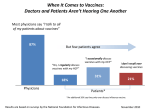
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Vectors in gene therapy wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Focal infection theory wikipedia , lookup
Non-specific effect of vaccines wikipedia , lookup
Public health genomics wikipedia , lookup
Marburg virus disease wikipedia , lookup
Canine parvovirus wikipedia , lookup
Herpes simplex research wikipedia , lookup
Multiple sclerosis research wikipedia , lookup
July 2007, Volume 20, Issue 3 ,pp. 391-532 Reviews: John Turnidge and David L. Paterson Setting and Revising Antibacterial Susceptibility Breakpoints Clin. Microbiol. Rev. 2007 20: 391-408. Elizabeth Foglia, Mary Dawn Meier, and Alexis Elward Ventilator-Associated Pneumonia in Neonatal and Pediatric Intensive Care Unit Patients Clin. Microbiol. Rev. 2007 20: 409-425. Alexandra Valsamakis Molecular Testing in the Diagnosis and Management of Chronic Hepatitis B Clin. Microbiol. Rev. 2007 20: 426-439. Anne Marie Queenan and Karen Bush Carbapenemases: the Versatile ß-Lactamases Clin. Microbiol. Rev. 2007 20: 440-458. Dale W. Griffin Atmospheric Movement of Microorganisms in Clouds of Desert Dust and Implications for Human Health Clin. Microbiol. Rev. 2007 20: 459-477. Susan Hariri and Matthew T. McKenna Epidemiology of Human Immunodeficiency Virus in the United States Clin. Microbiol. Rev. 2007 20: 478-488. Els N. T. Meeusen, John Walker, Andrew Peters, Paul-Pierre Pastoret, and Gregers Jungersen Current Status of Veterinary Vaccines Clin. Microbiol. Rev. 2007 20: 489-510. R. Fotedar, D. Stark, N. Beebe, D. Marriott, J. Ellis, and J. Harkness Laboratory Diagnostic Techniques for Entamoeba Species Clin. Microbiol. Rev. 2007 20: 511-532. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 391–408 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00047-06 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Setting and Revising Antibacterial Susceptibility Breakpoints John Turnidge1* and David L. Paterson2,3,4 Division of Laboratory Medicine, Women’s and Children’s Hospital, North Adelaide, Australia1; Division of Infectious Diseases, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania2; University of Queensland, Brisbane, Australia3; and Queensland Health Pathology Services, Royal Brisbane and Women’s Hospital, Brisbane, Australia4 INTRODUCTION .......................................................................................................................................................391 DEFINITIONS OF SUSCEPTIBILITY CATEGORIES ........................................................................................392 ORGANIZATIONS THAT SET BREAKPOINTS ..................................................................................................392 THE NATURE OF MICs...........................................................................................................................................392 DATA NEEDS FOR SETTING BREAKPOINTS...................................................................................................394 MIC Distributions and Wild-Type Cutoff Values...............................................................................................395 Phenotypic and Genotypic Resistance Markers .................................................................................................395 PK/PD Considerations ...........................................................................................................................................396 In vitro studies ....................................................................................................................................................396 Animal model studies .........................................................................................................................................397 Clinical studies of PD ........................................................................................................................................398 Estimation of target attainment .......................................................................................................................398 Outcome Data from Clinical Studies...................................................................................................................400 COMBINING CUTOFFS TO SET BREAKPOINTS.............................................................................................400 General Principles ..................................................................................................................................................400 Breakpoints by Infection Site................................................................................................................................401 Urinary tract........................................................................................................................................................401 Cerebrospinal fluid .............................................................................................................................................402 Breakpoint Setting before Resistance Has Emerged .........................................................................................402 Reevaluation of Breakpoints Years after the Commercial Release of an Antibacterial Agent....................402 SETTING ZONE DIAMETER BREAKPOINTS FOR DISK DIFFUSION TESTING.....................................403 UNANSWERED QUESTIONS AND FUTURE NEEDS .......................................................................................404 CONCLUSIONS .........................................................................................................................................................404 REFERENCES ............................................................................................................................................................405 the MIC for any given antibacterial that distinguishes wild-type populations of bacteria from those with acquired or selected resistance mechanisms (“wild-type breakpoints;” sometimes called microbiological breakpoints). Data for deriving this type of breakpoint are generated from moderate to large numbers of in vitro MIC tests, sufficient to describe the wild-type population. In this context, the wild-type strain is defined as a strain of a bacterium which does not harbor any acquired or selected resistance to the particular antibacterial being examined or to antibacterials with the same mechanism/site of action. The second are so-called clinical breakpoints, which refer to those concentrations (MICs) that separate strains where there is a high likelihood of treatment success from those bacteria where treatment is more likely to fail. In their simplest form, these breakpoints are derived from prospective human clinical studies comparing outcomes with the MICs of the infecting pathogen. The third use of the term “breakpoint” refers to antibacterial concentrations calculated from knowledge of a PD parameter and the dimension of that parameter that predicts efficacy in vivo. These are the pharmacokinetic/PD (PK/PD) breakpoints, where data that have been generated in an animal model are extrapolated to humans by using mathematical or statistical techniques. Recently, in an attempt to reduce confusion about the meaning of the term “breakpoint,” the European Committee on Antimicrobial Susceptibility Testing proposed the use of the term “epidemiolog- INTRODUCTION Breakpoints are an integral part of modern microbiology laboratory practice and are used to define susceptibility and resistance to antibacterials. Depending on the testing method, they are expressed as either a concentration (in mg/liter or g/ml) or a zone diameter (in mm). In general, all susceptibility testing methods require breakpoints, also known as interpretive criteria, so that the results of the tests can be interpreted as susceptible, intermediate, or resistant and reported as such to a broad range of clinicians. It is acknowledged that sophisticated prescribers may not require (or desire) breakpoints but rather utilize the MIC and knowledge of the pharmacodynamics (PD) of the antibacterial in question to optimize antibacterial selection and dosing. However, given the volume of specimens that a typical clinical microbiology laboratory receives and the diversity of clinicians that a laboratory serves, categorical interpretation of antibacterial susceptibility testing results is a practical necessity and is preferred by most clinicians. The term “breakpoint” has been used in a variety of ways in the literature (141). The first and most obvious one refers to * Corresponding author. Mailing address: Division of Laboratory Medicine, Women’s and Children’s Hospital, 72 King William Rd., North Adelaide, South Australia, Australia. Phone: 1 8 81616873. Fax: 61 8 81616189. E-mail: [email protected]. 391 392 TURNIDGE AND PATERSON ical (or wild-type) cutoff value” to replace the term “microbiological breakpoint” (82). We propose that the term “cutoff” be used more widely to describe the three types of “breakpoints” and that the term “breakpoint” be reserved for the final selected value to be applied in the clinical laboratory. Hence, we prefer the terms wild-type cutoff, PK/PD cutoff, and clinical cutoff to describe these entities. DEFINITIONS OF SUSCEPTIBILITY CATEGORIES Breakpoints are used to define susceptibility and resistance. (In this review, the term “susceptibility” is preferred over “sensitivity”). While these terms should be universally understood, they are frequently used ambiguously because they can refer to the direct interaction between the antibacterial agent and the organism or to the likelihood that the patient will respond to treatment. The first can be measured simply in vitro, while the second involves in vivo complexities such as the dose and dosing schedule, the site of infection, PK of the antibacterial in the individual, and a range of other factors, including the adequacy of host defenses. In some methods, breakpoints are set in such a manner as to create a third category, i.e., intermediate (susceptibility). This category has multiple purposes, including (i) providing a “buffer” between the resistant and susceptible categories to prevent serious interpretive errors and (ii) implying that the organism is susceptible if the antibacterial is concentrated at the site of infection (e.g., in urine) or suggesting that higher doses of antibacterial should be used where it is safe to do so to achieve efficacy. Two sets of category definitions are given below to accommodate the two types of meanings. In vitro definitions are as follows: susceptible, growth of the bacterial strain is inhibited by an antibacterial agent concentration in the range found for wild-type strains; resistant, growth of the bacterial strain is inhibited by an antibacterial agent concentration higher than the range seen for wild-type strains; and wild type, strains that harbor no acquired resistance mechanism to the antibacterial under question, specifically no resistance attributable to (i) mutation, (ii) acquisition of foreign DNA, (iii) up-regulation of an efflux pump, (iv) up-regulation of target production, or (v) any combination of these. PD and clinical definitions, currently listed in the newly developed international reference method ISO/DIS 20776-1 (78), are as follows: susceptible, the bacterial strain is inhibited by a concentration of an antibacterial agent that is associated with a high likelihood of therapeutic success; intermediate, the bacterial strain is inhibited by a concentration of an antibacterial agent that is associated with an uncertain therapeutic effect; and resistant, the bacterial strain is inhibited by a concentration of an antibacterial agent that is associated with a high likelihood of therapeutic failure. While intuitively appealing, these definitions do not capture all of the concepts embedded in susceptibility categories. A more encompassing set of definitions is provided by the Clinical and Laboratory Standards Institute (CLSI) (28), as follows. The “susceptible” category implies that isolates are inhibited by the usually achievable concentrations of antimicrobial agent when the recommended dosage (dosage regimen) is used for that site of infection. The “intermediate” category includes isolates with antimicrobial agent MICs that approach usually attainable blood and tissue levels and for which response rates may be lower than those CLIN. MICROBIOL. REV. for susceptible isolates. The intermediate category implies clinical efficacy in body sites where the drugs are physiologically concentrated (e.g., quinolones and -lactams in urine) or when a higherthan-normal dosage of a drug can be used (e.g., -lactams). The category also includes a buffer zone which should prevent small, uncontrolled technical factors from causing major discrepancies in interpretations, especially for drugs with narrow pharmacotoxicity margins. The “resistant” category implies that isolates are not inhibited by the usually achievable concentrations of the agent with normal dosage schedules and/or demonstrate MICs/zone diameters that fall in the range where specific microbial resistance mechanisms (e.g., -lactamases) are likely and that clinical efficacy against the isolate has not been shown reliably in treatment studies. ORGANIZATIONS THAT SET BREAKPOINTS The processes by which breakpoints are determined can vary widely between susceptibility testing methods. Frequently, these processes are not made explicit in the documentation of the method. For those methods that describe breakpoints without explanation of how the breakpoints are derived, it is assumed that the categories of susceptible, intermediate, and resistant are set using the wild-type cutoff. Methods with recently published breakpoints are outlined in Table 1. Only two international standard-setting groups, the Clinical and Laboratory Standards Institute (CLSI; formerly known as the NCCLS) and the European Union Committee on Antimicrobial Susceptibility Testing (EUCAST), have published guidelines on which data are required for, and how these data are applied to, breakpoint setting (24, 81). The U.S. Food and Drug Administration also sets breakpoints for antibacterials at the time of their approval for use. Unfortunately, breakpoints developed by various organizations may differ, creating confusion for clinical microbiologists, antibacterial susceptibility testing device manufacturers, and clinicians. Harmonization of breakpoints among these organizations should clearly be the aim, taking into account possible differences in doses and dosing schedules used in different parts of the world. THE NATURE OF MICs MICs, as currently measured, are presently the simplest estimates we have of the antibacterial effect in vitro. They are only semiquantitative (see below), yet they have significant utility. There is currently no better measure of antibacterial effect. All breakpoints are either MICs or zone diameter values correlated with MICs. As a consequence, an understanding of the nature of the MIC is fundamental to breakpoint setting. The central concept of an MIC is that it is a measurement of the activity of an antibacterial agent against an individual strain of an organism. It has become the reference measuring tool for susceptibility testing. The value of MIC measurement is frequently criticized because of the “unnatural” conditions under which it is performed, but that criticism misses the point. It is unnecessary for it to reflect exactly the conditions at the site of infection, and of course in most circumstances it cannot. Hence, the common practice of comparing MICs with levels measured in various body compartments is qualitative at best. VOL. 20, 2007 TABLE 1. Susceptibility testing methods with recently published breakpoints SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS Organization or test [method reference(s)] Arbeidsgruppen for antibiotikaspørsmål (Norwegian Working Group on Antibiotics [APA]) (18) British Society for Antimicrobial Chemotherapy (BSAC) (1, 11) Calibrated dichotomous sensitivity test (CDS; promulgated by a single laboratory in Sydney, Australia) (16, 17) Clinical and Laboratory Standards Institute (CLSI) (25, 26, 27, 28) and the U.S. Food and Drug Administration Commissie Richtlijnen Gevoeligheidsbepalingen (CRG) (30, 138, 139, 140) Method(s)—principal mediaa Disk diffusion—Mueller-Hinton or Iso-Sensitest Agar dilution, broth dilution, broth microdilution, disk diffusion—IsoSensitest agar and broth Disk diffusion—Sensitest agar For aerobic and facultative bacteria, broth dilution, broth microdilution, disk diffusion—Mueller-Hinton agar and broth; for anaerobic bacteria, agar dilution, broth microdilution—supplemented Brucella agar and broth Disk diffusion—Iso-Sensitest Comité de l’Antibiogramme de la Société Française de Microbiologie (CA-SFM) (29) Deutches Institut für Normung (DIN) (44, 45, 46, 49, 50, 51, 52, 53) Agar dilution, broth microdilution, disk diffusion—MuellerHinton Agar dilution, broth microdilution, disk diffusion—MuellerHinton EUCAST (65, 66, 67) Agar dilution, broth dilution, broth microdilution—MuellerHinton Japanese Society for Chemotherapy (JSC) (79, 80) Rosco Diagnostica (a commercial company based in Denmark) (114) Broth microdilution—Mueller-Hinton Mesa Española de Normalización de la Sensibilidad y Resistencia a los Antimicrobianos (MENSURA [Spain]) (121) Swedish Reference Group for Antibiotics (SRGA) (123) Disk diffusion, broth dilution, agar dilution—Mueller-Hinton a PDM, paper disk method. Disk (pressed tablet) diffusion—Mueller-Hinton, Iso-Sensitest, PDM, and Danish blood agar Agar dilution, disk diffusion, gradient diffusion—Iso-Sensitest The true value of an MIC is as a measuring tool that generates values to which other parameters, such as PD end points and clinical outcomes, can be reliably compared. This requires that MICs have a reasonable level of reproducibility, a subject that has not received a great deal of attention over the years. Indeed, it is frequently quoted that the “error” associated with measuring an MIC is “plus or minus one twofold dilution.” While this can work as a rule of thumb, results from so-called “tier 2 studies” described by the CLSI (24) for establishing quality control ranges show that precision of MIC measurements can be less than or greater than this, depending on the organism-antibacterial combination (131). The origins of the MIC can be traced back to the original Fleming paper on penicillin (strictly, on cultures of a Penicillium strain) (70). Introduced in this paper were the ideas of (i) serial twofold dilution of an antibacterial agent in broth to measure its activity against different species and (ii) reading the end point by “noting the opacity of the broth.” For decades, the conventional method of determining MICs was in normal test tubes containing 1 to 2 ml of broth, the so-called “macro method” (27). In the 1960s, the method was adapted to microtiter trays (23), and this has become the preferred method for performing MIC tests in broth. MICs can also be determined by agar dilution, where the antibacterial is incorporated into agar, again in a twofold dilution series, and the inoculum is spotted onto the agar surface prior to incubation (10, 27). The development and adoption of the serial twofold dilution 393 Breakpoint-setting parameters [reference(s)] Resistance markers, MIC distributions, PK/ PD, clinical and bacteriological outcomes (13, 14) PK and protein binding (formula), MIC distributions (95) Principally zone diameter distributions (16) MIC distributions, PK/PD, clinical/bacteriological outcome correlations (24) MIC distributions, PK/PD, clinical and bacteriological outcome correlates (31, 104) MIC distributions, PK, correlation with clinical and bacteriological outcome (95) MIC distributions, PK, correlation with clinical and bacteriological outcome (47, 48, 54) In vitro drug characteristics, MIC distributions, PK/PD, clinical outcome correlations (64) MIC-clinical outcome correlations (12, 115, 116) Zone diameters are calibrated against a range of different national and international MIC breakpoints as well as unique breakpoints for tests performed on Danish blood agar (114) MIC distributions, PK/PD, clinical and bacteriological outcomes (15, 98) “Pharmacological breakpoints” with speciesrelated adjustments series for MIC measurement, while originally done for convenience in the macro method, have serendipitously turned out to be valuable from at least one point of view. When the MICs of a particular antibacterial for a large number of strains of a single species are plotted on a histogram, it appears that the wild-type population follows a log-normal distribution (132) (Fig. 1). This means that wild-type MICs appear to be normally distributed on a logarithmic scale; the logarithm to base 2 is the simplest of these scales. Furthermore, strains with the same type of acquired resistance also have a log-normal distribution of MICs. For species in which a single resistance mechanism to an antibacterial predominates, it is therefore usual to see a bimodal distribution. Another important feature of the MIC as we currently measure it is that it actually represents a range of MICs. By way of example, Fig. 1 shows that 51,082 of 71,360 strains of Staphylococcus aureus have a vancomycin MIC of 1 mg/liter. In reality, this represents the individual MICs for those strains, each of which is ⬎0.5 mg/liter and ⱕ1 mg/liter. In other words, there are 51,082 strains whose MICs lie in the range of ⬎0.5 to ⱕ1 mg/liter. Indeed, it is quite possible to determine MICs between the two conventional twofold dilution series values by setting up such concentrations or by using gradient diffusion products (e.g., Etest [AB Biodisk, Solna, Sweden]). From the clinical and PD perspective, such a discriminatory ability may be quite useful. Indeed, in some settings, it would actually be preferable to have a more finely divided range of MICs than conventional twofold dilutions. For example, 394 TURNIDGE AND PATERSON CLIN. MICROBIOL. REV. FIG. 1. MIC distributions for four organism-antimicrobial pairs. In each case, the wild type appears as the log-normally distributed population at the lower MICs. COWT, calculated wild-type cutoff value. (Generated using data from http://217.70.33.99/Eucast2/ and reference 132.) “actual” MICs for amikacin for a Pseudomonas aeruginosa strain of ⬎4 to ⱕ8 g/ml will be recorded, using serial twofold dilution series, as 8 g/ml. Yet if the PD parameter that best predicts amikacin success for P. aeruginosa is a ratio of peak concentration achieved to MIC (103), a measured amikacin peak concentration of 40 g/ml and a recorded MIC of 8 g/ml will result in a ratio of 5. However, the true ratio may actually be closer to 10 if the “actual” MIC was just over 4 g/ml. No commercially available antibacterial susceptibility testing products give a more suitable finely divided range of MICs, and space limitations preventing long ranges of dilutions are an important reason for this. However, their development for research purposes is hindered by a misunderstanding of the precision of current MIC tests, often stated to be “plus or minus one twofold dilution,” as discussed above. The values generated by MIC tests will of necessity be influenced by the method employed (90). The results may differ by choice of technique (broth macrodilution, broth microdilution, agar dilution, or gradient diffusion), medium (MuellerHinton, Iso-Sensitest, or Sensitest medium, lot-to-lot variation, divalent cation concentrations, and the effects of additives, such as blood), inoculum size and concentration, incubation conditions (temperature and duration of incubation), and precision in the preparation of different concentrations of the antibacterial being used. Thus, an MIC is only meaningful when the methods and conditions of the test are known. It is for these reasons that the development of an interna- tional standard reference method for determining MICs was recently proposed. The recognition of the effectiveness of international standardization in other areas of scientific measurement has stimulated the development of an International Organization for Standards reference method for antibacterial susceptibility testing using broth microdilution and cation-adjusted Mueller-Hinton medium (78). It was published as an approved standard in late 2006. This is just the beginning, as the reference method will not work for all bacteria for which we might want to have MICs. Nevertheless, it is MICs generated using this reference methodology and its future enhancements that will become the standards for MICs in future. DATA NEEDS FOR SETTING BREAKPOINTS A range of data are used to assist breakpoint-setting organizations in selecting breakpoints, including in vitro microbiological data, animal and human PK/PD data, and clinical/bacteriological outcome data from prospective clinical studies. No single set of data provides all the information necessary to make decisions. As pointed out above, some methods concentrate mainly on in vitro data to establish breakpoints. Undoubtedly, human PK is factored into the decision, but how this occurs is not made explicit. We believe that the following four main types of data are necessary for the establishment of appropriate breakpoints: (i) MIC distributions and wild-type cutoffs; (ii) in vitro resistance markers, both phenotypic and ge- VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS notypic; (iii) PK/PD data from animal models and human studies; and (iv) outcome data, both clinical and bacteriological, from appropriate clinical studies and the MICs of the causative pathogens in those studies. For establishing zone diameter breakpoints, additional data establishing the relationship between zone diameters and MICs are required. The most important and difficult challenge in using these data is ensuring that there is an appropriate balance between the various forms of data, with consideration being given to the different pathogens and the types and severity of the infections associated with those pathogens. At present, there is no formula that can assist in deciding which data are more important in a particular circumstance. Instead, the final choice of emphasis on the different types of data is made by consensus among standard-setting committees and groups. Such an approach can be divisive, with individuals weighting different data types differently according to their training and skill base. It is essential that the membership of any breakpoint-setting committee include a mix of skills in all four main data areas. Since infections occur in different body compartments, e.g., blood, soft tissues, subarachnoid space, bladder, bone, lungs, or mucosal surfaces, breakpoints, at least in theory, should be developed for each infection syndrome, e.g., bloodstream infection, cellulitis, meningitis, lower urinary tract infection, osteomyelitis, pneumonia, or pharyngitis. This would vastly increase the complexity of breakpoint setting, and therefore almost all methods choose only one set of breakpoints or, sometimes, other sets of breakpoints adjusted to the type of infection in cases where drug concentrations are substantially different, such as urinary tract infection or meningitis. In general, bloodstream infections are considered most generally representative of serious infections, and therefore the PK of the drug in blood is used to set breakpoints. MIC Distributions and Wild-Type Cutoff Values Constructing MIC distributions for organism-antibacterial combinations is the first step in the development of breakpoints for that antibacterial. Broth and/or agar dilution MICs are determined for all organisms of interest, and histograms such as those in Fig. 1 are constructed. Inspection of a histogram gives an immediate picture of whether only wild-type strains are present or whether strains with abnormally elevated MICs are also included. Ideally, these histograms should be constructed on a species-by-species basis because it is unlikely that even related species will have exactly the same modal MIC or wild-type range of MICs (or same mean and standard deviation on the log-normal scale). Often, species are combined for convenience, such as coagulase-negative staphylococci or Enterobacter species, but in general it is better to avoid this where possible. It is also desirable that the full range of the wild-type MIC distribution is included in the antibacterial dilution series and that the MICs are not truncated at one or the other end of the distribution. From the full range of the wild-type MIC distribution, simple inspection will often allow one to estimate where the upper end of the wild-type distribution ends and thus to define wild-type cutoff values, with the wild type being defined as above. EUCAST has released tables and histograms showing a range of wild-type MIC distributions for many or- 395 ganisms and antibacterials. They are freely available on the Internet at www.eucast.org. The data are a collation of MICs collected from a wide array of national and international studies using defined methods. If the full span of wild-type MICs is available, it is also amenable to statistical analysis. Using this type of data, statistical techniques that can reliably determine wild-type cutoff values have recently been developed, thus eliminating the need to estimate cutoff values by inspection (92, 132). This is particularly useful for distributions where there is apparent overlap between the wild-type distribution and the distribution of abnormal strains, such as the example of Pseudomonas aeruginosa and gentamicin in Fig. 1. The first of these methods uses an iterative process to find the optimum fit of the cumulative distribution MICs and works readily on standard twofold MICs. The second method uses a reflection of the lower half of the wild-type distribution about the estimated mean and requires intermediate values between the standard twofold dilution series to be effective. It was adapted from an original method used to determine zone diameter ranges and interpretive criteria of susceptible strains (92, 91). In setting breakpoints, it is generally considered inappropriate to select values that fall inside the wild-type range. Placing breakpoints above or below wild-type distributions creates no problems with interpretation. However, when breakpoints fall inside wild-type distributions, it creates the somewhat anomalous splitting of strains into “susceptible” and “resistant” strains even though the latter do not have an acquired resistance mechanism. In some cases, such a split cannot be avoided, because the desire is to have as few breakpoints as possible for a given antibacterial agent and for some species the wild-type MIC distribution is elevated compared to that for most other “susceptible” species. A typical example of this type of splitting is seen with Stenotrophomonas maltophilia and ceftazidime, where the wild-type distribution ranges from values within reach of those achievable clinically to values well above those that can be achieved with maximum doses. Phenotypic and Genotypic Resistance Markers It is frequently possible to identify the existence of a resistance mechanism in a bacterial isolate by methods other than measuring the MIC (or zone diameter). These methods can be phenotypic or genotypic. Phenotypic methods include (i) direct detection of degrading enzymes (e.g., -lactamase testing); (ii) screening plates by using a concentration lower than the breakpoint (e.g., extended-spectrum -lactamase screening); (iii) medium modification to enhance resistance expression (e.g., use of brain heart infusion agar to detect vancomycin-resistant enterococci); (iv) modification of incubation conditions to enhance resistance expression (e.g., incubation at ⱕ30 to 35°C to detect methicillin resistance in staphylococci); (v) -lactamase confirmation by the use of specific inhibitors, such as clavulanate or EDTA; (vi) induction tests (e.g., macrolide induction of clindamycin resistance); (vii) direct detection of the protein conferring resistance (e.g., agglutination detection of penicillin-binding protein 2a in Staphylococcus aureus); and (vii) detection of resistance to high levels of an antibacterial (e.g., high-level aminoglycoside resistance in enterococci) (124). 396 TURNIDGE AND PATERSON Genotypic tests are usually reserved for confirmation of phenotypic resistance (112). The one common exception is the detection of mecA in staphylococci by PCR methods. This gene encodes the altered penicillin-binding protein 2a and is associated with methicillin “resistance.” This association holds strongly for S. aureus, where the detection of mecA correlates strongly with MICs that are greater than the wild-type cutoff value (oxacillin MIC, 2 mg/liter [CLSI]). Problems with the correlation in coagulase-negative staphylococci led to the significant lowering of the oxacillin breakpoint to 0.5 mg/liter by the CLSI to achieve greater correlation with the presence of mecA (28). This was precautionary, as there are no clinical data available to confirm or refute whether strains of coagulasenegative staphylococci with a MIC of 1 or 2 mg/liter will respond to treatment. If simple special phenotypic (i.e., other than the MIC itself) or genotypic methods can be developed to detect acquired resistance, then ideally these should be used to validate the choice of wild-type cutoff values. Strains with MICs at and to either side of the wild-type cutoff should be subjected to those special phenotypic and/or genotypic tests to ensure correlation. If necessary, the wild-type cutoff should be adjusted to that defined by the presence of the resistance mechanism. Most methods that recommend the use of special phenotypic and/or genotypic tests also recommend that the isolate be reported as resistant regardless of the conventional MIC-based susceptibility result. Put another way, the recommendation is that the isolate should be interpreted as resistant, the usual breakpoint does not apply, and the MIC result should be ignored. The validity of this approach has rarely been tested and would be difficult to test except in animal models. Instead, the conservative view is taken that in vitro detection of resistance to an antibacterial by additional special phenotypic and/or genotypic tests should preclude the use of that antibacterial (see the example above with mecA and coagulase-negative staphylococci). Fortunately, for many organism-antibacterial combinations, the relationship between special phenotypic and/or genotypic tests, elevated MICs, and poor treatment outcomes appears convincing. There are some notable exceptions, such as Streptococcus pneumoniae and penicillin, where the resistance phenotype and genotype, that is, the presence of altered penicillin-binding proteins and/or the genes encoding them, do not appear to correlate well with outcomes (68, 108, 142), except in cases of meningitis. This situation provides a conundrum when breakpoints have already been set for a particular organism-antibacterial combination. New resistance mechanisms are frequently detected only after an antibacterial has been in clinical use for some time. These may represent examples of “hidden resistance” whereby resistance mechanisms are detected genotypically in organisms that have MICs within the “susceptible” range. Examples of “hidden resistance” which have not been addressed completely by organizations which set breakpoints include quinolone target mutations in gram-negative bacilli (42) and Streptococcus pneumoniae (43), extended-spectrum beta-lactamases in organisms that also constitutively produce AmpC (125), and metallo-beta-lactamase production by gram-negative bacilli (73). At the local level, clinical laboratories may be faced with a dilemma whereby resistance mechanisms are confirmed to be occurring by genotypic methods yet no review of CLIN. MICROBIOL. REV. breakpoints has been made by CLSI or EUCAST. In some circumstances, phenotypic methods may have been described to assist in detection of these resistance mechanisms. However, we caution that such methods are often based on organisms from a limited geographic area and may not be universally applicable. Their greatest use may be for epidemiologic purposes rather than for communication to prescribers. PK/PD Considerations With the development of our understanding in the PD of antibacterials, one of the major benefits has been its application to the setting of breakpoints (4, 60, 105). PD is the study of drug effects over time and is therefore intimately linked with the changes in drug concentrations over time, namely, PK. The terms are usually linked to create the term PK/PD. For antibacterials, PK/PD is the study of the relationship between PK variables and microbial inhibition or killing in vivo and, by extension, clinical outcome (41, 58). Extensive studies in animal and in vitro PK models over the last 20 years have led to a clear understanding of PK/PD relationships of many classes of antibacterials, including the -lactams, aminoglycosides, quinolones, macrolides, lincosamides, tetracyclines, and glycopeptides. Clinical studies supporting the evidence generated in animal models have been conducted for -lactams, aminoglycosides, and fluoroquinolones (2). In vitro studies. The PD properties of an antibacterial agent are initially explored in vitro. Bacterial inhibition and killing are initially examined by exposing species of interest to a range of fixed drug concentrations, e.g., different multiples of the MIC, and measuring viable counts over a number of hours (33). The following two major patterns are found: concentration-dependent killing, where killing becomes more rapid and profound with increasing drug concentrations; and concentration-independent or time-dependent killing, where no further increase in the rate of killing is seen for concentrations much above the MIC (33). The second in vitro phenomenon of importance is the so-called postantibiotic effect (PAE). This is a period of delayed regrowth, following drug removal, after brief periods of exposure to an antibacterial in vitro. Again, two major patterns emerge, as follows: a moderate to long delay in regrowth (prolonged PAE) and immediate regrowth or only a short delay (minimal PAE) (40). Other persistent antibacterial effects have been described, such as postantibiotic leukocyte enhancement (97) and postantibiotic sub-MIC effects (106). None of these other effects have added greatly to our understanding of the action of antibacterials in vivo or provided information that would be predictive of clinical efficacy. Demonstration of in vitro PAE requires validation in an animal model, because the persistent effect observed in vitro may occasionally be absent in vivo (1, 134). However, in most circumstances, a combination of the bacterial killing and PAE patterns is sufficient to describe in vitro PD properties and to make predictions about in vivo PK/PD properties, as summarized in Table 2. In vitro studies can be extended to sophisticated PD models, where the PK of drugs in humans are simulated and the effects on bacterial killing measured (96). Most studies using this technique have focused on addressing specific questions rather than defining the fundamental PD properties of drugs. In other VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS 397 TABLE 2. PD properties of various antibacterial classes Killing/inhibition pattern Time dependent Concentration dependent a PAE In vivo PK/PD parameter predicting efficacy Antibiotic class(es)a Minimal Minimal Prolonged Prolonged Minimal Prolonged %T⬎MIC AUC/MIC ratio AUC/MIC ratio Cmax/MIC ratio AUC/MIC ratio AUC/MIC ratio and/or Cmax/MIC ratio -Lactams Linezolid* Macrolides, lincosamides, tetracyclines Glycopeptides* Polymyxins Aminoglycosides, quinolones, streptogramins, ketolides, daptomycin *, the PK/PD parameter predicting efficacy varies with the organism under study and the type of animal model (1, 88, 89). cases, they have confirmed earlier findings from animal models. In vitro PD model studies have been helpful in one area, namely, defining the importance of a regimen in preventing the emergence of resistant subpopulations (19, 69). For a small number of bacterium-drug interactions, selection of resistant subpopulations present in the original bacterial population is a problem. The most notable of these is aminoglycosides and Pseudomonas aeruginosa (76). Animal model studies. Studies with animal models have been critical to our understanding of PK/PD relationships. The earliest work, by Eagle and colleagues, showed the importance of the dosing interval in determining the efficacy of penicillin (61). It took another 35 years before progress was made in this area, when the neutropenic mouse thigh and pneumonia models were employed for the first time to define possible PD parameters predicting efficacy (94, 135). These studies were the first to clearly define the relationship between PK parameters and bacterial killing in tissues by using PD principles. They successfully showed that different classes of antibacterials could have different predictors of efficacy. The principal value of these two models has been to assist in defining PK/PD parameters for the different drug classes, as they were not designed to directly reflect the clinical picture in humans. The three PK/PD predictors of efficacy for which relationships have been shown are (i) time above the MIC (T⬎MIC), usually expressed as a percentage of the dosing interval; (ii) the ratio of the area under the curve over 24 h to the MIC (AUC24/ MIC); and (iii) the peak level-to-MIC ratio (Cmax/MIC). In this context, the AUC is the area found under the plasma concentration-time profile, and the Cmax is the maximum plasma concentration after each dose. The choice of the three PK components T⬎MIC, AUC, and Cmax rather than other PK components relates to the fact that these components have relatively low interdependence, i.e., they can vary significantly without greatly affecting each other. Thus, dosing regimens can be varied in animal model studies so widely that it is possible to minimize the effect of interdependence and thereby maximize the chance that only one of the components will be highly statistically correlated. An example of this is shown in Fig. 2, showing significant scatter and poor correlation between AUC24/MIC or Cmax/MIC and killing but excellent correlation to T⬎MIC for one organism-antibacterial pair in the mouse pneumonia model. Note that “MIC” appears in each of the PK/PD parameters. For the utility of PK/PD data in setting breakpoints, this was a pivotal finding. When the PK/PD parameters are “controlled for” by using MIC in the denominator, it is possible to show that similar agents have similar parameter magnitudes for the same degree of killing when MIC differences between these agents are factored into the analysis (36, 37, 137). Another important feature of the PK/PD parameters is that correspondences between drugs are best when protein binding is taken into account (1, 37). For instance, the T⬎MIC percentages for maximum killing of Staphylococcus aureus in the mouse thigh model are identical for cefotaxime and ceftriaxone, but only when free drug concentrations are used in the analysis (37). This is strong evidence that protein binding must be taken into account when applying PK/PD findings to the setting of breakpoints. Questions arise regarding the applicability of PK/PD parameter data generated in animal models such as the mouse thigh model, where the end point is bacteriostasis or bacterial killing after 24 h. Some confidence about the parameters and their FIG. 2. Relationship between PK/PD parameters and killing of Klebsiella pneumoniae by cefotaxime in the mouse pneumonia model. (Reprinted from reference 37 with permission from Elsevier.) 398 TURNIDGE AND PATERSON CLIN. MICROBIOL. REV. TABLE 3. Prospective clinical studies of PK/PD parameters Agent -Lactams Cefmenoxime Cefepime Penicillins and cephalosporinsb Fluoroquinolones Ciprofloxacin Levofloxacin Gatifloxacin and levofloxacin Grepafloxacin Garenoxacin Levofloxacin Ciprofloxacin Other agents Gentamicin Gentamicin and tobramycin Vancomycin PK/PD parameter selected Magnitude of parameter for maximum efficacy (clinical cure rate)f Nosocomial pneumonia Hospitalized, various Otitis media caused by Streptococcus pneumoniae or Haemophilus influenzae Time above DRCa Time above 4.3⫻ MIC Time above MIC 70–100% 100% 60% 118 126 38 Mainly nosocomial pneumonia Serious community-acquired infection Streptococcus pneumoniae communityacquired pneumonia and AECB AECB Community-acquired pneumonia, AECB, and sinusitis Nosocomial pneumonia Pseudomonas aeruginosa bacteremia AUC24/MIC ratio Cmax/MIC ratio fuAUC24/MIC ratiod ⱖ125 ⱖ12.2 ⱖ33.7 71 111 1 AUC24/MIC ratio Nonee ⱖ175 72 133 AUC24/MIC ratio Cmax/MIC ratio ⱖ87 ⱖ8 59 143 Nosocomial pneumonia Pseudomonas aeruginosa bacteremia Cmax/MIC ratio Cmax/MIC ratio ⱖ10 ⱖ8 85 143 Staphylococcus aureus lower respiratory tract infection AUC24/MIC ratio Infection(s)c ⱖ350 Reference(s) 101, 102 a DRC was used as a surrogate of the MIC per reference 128. This was an analysis of data from multiple clinical studies. c AECB, acute exacerbations of chronic bronchitis. d fu, fraction unbound. e Due to the very high activity of the agent, fuAUC24/MIC ratios were ⬎200 in more than 90% of patients. f Magnitudes are expressed in terms of total drug, rather than unbound drug, unless stated otherwise. b magnitudes can be taken from analyses that have compared them with mortality in a range of animal models (39) and across different organisms (41). The magnitudes of the relevant parameters predicting efficacy are thought to vary depending on a range of factors, with the most important being the class of drug and the infection compartment. T⬎MIC percentages to achieve bacteriostasis (no net growth or killing at the site of infection in the animal model) vary between -lactam classes, with cephalosporins requiring the highest percentage and carbapenems the lowest. Variation between bacterial species is sometimes seen. Clinical studies of PD. There are few clinical studies that have been conducted deliberately to confirm the findings of in vitro and animal model PD. This is understandable because in order to confirm the choice of PK/PD parameters, it is necessary to have both clinical and microbiological failures, as well as sufficient variation in dosing regimens, to separate out the correct PK/PD parameter. Planning for failures in studies is generally considered unethical, and wide variation in dosing regimens is impractical. Hence, most studies in this area have been retrospective, although a few key ones have analyzed outcomes prospectively based on PK/PD parameters (2). Those that have been conducted prospectively have confirmed the analyses made in vitro and with animal PK/PD models. A number of clinical studies have shown a relationship between MIC and treatment outcome. Conditions and agents studied include suspected gram-negative bacteremia and cefoperazone (56), multiple different infections and cefoperazone (36) or cefotaxime (55), acute otitis media and cefu- roxime axetil (75), Bacteroides fragilis and cefoxitin (120), methicillin-resistant Staphylococcus aureus bacteremia and vancomycin (117), and extended-spectrum -lactamase-producing gram-negative bacteremia and third-generation cephalosporins (83). By itself, this relationship is informative about the importance of the MIC but otherwise unhelpful because it is not possible to know which of the three PK/PD parameters is relevant. This comes about because dosage regimens in these studies are usually fixed or vary little, making it impossible to control for the level of interdependence of the PK/PD variables. Furthermore, the MIC of the infecting pathogen and the dosing schedule of the antibacterial agent are not the only determinants of efficacy, and the host response can be important or even dominant in the resolution of infection. Most importantly, we cannot determine from such studies whether the dosing schedules used were optimal. If the PK/PD parameters are known, the effects of changes in the dosing schedule can be predicted and dosing schedules optimized. Fortunately, there are some studies that have been able to examine which of the PK/PD parameters are important. These are listed in Table 3. In each case, the selected parameter and its magnitude correlate reasonably well with those found in animal models. These data strengthen the case for using PK/PD parameters and their magnitudes for estimation of breakpoints that have clinical meaning. Estimation of target attainment. Initial attempts at using PK/PD data to estimate breakpoints used average PK values (129). This is unsatisfactory because of significant intersubject VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS 399 FIG. 3. (A) Fractional probability of target attainment (expressed as a percentage) of intravenous levofloxacin at 500 mg daily, based on PK in healthy volunteers. (Generated using data from reference 22.) (B) Fractional probability of target attainment of intravenous levofloxacin at 750 mg daily, based on PK in a clinical efficacy study, compared to MIC distributions of two major nosocomial pneumonia pathogens. (Reprinted from reference 59 with permission. © 2004 by the Infectious Diseases Society of America. All rights reserved.) variability in PK, even in young healthy subjects (58). PK values can vary even more during illness and as a consequence of underlying diseases. Both kinds of variation need to be included in any estimation of breakpoints based on PK/PD data. The method for factoring in variation is known as Monte Carlo simulation and was first successfully employed in antibacterial PD by Drusano et al. (57, 58). Monte Carlo simulation is a statistical technique whereby a population of values of interest is simulated using existing data, such as the mean and standard deviation from a standard (small) PK/PD study, and then randomly generating a large number of patient values according to an underlying statistical distribution, such as the normal (Gaussian) or, sometimes, log-normal distribution. In this way, the variation in time above a certain value, the AUC24, and Cmax, as found for patients given a defined dosage regimen, can be “created” and used to estimate the probabilities of reaching certain values when large numbers of patients are treated with that regimen. In the context of antibacterials, these “certain values” will be determined by the magnitude of the PK/PD parameter predicting maximum efficacy, for example, 50% for %T⬎MIC of a -lactam, 100 for an AUC24/ MIC ratio of a fluoroquinolone, or 10 for a Cmax/MIC ratio of an aminoglycoside. With these values, it is possible to find the probability that these magnitudes will be reached at different MICs. This whole process is called target attainment analysis. When target attainment rates fall below 90%, the probability of that dosing regimen being effective is significantly diminished, and the PK/PD cutoff becomes the highest MIC for which the target attainment exceeds 90%. The figure of 90% has been chosen arbitrarily and has not been validated clinically but is widely used in target attainment analysis. By way of example, using healthy volunteer PK/PD data for intravenous levofloxacin at a dose of 500 mg daily (22), the 400 TURNIDGE AND PATERSON CLIN. MICROBIOL. REV. fractional probabilities of achieving an AUC24/MIC ratio of at least 100 for different MICs are shown in Fig. 3A. Improvements on this basic process can be made when (i) PK data from prospective clinical studies are used, rather than conventional PK data from volunteer studies (58); and (ii) MIC distributions, as described above, are factored into the analysis (59). These improvements are shown in Fig. 3B, from a study of intravenous levofloxacin (750 mg once daily) for treatment of nosocomial pneumonia (59). Monte Carlo simulation is being used increasingly to assist in developing clinical breakpoints (5, 105). It is now recognized that including between-patient variation in PK and betweenorganism variation in MICs is an essential component that must be taken into account when setting breakpoints. It is no longer appropriate to use average PK/PD values or susceptibility measures such as the MIC90 (1). Results of Monte Carlo simulation may vary according to the input variables and underlying assumptions used. The input variables, as mentioned above, include a variety of PK parameters and the extent of protein binding of the antibacterial. Clearly, such variables may differ from source study to study. The adequacy of these studies may influence the outcome of the Monte Carlo simulation. Even the use of apparently equivalent parameters, such as half-life versus clearance, in different models may greatly alter the results. The number of patient simulations is also of relevance; typically, 5,000 to 10,000 such simulations are used. A number of different programs are used to conduct Monte Carlo simulation (such as Adapt, Crystal Ball, etc.), but it is unlikely that the use of different programs will substantially alter the results of target attainment analysis. We believe that standard, predefined methodologies for Monte Carlo simulation should be used by organizations setting breakpoints and that these methods should be internationally harmonized. pathogens likely to be involved in the infection under study. (ii) As a consequence of point i and the nature of MIC distributions, it is likely that for some species, there will be few infections included in the study caused by strains at the top end of the distribution, especially because sample sizes for an individual species are likely to be small. This greatly enhances the chance of error in estimating a clinical breakpoint. (iii) There is little consensus on which rates of cure and/or eradication are acceptable. From a regulatory standpoint, such studies typically test noninferiority compared to a drug that has previously received regulatory approval. It is not clear whether this is always the appropriate end point for assessment of breakpoints. Indeed, regulators have recently recognized the limitations of comparative noninferiority studies and in some cases are now requesting studies designed to show superiority or to be placebo controlled. (iv) No numerical account is taken of natural response rates, which can be quite high for some common bacterial infections. (v) Not all species of interest in a particular infection necessarily get included in prospective studies. Thus, breakpoints may be extrapolated inappropriately from commoner species. Ideally, clinical studies would recruit significant numbers of cases where the infecting pathogen has an MIC on either side of the wild-type and PK/PD cutoff values to assist in the clinical validation of final breakpoints. However, this is unlikely to happen for a range of reasons, including point ii above and the fact that the only studies with sufficient recruitment are those conducted on new, potent antibacterial agents for regulatory registration purposes. Of interest, the use of clinical outcome data to define breakpoints has been popular in the past in Japan. Examination of outcomes by MIC has led to the selection of a breakpoint estimated to be the lowest MIC where maximum or near-maximum efficacy has been achieved (115,116). Outcome Data from Clinical Studies COMBINING CUTOFFS TO SET BREAKPOINTS Some organizations give the greatest weight to prospective clinical studies to define breakpoints, especially those trials done for regulatory purposes. Such trials are typically randomized controlled trials with predefined dosage regimens, clinical end points, and bacteriologic end points. An advantage of such studies is that concomitant antibacterial use is typically minimal so that the effects of the antibacterial under study can be studied independently. Most often, the appropriate correlation in such studies is clinical and/or bacteriological outcome versus the MIC for the infecting pathogens. Clinical studies may have their greatest value in providing a “reality check” for cutoffs derived from microbiologic and PK/PD studies. Clinical failure rates in excess of those predicted by microbiological and PK/PD data should trigger a reassessment of breakpoints prior to an antibacterial’s commercial introduction. It is important to note that there is a range of problems with regulatory clinical studies that limit our ability to always correctly interpret these data for breakpoint setting. These limitations are as follows. (i) The great majority of studies predefine “resistance” and exclude/withdraw patients with “resistant” strains. The way “resistance” is predefined is often not clear to the investigators but often appears to be based, at least in part, on MIC distributions, i.e., the wild-type cutoff for General Principles The most comprehensive process for setting MIC breakpoints involves a comparison between PK/PD cutoffs, clinical cutoffs, and wild-type cutoffs. Ideally, it is based on a full set of data, as outlined in Table 4. Combining cutoffs into a single breakpoint is still largely a matter of judgment. The process, unfortunately, can be swayed by the composition of skills and biases of the group making the breakpoint decisions. It is largely a question of how to merge the three cutoffs in such a way as to ensure that (i) strains that are likely to respond to treatment with the chosen dosage schedule at the likely site of infection are classified as susceptible and (ii) strains that are unlikely to respond to treatment with the chosen dosage schedule at the likely site of infection are classified as resistant. It is our opinion that the initial step in breakpoint determination should be an assessment of the MIC distribution for a contemporary collection of isolates obtained from global sources. The PK/PD cutoff should be applied to this collection. We believe that the PK/PD cutoff provides the greatest amount of “value” in this situation because it includes much of the relevant data in its construction, including (i) the MIC as it is measured in vitro; (ii) the relevant PD parameter and its mag- VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS 401 TABLE 4. Preferred data sets for establishing breakpoints of an antibacterial Area of investigation Data item In vitro activity .........................................Construct MIC distributions of bacterial species of interest Determine wild-type cutoffs for individual species or closely related species If possible, determine resistance mechanisms if there is evidence of acquired resistance in any species and correlate them with wild-type cutoffs In vitro PD................................................Determine if killing in vitro is concentration or time dependent Determine duration of the PAE Measure protein binding in animals used in the animal model for PK/PD and in humans Animal model PD ....................................Determine PK in animal model Determine PD parameter that best predicts efficacy (bacterial killing) in animal model (e.g., mouse thigh), i.e., either %T⬎MIC, AUC24/MIC, or Cmax/MIC Estimate the magnitude of the PD parameter based on unbound drug (target) that produces bacteriostasis or near-maximum killing Human PD................................................Determine PK in humans (usually with volunteer studies), including nature of population kinetic model (normal or log-normal) and means and standard deviations of volumes of distribution, elimination half-lives, AUCs, and Cmax values (depending on the relevant PD parameter) Using Monte Carlo simulation, calculate target attainment rates for unbound drug at different MICs for bacteriostasis and near-maximum killing Set PD cutoff at the highest MIC where target attainment exceeds 90% Clinical outcome studies .........................Collect outcome data from prospective clinical studies by infection type and by bacterial species (clinical efficacy and bacteriological efficacy) Perform PK studies with at least a subset of patients and compare results with those of previous volunteer studies (as described for human PD studies) Tabulate both types of outcomes by infection type, by bacterial species, and by MIC Select clinical cutoff (by species and infection type if necessary) as the highest MIC giving maximum efficacy Disk diffusion zone diameters ................Construct scattergrams of zone diameters versus MICs of closely related species by species nitude, which predicts in vivo efficacy; (iii) human PK and its intersubject variation; and (iv) the dosing regimen. PK/PD breakpoints are sometimes criticized on the basis that they are established using PK/PD data from plasma/serum, which do not necessarily reflect what is happening to the drug in tissues where infections are located. This criticism misses the point. When the PD parameter that predicts efficacy is determined with an animal model, it is by using the plasma levels as a surrogate for what is happening at the site of infection. If a robust relationship can be found between bacterial inhibition and killing and plasma PK/PD (which is almost always true), then that is all that is needed to validate the model. This is true for both bactericidal and bacteriostatic agents. Clinical cutoffs should be considered a validation tool for PK/PD cutoffs and, because of the limitations stated above, should not necessarily receive greater weighting. Clinical cutoffs are most important when they fall below PK/PD cutoffs. If this is the case, it suggests that further PK/PD work is required to understand the relationship between PD and outcomes. Different results arising from application of clinical versus PK/PD cutoffs may arise when differing dosage regimens of the drug are licensed or used in clinical practice. PK/PD cutoffs apply to specific dosage regimens. The most conservative approach is to generate PK/PD cutoffs based on the lowest approved dosage regimen for the drug. It would be logical to apply different breakpoints to different dosage regimens—unfortunately, communication of such information by clinical microbiology laboratories to prescribers may be difficult. Ideally, authorities setting breakpoints for susceptibility testing should specify the dosage regimen on which their breakpoints are based. The principal application of wild-type cutoff values is to examine whether the PK/PD and clinical cutoffs fall below them and inside the wild-type MIC distribution. If this does occur, then problems will be encountered in testing and interpretation, as some wild-type strains will be “susceptible” and others “intermediate” or “resistant,” and because of day-today variations in testing results, some strains could readily end up in any of the three categories. A general solution to the possible breakpoint splitting of wild-type distributions has not been agreed upon yet. In practice, if the split is at the high end of the distribution, then the breakpoint is not modified and it is accepted that a small number of strains with wild-type MICs will test as “intermediate” or “resistant.” If the split occurs in the center or lower end of the MIC distribution, the conservative approach is to nominate that species as intermediate or resistant. Breakpoints by Infection Site Breakpoints are usually established on the basis that they are relevant at all sites of infection. However, there are body compartments where antibacterials can be concentrated or have restricted penetration. Urinary tract. Many antibacterials are excreted primarily in urine and achieve concentrations substantially higher than those seen in plasma. In many methods, it is conventional not to adjust breakpoints for urinary tract infections for the ma- 402 TURNIDGE AND PATERSON jority of agents in this category in order to reduce the complexity of multiple interpretative criteria. Whether this is warranted is unclear. Conversely, for those methods that do supply different breakpoints for urinary tract isolates causing lower urinary tract infection, it is usually not possible for the laboratory to distinguish isolates associated with lower versus upper urinary tract infection. Previously, it was believed that urinary concentrations predicted the outcome better than did serum concentrations, at least for lower urinary tract infection (122). Prospective clinical data on the outcome of treating urinary tract infections caused by organisms with various levels of susceptibility and resistance are scarce. Those studies that do examine this have found that the current CLSI breakpoints for trimethoprim-sulfamethoxazole, which are based on systemic PK, effectively separate clinical and microbiological successes and failures or relapse (20, 77, 113) and that failure rates with strains defined as resistant using systemic PK are the same as those seen with placebo (100). Recent attempts to examine the PD of urinary tract infection treatment have shown a modest relationship between the total time that aminopenicillins exceed the MIC in plasma and treatment outcomes in clinical studies (74). Maximum efficacy is achieved when the total time is ⱖ30 h. Further studies with a mouse model of ascending urinary tract infection have been able to show a correlation between the MIC and bacterial killing for sulfamethizole but not amdinocillin (87). Despite all the uncertainties, some methods, such as that of the British Society for Antimicrobial Chemotherapy, do provide higher breakpoints for more than 10 drugs used for urinary tract infection (95). Other authorities, such as the CLSI and Comité de l’Antibiogramme de la Société Française de Microbiologie, provide “urinary” breakpoints for agents whose primary role is the treatment of urinary tract infection, for instance, nitrofurantoin, sulfonamides, trimethoprim, norfloxacin and some other quinolones, fosfomycin, and amdinocillin (28). More work will be required in the area of urinary tract levels, PD, and outcomes. Cerebrospinal fluid. Although the penetration of many drugs into cerebrospinal fluid is known to be restricted, even in the presence of inflammation, breakpoints are not usually adjusted to take the specialized PK of antibacterials in the subarachnoid space into account, principally because it is standard practice to use much higher doses for bacterial meningitis to compensate for restricted penetration. Almost all of the information relevant to breakpoint setting has been generated by observing treatment failures, mostly to expanded-spectrum cephalosporins, of pneumococcal meningitis (84, 86, 107). A number of organizations have adjusted their breakpoints for pneumococci when they are associated with meningitis, but mainly for expanded-spectrum cephalosporins. Most methods already had low breakpoints for penicillin, and these did not require adjustment when strains with reduced susceptibility emerged. Breakpoint Setting before Resistance Has Emerged When antibacterial agents with novel mechanisms of action are developed, it is usual for there to be no resistance among the bacteria within their spectrum. It is therefore not possible to be confident of a correlation between susceptibility and CLIN. MICROBIOL. REV. treatment outcome from clinical studies, as no patients will have been infected with “resistant” strains. Under these circumstances, in vitro data, animal PD data, and human PK data (with Monte Carlo simulation) are used to define breakpoints. If the calculated PK/PD cutoff is substantially greater than the wild-type cutoff for any species, the conservative decision is usually to set the MIC breakpoint only one or two doubling dilutions above the wild-type cutoff value. Some authorities, such as the CLSI, will choose to set only a single breakpoint, with that being “susceptible.” When resistance has emerged, it is then possible to reexamine the tentative breakpoint and to establish a resistance breakpoint and, if necessary, an intermediate range. Reevaluation of Breakpoints Years after the Commercial Release of an Antibacterial Agent A difficult situation arises when new mechanisms of bacterial antibacterial resistance are detected a significant time after breakpoints were initially determined. Such a situation has occurred with the discovery of -lactamase types such as extended-spectrum -lactamases (109) and metallo--lactamases (136). When these new mechanisms of resistance are found in organisms susceptible to an antibacterial potentially subject to these resistance mechanisms, a case may be made for reevaluating breakpoints. Such a signal to reevaluate breakpoints is often hastened by clinical case reports describing failure of the antibacterial in question when used in treatment of an organism harboring the new mechanism of resistance. In other circumstances, a renewed understanding of the PK/PD of an antibacterial may serve as the trigger for breakpoint reevaluation (7). Breakpoint organizations in the United States, in particular CLSI, do reevaluate breakpoints when it is deemed necessary. However, their implementation is more difficult because of the role of the regulatory authority in setting and altering breakpoints. Additionally, there may be commercial reluctance to invest funds in the provision of new clinical data assessing the need to reevaluate breakpoints. In particular, it is highly unlikely that prospective clinical studies will be performed in an environment similar to that in which regulatory studies are conducted. Finally, antibacterials undergoing reevaluation may be generically available; in this situation, there is unlikely to be any commercial support for provision of new clinical data. It is our opinion that the discovery of new antibacterial resistance mechanisms occurring in organisms which are “susceptible” to an antibacterial using previously established breakpoints should be the prompt that necessitates reevaluation of antibacterial breakpoints. It is our belief that the regulatory authorities should mandate that the manufacturer of the drug undergo collection of PK/PD and clinical data relevant to the breakpoint reevaluation. The PK/PD data should be of the standard we have previously defined. It is impractical to suggest that the clinical data should come from new prospective randomized trials. Rather, such clinical data should come from large data sets for consecutive patients treated with the relevant antibacterial. In general and where relevant, bloodstream infection is the most useful infection type in such a reevaluation since there can be little debate as to the clinical relevance of such an infection. Clinical and bacteriologic end VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS 403 FIG. 4. “Scattergram” of MICs versus zone diameters. Numbers represent the number of isolates at each MIC/zone diameter pair (e.g., there were 17 isolates whose MICs were ⱖ256 mg/liter and whose zone diameters were ⱕ6 mm). (Reprinted from reference 130 with permission of the publisher.) points (i.e., rates of cure, improvement and failure, or eradication and persistence) should be predefined rather than determined after preliminary exploration of the data. Potential confounders to an association with clinical outcome include the dosing regimen, organism type, portal of entry of the organism, comorbid illnesses, severity of illness, presence of immunosuppression, and concomitant antibacterial therapy. The clinical data set should be of sufficient size that the assessment is adequately powered to determine statistically significant differences in outcome from infections due to organisms with different MICs. A further layer of substantiation of clinical outcome may come from comparison of outcomes from infections with the target organism/MIC but with different antibacterials used for treatment. An example is provided in order to illustrate such an approach. A variety of -lactamases which can hydrolyze cefepime yet occur in organisms with cefepime MICs within the susceptible range have been detected (110). PK/PD analysis suggests that target attainment may be suboptimal using regulator-approved doses of cefepime for some MICs previously regarded as susceptible (127). A data set of consecutive patients with gram-negative bacteremia treated with cefepime would be collected, including the acquisition of data on confounding variables. The outcomes for patients infected with organisms of different MICs would be compared and analyzed using logistic regression or other multivariate analysis. If statistically significant differences in outcomes at different MICs occur, this would provide strong support for a change in breakpoint. Case reports or small case series (particularly of nonconsecutive patients) may be a “signal” to reevaluate breakpoints but should not themselves be regarded as satisfactory evidence when breakpoints are being reevaluated. If inadequate clinical data are present to support or refute breakpoint revisions based on PK/PD analysis, either the breakpoints based on PK/PD analysis should be accepted or phenotypic screening and confirmatory tests for the resistance mechanism might be developed. It is appreciated that the latter approach is more “conservative,” but many involved in breakpoint setting may prefer to “play it safe” and avoid risking patient safety by properly categorizing a potentially compromised antibacterial agent. In many cases, development of screening and confirmatory tests for new resistance mechanisms can be undertaken prior to acquisition of solid clinical data relevant to breakpoint assessment. The downside of this approach is that broaderspectrum agents are often inadvertently “promoted” by the testing laboratory. SETTING ZONE DIAMETER BREAKPOINTS FOR DISK DIFFUSION TESTING Susceptibility testing by the disk diffusion method rather than the MIC-based method is still very widely used. Although there have been attempts to establish disk diffusion interpretive criteria, usually by defining resistance as zone diameters smaller than those of the wild type (91), the only valid method is to correlate zone diameters with MICs as described below. Hence, once MIC breakpoints have been set, zone diameter breakpoints can be developed. The simplest approach is to plot a “scattergram” of zone diameters versus MICs for strains tested by both methods (130) (Fig. 4). Scattergrams allow visual inspection of the correlation between zone diameters and MICs. Originally, it was considered appropriate to combine similar species on a single scattergram and fit a regression line through the data points (63). However, this does not readily lead to the setting of criteria to discriminate between susceptible and resistant strains. Furthermore, MIC and zone diameter data are not evenly distributed along a continuum for a species or related species, but tend to cluster. Hence, the validity of applying regression is questionable. The first effective statistical method for setting zone diameter interpretive criteria based on scattergram data was developed by Metzler and DeHaan (99). They developed the so-called error-rate-bounded method, which involves the selection of zone diameter values defining resistance and susceptibility from predefined acceptable rates of error. Tolerance of “very major” errors, where strains known to be resistant on MIC testing but whose zone diameter criterion would define them as susceptible, was set very low, at ⱕ1% of all MIC-zone diameter pairs (Fig. 4). This was justified because calling resistant strains susceptible in a laboratory test could cause serious adverse consequences for the patient. Tolerance of “ma- 404 TURNIDGE AND PATERSON jor” errors, i.e., susceptible by MIC data but resistant by zone diameter data, was accepted at a level of ⱕ5%. These authors did not comment on acceptable rates of “minor” errors. Their analysis was also based on the use of a single MIC breakpoint defining susceptibility and resistance only on the basis of MICs, an infrequent situation for most susceptibility testing methods. The error-rate-bounded method has been enhanced through the work of Brunden et al. (21). They adapted the method to two MIC breakpoints defining intermediate susceptibility as well as susceptibility and resistance and introduced an accepted “minor” error rate of ⱕ5%. Minor errors are zone diameter results that categorize either resistant or susceptible MIC results as intermediate or intermediate MIC results as resistant or susceptible (Fig. 4). They also introduced the concept of iteration to find the best fit of zone diameters that would minimize errors overall. This was achieved through the development of an index (index ⫽ [possible susceptible zone diameter ⫺ possible proposed resistant zone diameter]/percentage error). By trying different possible zone diameter pairs to define susceptible and resistant, the aim is to maximize the value of the index. While their accommodation of an intermediate range of MICs is now widely accepted, the iterative method of fitting has yet to be widely adopted. An alternative method, again based on the concept of error rate bounding, has been proposed by the CLSI (24). In this system, discrepancy rates for very major, major, and minor errors are established for three different bands within the MIC range, whose widths vary according to the range of the intermediate MIC category. For example, in Fig. 4, the bands are ⱕ2 mg/liter, 4 to 16 mg/liter, and ⱖ32 mg/liter. Adjustments to discrepancy rates are made if there is only a single MIC breakpoint with no intermediate range. Readers are referred to the CLSI M23 document for further explanation about the method and its application (24). The method offers greater flexibility in establishing zone diameter interpretive criteria, but possibly at the cost of reduced correlation with MICs. Kronvall et al. proposed a different approach altogether in setting zone diameter breakpoints (91, 92). They noted the resemblance of zone diameter distributions of the wild-type population to a normal distribution and developed a proprietary method for defining the distribution and setting a single zone diameter breakpoint. The method takes advantage of the fact that the upper end of the normal distribution is easily recognized graphically, and the data can be used statistically to define the lower end of the wild-type distribution by, in a sense, “reflecting” the upper end. MICs or breakpoints are not used in the analysis, and it cannot be used to determine intermediate zone diameter breakpoints. An entirely different alternative to establishing zone diameter interpretive criteria has been proposed by Craig (32). The method is based on detailed statistical modeling of the spread and error of MICs and zone diameters and was designed to reduce the sometimes arbitrary choice of zone diameter breakpoints which can still occur when error-rate-bounded methods are used. Although this is the most sophisticated of the methods developed so far, it has yet to become adopted by any standard-setting body. In summary, there is no current consensus on the optimum method for determining zone diameter breakpoints. While the method of Craig is the most robust statistically, it is not in a CLIN. MICROBIOL. REV. form, such as a computer program, that can be used readily by those charged with determining these breakpoints. One or another error-rate-bounded method will continue to be the standard for the immediate future. That recommended by Brunden et al. would serve the breakpoint setting community well for the foreseeable future. UNANSWERED QUESTIONS AND FUTURE NEEDS In spite of significant progress being made in recent years, breakpoint setting is still not an exact science. There are quite a few questions whose answers will improve the decision-making process and increase the interpretive value of breakpoints, including the following. (i) Should strains possessing an acquired resistance mechanism but having MICs below the PK/PD breakpoint be considered resistant and reported as such, regardless of the MIC? (ii) What are the best magnitudes to choose for the various PK/PD parameters? (iii) Do the magnitudes of the PK/PD parameters apply to all infection sites, or do they vary by site? (iv) Do the magnitudes of the PK/PD parameters apply to all bacteria, or do they vary by species? (v) Should breakpoints be linked to a specific dosage regimen? (vi) Is there utility in considering the MIC in conjunction with the breakpoint (e.g., the “breakpoint quotient,” or the MIC of the infecting pathogen divided by the breakpoint MIC), similar to the method proposed long ago for comparing blood or tissue levels with the MIC of the infecting pathogen (62)? (vii) If not all data are available, which data would be considered a minimum for setting a breakpoint? (viii) What systems need to be established to ensure the timely review of breakpoints as resistance emerges? (ix) Can MIC measurements be improved to give greater precision and reproducibility? We look forward to the work of our colleagues to provide answers to these and other questions so that the art and science of breakpoint setting might converge in the not too distant future. CONCLUSIONS Breakpoints can be used in several ways. They allow communication from the clinical laboratory to the prescriber regarding the likelihood that a particular antibacterial regimen will be clinically useful in the treatment of patients with infections. They also allow the epidemiologic study of changing resistance patterns in a defined institution or geographic area. Breakpoints should be set prior to an antibacterial being used clinically and must be reviewed when mechanisms of resistance to the antibacterial become apparent. Breakpoint setting is not an exact science. It requires knowledge of the wild-type distribution of MICs, assessment of the PK/PD of the antibacterial, and study of the clinical outcome of infections when the antibacterial is used. Our current state of knowledge about each of these facets of breakpoint setting is imperfect. Breakpoint-setting organizations must utilize experts in microbiology, PD, and clinical infectious diseases in order to come to a consensus regarding the most appropriate breakpoint to be utilized. If appropriately developed and revised, breakpoints have greater relevance to the prescriber than does phenotypic VOL. 20, 2007 SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS detection of resistance mechanisms. However, breakpointsetting organizations may also play a role in developing phenotypic tests for detection of resistance mechanisms, as this information may have epidemiologic and clinical importance that complements use of the breakpoint. REFERENCES 1. Ambrose, P. G., and R. Quintiliani. 2000. Limitations of single point pharmacodynamic analysis. Pediatr. Infect. Dis. J. 19:769. 2. Ambrose, P. G., S. M. Bhavnani, C. M. Rubino, A. Louie, T. Gumbo, A. Forrest, and G. Drusano. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it’s not just for mice anymore. Clin. Infect. Dis. 44:79–86. 3. Ambrose, P. G., D. M. Grasela, T. H. Grasela, J. Passarell, H. B. Mayer, and P. F. Pierce. 2001. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 45:2793–2797. 4. Ambrose, P. G. 2005. Antimicrobial susceptibility breakpoints: PK-PD and susceptibility breakpoints. Treat. Respir. Med. 4(Suppl. 1):5–11. 5. Ambrose, P. G. 2006. Monte Carlo simulation in the evaluation of susceptibility breakpoints: predicting the future: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy 26:129–134. 6. Amsterdam, D. 2005. Susceptibility testing of antimicrobials in liquid media, p. 61–143. In V. Lorian (ed.), Antibiotics in laboratory medicine, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. 7. Andes, D., and W. A. Craig. 2005. Treatment of infections with ESBLproducing organisms: pharmacokinetic and pharmacodynamic considerations. Clin. Microbiol. Infect. 11(Suppl. 6):10–17. 8. Andes, D., R. Walker, S. Ebert, and W. Craig. 1994. Increasing protein binding of cefonicid enhances its in vivo activity in an animal model of infection, abstr. A93. Abstr. 34th Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. 9. Andes, D., M. L. van Ogtrop, J. Peng, and W. A. Craig. 2002. In vivo pharmacodynamics of a new oxazolidinone (linezolid). Antimicrob. Agents Chemother. 46:3484–3489. 10. Andrews, J. 2001. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 48(Suppl. S1):5–16. 11. Andrews, J. January 2005, posting date. BSAC disc diffusion method for antimicrobial susceptibility testing, version 4, January 2005. British Society for Antimicrobial Chemotherapy, Birmingham, United Kingdom. www .bsac.org.uk. Accessed 5 February 2006. 12. Arakawa, S., T. Matsui, S. Kamidono, Y. Kawada, H. Kumon, K. Hirai, T. Hirose, T. Matsumoto, K. Yamaguchi, T. Yoshida, K. Watanabe, K. Ueno, A. Saito, and T. Teranishi. 1998. Derivation of a calculation formula for breakpoints of antimicrobial agents in urinary tract infections J. Infect. Chemother. 4:97–106. 13. Arbeidsgruppen for Antibiotikaspørsmål. 2006. AFAs brytningspunkter for bakteriers antibiotikafølsomhet, versjon 1.9. Arbeidsgruppen for Antibiotikaspørsmål, Tromso, Norway. www.unn.no/category10274. Accessed 1 November 2006. 14. Arbeidsgruppen for Antibiotikaspørsmål. 29 May 2005, posting date. Nødvendig bakgrunnsmateriale og prosedyrer for etablering og justering av mm-brytningssoner. Arbeidsgruppen for Antibiotikaspørsmål, Tromso, Norway. www.unn.no/article19775-927.html. 15. Baquero, F., J. Martı́nez-Beltrán, R. Cánton, and the remaining members of the Mesa Española de Normalización de la Sensibilidad y Resistencia a los Antimicrobianos (MENSURA). 1997. Criterios del grupo MENSURA para la definiciónde los puntos crı́ticos de sensibilidad a los antibióticos. Rev. Esp. Quimioter. 10:103–313. 16. Bell, S. M. 1975. The CDS disc method of antibiotic sensitivity testing (calibrated dichotomous sensitivity test). Pathology 7(Suppl.):1–48. 17. Bell, S. M., B. J. Gatus, J. N. Pham, and D. L. Rafferty. Accessed 1 November 2006. Antibiotic susceptibility testing by the CDS method. The CDS Users Group, Randwick, Australia. www.med.unsw.edu.au /pathology-cds. 2004. 18. Bergan, T., J. N. Bruun, A. Digranes, E. Lingaas, K. K. Melby, and J. Sander. 1997. Susceptibility testing of bacteria and fungi. Scand. J. Infect. Dis. 103(Suppl.):1–36. 19. Blaser, J., B. B. Stone, M. C. Groner, and S. H. Zinner. 1985. Impact of netilmicin regimens on the activities of ceftazidime-netilmicin combinations against Pseudomonas aeruginosa in an in vitro pharmacokinetic model. Antimicrob. Agents Chemother. 28:64–68. 20. Brown, P. D., A. Freeman, and B. Foxman. 2002. Prevalence and predictors of trimethoprim-sulfamethoxazole resistance among uropathogenic Escherichia coli isolates in Michigan. Clin. Infect. Dis. 34:1061–1066. 21. Brunden, M. N., G. E. Zurenko, and B. Kapik. 1992. Modification of the error-rate bounded classification scheme for use with two MIC break points. Diagn. Microbiol. Infect. Dis. 15:135–140. 22. Chien, S.-C., M. C. Rogge, L. G. Gibson, C. Curtin, F. Wong, J. Natarajan, R. R. Williams, C. L. Fowler, W. K. Cheung, and A. Chow. 1997. Pharma- 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 405 cokinetic profile of levofloxacin following once-daily 500-milligram oral and intravenous doses. Antimicrob. Agents Chemother. 41:2256–2260. Chitwood, L. A. 1969. Tube dilution antimicrobial susceptibility testing: efficacy of a microtechnique applicable to diagnostic laboratories. Appl. Microbiol. 17:707–709. Clinical and Laboratory Standards Institute. 2001. Development of in vitro susceptibility testing criteria and quality control parameters; approved guideline—2nd ed. CLSI document M23-A2. CLSI, Wayne, PA. Clinical and Laboratory Standards Institute. 2004. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard—6th ed. CLSI document M11-A6. CLSI, Wayne, PA. Clinical and Laboratory Standards Institute. 2006. Performance standards for antimicrobial disk susceptibility tests; approved standard—9th ed. CLSI document M2-A9. CLSI, Wayne, PA. Clinical and Laboratory Standards Institute. 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—7th ed. CLSI document M7-A7. CLSI, Wayne, PA. Clinical and Laboratory Standards Institute. 2007. Performance standards for antimicrobial susceptibility testing; 16th informational supplement. CLSI document M100-S17. CLSI, Wayne, PA. Comité de l’Antibiogramme de la Société Française de Microbiologie. February 2006, accession date.Communiqué 005. Société Française de Microbiologie, Paris, France. www.sfm.asso.fr. Commissie Richtlijnen Gevoelighheidsbepalingen. 1996. Standaadisatie van gevoelighheidsbepalingen. Ned. Tijschr. Med. Microbiol. 4:104–106. Commissie Richtlijnen Gevoelighheidsbepalingen. 2000. Interpretie van gevoelighheidsonderzoek en gevoelighheidscriteria voor antibacteriële middelen in Nederland. Ned. Tijdschr. Med. Microbiol. 8:79–81. Craig, B. A. 2000. Modeling approach to diameter breakpoint determination. Diagn. Microbiol. Infect. Dis. 36:193–202. Craig, W. A., and S. C. Ebert. 1991. Killing and regrowth of bacteria in vitro: a review. Scand. J. Infect. Dis. 74(Suppl.):63–70. Craig, W. A., Y. Watanabe, and S. Ebert. 1992. AUC/MIC ratio is the unifying parameter for comparison of the in vivo activity among aminoglycosides, abstr. 49. Abstr. 32nd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. Craig, W. A., S. Ebert, and Y. Watanabe. 1993. Differences in time above MIC required for efficacy of beta-lactams in animal infection models, abstr. 86. Abstr. 33rd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. Craig, W. A. 1993. Qualitative susceptibility tests versus quantitative MIC tests. Diagn. Microbiol. Infect. Dis. 16:231–236. Craig, W. A. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 22:89–96. Craig, W. A., and D. Andes. 1996. Pharmacokinetics and pharmacodynamics of antibiotics in otitis media. Pediatr. Infect. Dis. J. 15:255–259. Craig, W. A. 1996. Antimicrobial resistance issues of the future. Diagn. Microbiol. Infect. Dis. 25:213–217. Craig. W. A., and S. Gudmundsson. 1996. Postantibiotic effect, p. 296–329. In V. Lorian (ed.), Antibiotics in laboratory medicine, 4th ed. Williams and Wilkins, Baltimore, MD. Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1–12. Crump, J. A., T. J. Barrett, J. T. Nelson, and F. J. Angulo. 2003. Reevaluating fluoroquinolone breakpoints for Salmonella enterica serotype Typhi and for non-Typhi salmonellae. Clin. Infect. Dis. 37:75–81. Davies, T. A., A. Evangelista, S. Pfleger, K. Bush, D. F. Sahm, and R. Goldschmidt. 2002. Prevalence of single mutations in topoisomerase II genes among levofloxacin-susceptible clinical strains of Streptococcus pneumoniae isolated in the United States in 1992 to 1996 and 1999 to 2000. Antimicrob. Agents Chemother. 46:119–124. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 3. Agar diffusion test. DIN document 58940-. Beuth Verlag GmbH, Berlin, Germany. Deutches Institut für Normung. 2000. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 3. Agar diffusion test—data for the interpretation of inhibition zone diameters. DIN document 58940supplement 1. Beuth Verlag GmbH, Berlin, Germany. Deutches Institut für Normung. 1989. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 3. Agar diffusion test—zone diameters with control strains. DIN document 58940-supplement 2. Beuth Verlag GmbH, Berlin, Germany. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 4. Evaluation classes of the minimum inhibitory concentration. DIN document 58940-. Beuth Verlag GmbH, Berlin, Germany. Deutches Institut für Normung. 2004. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 4. Evaluation classes of the minimum inhibitory concentration—MIC breakpoints of antibacterial agents. DIN document 58940-supplement 1. Beuth Verlag GmbH, Berlin, Germany. 406 TURNIDGE AND PATERSON 49. Deutches Institut für Normung. 2003. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 6. Determination of the minimum inhibitory concentration (MIC) with the agar dilution method. DIN document 58940-. Beuth Verlag GmbH, Berlin, Germany. 50. Deutches Institut für Normung. 1989. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 6. Determination of the minimum inhibitory concentration (MIC) with the agar dilution method—MICs of agents with control strains for the agar dilution method. DIN document 58940-supplement 1. Beuth Verlag GmbH, Berlin, Germany. 51. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 81. Microdilution—special requirements for testing non-fastidious bacteria. DIN document 58940-1 1. Beuth Verlag GmbH, Berlin, Germany. 52. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 82. Microdilution—special requirements for testing fastidious bacteria. DIN document 58940-1. Beuth Verlag GmbH, Berlin, Germany. 53. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 83. Microdilution—special requirements for testing strictly anaerobic bacteria. DIN document 58940-3. Beuth Verlag GmbH, Berlin, Germany. 54. Deutches Institut für Normung. 2002. Medical microbiology—susceptibility testing of pathogens to antimicrobial agents. 10. Criteria for the evaluation of the in vitro efficacy and the admission of new antimicrobial agents. DIN document 58940-10. Beuth Verlag GmbH, Berlin, Germany. 55. Doern, G. V. 1995. The in vitro activity of cefotaxime versus bacteria involved in selected infections of hospitalized patients outside the intensive care unit. Diagn. Microbiol. Infect. Dis. 22:13–17. 56. Drusano, G. L. 1988. Role of pharmacokinetics in the outcome of infections. Antimicrob. Agents Chemother. 32:289–297. 57. Drusano, G. L., S. L. Preston, C. Hardalo, R. Hare, C. Banfield, D. Andes, O. Vesga, and W. A. Craig. 2001. Use of preclinical data for selection of a phase II/III dose for evernimicin and identification of a preclinical MIC breakpoint. Antimicrob. Agents Chemother. 45:13–22. 58. Drusano, G. L. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2:289–300. 59. Drusano, G. L., S. L. Preston, C. Fowler, M. Corrado, B. Weisenger, and J. Kahn. 2004. Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in eradication of the infecting pathogen, in patients with nosocomial pneumonia. J. Infect. Dis. 189:1590–1597. 60. Dudley, M. N., and P. G. Ambrose. 2000. Pharmacodynamics in the study of drug resistance and establishing in vitro susceptibility breakpoints: ready for prime time. Curr. Opin. Microbiol. 3:515–521. 61. Eagle, H., R. Fleischman, and M. Levy. 1953. Continuous versus discontinuous therapy with penicillin: the effect of interval between injections on therapeutic efficacy. N. Engl. J. Med. 248:481–488. 62. Ellner, P. D., and H. C. Neu. 1981. The inhibitory quotient. A method for interpreting minimum inhibitory concentration data. JAMA 246:1575–1578. 63. Ericsson, H., and J. C. Sherris. 1971. Antibiotic sensitivity testing. Report of an international collaborative study. Acta Pathol. Microbiol. Scand. 217(Suppl. B):1–90. 64. European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). 2000. Determination of antimicrobial susceptibility test breakpoints. Clin. Microbiol. Infect. 6:570–572. 65. European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). 2000. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin. Microbiol. Infect. 6:509–515. 66. European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). 2000. Terminology relating to methods for the determination of susceptibility of bacteria to antimicrobial agents. Clin. Microbiol. Infect. 6:503–508. 67. European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). 2000. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin. Microbiol. Infect. 9:1–7. 68. Falcó, V., B. Almirante, Q. Jordano, L. Calonge, O. del Valle, C. Pigrau, A. M. Planes, J. Gavaldà, and A. Pahissa. 2004. Influence of penicillin resistance on outcome in adult patients with invasive pneumococcal pneumonia: is penicillin useful against intermediately resistant strains? J. Antimicrob. Chemother. 54:481–488. 69. Firsov, A. A., S. N. Vostrov, I. Y. Lubenko, K. Drlica, Y. A. Portnoy, and S. H. Zinner. 2003. In vitro pharmacodynamic evaluation of the mutant selection window hypothesis using four fluoroquinolones against Staphylococcus aureus. Antimicrob. Agents Chemother. 47:1604–1613. CLIN. MICROBIOL. REV. 70. Fleming, A. 1929. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 10:226–236. 71. Forrest, A., D. E. Nix, C. H. Ballow, T. F. Goss, M. C. Birmingham, and J. J. Schentag. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 37:1073–1081. 72. Forrest, A., S. Chodosh, M. A. Amantea, D. A. Collins, and J. J. Schentag. 1997. Pharmacokinetics and pharmacodynamics of oral grepafloxacin in patients with acute bacterial exacerbations of chronic bronchitis. J. Antimicrob. Chemother. 40(Suppl. A):45–57. 73. Franklin, C., L. Liolios, and A. Y. Peleg. 2006. Phenotypic detection of carbapenem-susceptible metallo--lactamase-producing gram-negative bacilli in the clinical laboratory. J. Clin. Microbiol. 44:3139–3144. 74. Frimodt-Møller, N. 2002. Correlation between pharmacokinetic/pharmacodynamic parameters and efficacy for antibiotics in the treatment of urinary tract infection. Int. J. Antimicrob. Agents 19:546–553. 75. Gehanno, P., G. Lenoir, and P. Berche. 1995. In vivo correlates for Streptococcus pneumoniae penicillin resistance in acute otitis media. Antimicrob. Agents Chemother. 39:271–272. 76. Gerber, A. U., A. P. Vastola, J. Brandel, and W. A. Craig. 1982. Selection of aminoglycoside-resistant variants of Pseudomonas aeruginosa in an in vivo model. J. Infect. Dis. 146:691–697. 77. Gupta, K., T. M. Hooton, and W. E. Stamm. 2001. Increasing antimicrobial resistance and the management of uncomplicated community-acquired urinary tract infections. Ann. Int. Med. 135:41–50. 78. International Organization for Standards. 15 November 2006, posting date. Susceptibility testing of infectious agents and evaluation of performance of antimicrobial susceptibility testing devices. 1. Reference method for testing the in vitro activity of antimicrobial agents against rapidly growing anaerobic bacteria involved in infectious diseases. ISO 20776-1. International Organization for Standards, Geneva, Switzerland. http://www.iso .org/iso/en/CatalogueDetailPage.CatalogueDetail?CSNUMBER⫽41630. 79. Japanese Society for Chemotherapy. 1990. Method for the determination of minimum inhibitory concentration (MIC) of aerobic bacteria by microdilution method. Chemotherapy (Tokyo) 38:102–105. 80. Japanese Society for Chemotherapy. 1993. Method for the determination of minimum inhibitory concentration (MIC) of fastidious bacteria and anaerobic bacteria by microdilution method. Chemotherapy (Tokyo) 41:183–189. 81. Kahlmeter, G. 16 October 2006, accession date. EUCAST procedure for harmonising and defining breakpoints. EUCAST, Basel, Switzerland. http: //www.srga.org/EUCAST/bpsetting.htm. 82. Kahlmeter, G., D. F. J. Brown, F. W. Goldstein, A. P. MacGowan, J. W. Mouton, A. Österland, A. Rodloff, M. Steinbakk, P. Urbaskova, and A. Vatopoulos. 2003. European harmonisation of MIC breakpoints for antimicrobial susceptibility testing of bacteria. J. Antimicrob. Chemother. 52: 145–148. 83. Kang, C.-I., S.-H. Kim, W. B. Park, K.-D. Lee, H.-B. Kim, E.-C. Kim, M.-Y.Oh, and K.-W. Choe. 2004. Bloodstream infections due to extendedspectrum -lactamase-producing Escherichia coli and Klebsiella pneumoniae: risk factors for mortality and treatment outcome, with special emphasis on antimicrobial therapy. Antimicrob. Agents Chemother. 48: 4574–4581. 84. Kaplan, S. L. 2002. Management of pneumococcal meningitis. Pediatr. Infect. Dis. J. 21:589–592. 85. Kashuba, A. D. M., A. N. Nafziger, G. L. Drusano, and J. S. Bertino. 1999. Optimizing aminoglycoside therapy for nosocomial pneumonia. Antimicrob. Agents Chemother. 43:623–629. 86. Kellner, J. D., D. W. Scheifele, S. A. Halperin, M. C. Lebel, D. Moore, N. Le Saux, E. L. Ford-Jones, B. Law, W. Vaudry, and Other Members of the Canadian Paediatric Society/Centre for Infectious Disease Prevention and Control, Immunization Monitoring Program. 2002. Outcome of penicillinnonsusceptible Streptococcus pneumoniae meningitis: a nested case-control study. Pediatr. Infect. Dis. J. 21:903–909. 87. Kerrn, M. B., N. Frimodt-Møller, and F. Espersen. 2003. Effects of sulfamethizole and amdinocillin against Escherichia coli strains (with various susceptibilities) in an ascending urinary tract infection mouse model. Antimicrob. Agents Chemother. 47:1002–1009. 88. Knudsen, J. D., K. Fuursted, F. Espersen, and N. Frimodt-Møller. 1997. Activities of vancomycin and teicoplanin against penicillin-resistant pneumococci in vitro and in vivo and correlation to pharmacokinetics parameters in the mouse peritonitis model. Antimicrob. Agents Chemother. 41:1910–1915. 89. Knudsen, J. D., K. Fuursted, S. Raber, F. Espersen, and N. FrimodtMøller. 2000. Pharmacodynamics of glycopeptides in the mouse peritonitis model of Streptococcus pneumoniae or Staphylococcus aureus infection. Antimicrob. Agents Chemother. 44:1247–1254. 90. Koeth, L. M., A. King, H. Knight, J. May, L. A. Miller, I. Phillips, and J. A. Poupard. 2000. Comparison of cation-adjusted Mueller-Hinton broth with Iso-Sensitest broth for the NCCLS broth microdilution method. J. Antimicrob. Chemother. 46:369–376. 91. Kronvall, G., G. Kahlmeter, E. Myrne, and M. F. Galas. 2003. A new VOL. 20, 2007 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. SETTING ANTIBACTERIAL SUSCEPTIBILITY BREAKPOINTS method for normalized interpretation of antimicrobial resistance from disk test results for comparative purposes. Clin. Microbiol.Infect.9:120–132. Kronvall, G. 2003. Determination of the real standard distribution of susceptible strains in zone histograms. Int. J. Antimicrob. Agents 22:7–13. Kronvall, G., I. Karlsson, M. Walder, M. Sörberg, and L. E. Nisson. 2006. Epidemiological MIC cut-off values for tigecycline calculated from Etest values using normalized resistance interpretation. J. Antimicrob. Chemother. 57:498–505. Leggett, J. E., B. Fantin, S. Ebert, K. Totsuka, B. Vogelman, W. Calame, H. Mattie, and W. A. Craig. 1989. Comparative antibiotic dose-effect relations at several dosing intervals in murine pneumonitis and thigh-infection models. J. Infect. Dis. 159:281–282. MacGowan, A., and R. Wise. 2001. Establishing MIC breakpoints and the interpretation of in vitro susceptibility tests. J. Antimicrob. Chemother. 48(Suppl. S1):17–28. MacGowan, A., C. Rogers, and K. Bowker. 2003. In vitro models, in vivo models, and pharmacokinetics: what can we learn from in vitro models? Clin. Infect. Dis. 33(Suppl. 3):S214–S220. McDonald, P. J., B. H. Wetherall, and H. Pruul. 1981. Postantibiotic leukocyte enhancement: increased susceptibility of bacteria pretreated with antibiotics to activity of leukocytes. Rev. Infect. Dis. 3:38–44. Mesa Española de Normalización de la Sensibilidad y Resistencia a los Antimicrobianos (MENSURA). 2000. Recommedaciones del grupo MENSURA para la selecciónde antimicrobianos en el studio de la sensibilidad y criterios para la interpretacióndel antibiograma. Rev. Esp. Quimioter. 13:73–86. Metzler, C. M., and R. M. DeHaan. 1974. Susceptibility tests of anaerobic bacteria: statistical and clinical considerations. J. Infect. Dis. 30:88–594. Miller, L. G., and A. W. Tang. 2004. Treatment of uncomplicated urinary tract infections in an era of increasing antimicrobial resistance. Mayo Clin. Proc. 79:1048–1054. Moise, P. A., A. Forrest, S. M. Bhavnani, M. C. Birmingham, and J. J. Schentag. 2000. Area under the inhibitory curve and a pneumonia scoring system for predicting outcomes of vancomycin therapy for respiratory infections by Staphylococcus aureus. Am. J. Hosp. Pharm. 57(Suppl. 2):S4–S9. Moise-Broder, P. A., A. Forrest, M. C. Birmingham, and J. J. Schentag. 2004. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin. Pharmacokinet. 43:925–942. Moore, R. D., P. S. Lietman, and C. R. Smith. 1987. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimum inhibitory concentration. J. Infect. Dis. 155:93–99. Mouton, J. W., J. E. Degener, B. van Klingeren, A. J. de Neeling, and Commissie Richtlijnen Gevoelighheidsbepalingen. 2000. Het vastellen van gevoelighehidscriteria voor antibcateriële middelen in Nederland: verleden, heden en toekomst. Ned. Tijdschr. Med. Microbiol.8:73–78. Mouton, J. W. 2003. Impact of pharmacodynamics on breakpoint selection for susceptibility testing. Infect. Dis. Clin. N. Am. 17:579–598. Odenholt Tornqvist, I., S. E. Holm, and O. Cars. 1991. Pharmacodynamic effects of subinhibitory antibiotic concentrations. Scand. J. Infect. Dis. 74(Suppl.):94–101. Olivier, C., R. Cohen, P. Begué, and D. Floret. 2000. Bacteriologic outcome of children with cefotaxime- or ceftriaxone-susceptible and -nonsusceptible Streptococcus pneumoniae meningitis. Pediatr. Infect. Dis. J. 19:1015–1017. Pallares, R., J. Liñares, M. Vadillo, C. Cabellos, F. Manresa, P. F. Viladrich, R. Martin, and F. Gudiol. 1995. Resistance to penicillin and cephalosporin and mortality from severe pneumococcal pneumonia in Barcelona, Spain. N. Engl. J. Med. 333:474–480. Paterson, D. L., and R. A. Bonomo. 2005. Extended-spectrum beta-lactamases: a clinical update. Clin. Microbiol. Rev. 18:657–686. Paterson, D. L., W.-C. Ko, A. Von Gottberg, J. M. Casellas, L. Mulazimoglu, K. P. Klugman, R. A. Bonomo, L. B. Rice, J. G. McCormack, and V. L. Yu. 2001. Outcome of cephalosporin treatment for serious infections due to apparently susceptible organisms producing extended-spectrum beta-lactamases: implications for the clinical laboratory. J. Clin. Microbiol. 39:2206– 2212. Preston, S. L., G. L. Drusano, A. L. Berman, C. L. Fowler, A. T. Chow, B. Dornseif, V. Reichl, J. Natarajan, and M. Corrado. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125– 129. Rasheed, J. K., and F. C. Tenover. 2003. Detection and characterization of antimicrobial resistance genes in bacteria, p. 1196–1212. In P. R. Murray, E. J. Baron, J. H. Jorgensen, M. A. Pfaller, and R. H. Yolken (ed.), Manual of clinical microbiology, 8th ed., vol. 1. ASM Press, Washington, DC. Raz, R., B. Chazan, Y. Kennes, R. Colodner, E. Rottensterrich, M. Dan, W. Stamm, and the Israeli Urinary Tract Infection Group. 2002. Empiric use of trimethoprim-sulfamethoxazole (TMP-SMX) in the treatment of women with uncomplicated urinary tract infections, in a geographical area with a high prevalence of TMP-SMX-resistant uropathogens. Clin. Infect. Dis. 24:1165–1169. Rosco Diagnostica. 2005–2006. User’s guide. Neo-Sensitabs susceptibility testing, 17th ed. Rosco Diagnostica A/C, Taastrup, Denmark. 407 115. Saito, A. 1995. Clinical breakpoints for antimicrobial agents in pulmonary infections and sepsis: report of the Committee for Japanese Standards for Antimicrobial Susceptibility Testing of Bacteria. J. Infect. Chemother. 1:83–88. 116. Saito, A., T. Inamatsu, J. Okada, T. Oguri, H. Kanno, N. Kusano, H. Kumon, K. Yamaguchi, A. Watanabe, and K. Watanabe. 1999. Clinical breakpoints in pulmonary infections and sepsis: new antimicrobial agents and supplemental information for some agents already released. J. Infect. Chemother. 5:223–226. 117. Sakoulas, G., P. A. Moise-Broder, J. Schentag, A. Forrest, R. C. Moellering, Jr., and G. M. Eliopoulos. 2004. Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 42:2398–2402. 118. Schentag, J. J., I. L. Smith, D. J. Swanson, C. DeAngelis, J. E. Fracasso, A. Vari, and J. W. Vance. 1984. Role of dual individualization with cefmenoxime. Am. J. Med. 77:43–50. 119. Sirot, J., P. Courvalin, and C.-J. Soussy. 1996. Definition and determination of in vitro antibiotic susceptibility breakpoints for bacteria. Clin. Microbiol. Infect. 2(Suppl. 1):S5–S10. 120. Snydman, D. R., G. J. Cuchural, L. McDermott, and M. Gill. 1992. Correlation of various in vitro testing methods with clinical outcomes in patients with Bacteroides fragilis group infections treated with cefoxitin: a retrospective study. Antimicrob. Agents Chemother. 36:540–544. 121. Sociedad Españolade Enfermedades Infecciosas y Microbiologı́a Clı́nica. 2000. Procedimientos en microbiologı́a clı́nica: métados básicos para el estudio de la sensibilidad a los antimicrobianos. Sociedad Españolade Enfermedades Infecciosas y Microbiologı́a Clı́nica, Madrid, Spain. http://www .seimc.org/protocolos/microbiologia/. 122. Stamey, T. A., W. R. Fair, M. M. Timothy, M. A. Millar, G. Mihara, and Y. C. Lowery. 1974. Serum versus urinary antimicrobial concentrations in cure of urinary-tract infections. N. Engl. J. Med. 291:1159–1163. 123. Swedish Reference Group on Antibiotics. 11 February 2004, revision date. The principles behind SRGA breakpoints. Swedish Reference Group on Antibiotics, Vaxjo, Sweden. www.srga.org. Accessed 5 February 2006. 124. Swenson, J. M., J. F. Hindler, and J. H. Jorgensen. 2003. Special phenotypic methods for detecting antibacterial resistance, p. 1178–1195. In P. R. Murray, E. J. Baron, J. H. Jorgensen, M. A. Pfaller, and R. H. Yolken (ed.), Manual of clinical microbiology, 8th ed., vol. 1. ASM Press, Washington, DC. 125. Szabó, D., R. A. Bonomo, F. Silveira, A. W. Pasculle, C. Baxter, P. K. Linden, A. M. Hujer, K. M. Hujer, K. Deeley, and D. L. Paterson. 2005. SHV-type extended-spectrum beta-lactamase production is associated with reduced cefepime susceptibility in Enterobacter cloacae. J. Clin. Microbiol. 43:5058–5064. 126. Tam, V. H., P. S. McKinnon, R. L. Akins, M. L. Ryback, and G. L. Drusano. 2002. Pharmacodynamics of cefepime in patients with gram-negative infections. J. Antimicrob. Chemother. 50:425–428. 127. Tam, V. H., P. S. McKinnon, R. L. Akins, G. L. Drusano, and M. L. Ryback. 2003. Pharmacokinetics and pharmacodynamics of cefepime in patients with various degrees of renal function. Antimicrob. Agents Chemother. 47:1853–1861. 128. Turnidge, J. D. 1998. The pharmacodynamics of -lactams. Clin. Infect. Dis. 27:10–22. 129. Turnidge, J. 1999. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Drugs 58(Suppl. 2):29–36. 130. Turnidge, J. D., and J. M. Bell. 2005. Antimicrobial susceptibility on solid media, p. 8–60. In V. Lorian (ed.), Antibiotics in laboratory medicine, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. 131. Turnidge, J., and G. Bordash. 2007. Statistical methods for establishing quality control ranges for antibacterial agents in Clinical and Laboratory Standards Institute susceptibility testing. Antimicrob. Agents Chemother. 51:2483–2488. 132. Turnidge, J., G. Kahlmeter, and G. Kronvall. 2006. Statistical characterisation of bacterial wild-type MIC value distributions and the determination of epidemiological cut-off values. Clin. Microbiol. Infect. 12:418–425. 133. Van Wart, S., L. Phillips, E. A. Ludwig, R. Russo, D. A. Gajjar, A. Bello, P. G. Ambrose, C. Costanzo, T. H. Grasela, R. Echols, and D. M. Grasela. 2004. Population pharmacokinetics of garenoxacin in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 48:4766–4777. 134. Vogelman, B., S. Gudmundsson, J. Turnidge, J. Leggett, and W. A. Craig. 1988. In vivo postantibiotic effect in a thigh infection in neutropenic mice. J. Infect. Dis. 157:287–298. 135. Vogelman, B., S. Gudmundsson, J. Leggett, J. Turnidge, S. Ebert, and W. A. Craig. 1988. Correlation of antimicrobial pharmacokinetic parameters with therapeutic efficacy in an animal model. J. Infect. Dis. 158:831–847. 136. Walsh, T. R., M. A. Toleman, L. Poirel, and P. Nordmann. 2005. Metallo-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 18:306–325. 137. Watanabe, Y., S. Ebert, and W. Craig. 1992. AUC/MIC ratio is the unifying parameter for comparison of the in-vivo activity among fluoroquinolones, 408 138. 139. 140. 141. TURNIDGE AND PATERSON abstr. 42. Abstr. 32nd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. Werkgroep Richtlijnen Gevoelighheidsbepalingen. 1981. Standaadisatie van gevoelighheidsbepalingen. RIVM, Bilthoven, The Netherlands. Werkgroep Richtlijnen Gevoelighheidsbepalingen. 1990. Standaadisatie van gevoelighheidsbepalingen, supplement e. RIVM, Bilthoven, The Netherlands. Werkgroep Richtlijnen Gevoelighheidsbepalingen. 1991. Standaadisatie van gevoelighheidsbepalingen, supplement e, bijlage. RIVM, Bilthoven, The Netherlands. Wikler, M. A., and P. G. Ambrose. 2005. The breakpoint, p. 1–7. In V. CLIN. MICROBIOL. REV. Lorian (ed.), Antibiotics in laboratory medicine, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA. 142. Yu, V. L., C. C. C. Chiou, C. Feldman, A. Ortqvist, J. Rello, A. J. Morris, L. M. Baddour, C. M. Luna, D. R. Snydman, M. Ip, W. C. Ko, B. F. Chedid, A. Andremont, and K. Klugman. 2003. An international prospective study of pneumococcal bacteremia: correlation with in vitro resistance, antibiotics administered, and clinical outcome. Clin. Infect. Dis. 37:320–327. 143. Zelenitsky, S. A., G. K. Harding, S. Sun, K. Ubhi, and R. E. Ariano. 2003. Treatment and outcome of Pseudomonas aeruginosa bacteraemia: an antibiotic pharmacodynamic analysis. J. Antimicrob. Chemother. 52: 668–674. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 409–425 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00041-06 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Ventilator-Associated Pneumonia in Neonatal and Pediatric Intensive Care Unit Patients Elizabeth Foglia, Mary Dawn Meier, and Alexis Elward* Division of Infectious Diseases, Department of Pediatrics, Washington University School of Medicine, St. Louis, Missouri INTRODUCTION .......................................................................................................................................................409 OUTCOMES................................................................................................................................................................410 Recommendations for Current Practice and Future Research ........................................................................411 DIAGNOSIS ................................................................................................................................................................411 Clinical Criteria ......................................................................................................................................................411 Invasive Testing in Adults .....................................................................................................................................412 Invasive Testing in Children .................................................................................................................................413 Computerized Surveillance ....................................................................................................................................413 Recommendations for Current Practice and Future Research ........................................................................414 MICROBIOLOGY ......................................................................................................................................................414 RISK FACTORS FOR VAP IN NICU PATIENTS ................................................................................................414 RISK FACTORS FOR VAP IN PICU PATIENTS.................................................................................................415 Recommendations for Current Practice and Future Research ........................................................................415 PREVENTION.............................................................................................................................................................416 Head-of-Bed Elevation............................................................................................................................................416 In-Line Suctioning ..................................................................................................................................................416 H2 Blockers/Sucralfate ...........................................................................................................................................416 Hand Hygiene ..........................................................................................................................................................417 Selective Decontamination.....................................................................................................................................417 Oral Hygiene............................................................................................................................................................419 The Bundle Approach ............................................................................................................................................419 Educational Interventions......................................................................................................................................419 Recommendations for Current Practice and Future Research ........................................................................420 VAP TREATMENT .....................................................................................................................................................420 Empirical Therapy ..................................................................................................................................................420 Specific Treatment ..................................................................................................................................................422 Duration of Therapy...............................................................................................................................................423 Recommendations for Current Practice and Future Research ........................................................................423 CONCLUSION............................................................................................................................................................423 REFERENCES ............................................................................................................................................................423 The most recent National Nosocomial Infection Surveillance (NNIS) data from 2002 to 2004 show NICU VAP rates ranging from 1.4 to 3.5 per 1,000 ventilator days (68). In 1998, a crosssectional study of hospital-acquired infections in 50 children’s hospitals was performed by the Pediatric Prevention Network (88). Of 43 children’s hospitals that returned questionnaires reporting NICU and PICU surveillance data, the VAP rate by device days was reported by 19 hospitals, and 12 hospitals provided VAP rates stratified by birth weight groups (Table 1). In this cross-sectional survey, VAP rates were highest for the 1,001- to 1,500-g and ⬍1,000-g birth weight categories. Differences in study methodology and case mix can influence the reported incidence of VAP (6). Surveillance methods differ across study institutions, introducing variability into the reported incidence of VAP. The influence of surveillance intensity on the reported prevalence of nosocomial infections is illustrated by a 41-month surveillance study in a children’s hospital. Infection control surveillance was conducted twice a week for the first 2 years of the study and then daily for the second 2 years of the study through a nursing sentinel sheet. Those investigators found a 50% increase in the incidence of INTRODUCTION Ventilator-associated pneumonia (VAP) is pneumonia in mechanically ventilated patients that develops later than or at 48 h after the patient has been placed on mechanical ventilation. VAP is the second most common hospital-acquired infection among pediatric and neonatal intensive care unit (ICU) (NICU) patients (41, 43). Overall, VAP occurs in 3 to 10% of ventilated pediatric ICU (PICU) patients (1, 28). Surveillance studies of nosocomial infections in NICU patients indicate that pneumonia comprises 6.8 to 32.3% of nosocomial infections in this setting (26, 39, 48). The incidence of VAP is higher in adult ICU patients, ranging from 15 to 30% (8, 31, 50, 70, 90). NICU VAP rates vary by birth weight category as well as by institution. Two large studies are summarized in Table 1. * Corresponding author. Mailing address: Pediatric Infectious Diseases, Washington University School of Medicine, Box 8116, St. Louis Children’s Hospital, One Children’s Place, St. Louis, MO 63110. Phone: (314) 454-6050. Fax: (314) 454-2836. E-mail: Elward_A@kids .wustl.edu. 409 410 FOGLIA ET AL. CLIN. MICROBIOL. REV. TABLE 1. VAP rates stratified by birth weighta Range (median) of VAP rates by birth weight (g) of: Study b NNIS Stoverc a b c Source 86–102 high-risk nurseries 12 NICUs ⱕ1,000 1,001–1,500 1,501–2,500 ⬎2,500 0.0–8.5 (2.4) 0.0–21.2 (3.5) 0.0–8.0 (0.0) 0.0–34.5 (4.9) 0.0–6.1 (0.0) 0.0–13.7 (1.1) 0.0–3.2 (0.0) 0.0–6.0 (0.9) Range given is the 10th to 90th percentile for NNIS data. See reference 68. See reference 88. reported nosocomial infections following the introduction of daily surveillance (39). NNIS definitions for VAP were revised in 2002, resulting in a more stringent definition of VAP. Studies of VAP incorporating these revised definitions reported lower rates of VAP, making it difficult to know if VAP was previously overdiagnosed or is now underdiagnosed. The revised definitions must also be considered when VAP rates are compared over time. Applying the Centers for Disease Control and Prevention (CDC) definitions for VAP in low-birth-weight infants introduces additional complexity in defining the incidence of VAP. CDC definitions for VAP exist for infants ⬍1 year of age, but there are no specific definitions for low- or very-lowbirth-weight infants. These patients often have comorbidities such as bronchopulmonary dysplasia, hyaline membrane disease, bloodstream infections (BSIs), and necrotizing enterocolitis that obscure clinical, laboratory, and radiographic evidence of VAP. OUTCOMES VAP is associated with increased morbidity in PICU patients, specifically, a longer duration of mechanical ventilation. Fischer et al. (34) performed a prospective cohort study to determine the delay of extubation attributable to VAP among neonates and children undergoing repair of congenital heart disease. Twenty-six of the 272 patients enrolled over a 22month period developed VAP (9.6%). VAP diagnosis was made when the following criteria were met: fever exceeding 38.5°C, tachypnea and/or otherwise unexplained increased oxygen requirement, elevated white blood cell count (⬎15 ⫻ 109 cells/liter), a cultured pathogen from tracheal aspirate together with a positive gram stain, and increased leukocyte contents, plus an infiltrate on chest radiographs persisting for 48 h or more (29). Using a Cox proportional hazards model to control for complexity of surgery, other respiratory complications, and secondary surgeries, those investigators found that the median delay of extubation attributable to VAP was 3.7 days (average of 5.2 versus 1.5 for patients with and without VAP, respectively). VAP rates increased dramatically for patients intubated for long periods of time. Among patients extubated within the first 3 days of surgery, only 4% developed VAP, compared to 40% of postoperative cardiothoracic surgery patients intubated longer than 30 days (34). Of the 26 VAP cases, 19 occurred within the first 3 to 6 days after surgery. Presumed VAP is also associated with additional resource utilization with respect to antibiotic administration. VAP is the most common reason for the initiation of empirical antibiotics among PICU patients. A prospective cohort study at an aca- demic tertiary care center performed in a PICU (n ⫽ 456) found that over half (56.6%) of all patients (n ⫽ 258) received antibiotics (33). Treatment for suspected VAP comprised 616 of 1,303 (47%) of the antibiotic treatment days. Those authors reviewed medical records to determine whether patients had evidence of an alternative explanation for the symptoms attributed to VAP, such as a viral infection. For 40% of the antibiotic days (552/1,303 treatment days), patients were classified as having no infection (i.e., did not meet clinical criteria as defined by the CDC) or as having a viral infection. Those authors concluded that an intervention targeted at decreasing antibiotic use for VAP would have the greatest impact on antibiotic use. In pediatric populations, the published data are unmatched for severity of illness and univariate but suggest that pediatric patients with VAP may have excess mortality and length of PICU and NICU stay. The European Multicenter Trial examined the epidemiology of hospital-acquired infections in 20 units (5 PICUs, 7 neonatal units, 2 hematology-oncology units, and 8 general pediatric units) in eight countries, with a total of 14,675 admissions (710 admission in PICUs) (77). Those investigators found the infected patients had a longer mean length of stay in the PICU (26.1 ⫾ 17.3 versus 10.6 ⫾ 6 days; P ⬍ 0.001) than uninfected patients. The mortality rate was 10% for PICU patients with nosocomial infections. The mortality and length of stay associated specifically with VAP were not reported, although VAP accounted for 53% of the nosocomial infections in PICU patients. Mortality among uninfected PICU patients was not reported. Similarly, PICU length of stay in a 9-month prospective cohort study in an academic tertiary care center revealed that patients with VAP (n ⫽ 30) had a mean PICU length of stay of 27 days versus 6 days for uninfected patients (n ⫽ 595) (P ⫽ 0.001) (28). In that same study, the mortality rates with and without VAP were 20% and 7%, respectively (P ⫽ 0.065). Outcomes between patients on mechanical ventilation for more than 8 days with VAP (n ⫽ 30) and those without VAP (n ⫽ 62) were also compared. PICU length of stay was longer for VAP patients (27.53 ⫾ 20.09 versus 18.72 ⫾ 35 days), as was hospital length of stay (52.63 ⫾ 37.43 versus 33.77 ⫾ 49.51 days), but no differences in mortality rates for VAP (20%) or uninfected patients (21%) were found. Almuneef et al. (1) determined in a prospective cohort study (n ⫽ 361) that PICU lengths of stay with (n ⫽ 37) and without (n ⫽ 324) VAP were longer for patients with VAP (33.70 ⫾ 30.28 versus 14.66 ⫾ 17.34 days; P ⫽ 0.001). Statistically significant differences in mortality rates between patients with VAP and those without VAP were not found (P ⫽ 0.362). Both of those studies performed only univariate analyses to compare mortality rates among patients with and with- VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS out VAP. Multivariate analysis of predictors of mortality among PICU patients with sufficient numbers of VAP controlling for severity of illness both at admission and at the time of VAP as well as other potential predictors of death is necessary to determine the true attributable mortality of VAP in pediatric patients. VAP has also been shown to increase hospital costs. The cost of VAP was analyzed in a 2-year study of PICU patients (n ⫽ 1919) with a single admission (38). The direct cost for patients with VAP (n ⫽ 56) was $38,614, and that for patients without VAP was $7,682. In a multivariate analysis controlling for other predictors of cost including age, severity of illness, underlying disease, and ventilator days, VAP was independently associated with a direct cost of $30,931 (95% confidence interval [CI], $18,349 to $82,638) (38). This is a single study from an academic tertiary care center; further studies are needed to determine whether the results from this single center are generalizable. Recommendations for Current Practice and Future Research Differences in the incidence of VAP occur as a result of the definitions used, persons doing surveillance, and frequency of surveillance. Standardization of surveillance methodology and validation of current definitions against a histopathologic or microbiologic “gold standard” would make interinstitutional comparisons meaningful, particularly in light of mandatory reporting of health care-associated infections. Recent literature suggests that pediatric VAP is associated with increased morbidity, antibiotic use, PICU cost, and PICU and hospital length of stay. Prospective studies using consistent definitions of VAP and controlling for severity of illness both at admission and at the time of VAP as well as other possible predictors of death and length of stay are necessary to define the true attributable mortality and cost associated with VAP in pediatric patients. DIAGNOSIS Clinical Criteria The lack of a gold standard for the diagnosis of VAP in both adults and children makes an interpretation of the literature complex. The clinical criteria for the diagnosis of VAP have been established by the NNIS and the CDC (22). Patients who are mechanically ventilated for more than or equal to 48 h must have two or more abnormal chest radiographs with at least one of the following symptoms: new or progressive and persistent infiltrate, consolidation, cavitation, and/or pneumatoceles (in infants ⱕ1 year of age). However, in patients without underlying pulmonary or cardiac disease (respiratory distress syndrome, bronchopulmonary dysplasia, pulmonary edema, or chronic obstructive pulmonary disease), one definitive chest radiograph is acceptable. In addition to abnormal chest radiographs, a patient must have at least one of the following symptoms: fever (⬎38°C) with no other recognized cause, leukopenia (⬍4,000 white blood cells [WBC]/mm3) or leukocytosis (ⱖ12,000 WBC/mm3), and at least two of the following criteria: new onset of purulent sputum, change in 411 character of sputum, increased respiratory secretions, or increased suctioning requirements; new onset of or worsening cough, dyspnea, or tachypnea; rales or bronchial breath sounds; and worsening gas exchange (e.g., O2 desaturations [e.g., PaO2/ FiO2 levels of ⱕ240], increased oxygen requirements, or increased ventilation demand). The criteria described above may be used to diagnose VAP in children; however, specific diagnostic criteria for VAP have been developed for infants ⱕ1 year of age and children ⬎1 and ⱕ12 years of age. Infants that are ⱕ1 year old must have worsening gas exchange (oxygen desaturations, increased oxygen requirements, or increased ventilator demand) and at least three of the following criteria: temperature instability with no other recognized cause; new onset of purulent sputum, change in character of sputum, increased respiratory secretions, or increased suctioning requirements; apnea, tachypnea, nasal flaring with retraction of chest wall, or grunting; wheezing, rales, or rhonchi; cough; and bradycardia (⬍100 beats/min) or tachycardia (⬎170 beats/min). Children ⬎1 and ⱕ12 years of age must meet at least three of the following criteria: fever (⬎38.4°C or ⬎101.1°F) or hypothermia (⬍37°C or 97.7°F) with no other recognized cause; leukopenia (⬍4,000 WBC/mm3) or leukocytosis (ⱖ15,000 WBC/mm3); new onset of purulent sputum, change in character of sputum, increased respiratory secretions, or increased suctioning requirements; rales or bronchial breath sounds; and worsening gas exchange (O2 desaturations [pulse oximetry of ⬍94%], increased oxygen requirements, or increased ventilation demand). NNIS/CDC criteria do not require microbiologic confirmation to diagnose pneumonia. In summary, many of the diagnostic criteria are similar for the ⱕ1-year-old and ⬎1- or ⱕ12-year-old age groups. Temperature instability is a diagnostic criterion for the ⱕ1-year-old age group; either temperature elevation or hypothermia is a criterion for the ⬎1- and ⱕ12-year-old age group. For the ⬍1-year-old group, cough, bradycardia, tachycardia, nasal flaring, grunting, and wheezing are diagnostic criteria not listed for the ⬎1- or ⱕ12-year-old age groups, although for the older age group, dyspnea without further specific definition is a diagnostic criterion. Worsening gas exchange, change in character or amount of sputum, cough, rales, or bronchial breath sounds are criteria for diagnosis in all three age groups. We suggest that a consistent use of the age-specific definitions are preferred, although we were unable to find any published studies directly comparing the sensitivity and specificity of the agespecific definitions to those for any age group. Clinical definitions for VAP may be applied inconsistently, and the lack of specific definitions of components of the clinical definition such as worsening gas exchange, oxygen desaturations, increased oxygen requirements, and increased ventilator demand may exacerbate this. Cordero et al. (19) determined differences in the application of the CDC definitions using NICU patients (n ⫽ 37) diagnosed with VAP by interpretation of CDC definitions by infection control practitioners (ICPs) and a positive tracheal aspirate culture. A panel of neonatologists reviewed the clinical and laboratory evidence as well as the radiographs. The neonatologists diagnosed VAP in only seven patients. The neonatologists categorized the other patients as having asymptomatic airway colonization (n ⫽ 12), BSI (n ⫽ 7), and airway colonization with equivocal signs of infection (n ⫽ 11). Among 8 of the 11 patients with equivocal signs of infection, the general radiologist report 412 FOGLIA ET AL. CLIN. MICROBIOL. REV. TABLE 2. Accuracy of invasive diagnostic techniques for the diagnosis of VAP in adults and childrena Age group and source (reference) No. of patients Adults Rouby et al. (80) Chastre et al. (13) Fabregas et al. (30) 26 26 25 Protected mini-BAL PSB TBA Protected BAL BAL PSB Any invasive diagnostic technique Children Gauvin et al. (42) 10 BAL (104 CFU/ml) 29 Bacterial index ⬎5 ICB Endotracheal cultures BAL (104 CFU/ml) Labenne et al. (60) SE (%) SP (%) Histopathology, lung tissue culture Histopathology, lung tissue culture Histopathology, lung tissue culture 70 100 69 39 77 62 85 69 60 92 100 58 75 50 Expert opinion; 2/3 blinded to BAL and PSB results 50 80 78 30 90 72 86 95 40 88 69 79 90 95 88 88 Diagnostic technique Gold standard(s) (i) Positive pleural fluid culture, (ii) computed tomography scan with abscess, (iii) histopathology, (iv) lung tissue culture, (v) blood and tracheal aspirate positive for same organism without other source, (vi) expert opinion; 2/3 blinded to BAL/PSB PSB ICB on gram stain and ⫹ BAL ICB on gram stain and ⫹ BAL and ⫹ PSB a SE, sensitivity; SP, specificity; TBA, tracheobronchial aspirates; ICB, intracellular bacteria. stated that the radiographic changes were suggestive of VAP; the neonatologist panel, reviewing the same radiographs, concluded that VAP was unlikely in these patients. Those authors concluded that an isolated positive tracheal aspirate does not distinguish between airway colonization and VAP and that routine radiology reports without definitive clinical and laboratory evidence may be misleading. Invasive Testing in Adults NNIS/CDC criteria for VAP do not require microbiologic confirmation. A brief review of invasive testing to confirm the diagnosis of VAP in adults will be performed, given the paucity of literature regarding invasive testing in children. It is unclear whether the adult experience can be extrapolated to children. Several studies have examined the accuracy of invasive testing for the diagnosis of VAP in critically ill adults (Table 2). Microbiologic examination of specimens obtained from bronchoalveolar lavage (BAL) or protected specimen brush (PSB) have an estimated 70% sensitivity and 77% specificity compared to histopathology and/or lung tissue culture (13, 30, 80). In one of the most comprehensive studies of VAP in mechanically ventilated adults, Rouby et al. (80) sought to describe the histologic and bacteriologic characteristics of human nosocomial pneumonia and to evaluate the accuracy of protected mini-BAL for the diagnosis of VAP compared to lung tissue cultures and lung histology in patients who died while on mechanical ventilation. Twenty-six patients had both positive pathology and lung tissue culture; in 20 of these patients, the BAL and lung tissue culture results were concordant (Fig. 1). Chastre (13) compared the accuracy of the bronchoscopic PSB to that of histologic and bacteriologic examinations of pulmonary specimens in adults (n ⫽ 26). PSB and lung cultures were highly correlated (r ⫽ 0.65; n ⫽ 28; P ⬍ 0.001) and higher in patients not on antibiotics within 1 week before death than in patients on antibiotics before death (r ⫽ 0.55; n ⫽ 33; P ⬍ 0.001). Pneumonia was not found by histology or lung tissue culture when PSB culture organisms were ⬍103 CFU per ml. PSB cultures with ⱖ103 CFU/ml identified all patients with histologically proven pneumonia. In patients treated with antibiotics, four patients had microorganisms isolated by PSB with concentrations of ⬎103 CFU/ml not found in the lung tissue cultures. Fabregas and colleagues (30) sought to determine the accu- FIG. 1. Comparison of protected minibronchoalveolar lavage with histopathology and bacteriology for diagnosing VAP. VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS racy of clinical criteria and microbiologic testing for the diagnosis of VAP. The clinical pulmonary infection score (CPIS) was used to compare microbiological criteria, clinical criteria, and sampling techniques. Lung biopsies were performed for 25 mechanically ventilated patients immediately after death. The reference standard was the presence of positive histology for pneumonia or positive lung cultures. Chest X-ray infiltrates and at least two of three clinical criteria achieved sensitivity and specificity of 69% and 75%, respectively. The CPIS sensitivity and specificity were 77% and 42%, respectively. Noninvasive and invasive techniques achieved similar results. All diagnostic techniques combined (PSB, BAL fluid, and protected BAL fluid) achieved sensitivity and specificity of 85% and 50%, respectively. A meta-analysis done in 2005 (n ⫽ 628) determined whether invasive testing altered the management and mortality of VAP in critically ill adults (84). VAP was confirmed bronchoscopically in 44 to 69% of the patients. Overall, antibiotics were almost three times as likely to be changed if a bronchoscopy was performed. In a separate pooled analysis of prospective uncontrolled trials, alteration in antibiotic prescription occurred 50% (36 to 65%) of the time. To our knowledge, no similar meta-analyses exist for pediatric populations. Invasive Testing in Children A few studies have examined the sensitivity and specificity of lower airway sampling in PICU patients and found the sensitivity and specificity of BAL (104 CFU/ml) to be 50 to 72% and 80 to 88%, respectively (42, 60) (Table 2). The nonbronchosopic BAL (NB-BAL) has been used in children as an alternative to fiberoptic BAL to diagnose pneumonia. This procedure is performed by placing a suction catheter into the endotracheal tube until resistance is met and then placing and suctioning back a small amount of sterile normal saline from the lower airway. The presence of bubbles in the returned fluid is suggestive that a lower airway sample containing surfactant has been obtained. Quantitative cultures of the fluid are then performed by the microbiology laboratory. This procedure may be advantageous compared to fiberoptic bronchoscopic techniques because bronchoscopic equipment is not required, a trained bronchoscopist is not necessary, and the diagnostic accuracy is comparable to that of fiberoptic techniques (36, 37, 71). Gauvin et al. (42) performed a 27-month prospective cohort study of PICU patients suspected of having VAP in a tertiary academic care center. Of 30 patients, 10 were diagnosed with VAP and 9 were diagnosed with ventilator-associated tracheitis by an expert panel. The expert panel was used as the reference standard; they were given clinical, radiographic, and microbiologic data but were blinded to the BAL results. A bacterial index (sum of the log of all species obtained from BAL) of ⬎5 had the highest correlation with the reference standard (concordance, 83%; kappa ⫽ 0.61), a sensitivity of 78%, a specificity of 86%, a positive predictive value of 70%, and a negative predictive value of 90% (42). Intracellular bacteria and gram stain from BAL were specific (95% and 81%, respectively) but not sensitive (30% and 50%, respectively) for the diagnosis of pediatric VAP, whereas clinical criteria and endotracheal cultures were sensitive (100% and 90%, respec- 413 tively) but not specific (15% and 40%, respectively). That study concluded that blind BALs with a bacterial index of ⬎5 are the most reliable method for diagnosing VAP in mechanically ventilated children. The study did not describe what proportion of patients were on antibiotics or the duration of antibiotic exposure prior to BAL. Labenne et al. (60) also investigated the sensitivity and specificity of PSB and BAL in PICU patients with suspected VAP. The gold standards used by those investigators were a positive pleural fluid culture, computed tomography scan with pulmonary abscesses, histopathological evidence, positive lung biopsy (⬎104 CFU/gram), the same bacteria isolated in blood and endotracheal aspirate without another source, or clinical diagnosis using CDC guidelines established independently by two investigators blinded to PSB/BAL culture results. Of 103 patients, 29 were diagnosed with VAP, 10 were labeled as “uncertain,” and 64 were classified as not having VAP. Thirteen of 64 patients with negative PSB and BAL cultures had antibiotics stopped after 48 h, 25 of 64 had negative cultures, and antibiotics were not used at all, and 28 of 38 had a positive tracheal aspirate culture but negative PSB and BAL, so antibiotics were discontinued prior to the standard 7-day treatment in that center. The sensitivity and specificity for BAL fluid culture were 72% and 88%, respectively. The intracellular bacteria and the BAL combined had sensitivity and specificity of 79% and 88%, respectively. Use of PSB culture results in combination with intracellular bacteria and BAL further increased the sensitivity and specificity to 90% and 88%, respectively. The PSB and BAL are effective methods of collecting distal samples and were helpful in diagnosing VAP. However, a combined diagnostic approach was superior to either one alone. The safety of the NB-BAL in children has also been determined by several studies; few adverse experiences (n ⫽ 18) have been reported (12, 42, 60, 66, 79). The types of adverse events were sustained oxygen desaturation requiring increased ventilatory support (n ⫽ 11), pneumothorax (n ⫽ 4), hypotension (n ⫽ 2), and significant increase in intracranial pressure (n ⫽ 1). Of the 11 patients who experienced sustained oxygen desaturations, 7 patients were diagnosed with acute respiratory distress syndrome and saturations in the low 80s before the procedure was performed. Pneumothorax occurred in patients less than 1 month of age (n ⫽ 4). Hypotension occurred in patients requiring dopamine before the NB-BAL procedure began. The safety of NB-BAL and BAL have been examined in a study evaluating the diagnosis of infectious and interstitial lung disease in children (n ⫽ 32) (79). That study found that both NB-BAL and the BAL were safe, as respiratory rate, heart rate, and oxygen saturation were monitored during the procedure and a minimum of 6 h afterwards. Patients did not require increased supplemental oxygen after the procedure, and no major airway bleeding occurred. Computerized Surveillance Recently, a retrospective study was performed to determine the accuracy of computerized surveillance to detect nosocomial pneumonia in two NICUs over a 2-year period (n ⫽ 2,932 patients) (46). The automated monitoring system was a natural language processor, referred to as the Medical Language Ex- 414 FOGLIA ET AL. traction and Encoding system, that converted the electronic narrative reports to coded descriptions to identify patients with pneumonia. The automated monitoring system was compared to diagnosis of VAP by an ICP who performed prospective surveillance for pneumonia using NNIS definitions. A total of 1,277 patients had chest radiographs. In NICU 1, seven cases of VAP were identified by the ICP prospective surveillance; five of the seven cases found by the ICP were also identified by automated computer surveillance, which flagged an additional 61 patients with possible nosocomial pneumonia. Nine were considered to be inappropriately flagged by a second independent ICP review. The sensitivity and specificity of the computerized surveillance were 71% and 99.8%, respectively. The positive predictive value was 7.9%, and the negative predictive value was ⬎99%. In NICU 2, 10 cases of VAP were identified by the ICP; only 2 of these were flagged by the computer. Further investigation revealed that 7 of the original 10 cases were not flagged by the computer because the original chest radiograph reports could not be found. Eight hundred thirtysix patients had chest radiographs performed; 84 were flagged by the computer as VAP. The sensitivity and specificity for NICU 2 could not be calculated. The findings of that study indicate that computerized surveillance may be useful in streamlining the identification of patients with possible VAP who require a more time-consuming chart review by an ICP. This system was not linked with microbiology reports. An unstudied area is computerized surveillance linking both radiology and microbiology reports. Recommendations for Current Practice and Future Research The lack of a gold standard plagues all literature regarding VAP in both adults and children. The current literature suggests that NB-BAL, BAL, and PSB are safe in older children who do not have severe hypoxemia, increased intracranial pressure, hemodynamic instability, or bleeding problems and that invasive testing is sensitive and specific compared to a reference of expert opinion. Comparison of the clinical definitions, including the age-specific definitions, with and without invasive testing against histopathology and/or lung tissue culture would be a valuable addition to the literature. The feasibility of using NB-BAL in a general patient population and the effect on antibiotic use remain to be determined. Computerized surveillance has the potential for considerable time savings, particularly if electronic surveillance of the radiographic reports could be combined with that of microbiology and vital signs and validated against the current practical gold standard of application of CDC/NNIS definitions by an experienced clinician who has reviewed the complete medical record. Additional computerized surveillance studies are necessary to help further understand the impact that computerized surveillance may have on diagnosing pneumonia. MICROBIOLOGY Understanding the microbiology of VAP is critical for guiding decisions regarding empirical antibiotic therapy. A retrospective cohort study of the microbiologic etiology of VAP in the ICU setting was performed in three hospital settings: a CLIN. MICROBIOL. REV. large teaching hospital, a community hospital, and a children’s hospital (5). The most commonly isolated organisms were similar across adult and pediatric hospitals: Staphylococcus aureus (28.4%), Pseudomonas aeruginosa (25.2%), and other gramnegative bacilli (26.6%). The microbiologies of early-onset and late-onset infections differed in the adult populations, but this was not the case in the children’s hospital. Pseudomonas aeruginosa and Staphylococcus aureus were the most commonly isolated organisms in the children’s hospital. Pseudomonas aeruginosa was more common in the PICU than in the NICU (33.3% versus 17%; P ⫽ 0.01), while Staphylococcus aureus was more common in the NICU than in the PICU (38% versus 17.6%; P ⬍ 0.001). A prospective cohort study of VAP in the same NICU was performed. Most of the tracheal isolates from patients with VAP grew polymicrobial cultures; the organisms most commonly isolated included Staphylococcus aureus (23%), Pseudomonas aeruginosa (38.4%), Enterobacter spp. (38.4%), and Klebsiella spp. (23%) (3). A limitation of these studies is that the vast majority of isolates were from endotracheal aspirates rather than from invasive sampling of the lower airway, and thus, the results may represent oropharyngeal flora. Two studies reported differences in the microbiologies of early-onset and late-onset nosocomial pneumonia among children. Group B streptococci were most commonly isolated from infants with maternally acquired pneumonia (31.8%), while these organisms were rarely isolated in cases of late-onset pneumonia (1.3%). The frequency of Staphylococcus aureus increased from 2.4% of maternally acquired cases to 18.7% of non-maternally-acquired cases of pneumonia, and Pseudomonas aeruginosa frequency increased from 2.9% of maternally acquired cases to 12.9% of non-maternally-acquired cases of pneumonia (43). A 41-month prospective surveillance study of nosocomial infections in a NICU divided pneumonia into early-onset (onset of symptoms within first 48 h of life) and late-onset (onset of symptoms more than 48 h after birth) infections (98). There were 35 cases of definite or probable early-onset pneumonia. In 26 of these cases, potential pathogens were identified: 18 (76.9%) group B streptococci, 1 (3.8%) group F streptococcus, 3 (11.5%) Streptococcus pneumoniae isolates, and 2 (7.7%) nontypeable Haemophilus influenzae infections. Late-onset pneumonia occurred in 36 of 358 (10%) neonates who were ventilated for over 24 h. Cultures were taken from endotracheal tubes or nasopharyngeal secretions for 41 episodes of late-onset nosocomial pneumonia. The most commonly isolated organisms were coliform spp. (n ⫽ 18; 43.9%), Pseudomonas aeruginosa (n ⫽ 14; 34.1%), and Staphylococcus aureus (n ⫽ 6; 14.6%). RISK FACTORS FOR VAP IN NICU PATIENTS In pediatric populations, the pathogenesis of VAP is not well studied. In adult patients, aspiration of oropharyngeal secretions, inhalation of aerosols containing bacteria, hematogenous spread, and bacterial translocation from the gastrointestinal tract are all considered to be mechanisms of the development of VAP (89). Neonates have unique characteristics predisposing them to nosocomial infections. These patients’ immature immune sys- VOL. 20, 2007 tems place them at increased risk for infection (24). Skin and mucous membranes are more permeable and are less effective barriers to infection (47). Abnormal granulocyte migration and bacterial digestion in these patients have been demonstrated. Additionally, decreased activity of complement, particularly complement opsonization, occurs in newborns (40). Lastly, hypogammaglobulinemia occurs in premature newborns. Maternal immunoglobulin G (IgG) is transported to the fetus in the second and last trimesters of pregnancy, and fetal IgG levels reach maternal levels by term (58). Levels of IgG are lower in premature newborns, as maternal levels have not yet been attained. In the initial months following birth, maternal IgG levels drop, and it takes the infant months to produce ample levels of IgG and other immunoglobulins. Low birth weight has been shown to be a risk factor for the development of nosocomial pneumonia. A 41-month surveillance study demonstrated a significant association between a birth weight of ⬍1,500 g and a higher rate of nosocomial pneumonia (48). However, low birth weight may be a marker for an increased duration of mechanical ventilation. That study was limited by the lack of a specific control for the duration of mechanical ventilation. Apisarnthanarak et al. (3) focused on estimated gestational age (EGA) rather than birth weight in their 10-month-long case control study of 211 intubated NICU patients. VAP rates were much higher in babies with an EGA of ⬍28 weeks (19 VAP cases) than in babies with an EGA of ⱖ28 weeks (5 VAP cases) (P ⬍ 0.001) (3). The VAP rate per 1,000 ventilator days was also higher in babies with an EGA of ⬍28 weeks (6.5/1,000 ventilator days) than in babies with an EGA of ⱖ28 weeks (4.0/1,000 ventilator days) but was not statistically significant (P ⫽ 0.34) (3). Not all investigators found an inverse relationship between birth weight and frequency of nosocomial pneumonia. A prospective surveillance study of nosocomial infections in seven Brazilian NICUs found that the rate of nosocomial pneumonia was actually higher in neonates with birth weights of ⬎1,500 g than in babies with birth weights of ⱕ1,500 g (4.4/1,000 patient days versus 2.8/ 1,000 patient days) (72). Prior BSIs have been identified as a being a risk factor for VAP in NICU patients. In babies with an EGA of ⬍28 weeks, history of a prior BSI was the only significant risk factor for the development of VAP in multivariate analyses after controlling for the duration of mechanical ventilation (P ⫽ 0.03). Although none of the cases of VAP were caused by the same organism as that which caused the BSI, those authors suggested that prior BSI may serve as a surrogate for severity of illness rather than actually contributing to VAP (3). The design of the NICU may also have an effect on the incidence of nosocomial infections and specifically VAP. A 5-year prospective study of nosocomial infections in a NICU was performed (44). Midway through that study, the NICU location was moved from cramped quarters adjacent to a busy medical ward to a new facility. The new nursery had a 50% increase in staffing and improved infection control features. In the old nursery, 16 of 492 patients had pneumonia, whereas in the new nursery, only 1 patient of 419 had pneumonia. While the new nursery had improved structural infection control measures such as more space per patient, a large number of sinks, and a separate isolation room, it is not clear if other practices of care, such as head-of-bed elevation or suctioning, VAP IN PEDIATRIC PATIENTS 415 changed after the move to the new unit. Those authors did not report any changes in infection control surveillance or diagnosis in the new nursery. RISK FACTORS FOR VAP IN PICU PATIENTS Several prospective cohort studies described risk factors for pediatric VAP. In a prospective cohort study at a tertiary care center, genetic syndrome (odds ratio [OR], 2.37; 95% CI, 1.01 to 5.46), transport out of the PICU (OR, 8.90; 95% CI, 3.82 to 20.74), and reintubation (OR, 2.71; 95% CI, 1.18 to 6.21) were all independent predictors of pediatric VAP (28). That study also found that primary BSIs were associated with the development of VAP, as five of the nine patients with primary BSIs and VAP had the BSI first. Another prospective cohort study identified prior antibiotic use (OR, 2.45; 95% CI, 1.112 to 5.405), continuous enteral feeding (OR, 2.29; 95% CI, 1.093 to 4.798), and bronchoscopy (OR, 5.04; 95% CI, 1.665 to 15.266) as being independent predictors of pediatric VAP (1). Immunosuppressant drugs (OR, 4.8; P ⫽ 0.04), immunodeficiency (OR, 6.9; P ⫽ 0.06), and neuromuscular blockade (OR, 11.4; P ⫽ 0.002) were also found to be independent predictors in another prospective cohort study (32). Torres et al. (94) identified several factors associated with increased risk of developing VAP: reintubation (OR, 4.95; 95% CI, 3.48 to 7.04; P ⫽ 0.000012), gastric aspiration (OR, 5.05; 95% CI, 3.28 to 7.77; P ⫽ 0.00018), mechanical ventilation for ⬎3 days (OR, 1.17; 95% CI, 1.15 to 1.19; P ⫽ 0.015), chronic obstructive pulmonary disease (OR, 1.89; 95% CI, 1.38 to 2.59; P ⫽ 0.048), and positive end-expiratory pressure (OR, 1.85; 95% CI, 1.30 to 2.64; P ⫽ 0.092). In nosocomial pneumonia patients, factors associated with increased mortality risk were a rapidly fatal underlying condition (OR, 8.84; 95% CI, 3.52 to 22.22; P ⫽ 0.0018), worsening acute respiratory failure from developing pneumonia (OR, 11.94; 95% CI, 4.75 to 30; P ⫽ 0.0096), septic shock (OR, 2.83; 95% CI, 1.41 to 5.78; P ⫽ 0.016), inappropriate antibiotic treatment (OR, 5.81; 95% CI, 2.70 to 12.48; P ⫽ 0.02), and non-cardiac-surgery ICU patients (OR, 3.38; 95% CI, 1.70 to 6.71; P ⫽ 0.08) (78). Medications associated with the development of VAP are NADP, steroids, and histamine type 2 receptor blockers (28). Recommendations for Current Practice and Future Research Several factors have been identified as being risk factors for VAP in NICU and PICU patients. Many of these factors reflect a risk for aspiration such as that which may occur during reintubation, physical movement out of the ICU, and bronchoscopy. In addition, neuromuscular weakness and immunodeficiency may predispose a patient to VAP, as does prolonged mechanical ventilation. A risk stratification system incorporating preventable and nonpreventable risk factors for pediatric VAP might assist intensivists in the development of pediatric VAP prevention bundles and methods for identifying meaningful indicators as a measure of an institution’s success at VAP prevention. Larger, multicenter, randomized controlled trials using a standard reference definition of VAP to test interventions to prevent aspiration in children would be useful. Testing the efficacy of a standardized assessment of readiness 416 FOGLIA ET AL. to wean mechanical ventilatory support would also be useful in this patient population, as would a standardized assessment of pain and the need for sedation and neuromuscular blockade. PREVENTION Several recommendations have been given to decrease VAP. The CDC and Healthcare Infection Control Practices Advisory Committee suggest using orotracheal tubes (instead of nasotracheal tubes) when patients require mechanical ventilation, changing breathing circuits of ventilators only if they malfunction or if they are visibly contaminated, and using endotracheal tubes with dorsal lumens to allow respiratory secretions to drain (89). There are no recommendations for the preferential use of sucralfate, histamine 2 receptor antagonists, or antacids for stress bleeding prophylaxis (89). Head-of-Bed Elevation Supine position has been associated with VAP in adult patients, which is thought to be related to an increase in gastroesophageal reflux and aspiration. Semirecumbent positioning has been demonstrated to decrease surrogate outcomes such as aspiration and gastroesophageal reflux in adults (16), and one clinical trial demonstrated a dramatic decrease in the incidence of confirmed VAP in patients with head-of-bed elevation (5% versus 23%; OR, 6.8; 95% CI, 1.7 to 26.7) (25). The efficacy of semirecumbent positioning in preventing VAP in children has not been established. One age- and sex-matched case control study of nosocomial pneumonia in children found that head-of-bed elevation did not differ between cases and controls. That study was limited by small numbers of cases and controls (n ⫽ 9 for each group) (10). Additionally, sizerelated factors must be considered in the utility of semirecumbent positioning in children. For instance, elevating the head ⬎30° is logistically challenging for small pediatric patients such as infants and toddlers. In-Line Suctioning Endotracheal suctioning is used for eliminating bronchopulmonary secretions from the airway. Traditional open endotracheal suction requires disconnection from the ventilator. This process has been shown to result in increased intracranial pressure, increased mean blood pressure, and hypoxia in mechanically ventilated children (27, 57). The introduction of a closed multiuse suction catheter in the 1980s allowed endotracheal suctioning without disconnection from the ventilator. In critically ill adult populations, closed suction systems have been shown to result in fewer physiologic disruptions such as arterial and venous desaturations and arrhythmias (54). Closed endotracheal suction systems present the potential for bacterially contaminated secretions to pool in the lumen of the tube, with reinoculation of the respiratory tract with each repeated suctioning. On the other hand, a closed system could potentially decrease environmental contamination of the respiratory device. Many studies of critically ill adults have compared the incidence of airway colonization and nosocomial pneumonia in patients on a closed multiuse system to that in patients on a single-use open suction system. The frequency of CLIN. MICROBIOL. REV. airway colonization has been shown to be significantly more frequent in patients on the closed suction system (23). However, studies have not demonstrated an increased frequency of nosocomial pneumonia in patients on the closed suction system (23, 54). Indeed, a more recent prospective randomized study of 102 ventilated adults demonstrated an increased risk of VAP for patients with an open suction system compared to a closed suction system (adjusted risk, 3.5; 95% CI, 1.0 to 12.33) (17). There are currently no CDC recommendations regarding the preferential use of closed or open suction systems, nor are there recommendations regarding the frequency of change for multiuse closed suctioning systems in a single patient (89). A single study has compared open and closed suction systems in critically ill children. Cordero et al. (20) monitored 133 ventilated NICU patients who were alternately assigned to a closed or open suction system for bacterial airway colonization, nosocomial pneumonia, BSI, and bronchopulmonary dysplasia. A definition of nosocomial pneumonia required radiographic evidence of “probable” pneumonia (new airspace disease or a parenchymal process) and positive blood cultures and tracheal culture for a respiratory pathogen. Colonization patterns from tracheal cultures were comparable between groups, with gram-positive colonization occurring by the second week of intubation and gram-negative colonization occurring after the third week of ventilation. There were no significant differences in the incidences of VAP or BSIs or mortality between patient groups. Additionally, the numbers of endotracheal suctions per day, the numbers of reintubations, the incidences and severities of bronchopulmonary dysplasia, and the numbers of infants discharged on supplemental oxygen were similar between groups. Finally, 40 of 44 (91%) NICU nurses judged the closed suction system to be easier to use, less time-consuming, and better tolerated by NICU patients. H2 Blockers/Sucralfate The acidification of gastric contents is thought to decrease colonization with potentially pathogenic bacteria. Stress ulcer prophylactic medications that increase gastric pH, like H2antagonists and antacids, may increase colonization with pathogenic organisms and increase the risk of VAP (18). Sucralfate is an alternative stress ulcer prophylactic agent that does not alter gastric pH, and this medication may lower the risk of VAP while maintaining stress ulcer prophylaxis. Over 20 clinical trials with adults have investigated the risk of VAP associated with these medications. Of seven meta-analyses of these clinical trials, four found a significant reduction in the incidence of VAP in patients treated with sucralfate compared to patients receiving H2 antagonists. The same effect occurred in the other three analyses but did not reach statistical significance. Three of these meta-analyses demonstrated a significant reduction in mortality associated with sucralfate therapy (16). Two clinical trials compared the risk of VAP with various methods of stress ulcer prophylaxis in pediatric patients. A retrospective study included 155 PICU patients who had a nasogastric tube in place and were mechanically ventilated for ⬎48 h: 54 were given ranitidine, 53 were give sucralfate, and 48 were not on stress ulcer prophylaxis (62). There was no signif- VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS icant difference in the incidences of VAP between patients treated with ranitidine and patients treated with sucralfate (11.1% versus 7.5%; 2 ⫽ 0.40; P ⫽ 0.52). That study had several limitations. Patients were not randomized into study groups, and patient characteristics differed between patients given stress ulcer prophylaxis and those who were not given prophylaxis. The retrospective nature of the study may have resulted in errors in diagnosing VAP. A prospective study was performed to study the incidence of VAP and associated mortality among patients randomized to one of four groups for stress ulcer prophylaxis in Turkey (101). That study included 160 PICU patients: 38 received sucralfate, 42 received ranitidine, 38 received omeprazole, and 42 did not receive prophylaxis. VAP occurred in 70 of 160 (44%) patients, ranging from 41 to 48% in individual treatment groups. There was no difference in the incidence of VAP across treatment groups. The overall mortality rate was 35 of 160 (22%) and did not differ significantly among treatment groups, ranging from 21 to 23% across groups. The overall incidence of VAP (44%) in this study was much higher than that reported in other pediatric studies from referral hospitals (5.1% to 10.2%) (1, 28). It is possible that VAP was overdiagnosed in that study, although diagnostic criteria used in that study were similar to criteria used in this country. If VAP was overdiagnosed, this effect would likely be distributed throughout all study groups. That study may also have been underpowered to detect differences in the incidences of VAP among these patient groups. Both of those studies failed to demonstrate a difference in the incidence of VAP in patients treated with sucralfate compared to those treated with agents that alter gastric pH. Additionally, neither study demonstrated an increased risk of VAP in patients treated with agents that alter gastric pH compared to that in patients with no treatment. The microbiologies of infections were similar across treatment groups, and many infections were caused by organisms that are not likely to be affected by stress ulcer prophylaxis. It is possible that the study sizes presented were simply too small to appreciate a significant difference in the incidence of VAP, or it is possible that stress ulcer prophylaxis is not associated with VAP in the pediatric population. Larger prospective randomized studies of children are needed to asses the impact of stress ulcer prophylaxis on VAP and whether sucralfate has a protective effect compared to medications that decrease gastric acidity. Hand Hygiene Efforts at reducing person-to-person transmission of bacteria are crucial for preventing nosocomial infections. Significant bacterial contamination of hospital employees’ hands during routine patient care has been demonstrated (75). The concept that routine hand washing by health care workers reduces nosocomial infections is not new, but the first study investigating the impact of hand hygiene on the rate of hospital-acquired infections in NICU patients was recently performed (99). A 2-year-long multimodal intervention was instigated, which consisted of formal lectures, written and posted instruction regarding proper hand hygiene technique, covert observation, financial incentives, and regular feedback of observed hand hygiene rates. Surveillance of hand washing compliance and nosocomial infections from the pre- and postintervention periods 417 were compared. The rate of hand hygiene compliance increased from 43% at baseline to 80% during the intervention, and the rate of respiratory infections decreased from 3.35 to 1.06 per 1,000 patient days (P ⫽ 0.002) in the pre- and postintervention periods. The two parameters were statistically correlated (r ⫽ ⫺0.385; P ⫽ 0.014). That study is helpful in demonstrating an association of hand hygiene and prevention of nosocomial pneumonia, but it has limitations. The beforeafter design of the study makes it difficult to assess if a reduction in the rate of pneumonia is attributable exclusively to the increase in hand hygiene. An intervention of this magnitude may have altered other clinical practices related to the spread of bacterial contamination, as employees’ awareness of preventing nosocomial infections was increased. Those authors did point out that no changes in the use of surfactant or suction procedures occurred during the study period but did not comment on other procedural changes that may have occurred such as head-of-bed elevation, stress ulcer prophylaxis, or oral hygiene changes. Additionally, while rates of hand hygiene compliance remained at 81% during the 16-month postintervention period, it is unclear how sustainable this effect was after observations were discontinued. A prospective study of a 3-month-long implementation of an intervention to decrease rates of nosocomial infection in NICU patients was undertaken (69). The intervention consisted of three parts: (i) grouping of all blood-taking tasks to reduce the number of daily blood draws, (ii) reducing the frequency of blood investigations after stabilization of acute illness, and (iii) using an aseptic delivery system of drugs though a central venous catheter to reduce peripheral intravenous access. The incidences of nosocomial infection in the NICU between the 1-year preintervention period and the 1-year postintervention period were compared. VAP rates declined from 3.3/1,000 ventilator days to 1.0/1,000 ventilator days after the intervention. Again, that study was limited by the before-after nature of the design. Those authors acknowledged that practices regarding mechanical ventilation also changed during the study period, as patients were weaned from the ventilator more aggressively and as soon as possible. Earlier weaning may have contributed to lowering the VAP rates, as prolonged intubation is a risk factor for VAP in children (62). The importance of hand hygiene in preventing horizontal transmission of pathogens among mechanically ventilated patients was highlighted by a study performed by Sole et al. (86) to evaluate the proportion of suctioning devices colonized with pathogenic bacteria and to correlate the bacteria found on respiratory equipment with those found in patients’ mouths and sputum. Those investigators found that within 24 h of changing to new suctioning equipment, 94% of tonsil suction tubing, 83% of in-line suction tubing, and 61% of distal suction connectors were colonized with pathogenic bacteria similar to those found in the patients’ oropharynx and sputum (86). Selective Decontamination The impact of using topical antibiotics on tracheostomy sites on exogenous colonization or infection of the lower airways has been studied. A 2-year-long prospective observational cohort study was performed with 23 children who were treated with 2% paste of polymyxin E and tobramycin on the trache- 418 FOGLIA ET AL. ostoma four times a day for the first two postoperative weeks (65). Only 1 of 23 (4%) patients developed exogenous colonization or infection of the lower respiratory tract, which was lower than that in historical controls (6/22 [27%]). While topical antibiotics may be useful in preventing exogenous colonization or infection of the lower airways in children with tracheostoma, the risk of endogenous colonization remains high. Endogenous colonization or infection occurred in 15 episodes in 14 of 23 (61%) patients during the 2-week postoperative period. Many investigators have studied the efficacy of selective digestive tract decontamination (SDD) in preventing VAP. SDD traditionally consists of a regimen of topical antimicrobials applied to the oropharynx and through a nasogastric tube, with the aim of reducing the burden of pathogenic bacteria in aspirated secretions. While the majority of trials have focused exclusively on the use of topical antimicrobials, many have also used a short course of intravenous antimicrobial therapy. Seven meta-analyses of randomized, controlled trials of SDD in adults all showed a significant reduction in the risk of VAP, and four of those analyses also demonstrated a significant reduction in mortality in patients treated with SDD (16). One recent meta-analysis divided trials into those that used topical antibiotics alone and those that used a combination of topical and systemic antimicrobials for the prevention of nosocomial respiratory infections (61). That analysis included 32 randomized, controlled trials including a total of 5,185 adult patients. A protective effect was demonstrated in trials comparing patients on a combination of systemic and topical antibiotics with controls (OR, 0.35; 95% CI, 0.29 to 0.41) and in trials comparing patients on topical antibiotics alone with controls (OR, 0.52; 95% CI, 0.43 to 0.63). A significant reduction in mortality was seen only in trials that used a combination of topical and systemic therapy (OR, 0.78; 95% CI, 0.68 to 0.89). Mortality from VAP was not reduced when topical therapy alone was used (OR, 0.97; 95% CI, 0.81 to 1.16). Recent evidence suggests that results from some of those trials may be overly optimistic. A meta-analysis of 32 primary trials of SDD was performed to assess the impact of study methodology on results (97). Study methodology was evaluated based on allocation and concealment, patient selection, patient characteristics, blinding, and definition of nosocomial pneumonia. That analysis found an inverse relationship between the methodologic quality and benefit of SDD on the incidence of pneumonia, suggesting that the benefit of SDD for the prevention of VAP may be overestimated by many clinical trials (97). Studies focusing on the use of SDD to prevent VAP in children have conflicting results. A prospective study of SDD in 226 PICU patients randomized study subjects into a treatment (n ⫽ 116) or control (n ⫽ 110) group (81). The treatment group received colistin, tobramycin, and nystatin orally or through a nasogastric tube every 6 h, and patients were monitored for the development of nosocomial infection in any body site. There were 87 episodes of any nosocomial infection in 65 of 226 (28.8%) patients. The most common nosocomial infections were catheter-related bacteremia, sepsis, pneumonia, and urinary tract infection. The overall incidence of nosocomial infection across all sites did not differ between treatment and control groups. However, when infections were studied by CLIN. MICROBIOL. REV. body site, patients in the treatment group had a significantly lower frequency of pneumonia (2.6% versus 7.2%). In multivariate analyses, SDD retained a protective effect against pneumonia (OR, 0.21; 95% CI, 0.06 to 0.8). There was no significant difference in overall mortality between the treatment and control groups (six versus five patients). Patients in that study were randomized and were well matched for most variables with the exception of severity of illness; the treatment group had more severely ill patients. This difference in severity of illness would be expected to skew the results toward the null hypothesis, but there were actually fewer cases of pneumonia in the more severely ill group who were treated with SDD. A prospective, randomized, double-blinded study was performed to determine the efficacy of SDD in preventing nosocomial infections in severely burned (⬎30% total body surface area) PICU patients (7). Patients were randomized to the treatment group (n ⫽ 11) or control group (n ⫽ 12). The treatment group was given a mixture of polymyxin E, tobramycin, and amphotericin B four times daily by nasogastric tube. No significant differences regarding demographics, underlying conditions, inhalation, injury, or percent of surface area burned between patient and control groups existed. There was no difference in the proportion of patients with colonization of wounds, feces, nasogastric aspirates, or sputum between groups at the start of the study or throughout the study. No significant differences between groups were noted with regard to the serious complications measured: sepsis, pneumonia, gastrointestinal bleeding, respiratory distress syndrome, and mortality. The group treated with SDD had a higher incidence of diarrhea than the control group (82% versus 17%; P ⫽ 0.003). Results from that study suggest that SDD may not prevent nosocomial infections in pediatric patients. However, that study was limited by a small sample size (n ⫽ 23). Additionally, results of that study may not be generalizable to all PICU patients, as that study was restricted to burn patients. A prospective nonrandomized cohort study was performed to determine the impact of SDD on nosocomial infections in NICU patients (49). The decision to administer decolonization was left to attending physicians. Investigators later determined if patients had received well-performed decolonization (decolonization within the first 5 days with oral polymyxin E, tobramycin, and nystatin), incorrect decolonization (started after 5 days or less than three drugs used), or no decolonization. The incidence of nosocomial respiratory infection was lower in patients given well-performed (2.5%) or incorrect (7%) decolonization (P value not given). Interestingly, the incidence of nosocomial respiratory infection was lowest in patients who were not decolonized (1%). However, because patients were not randomized into treatment groups, significant underlying differences between groups, including gestational age, birth weight, NICU length of stay, exposure to central catheters, and respiratory support, existed. To control for these differences, investigators performed logistic regression and found that well-performed selective intestinal decolonization exerted a protective effect toward nosocomial infections of intestinal origin (OR, 0.17; 95% CI, 0.03 to 0.83). This group of infections included respiratory tract infections, sepsis, surgical wound infections, and urinary tract infections. The investigators did not supply a separate analysis of the impact of SDD on respiratory infections alone. VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS Oral Hygiene The CDC suggests that health care facilities develop and implement a comprehensive oral hygiene program for patients in acute-care settings or residents in long-term care facilities who are at high risk for health care-associated pneumonia (89). Fitch et al. (35) demonstrated that an oral care protocol and scores developed by a dental hygienist could be used by ICU nurses to improve oral health in critically ill adult patients. Mean oral inflammation scores were significantly lower after the implementation of a standard oral care protocol using toothpaste, antibacterial mouthwash, and oral gel (3.9 [standard error of the mean, 3.0] versus 12.4 [standard error of the mean, 2.2]; P ⫽ 0.03). Those investigators also noted lower mean scores for oral candidiasis, purulence, bleeding, and plaque, but the differences were not statistically significant. The dental hygienist and nurses’ assessments had a high degree of interrater reliability (kappa ⫽ 0.64). The scores used in that study were developed by one of the investigators and reviewed by other dental faculty members but were not validated in other patient populations. In addition, those investigators did not examine the effect of the standard oral care protocol on the incidence of VAP or bacterial oropharyngeal colonization. Bergmans et al. (9) performed a prospective, randomized, placebo-controlled, double-blind study in adult ICU patients to determine if VAP was preventable by the modulation of bacterial flora in the oropharynx. Those investigators compared topical prophylaxis to the buccal cavities with 2% each gentamicin, colistin, and vancomycin (n ⫽ 87) to an Orabase placebo group (n ⫽ 78) (group A) and a second control group of patients admitted to an ICU where no topical preparation was used (n ⫽ 61) (group B). Topical prophylaxis eradicated a significantly higher proportion of organisms present on admission in the oropharynx in the treatment group than in either control group (75% of the treatment group versus 0% in the placebo group and 9% in the no-preparation ICU group; P ⬍ 0.00001). Topical prophylaxis was also effective in eradicating organisms from the trachea (treatment group, 52%; group A, 22%; group B, 7% [P ⱕ 0.03]). The incidence of VAP was also lower in the treatment group (10%) than in the controls (group A, 31% [P ⫽ 0.001]; group B, 23% [P ⫽ 0.04]). That study concluded that preventing oropharyngeal colonization is protective against VAP, with an absolute risk reduction of 0.21 (95% CI, 0.09 to 0.33); treating five patients with topical antibiotics would prevent one case of VAP. VAP was defined prospectively using CDC definitions and confirmed with BAL or PSB. However, it is unclear whether the person who determined whether the patients had VAP was blinded to the treatment group. In addition, the treatment group received enteral feeds more frequently than controls, which could alter oropharyngeal flora. The placebo group (group A) was significantly more likely than the treatment group to receive sucralfate, another potential confounder of oropharyngeal colonization. Pineda et al. (73) performed a meta-analysis to determine if oral chlorhexidine treatment reduced the incidence of VAP. Four randomized controlled trials including 1,202 patients met inclusion criteria for the meta-analysis. Patients in the chlorhexidine treatment group were less likely to develop VAP than those in the control group (4% [24 of 587] versus 7% [41 of 615]), although the difference did not reach statistical signifi- 419 cance (OR, 0.42; 95% CI, 0.16 to 1.06; P ⫽ 0.07). ICU length of stay and duration of mechanical ventilation did not differ between the groups. Mortality was not significantly different between the two groups (OR, 0.77; 95% CI, 0.28 to 2.11; P ⫽ 0.6). The magnitude of the protective OR is striking, as is the proximity of the CIs to statistical significance, suggesting that additional studies with larger sample sizes might demonstrate a significant protective effect from oral chlorhexidine rinses. Of note, patients in those studies received either a 0.12% chlorhexidine rinse twice a day (n ⫽ 914) or 0.2% chlorhexidine gel three times a day (n ⫽ 288). A meta-analysis of seven randomized controlled trials (n ⫽ 1,650 patients) performed by Chlebicki and Safdar (15) revealed a similar protective effect with a relative risk (RR) of 0.74 (95% CI, 0.56 to 0.96; P ⫽ 0.02) using a fixed-effects model and a RR of 0.70 (95% CI, 0.47 to 1.04; P ⫽ 0.07) using a random-effects model for patients treated orally with chlorhexidine. The risk reduction was even higher in cardiac surgery patients (RR, 0.41; 95% CI, 0.17 to 0.98; P ⫽ 0.04) (15). The Bundle Approach In December 2004, the Institute for Healthcare Improvement (IHI) challenged hospitals to save 100,000 lives by June 2006 (21). One of the six evidence-based guidelines to be implemented was the prevention of VAP. The VAP bundle for adults is to (i) avoid/decrease endotracheal intubation and duration of mechanical ventilation whenever possible, (ii) use orotracheal and orogastric tubes to decrease the risk of hospital-acquired sinusitis, (iii) avoid heavy sedation and neuromuscular blockade with depression of cough reflexes, (iv) maintain endotracheal cuff pressures to greater than 20 cm water, (v) prevent condensate in tubing from entering the lower respiratory tract, (vi) maintain head-of-bed elevation at 30° to 45°, (vii) maintain oral care, and (viii) maintain hand hygiene (21, 67). The team approach using the IHI bundle has been shown to be successful in reducing VAP (21). The bundle approach has been used at the Children’s Hospital in Boston and at Vanderbilt Children’s Hospital. In the latter, an education and intervention termed “ZAP VAP” was put into practice, with their efforts emphasizing the IHI bundle (21). Prevention included hand washing, elevating the head of the bed 30° to 45°, monitoring gastric residuals every 4 h to prevent aspiration, providing aggressive oral care (and documentation) every 2 h, managing hypopharyngeal secretions, providing in-line endotracheal suction, and providing equipment care. During the first 6 months of implementation, the time between VAP occurrences has nearly tripled. Educational Interventions Identifying effective measures for preventing VAP is only as useful as the proper implementation of these measures in the clinical setting. Many studies have shown a reduction in rates of VAP following initiatives to educate health care workers about the epidemiology of VAP and infection control measures used to prevent VAP (89). Most of those studies were performed in the adult ICU setting. A recent educational intervention was performed in an integrated health system, with 420 FOGLIA ET AL. results compared across a large adult teaching hospital, two community hospitals, and a pediatric teaching hospital (4). The targeted health care workers were respiratory care practitioners and nursing staff working in the ICU setting. This intervention centered on a 10-page self-study module that focused on multiple aspects of VAP and also included posters, fact sheets, and in-services for nursing staff and respiratory therapists. VAP rates between the 12-month preintervention period and the 18-month postintervention period were compared. Nursing compliance rates were highest among nurses at the pediatric hospital (100%) and one of the community hospitals (98.9%). The adult teaching hospital and the other community hospital had significantly lower compliance rates among nurses (64.9% and 44.2%; P ⬍ 0.001). Three hospitals had a significant drop in the VAP rates from the preintervention period to the postintervention period. The VAP rate at the pediatric hospital fell 38%, from 7.9 episodes to 4.9 episodes per 1,000 ventilator days (P ⬍ 0.001). The community hospital with no change in the rate of VAP had the lowest compliance of respiratory therapists compared to the other three hospital combined (56.3% versus 95.2%; P ⬍ 0.001). Not all lapses in infection control measures result from a lack of knowledge. A survey of NICU health care workers was performed to investigate the knowledge, beliefs, and practices regarding nosocomial infections and infection control measures (56). The survey revealed some areas in which health care workers’ actions arose from unawareness of data related to infection control. For instance, few participants believed that nosocomial infections were related to health care workers’ rings (40%), artificial fingernails (61%), or long fingernails (48%). However, that study also revealed some disconnects between knowledge and practice. Although 96% of respondents believed that using sterile techniques for catheter insertion and care reduces a patient’s risk for BSI, only 67% reported using full sterile barriers at least 76% of the time when participating in inserting a line. Likewise, 91% of participants believed gloves are important for preventing the spread of nosocomial infections, but only 53% reported changing their gloves in all indicated situations. That study demonstrated the need for increased educational efforts to bridge the gaps in knowledge of infection control recommendations. Additionally, the study demonstrated that a lack of knowledge alone does not account for the lapses in infection control practices in the NICU studied. The most common barriers to infection control perceived by respondents included logistics (54%), time (48%), and lack of supplies (47%). Interventions that lower rates of VAP may have temporary effects, with VAP rates eventually rising following the conclusion of the intervention, indicating the need for continuous reinforcement of interventional measures (55). Factors associated with noncompliance with hand hygiene exist at the individual, group, and institutional levels (74). A proposed framework for the promotion of hand hygiene includes 12 factors: (i) education, (ii) routine observation and feedback, (iii) engineering controls, (iv) patient education, (v) reminders in the workplace, (vi) administrative sanctions and rewards, (vii) change in hand hygiene agents, (viii) promotion of workers’ skin care, (ix) active participation at the individual and institutional level, (x) maintenance of an institutional safety climate, (xi) enhancement of individual and institutional self- CLIN. MICROBIOL. REV. efficacy, and (xii) avoidance of overcrowding, understaffing, and excessive workload (74). The diversity of these factors emphasizes the need for a multipronged and continuous approach necessary to maintain high levels of compliance with infection control measures. A summary of interventional measures to decrease the incidence of VAP in children is provided in Table 3. Recommendations for Current Practice and Future Research There is scant literature regarding testing the efficacy of head-of-bed elevation, in-line suctioning, and preferential use of sucralfate over histamine type 2 receptor antagonists in pediatric VAP prevention. However, head-of-bed elevation and other measures to prevent aspiration, a consistent approach to oral hygiene, meticulous hand hygiene, and regular assessment of readiness to wean are biologically plausible as effective VAP prevention measures in children. Further studies documenting that head-of-bed elevation in children decreases aspiration and risk of pneumonia as well as determining the natural history of aerodigestive tract colonization and its relationship to gastric acidity in children may shed light on the risk/benefit ratio of sucralfate and/or H2 blockers and the number needed to treat to prevent pediatric VAP. VAP TREATMENT Treatment of suspected VAP is centered on an approach of initial empirical therapy with broad-spectrum antibiotics followed by de-escalation to specific antimicrobial therapy once culture results are known or discontinuation of antibiotics if VAP is no longer suspected. The American Thoracic Society and Infectious Disease Society of America have recently published an updated version of their evidence-based guidelines for the management of VAP in adults (2). Key recommendations in the new document include the use of early, appropriate, and broad-spectrum antibiotics for empirical therapy; utilization of empirical antibiotics from a different class than antibiotics that the patient has recently received; judicious use of combination therapy in hospital-acquired pneumonia; the potential use of linezolid as an alternative to vancomycin for VAP caused by methicillin-resistant Staphylococcus aureus (MRSA); the use of colistin for patients with VAP caused by carbapenem-resistant Acinetobacter species; the potential use of aerosolized antibiotics as adjunctive therapy for patients with VAP caused by certain antibiotic-resistant organisms; deescalation of antibiotics based on patients’ culture results and clinical improvement; and a shorter duration of antibiotics for patients with uncomplicated health care-associated pneumonia from bacteria other than nonfermenting gram-negative bacilli. These guidelines are based on data from clinical trials of hospital-acquired pneumonia in adult patients. There have been few clinical studies regarding the optimal treatment for VAP in children. Empirical Therapy The importance of prompt initiation of appropriate empirical therapy for suspected VAP has been demonstrated in VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS 421 TABLE 3. Summary of interventions to prevent VAPa Intervention and source (reference) Design Patient description Lopriore et al. (62) Prospective active surveillance Retrospective case control 361 PICU patients(37 with VAP, 324 without VAP) 155 PICU patients (13 with VAP, 142 without VAP) Cases had higher frequency of enteral feeds (48.6% vs 26.8%; OR, 2.58; P ⫽ 0.006) No significant difference between cases and controls (53.8% vs 43.6%) Motility agents Lopriore et al (62) Retrospective case control 155 PICU patients (13 with VAP, 142 without VAP) Cases had higher frequency of motility agent use (P ⬍ 0.05) (association not significant in logistic regression) Matched case control study 18 PICU patients (9 with VAP, 9 without VAP) No significant difference between cases and controls in head-of-bed elevation Prospective, randomized 133 NICU patients (67 closed, 66 open) No difference in diagnosis of nosocomial pneumonia between groups (n ⫽ 5 patients each) Prospective, randomized, double-blinded Prospective, randomized, nonblinded 23 burn patients (11 SDD, 12 placebo) 226 PICU patients (116 with SDD, 110 without SDD) Prospective cohort, nonrandomized 536 neonates (58 WP SID, 88 IP SID, 392 no SID) No difference in rates of pneumonia (1 case in SDD group, 0 in placebo group) SDD protective toward respiratory infection in logistic regression (OR, 0.21; 95% CI, 0.06–0.8) WP SID protective toward NI of intestinal origin in logistic regression (OR, 0.17; 95% CI, 0.03–0.83). Retrospective case control 155 PICU patients (54 ranitidine, 53 sucralfate, 48 no prophylaxis) Prospective, randomized, nonblinded 160 PICU patients (38 sucralfate, 42 ranitidine, 38 omeprazole, 42 no prophylaxis) Prospective study of hand hygiene campaign NICU patients admitted during hand hygiene campaign Prospective study of educational intervention PICU patients at pediatric teaching hospital Prospective surveillance 493 NICU patients (227 before period, 266 after period) Non-statistically-significant decrease in VAP rate from 3.3 to 1.0 per 1,000 ventilator days (P ⫽ 0.22) Meta-analysis, seven randomized controlled trials 1,650 patients total (n ⫽ 812 关topical chlorhexidine兴; n ⫽ 838 关comparator兴) Pineda et al. (73) Meta-analysis, four randomized controlled trials 1202 patients total (n ⫽ 587 关chlorhexidine group兴; n ⫽ 615 关control group兴) Bergmans et al. (9) Prospective, randomized, double-blinded, placebo controlled Longitudinal design, repeated measures 87 patients (treatment group), 78 patients, (control group A), and 61 patients (control group B) ICU nurses and dental hygienist Topical chlorhexidine reduced VAP incidence (RR, 0.74; P ⫽ 0.02); risk reduction was even higher in cardiac surgery patients (RR, 0.41; P ⫽ 0.04). Chlorhexidine group less likely to develop VAP (4%) compared to control group (7%)(P ⫽ 0.07); ICU length of stay, duration of mechanical ventilation, and mortality not significantly different VAP incidences were less in treatment group (10%) than in groups A (31%; P ⫽ 0.001) and B (23%; P ⫽ 0.04) Nurses following oral care protocols can help improve ICU patient oral health Enteral feeds Almuneef et al. (1) Head-of-bed elevation Black et al. (10) Closed vs open suctioning Cordero et al. (20) SDD Barret et al. (7) Ruza et al. (81) Herruzo-Cabrera et al. (49) Stress ulcer prophylaxis Lopriore et al. (62) Yildizdas et al. (101) Infection control interventions Won et al. (99) Babcock et al. (4) Nursing practice (decreasing peripheral intravenous access) Ng et al. (69) Oral hygiene Chlebicki and Safdar (15) Fitch et al. (35) a Outcome No significant difference in upper airway colonization with GNB, no significant difference in incidence of VAP No significant difference in VAP between patient groups Rate of respiratory infections dropped from 3.35 to 1.06 per 1,000 patient days (P ⫽ 0.002) 38% reduction in VAP rate from 7.9 to 4.9 episodes per 1,000 ventilator days (P ⬍ 0.001) WP, well performed; IP, incorrectly performed; GNB, gram-negative bacilli; NI, nosocomial infection. adults, with many studies describing higher mortality in patients who received delayed appropriate treatment for VAP (51, 59, 63). However, inappropriate use and overuse of antibiotics can lead to increased hospital expenditures and could potentially promote antibiotic resistance (33, 83). Empirical antibiotic therapy for suspected VAP accounts for a major proportion of inappropriate antibiotic use in pediatric patients, with up to 33% of patients receiving unwarranted antimicro- 422 FOGLIA ET AL. bial therapy for suspected VAP (33). Prescribing patterns have also shifted toward more expensive and broader-spectrum antibiotics in hospitalized children in recent years, with the proportion of total antibiotic expenditure used for vancomycin increasing from 0.2% in 1984 to 17.2% in 1994. Additionally, broad-spectrum cephalosporins accounted for 17.7% of antibiotic expenditures in 1984 and 49.6% in 1994 (96). Thus, prescribing patterns for empirical therapy for suspected VAP should maintain a balance between adequately covering patients who are potentially infected and minimizing unnecessary and prolonged exposure to antimicrobials. Infection with potentially antibiotic-resistant organisms accounts for a large proportion of VAP in adults (95). When selecting empirical therapy, physicians should be aware of the patient’s risk factors for infection with multidrug-resistant (MDR) bacteria, the antibiotics that the patient has recently received, and the local antibiotic resistance patterns. Risk factors for VAP with MDR pathogens in adults include mechanical ventilation for at least 7 days, prior antibiotic use, and prior exposure to broad-spectrum antibiotics (imipenem, broadspectrum cephalosporins, or fluoroquinolones) (95). In children, it has been postulated that patients may be colonized with organisms from their own preexisting endogenous flora in response to antibiotic pressure (92). Risk factors for colonization with antibiotic-resistant gram-negative organisms in PICU patients include younger age, increasing PRISM (pediatric risk of mortality) score, previous PICU admissions, intravenous antibiotic use in the past 12 months, and exposure to chronic care facilities (91, 93). Additional special considerations for the pediatric population include premature infants with an increased risk of Staphylococcus epidermidis infections and immunocompromised patients with an additional need for empirical antifungal therapy (52). Monotherapy for empirical coverage is recommended for adult patients with early-onset VAP without risk factors for infection with MDR pathogens, while combination therapy should be used for coverage of potential infection with MDR organisms or late-onset VAP based on a local antibiogram. Additionally, patients should be treated with antibiotics differing in class from those that they have recently received in case colonizing bacteria have developed antibiotic resistance from previous exposures (2). No consensus guidelines for empirical coverage of suspected VAP in children exist. Empirical therapy should be discontinued or altered based on culture results and clinical status. The fear that negative culture results may have missed an infection in critically ill children often leads to prolonged empirical antimicrobial therapy in neonates (87). A study of late-onset sepsis evaluations in neonates was undertaken to determine a sufficient time point for the discontinuation of empirical therapy. Those investigators found that 99% of blood cultures were positive within 48 h, and investigators used this finding as a basis for decreasing empirical coverage from 72 h to 48 h for suspected late-onset sepsis in neonates. That study examined cultures from sterile body sites only. Cultures from the respiratory tract and the vagaries of diagnosing VAP may not lend themselves to an easily defined time point for the discontinuation of empirical therapy in suspected VAP. Singh et al. (85) used the CPIS to guide the duration of empirical therapy in adults. Intensive care patients with newonset pulmonary infiltrate who were suspected of having pneu- CLIN. MICROBIOL. REV. monia were evaluated at baseline with five of the seven CPIS items on a scale of 0 to 2 each (temperature, blood leukocytes, tracheal secretions, oxygenation, and pulmonary radiography). Patients with a total CPIS of ⱕ6 were considered to have a low likelihood of pneumonia and were randomized to receive standard care as determined by their attending physician or experimental therapy with ciprofloxacin monotherapy. All patients were reevaluated at 3 days, and of those with CPIS remaining at ⱕ6, 100% of patients in the experimental arm had a cessation of antimicrobial therapy, compared to 4% of patients in the standard therapy arm. There was no difference in mortality or length of stay between treatment groups; antibiotic resistance, superinfections, or both occurred more frequently in patients receiving standard therapy than patients receiving experimental therapy (35% versus 15%; P ⫽ 0.017). Replication of these results in children could offer clinical guidelines for the rapid cessation of empirical antimicrobial therapy for suspected VAP. Specific Treatment Combination therapy exposes patients to multiple antibiotics and is more expensive; a prompt de-escalation of empirical therapy to more specific antimicrobial therapy should occur once culture results and susceptibilities are known. Monotherapy is recommended for patients who are not at risk for infection with MDR pathogens and for patients infected with gram-positive pathogens including MRSA. Additionally, patients with severe VAP should initially be treated with combination therapy but may be changed to monotherapy if lower respiratory tract cultures do not identify an antibiotic-resistant pathogen (2). Agents that have demonstrated efficacy for monotherapy in adult patients infected with susceptible organisms include fluoroquinolones, carbapenems, cefepime, and piperacillin-tazobactam (2). Recent interest surrounding the substitution of linezolid for vancomycin in the treatment of VAP caused by MRSA has emerged. A meta-analysis of two prospective, randomized, double-blind, multicenter, multinational trials in adults with nosocomial pneumonia comparing clinical cure from linezolid to vancomycin was performed (100). Patients with MRSA pneumonia who were treated with linezolid had survival that was significantly higher than that of patients treated with vancomycin (80.0% versus 63.5%; P ⫽ 0.03). Clinical cure rates were also higher for patients treated with linezolid than those treated with vancomycin (59.0% versus 35.5%; P ⬍ 0.01). Both of these effects remained significant in logistic regression models. A prospective, randomized, open-label, multicenter, multinational trial was performed to compare the efficacy and safety of linezolid and vancomycin for antibiotic-resistant gram-positive bacteremia and nosocomial pneumonia in hospitalized children (53). Among patients with nosocomial pneumonia, patients randomized to the linezolid group were more often mechanically ventilated (63.6% versus 10.0%; P ⫽ 0.011) and had more multiple-lobe involvement (90.9% versus 50.0%; P ⫽ 0.038). No significant difference in clinical cure was seen between patients treated with linezolid and those treated with vancomycin. However, patients treated with linezolid appeared to experience a faster resolution of dyspnea/tachypnea/grunting than patients with vancomycin, with fewer than 40% of patients receiving linezolid and more than 80% of patients receiving vancomycin demonstrating these symptoms of VAP VOL. 20, 2007 VAP IN PEDIATRIC PATIENTS on day 3 of treatment (P value not given). Additionally, patients treated with linezolid experienced a shorter mean duration of total therapy (10.5 days versus 12.8 days; P ⫽ 0.03) and intravenous therapy (8.5 days versus 11.3 days; P ⫽ 0.004). The frequency of drug-related adverse events was similar between groups. That analysis was a subset of a larger study, and it analysis was not powered for nosocomial pneumonia or bacteremia. Other subanalyses of this same patient population have demonstrated the safety of linezolid compared to vancomycin in children (64, 82). Aerosolized antibiotics have been studied most extensively in children in the setting of cystic fibrosis. The FDA approval of tobramycin solution for inhalation is only for maintenance therapy in patients with cystic fibrosis known to be colonized with Pseudomonas aeruginosa (76). In two randomized controlled trials, a modest improvement in the forced expiratory volume in 1 min occurred after 24 weeks of therapy with tobramycin solution for inhalation; there were significant decreases in CFU/ml of Pseudomonas aeruginosa in sputum, decreases in hospital admission days, and decreases in numbers of parenteral antibiotic days for treatment of Pseudomonas aeruginosa infections. To our knowledge, there are no data showing that treatment of acute pulmonary infections with aerosolized antibiotics is beneficial in children. Inhaled aminoglycosides do produce low but measurable serum concentrations (1 to 4 g/ml). Sputum concentrations vary. No ototoxicity or nephrotoxicity has been associated with the use of inhaled aminoglycosides, although many of those studies excluded children with serum creatinine levels greater than 2. The potential disadvantages of use include bronchospasm, increased MICs of the targeted organism, increased isolation of Candida and Aspergillus species from sputum, nebulization of microorganisms, and antibiotic contamination of the environment (76). Finally, subpopulations of pediatric patients deserve special consideration. Neurologically impaired children are at increased risk for aspiration or tracheostomy-associated pneumonia. The mixed microbiology of these infections, often including anaerobic organisms, warrants specific antimicrobial therapy against likely pathogens. A retrospective study of 57 neurologically impaired children with aspiration or tracheostomy-related pneumonia was performed to evaluate the efficacy of various antimicrobial therapies (11). Children with either type of pneumonia had better clinical improvement and microbiological response when treated with agents effective against penicillin-resistant anaerobic bacteria (ticarcillin-clavulanate or clindamycin) than patients treated with ceftriaxone (P ⬍ 0.05 for both tracheostomy-related pneumonia and aspiration pneumonia groups). Although this was a small study within a focused population, it underscores the need to account for unique underlying conditions in children that may predispose them to infections with specific pathogens. Duration of Therapy No consensus exists regarding the appropriate duration of antimicrobial therapy for VAP in adults, and the appropriate duration for proven infections in critically ill children has not been established (45). A multicenter, randomized, controlled trial was performed using adults with VAP to compare patients treated with appropriate empirical therapy for 8 days to patients treated 423 for 15 days (14). There was no difference in mortality or recurrent infections between groups. Among patients infected with nonfermenting gram-negative bacilli, patients treated for 8 days did have a higher relapse rate (32.8% versus 19.0%; 13.8% risk difference; 90% CI, 7.8% to 19.7%). However, among patients who experienced recurrent infections, patients treated for 8 days were less likely to become infected with MDR pathogens (42.1% versus 62.0%; P ⫽ 0.04). These data suggest that limiting treatment of VAP in patients infected with pathogens other than nonfermenting gram-negative bacilli is safe and may decrease the incidence of reinfection with MDR pathogens. Recommendations for Current Practice and Future Research No consensus guidelines exist for empirical coverage of suspected VAP in children. Empirical therapy should be discontinued or altered based on clinical status and culture results, preferably from lower airway samples. Patients who are severely ill and/or with previous exposure to the health care system should be treated with combination therapy. Areas for future research in children include the efficacy of linezolid compared to vancomycin for VAP treatment, the duration of optimal antibiotic therapy for VAP, and validation of the CPIS in children as well as its use in treatment decisions. CONCLUSION VAP is the second most common hospital-acquired infection among PICU patients. Empirical therapy for VAP accounts for approximately 50% of antibiotic use in PICUs. VAP is associated with an excess of 3 days of mechanical ventilation among pediatric cardiothoracic surgery patients. The attributable mortality and excess length of ICU stay of VAP have not been defined in matched case control studies. VAP is associated with an estimated $30,000 in attributable cost. Surveillance for VAP is complex and usually performed using clinical definitions established by the CDC. Invasive testing via BAL increases the sensitivity and specificity of the diagnosis. The pathogenesis is poorly understood in children, but several prospective cohort studies suggest that aspiration and immunodeficiency are risk factors. In children, educational interventions and efforts to improve adherence to hand hygiene have been associated with decreased VAP rates. More consistent and precise approaches to the diagnosis of pediatric VAP are needed to better define the attributable morbidity and mortality, pathophysiology, and appropriate interventions to prevent this disease. REFERENCES 1. Almuneef, M., Z. A. Memish, H. H. Balkhy, H. Alalem, and A. Abutaleb. 2004. Ventilator-associated pneumonia in a pediatric intensive care unit in Saudi Arabia: a 30-month prospective surveillance. Infect. Control Hosp. Epidemiol. 25:753–758. 2. American Thoracic Society. 2005. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 171:388–416. 3. Apisarnthanarak, A., G. Holzmann-Pazgal, A. Hamvas, M. A. Olsen, and V. J. Fraser. 2003. Ventilator-associated pneumonia in extremely preterm neonates in a neonatal intensive care unit: characteristics, risk factors, and outcomes. Pediatrics 112:1283–1289. 4. Babcock, H. M., J. E. Zack, T. Garrison, E. Trovillion, M. Jones, V. J. Fraser, and M. H. Kollef. 2004. An educational intervention to reduce ventilator-associated pneumonia in an integrated health system: a comparison of effects. Chest 125:2224–2231. 5. Babcock, H. M., J. E. Zack, T. Garrison, E. Trovollion, M. H. Kollef, and 424 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. FOGLIA ET AL. V. J. Fraser. 2003. Ventilator-associated pneumonia in a multi-hospital system: differences in microbiology by location. Infect. Control Hosp. Epidemiol. 24:853–858. Baltimore, R. S. 1998. Neonatal nosocomial infections. Semin. Perinatol. 22:25–32. Barret, J. P., M. G. Jeschke, and D. N. Herndon. 2001. Selective decontamination of the digestive tract in severely burned pediatric patients. Burns 27:439–445. Bercault, N., and T. Boulain. 2001. Mortality rate attributable to ventilatorassociated nosocomial pneumonia in an adult intensive care unit: a prospective case-control study. Crit. Care Med. 29:2303–2309. Bergmans, D. C., M. J. Bonten, C. A. Gaillard, J. C. Paling, S. van der Geest, F. H. van Tiel, A. J. Beysens, P. W. de Leeuw, and E. E. Stobberingh. 2001. Prevention of ventilator-associated pneumonia by oral decontamination: a prospective, randomized, double-blind, placebo-controlled study. Am. J. Respir. Crit. Care Med. 164:382–388. Black, S. R., E. Lo, M. Madriaga, M. Zimmerman, and J. Segreti. 2002. Nosocomial pneumonia in the PICU. Abstr. 40th Intersci. Conf. Antimicrob. Agents Chermother., abstr. K-452. Brook, I. 1996. Treatment of aspiration or tracheostomy-associated pneumonia in neurologically impaired children: effect of antimicrobials effective against anaerobic bacteria. Int. J. Pediatr. Otorhinolaryngol. 35:171–177. Burmester, M., and Q. Mok. 2001. How safe is non-bronchoscopic bronchoalveolar lavage in critically ill mechanically ventilated children? Intensive Care Med. 27:716–721. Chastre, J., F. Viau, P. Brun, J. Pierre, M. C. Dauge, A. Bouchama, A. Akesbi, and C. Gibert. 1984. Prospective evaluation of the protected specimen brush for the diagnosis of pulmonary infections in ventilated patients. Am. Rev. Respir. Dis. 130:924–929. Chastre, J., M. Wolff, J. Y. Fagon, S. Chevret, F. Thomas, D. Wermert, E. Clementi, J. Gonzalez, D. Jusserand, P. Asfar, D. Perrin, F. Fieux, S. Aubas, and the PneumA Trial Group. 2003. Comparison of 8 vs 15 days of antibiotic therapy for ventilator-associated pneumonia in adults: a randomized trial. JAMA 290:2588–2598. Chlebicki, M. P., and N. Safdar. 2007. Topical chlorhexidine for prevention of ventilator-associated pneumonia: a meta-analysis. Crit. Care Med. 35: 595–602. Collard, H. R., S. Saint, and M. A. Matthay. 2003. Prevention of ventilatorassociated pneumonia: an evidence-based systematic review. Ann. Intern. Med. 138:494–501. Combes, P., B. Fauvage, and C. Oleyer. 2000. Nosocomial pneumonia in mechanically ventilated patients, a prospective randomized evaluation of the Stericath closed suctioning system. Intensive Care Med. 26:878–882. Cook, D. J., B. K. Reeve, G. H. Guyatt, D. K. Heyland, L. E. Griffith, L. Buckingham, and M. Tryba. 1996. Stress ulcer prophylaxis in critically ill patients. Resolving discordant meta-analyses. JAMA 275:308–314. Cordero, L., L. W. Ayers, R. R. Miller, J. H. Seguin, and B. D. Coley. 2002. Surveillance of ventilator-associated pneumonia in very-low-birth-weight infants. Am. J. Infect. Control 30:32–39. Cordero, L., M. Sananes, and L. W. Ayers. 2000. Comparison of a closed (Trach Care MAC) with an open endotracheal suction system in small premature infants. J. Perinatol. 3:151–156. Curley, M. A., E. Schwalenstocker, J. K. Deshpande, C. C. Ganser, D. Bertoch, J. Brandon, and P. Kurtin. 2006. Tailoring the Institute for Health Care Improvement 100,000 Lives Campaign to pediatric settings: the example of ventilator-associated pneumonia. Pediatr. Clin. N. Am. 53:1231– 1251. Department of Health and Human Services. 23 August 2006, accession date. Criteria for defining nosocomial pneumonia. http://www.cdc.gov /ncidod/hip/NNIS/members/pneumonia/Final/PneumoCriteriaV1.pdf. Deppe, S. A., J. W. Kelly, L. L. Thoi, J. H. Chudy, R. N. Longfield, J. P. Ducey, C. L. Truwit, and M. R. Antopol. 1990. Incidence of colonization, nosocomial pneumonia, and mortality in critically ill patients using a Trach Care closed-suction system versus an open-suction system: prospective, randomized study. Crit. Care Med. 18:1389–1393. Donowitz, L. G. 1989. Nosocomial infection in neonatal intensive care units. Am. J. Infect. Control 17:250–257. Drakulovic, M. B., A. Torres, T. T. Bauer, J. M. Nicolas, S. Nogué, and M. Ferrer. 1999. Supine body position as a risk factor for nosocomial pneumonia in mechanically ventilated patients: a randomised trial. Lancet 354: 1851–1858. Drews, M. B., A. C. Ludwig, J. U. Leititis, and F. D. Daschner. 1995. Low birth weight and nosocomial infection of neonates in a neonatal intensive care unit. J. Hosp. Infect. 30:65–72. Durand, M., B. Sangha, L. A. Cabal, T. Hoppenbrouwers, and J. E. Hodgman. 1989. Cardiopulmonary and intracranial pressure changes related to endotracheal suctioning in preterm infants. Crit. Care Med. 17:506–510. Elward, A. M., D. K. Warren, and V. J. Fraser. 2002. Ventilator-associated pneumonia in pediatric intensive care unit patients: risk factors and outcomes. Pediatrics 109:758–764. Ely, E. W., A. M. Baker, D. P. Dunagan, H. L. Burke, A. C. Smith, P. T. Kelly, M. M. Johnson, R. W. Browder, D. L. Bowton, and E. F. Haponik. CLIN. MICROBIOL. REV. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 1996. Effect on the duration of mechanical ventilation of identifying patients capable of breathing spontaneously. N. Engl. J. Med. 335:1864–1869. Fabregas, N., S. Ewig, A. Torres, M. El-Ebiary, J. Ramirez, J. P. de La Bellacasa, T. Bauer, and H. Cabello. 1999. Clinical diagnosis of ventilator associated pneumonia revisited: comparative validation using immediate post-mortem lung biopsies. Thorax 54:867–873. Fagon, J. Y., J. Chastre, A. J. Hance, P. Montravers, A. Novara, and C. Gibert. 1993. Nosocomial pneumonia in ventilated patients: a cohort study evaluating attributable mortality and hospital stay. Am. J. Med. 94:281–288. Fayon, M. J., M. Tucci, J. Lacroix, C. A. Farrell, M. Gauthier, L. Lafleur, and D. Nadeau. 1997. Nosocomial pneumonia and tracheitis in a pediatric intensive care unit: a prospective study. Am. J. Respir. Crit. Care Med. 155:162–169. Fischer, J. E., M. Ramser, and S. Fanconi. 2000. Use of antibiotics in pediatric intensive care and potential savings. Intensive Care Med. 26:959– 966. Fischer, J. E., P. Allen, and S. Fanconi. 2000. Delay of extubation in neonates and children after cardiac surgery: impact of ventilator-associated pneumonia. Intensive Care Med. 26:942–949. Fitch, J. A., C. L. Munro, C. A. Glass, and J. M. Pellegrini. 1999. Oral care in the adult intensive care unit. Am. J. Crit. Care 8:314–318. Flanagan, P. G. 1999. Diagnosis of ventilator-associated pneumonia. J. Hosp. Infect. 41:87–99. Flanagan, P. G., G. P. Findlay, J. T. Magee, A. Ionescu, R. A. Barnes, and M. Smithies. 2000. The diagnosis of ventilator-associated pneumonia using non-bronchoscopic, non-directed lung lavages. Intensive Care Med. 26: 20–30. Foglia, E., C. Hollenbeak, V. Fraser, and A. Elward. 2006. Costs associated with nosocomial bloodstream infections and ventilator-associated pneumonia in pediatric intensive care unit patients. Abstr. 16th Annu. Meet. Soc. Healthcare Epidemiol. America, abstr. 109. Ford-Jones, E. L., C. M. Mindorff, J. M. Langley, U. Allen, L. Navas, M. L. Patrick, R. Milner, and R. Gold. 1989. Epidemiologic study of 4684 hospital-acquired infections in pediatric patients. Pediatr. Infect. Dis. J. 8:668– 675. Forman, M. L., and E. R. Stiehm. 1969. Impaired opsonic activity but normal phagocytosis in low-birth-weight infants. N. Engl. J. Med. 281:926– 931. Garner, J. S., W. R. Jarvis, T. G. Emori, T. C. Horan, and J. M. Hughes. 1996. CDC definitions for nosocomial infections, p. A1–A19. In R. N. Olmsted (ed.), APIC infection control and applied epidemiology: principles and practice. Mosby, St. Louis, MO. Gauvin, F., C. Dassa, M. Chaibou, F. Proulx, C. A. Farrell, and J. Lacroix. 2003. Ventilator-associated pneumonia in intubated children: comparison of different diagnostic methods. Pediatr. Crit. Care Med. 4:437–443. Gaynes, R. P., J. R. Edwards, W. R. Jarvis, D. H. Culver, J. S. Tolson, W. J. Martone, and the National Nosocomial Infection Surveillance System. 1996. Nosocomial infections among neonates in high-risk nurseries in the United States. Pediatrics 98:357–361. Goldmann, D. A., J. Freeman, and W. A. Durbin, Jr. 1983. Nosocomial infection and death in a neonatal intensive care unit. J. Infect. Dis. 147: 635–641. Gordon, A., and D. Isaacs. 2004. Late-onset infection and the role of antibiotic prescribing policies. Curr. Opin. Infect. Dis. 17:231–236. Haas, J. P., E. A. Mendonca, B. Ross, C. Friedman, and E. Larson. 2005. Use of computerized surveillance to detect nosocomial pneumonia in neonatal intensive care unit patents. Am. J. Infect. Control 33:439–443. Harpin, V. A., and N. Rutter. 1983. Barrier properties of the newborn infant’s skin. J. Pediatr. 102:419–425. Hemming, V. G., J. C. Overall, Jr., and M. R. Britt. 1976. Nosocomial infections in a newborn intensive-care unit. Results of forty-one months of surveillance. N. Engl. J. Med. 294:1310–1316. Herruzo-Cabrera, R., J. I. Garcia Gonzalez, P. Garcia-Magan, and J. del Rey-Calero. 1994. Nosocomial infection in a neonatal intensive care unit and its prevention with selective intestinal decolonization: a multivariant evaluation of infection reduction. Eur. J. Epidemiol. 10:573–580. Heyland, D. K., D. J. Cook, L. Griffith, S. P. Keenan, C. Brun-Buisson, et al. 1999. The attributable morbidity and mortality of ventilator-associated pneumonia in the critically ill patient. Am. J. Respir. Crit. Care Med. 159:1249–1256. Iregui, M., S. Ward, G. Sherman, V. J. Fraser, and M. H. Kollef. 2002. Clinical importance of delays in the initiation of appropriate antibiotic treatment for ventilator-associated pneumonia. Chest 122:262–268. Jacobs, R. F. 1991. Nosocomial pneumonia in children. Infection 19:64–72. Jantausch, B. A., J. Deville, S. Adler, M. R. Morfin, P. Lopez, B. EdgePadbury, S. Naberhuis-Stehouwer, and J. B. Bruss. 2003. Linezolid for the treatment of children with bacteremia or nosocomial pneumonia caused by resistant gram-positive bacterial pathogens. Pediatr. Infect. Dis. J. 22:S164– S171. Johnson, K. L., P. A. Kearney, S. B. Johnson, J. B. Niblett, N. L. MacMillan, and R. E. McClain. 1994. Closed versus open endotracheal suctioning: costs and physiologic consequences. Crit. Care Med. 22:658–666. VOL. 20, 2007 55. Kelleghan, S. I., C. Salemi, S. Padilla, M. McCord, G. Mermilliod, T. Canola, and L. Becker. 1993. An effective continuous quality improvement approach to the prevention of ventilator-associated pneumonia. Am. J. Infect. Control 21:322–330. 56. Kennedy, A. M., A. M. Elward, and V. J. Fraser. 2004. Survey of knowledge, beliefs, and practices of neonatal intensive care unit healthcare workers regarding nosocomial infections, central venous catheter care, and hand hygiene. Infect. Control Hosp. Epidemiol. 25:747–752. 57. Kerem, E., I. Yatsiv, and K. J. Goitein. 1990. Effect of endotracheal suctioning on arterial blood gases in children. Intensive Care Med. 16:95–99. 58. Kohler, P. F., and R. S. Farr. 1966. Elevation of cord over maternal IgG immunoglobulin: evidence for an active placental IgG transport. Nature 210:1070–1071. 59. Kollef, M. H., G. Sherman, S. Ward, and V. J. Fraser. 1999. Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest 115:462–474. 60. Labenne, M., C. Poyart, C. Rambaud, B. Goldfarb, B. Pron, P. Jouvet, C. Delamare, G. Sebag, and P. Hubert. 1999. Blind protected specimen brush and bronchoalveolar lavage in ventilated children. Crit. Care Med. 27:2537– 2543. 61. Liberati, A., R. D’Amico, Pifferi, V. Torri, and L. Brazzi. 2004. Antibiotic prophylaxis to reduce respiratory infections and mortality in adults receiving intensive care. Cochrane Database Syst. Rev. 2004(1):CD000022. 62. Lopriore, E., D. G. Markhorst, and R. J. Gemke. 2002. Ventilator-associated pneumonia and upper airway colonisation with gram negative bacilli: the role of stress ulcer prophylaxis in children. Intensive Care Med. 28:763– 767. 63. Luna, C. M., P. Vujacich, M. S. Niederman, C. Vay, C. Gherardi, J. Matera, and E. C. Jolley. 1997. Impact of BAL data on the therapy and outcome of ventilator-associated pneumonia. Chest 111:676–685. 64. Meissner, H. C., T. Tonwsend, W. Wenman, S. L. Kaplan, M. R. Morfin, B. Edge-Padbury, S. Naberhuis-Stehouwer, and J. B. Bruss. 2003. Hematologic effects of linezolid in young children. Pediatr. Infect. Dis. J. 22:S186– S192. 65. Morar, P., Z. Makura, A. Jones, P. Baines, A. Selby, J. Hughes, and R. van Saene. 2000. Topical antibiotics on tracheostoma prevents exogenous colonization and infection of lower airways in children. Chest 117:513–518. 66. Morrow, B., and A. Argent. 2001. Risks and complications of nonbronchoscopic bronchoalveolar lavage in a pediatric intensive care unit. Pediatr. Pulmonol. 32:378–384. 67. Munro, C. L., and M. J. Grap. 2004. Oral health and care in the intensive care unit: state of the science. Am. J. Crit. Care 13:25–34. 68. National Nosocomial Infections Surveillance System. 2004. National Nosocomial Infections Surveillance (NNIS) System report, data summary from January 1992 through June 2004, issued October 2004. Am. J. Infect. Control 32:470–485. 69. Ng, S. P., J. M. Gomez, S. H. Lim, and N. K. Ho. 1998. Reduction of nosocomial infection in a neonatal intensive care unit (NICU). Singapore Med. J. 39:319–323. 70. Papazian, L., F. Bregeon, X. Thirion, R. Gregoire, P. Saux, J. P. Denis, G. Perin, J. Charrel, J. F. Dumon, J. P. Affray, and F. Gouin. 1996. Effect of ventilator-associated pneumonia on mortality and morbidity. Am. J. Respir. Crit. Care Med. 154:91–97. 71. Perkins, G. D., S. Chatterjee, S. Giles, D. F. McAuley, S. Quinton, D. R. Thickett, and F. Gao. 2005. Safety and tolerability of nonbronchoscopic lavage in ARDS. Chest 127:1358–1363. 72. Pessoa-Silva, C. L., R. Richtmann, R. Calil, R. M. Santos, M. L. Costa, A. C. Frota, and S. B. Wey. 2004. Healthcare-associated infections among neonates in Brazil. Infect. Control Hosp. Epidemiol. 25:772–777. 73. Pineda, L. A., R. G. Saliba, and A. A. El Solh. 2006. Effect of oral decontamination with chlorhexidine on the incidence of nosocomial pneumonia: a meta-analysis. Crit. Care 10:R35. 74. Pittet, D. 2000. Improving compliance with hand hygiene in hospitals. Infect. Control Hosp. Epidemiol. 21:381–386. 75. Pittet, D., S. Dharan, S. Touveneau, V. Sauvan, and T. V. Perneger. 1999. Bacterial contamination of the hands of hospital staff during routine patient care. Arch. Intern. Med. 159:821–826. 76. Prober, C. G., P. D. Walson, J. Jones, and the Committee on Infectious Diseases and Committee on Drugs. 2000. Technical report: precautions regarding the use of aerosolized antibiotics. Pediatrics 106:e89. 77. Raymond, J., Y. Aujard, and the European Study Group. 2000. Nosocomial infections in pediatric patients: a European, multicenter prospective study. Infect. Control Hosp. Epidemiol. 21:260–263. 78. Richards, M. J., J. R. Edwards, D. H. Culver, R. P. Gaynes, and the National Nosocomial Infections Surveillance System. 1999. Nosocomial infections in pediatric intensive care units in the United States. Pediatrics 103:e39. 79. Riedler, J., J. Grigg, and C. F. Robertson. 1995. Role of bronchoalveolar lavage in children with lung disease. Eur. Respir. J. 8:1725–1730. 80. Rouby, J. J., E. M. De Lassale, P. Poete, M. H. Nicolas, L. Bodin, V. Jarlier, Y. Le Charpentier, J. Grosset, and P. Viars. 1992. Nosocomial broncho- VAP IN PEDIATRIC PATIENTS 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 425 pneumonia in the critically ill. Histologic and bacteriologic aspects. Am. Rev. Respir. Dis. 146:1059–1066. Ruza, F., F. Alvarado, R. Herruzo, M. A. Delgado, S. Garcia, P. Dorao, and F. Goded. 1998. Prevention of nosocomial infection in a pediatric intensive care unit (PICU) through the use of selective digestive decontamination. Eur. J. Epidemiol. 14:719–727. Saiman, L., J. Goldfarb, S. A. Kaplan, K. Wible, B. Edge-Padbury, S. Naberhuis-Stehouwer, and J. B. Bruss. 2003. Safety and tolerability of linezolid in children. Pediatr. Infect. Dis. J. 22:S193–S200. Shlaes, D. M., D. N. Gerding, J. F. John, Jr., W. A. Craig, D. L. Bornstein, R. A. Duncan, M. R. Eckman, W. E. Farrer, W. H. Greene, V. Lorian, S. Levy, J. E. McGowan, Jr., S. M. Paul, J. Ruskin, F. C. Tenover, and C. Watanakunakorn. 1997. Society for Healthcare Epidemiology of America and Infectious Diseases Society of America Joint Committee on the Prevention of Antimicrobial Resistance: guidelines for the prevention of antimicrobial resistance in hospitals. Clin. Infect. Dis. 25:584–599. Shorr, A. F., J. H. Sherner, W. L. Jackson, and M. H. Kollef. 2005. Invasive approaches to the diagnosis of ventilator-associated pneumonia: a metaanalysis. Crit. Care Med. 33:46–53. Singh, N., P. Rogers, C. W. Atwood, M. M. Wagener, and V. L. Yu. 2000. Short-course empirical antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit: a proposed solution for indiscriminate antibiotic prescription. Am. J. Respir. Crit. Care Med. 162:505–511. Sole, M. L., F. E. Poalillo, J. F. Byers, and J. E. Ludy. 2002. Bacterial growth in secretions and on suctioning equipment of orally intubated patients: a pilot study. Am. J. Crit. Care 11:141–149. Squire, E. N., Jr., H. M. Reich, G. B. Merenstein, B. E. Favara, and J. K. Todd. 1982. Criteria for the discontinuation of antibiotic therapy during presumptive treatment of suspected neonatal infection. Pediatr. Infect. Dis. 1:85–90. Stover, B. H., S. T. Shulman, D. F. Bratcher, M. T. Brady, G. L. Levine, W. R. Jarvis, and the Pediatric Prevention Network. 2001. Nosocomial infection rates in US children’s hospitals’ neonatal and pediatric intensive care units. Am. J. Infect. Control 29:152–157. Tablan, O. C., L. J. Anderson, R. Besser, C. Bridges, R. Hajjeh, CDC, and Healthcare Infection Control Practices Advisory Committee. 2004. Guidelines for preventing health-care-associated pneumonia, 2003: recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee. Morb. Mortal. Wkly. Rep. Recomm. Rep. 53:1–36. Tejerina, E., F. Frutos-Vivar, M. I. Restrepo, A. Anzueto, F. Abroug, F. Palizas, M. Gonzalez, G. D’Empaire, C. Apezteguia, A. Esteban, et al. 2006. Incidence, risk factors, and outcome of ventilator-associated pneumonia. J. Crit. Care 21:56–65. Toltzis, P., C. Hoyen, S. Spinner-Block, A. E. Salvator, and L. B. Rice. 1999. Factors that predict preexisting colonization with antibiotic-resistant gramnegative bacilli in patients admitted to a pediatric intensive care unit. Pediatrics 103:719–723. Toltzis, P., and J. L. Blumer. 2001. Nosocomial acquisition and transmission of antibiotic-resistant gram-negative organisms in the pediatric intensive care unit. Pediatr. Infect. Dis. J. 20:612–618. Toltzis, P., T. Yamashita, L. Vilt, and J. L. Blumer. 1997. Colonization with antibiotic-resistant gram-negative organisms in a pediatric intensive care unit. Crit. Care Med. 25:538–544. Torres, A., R. Aznar, J. M. Gatell, P. Jimenez, J. Gonzalez, A. Ferrer, R. Celis, and R. Rodriguez-Roisin. 1990. Incidence, risk, and prognosis factors of nosocomial pneumonia in mechanically ventilated patients. Am. Rev. Respir. Dis. 142:523–528. Trouillet, J. L., J. Chastre, A. Vuagnat, M. L. Joly-Guillou, D. Combaux, M. C. Dombret, and C. Gibert. 1998. Ventilator-associated pneumonia caused by potentially drug-resistant bacteria. Am. J. Respir. Crit. Care Med. 157:531–539. van Houten, M. A., M. Laseur, and J. L. Kimpen. 1998. Shift in antibiotic prescribing patterns in relation to antibiotic expenditure in paediatrics. Eur. J. Pediatr. 157:479–481. van Nieuwenhoven, C. A., E. Buskens, F. H. van Tiel, and M. J. Bonten. 2001. Relationship between methodological trial quality and the effects of selective digestive decontamination on pneumonia and mortality in critically ill patients. JAMA 286:335–340. Webber, S., A. R. Wilkinson, D. Lindsell, P. L. Hope, S. R. Dobson, and D. Isaacs. 1990. Neonatal pneumonia. Arch. Dis. Child. 65:207–211. Won, S. P., H. C. Chou, W. S. Hsieh, C. Y. Chen, S. M. Huang, K. I. Tsou, and P. N. Tsao. 2004. Handwashing program for the prevention of nosocomial infections in a neonatal intensive care unit. Infect. Control Hosp. Epidemiol. 25:742–746. Wunderink, R. G., J. Rello, S. K. Cammarata, R. V. Croos-Dabrera, and M. H. Kollef. 2003. Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumonia. Chest 124:1789–1797. Yildizdas, D., H. Yapicioglu, and H. L. Yilmaz. 2002. Occurrence of ventilator-associated pneumonia in mechanically ventilated pediatric intensive care patients during stress ulcer prophylaxis with sucralfate, ranitidine, and omeprazole. J. Crit. Care 17:240–245. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 426–439 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00009-07 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Molecular Testing in the Diagnosis and Management of Chronic Hepatitis B Alexandra Valsamakis* Department of Pathology, Division of Clinical Microbiology, The Johns Hopkins Medical Institutions, Baltimore, Maryland INTRODUCTION .......................................................................................................................................................426 HBV PROTEINS AND REPLICATION ..................................................................................................................426 EPIDEMIOLOGY .......................................................................................................................................................426 HBV INFECTION.......................................................................................................................................................427 ANTIVIRAL AGENTS................................................................................................................................................429 MOLECULAR ASSAYS IN THE DIAGNOSIS AND MANAGEMENT OF HBV INFECTION .....................430 Quantitative HBV DNA Assays: Utility ...............................................................................................................430 Molecular Tests for HBV Quantification: Available Assays and Performance Characteristics ..................432 HBV Genotyping: Utility ........................................................................................................................................433 HBV Genotyping Methods .....................................................................................................................................433 Antiviral Resistance Testing: Utility and Assays ...............................................................................................434 Detection of Core Promoter/Precore Mutations in HBeAgⴚ CHB: Utility and Assays ................................435 FUTURE TRENDS IN MOLECULAR DIAGNOSTIC TESTING FOR CHRONIC HEPATITIS B ..............435 ACKNOWLEDGMENTS ...........................................................................................................................................436 REFERENCES ............................................................................................................................................................436 ated with clinical improvement of hepatitis (reduced HBV DNA, normalized serum aminotransferase levels, and quiescence of inflammation in the liver) (9). HBV replicates primarily in human hepatocytes, although viral DNA can be found in peripheral blood mononuclear cells. Entry is mediated by envelope binding to an unknown receptor. After entry and virion uncoating, nucleocapsids are translocated into the nucleus, where cellular DNA repair enzymes complete virion DNA synthesis. The resultant covalently closed circular DNA (cccDNA) is the template for viral mRNA transcription, which is mediated by host polymerase. Replication-competent nucleocapsids comprised of core protein, encapsidated full-length pregenomic RNA, and viral polymerase are assembled in the cytoplasm. Genomic DNA is synthesized by reverse transcription of pregenomic RNA by viral polymerase. Encapsidated, relaxed, open circular DNA can be transported to the nucleus to become cccDNA and additional mRNA template, or it can be released from the host cell via a process that requires cytosolic packaging (along with polymerase) by envelope glycoproteins, budding into endoplasmic reticulum, and release after Golgi transit. HBV infection is noncytolytic. Clearance of infected cells is believed to be mediated in part by the noncytolytic intracellular activity of cytokines secreted by T cells (23). Cytotoxic T lymphocytes also lyse infected hepatocytes and induce liver injury (23). INTRODUCTION Hepatitis B virus (HBV) causes a highly complex chronic infection that impacts a significant proportion of the world’s population. Diagnostics for chronic hepatitis B have evolved from the simple detection of HBsAg through the complex antibody response against individual viral proteins and to the detection and quantification of viral DNA. Implementation of increasingly sensitive methods of HBV DNA quantification has greatly aided the diagnosis and management of disease. Assays are also available to determine HBV genotypes and to detect the presence of viral mutants, including those that confer drug resistance and others that downregulate HBV e antigen. In this review, an overview of the virus and chronic hepatitis B infection is provided. The current utility of the different types of molecular diagnostic tests is discussed, and the performance characteristics of the available assays are described. HBV PROTEINS AND REPLICATION HBV is an enveloped virus containing a 3.2-kb, partially double-stranded, relaxed circular genome. The genomic coding scheme is extraordinarily efficient; every nucleotide is a part of at least one open reading frame. The major viral proteins (polymerase, core, envelope, X, and e antigen) and their activities are shown in Table 1. HBsAg and HBeAg are particularly important in the management of chronic hepatitis B. Detectable HBsAg in serum is a marker of chronic infection. HBeAg in serum is a marker of high viral replication levels in the liver. Loss of HBeAg in serum and emergence of antiHBeAg antibody (termed HBeAg seroconversion) is associ- EPIDEMIOLOGY The worldwide burden of HBV is enormous. The World Health Organization (WHO) currently estimates that 2 billion people have been infected with HBV and that 360 million are chronically infected (153). HBV is a significant contributor to morbidity worldwide. Current estimates suggest that it causes * Mailing address: Department of Pathology, The Johns Hopkins Hospital, 600 North Wolfe St., Meyer B1-193, Baltimore, MD 21287. Phone: (410) 955-5077. Fax: (410) 614-8087. E-mail: avalsam1@jhmi .edu. 426 VOL. 20, 2007 MOLECULAR TESTING FOR CHRONIC HEPATITIS B 427 TABLE 1. HBV proteins Protein Function Envelope proteins: small (HBsAg), medium, large........................Glycoproteins located on virion surface; bind unknown cellular receptor to initiate virion entry into host cell Core protein (HBcAg) ...............................Encapsidates pregenomic RNA and partially double-stranded DNA genome in cytoplasm e antigen (HBeAg) .....................................Synthesized as precursor protein; proteolytically cleaved in endoplasmic reticulum; ultimately secreted extracellularly and found in peripheral blood; has diverse activities, including immunomodulation (tolerogenic) (21) and replication inhibition (45, 76, 127) Polymerase...................................................Reverse transcriptase, RNase H (degrades pregenomic RNA template during reverse transcription), DNA polymerase X protein......................................................Transcriptional transactivator; cofactor for hepatocellular carcinoma (14) 30% of cirrhosis and approximately 50% of hepatocellular carcinoma (HCC) globally (121). HBV can be transmitted perinatally, percutaneously, and sexually. Routes of transmission are dependent on the regional prevalence of infection (131). In areas of high endemicity such as Asia and the South Pacific, where seroprevalence is ⱖ8%, most infections are acquired perinatally or horizontally during childhood. In regions of intermediate seroprevalence (2 to 7%), such as sub-Saharan Africa, Alaska, the Mediterranean, and India, infection is transmitted during childhood (perinatally or horizontally) and later in life (sexually, by intravenous drug use, or by unsafe health care-related injection practices). In low-prevalence regions (most of the developed world and parts of Central and South America; seroprevalence, ⬍2%), HBV is usually transmitted sexually and by intravenous drug use. Historically, the scheme for distinguishing HBV subtypes was based on surface antigen serotyping. The current scheme is genetically based, with a genotype defined as greater than 8% divergence of the complete genome sequence at the nucleotide level (110). Eight genotypes (A to H) have been defined (6, 105, 109, 110, 136). HBV genotypes have distinct geographic distributions (128) (Fig. 1). Multiethnic populations tend to have multiple genotypes (25). HBV INFECTION HBV can cause acute and chronic infection. The incubation period between exposure and clinical presentation of acute infection is 1 to 4 months. The likelihood of development of symptomatic acute infection is age related, with a small proportion of cases (approximately 10%) occurring in children younger than 4 years old and a greater proportion (approximately 30%) in adults older than 30 years (99). Symptoms of acute HBV infection are similar to other causes of acute hepatitis, including influenza-like syndrome and right upper-quadrant pain. The clinical syndrome of acute hepatitis B has been well described and is diagnosed through the detection of viral proteins and host anti-HBV antibodies (53, 70). There is currently no role for molecular testing in the diagnosis of acute hepatitis B other than in the detection of asymptomatic patients during pretransfusion screening of blood products (67, 71). In contrast, chronic hepatitis B has been evolving as a clinical concept, and various paradigms have been utilized and FIG. 1. Geographic distribution of HBV genotypes. Letter sizes indicate relative prevalences in regions with multiple genotypes. Lowercase letters indicate HBV subtypes. 428 VALSAMAKIS CLIN. MICROBIOL. REV. TABLE 2. Useful markers for distinguishing chronic hepatitis B phases Phase ALT HBsAg HBeAg HBeAg antibody HBV DNA (IU/ml)a Immune tolerance Usually normal Present Present Absent ⱖ20,000 Immune clearance Present Present Absent ⱖ20,000 Inactive HBsAg carrier Elevated; can be episodic Usually normal; can have flares Present Absent Present ⬍20,000* HBeAg⫺ CHB Periodic flares Present Absent Present Occult hepatitis B Can be elevated Absent Absent Present in recovered HBV infection ⬎20,000 or ⬍20,000 ⬍20,000† a Liver histology Usually normal; can have mild inflammation Active inflammation Degree of abnormality dependent on disease severity during clearance phase (mild inflammation to inactive cirrhosis) Active inflammation Ranges from normal to cirrhosis and HCC Symbols: *, can be undetectable by PCR; †, usually (often detectable only with highly sensitive molecular tests). discarded. Importantly, molecular assays have begun to play increasingly significant roles in chronic hepatitis B. The content of this review is therefore focused on chronic hepatitis B and the commercial molecular assays that are available for its diagnosis and management. The probability of progression from acute to chronic hepatitis B infection is in part dependent on when acute infection occurs (158). The highest rates of chronicity are observed as a consequence of perinatal acquisition (90%) and in children less than 5 years of age who are infected horizontally through household contacts (25 to 30%). The incidence of chronic infection is lowest after acute infection during adulthood (⬍10%). A paradigm of four phases of chronic infection, consisting of immune tolerance, immune clearance, inactive carrier, and HBeAg-negative chronic hepatitis B (HBeAg⫺ CHB), is now widely accepted; however, not all patients go through each of the four phases (158). Markers that are helpful in distinguishing these phases are depicted in Table 2. The first phase, immune-tolerant chronic HBV, is observed primarily in perinatally acquired infection; it is characterized by high levels of viral replication (HBeAg seropositivity, high levels of HBV DNA in peripheral blood) and normal to minimally elevated alanine aminotransferase (ALT) levels indicating mild hepatitis, if any. This phase can last for decades. It occurs variably when infection is acquired later in childhood or as an adult. The immune tolerance phase is fairly benign, with a low incidence of cirrhosis and HCC. In a longitudinal study of HBsAgpositive Taiwanese adults who likely acquired infection perinatally, the annual incidence of cirrhosis was 0.5%, the cumulative probability of cirrhosis after 17 years was approximately 13%, and no HCC was observed during a mean follow-up period of 10.5 years (27). The majority of patients (85%) underwent HBeAg seroconversion between the ages of 20 and 39. The second phase, immune clearance, is associated with HBeAg seropositivity, high or variable HBV DNA concentrations in peripheral blood, periodic abnormalities in liver enzymes, and histologic evidence of active inflammation (158). The overall risk of progression to cirrhosis and HCC is directly related to disease activity during this phase, including overall duration and the number and severity of flares (26). This inflammatory phase also leads commonly to HBeAg seroconver- sion and entry into inactive HBsAg carrier status (the third phase of chronic infection). Spontaneous HBeAg seroconversion has been observed in 66% to 98% of prospectively studied cohorts (26) and occurs at an estimated rate of 10% per year (10, 100). Patients in the inactive HBsAg carrier state have normal liver enzymes and are HBeAg negative and anti-HBe antibody positive. Most individuals have low HBV DNA levels in peripheral blood (⬍5.0 log10 copies/ml); a minority have undetectable viral loads by PCR (165). Biopsy findings can range from mild inflammation and minimal fibrosis to inactive cirrhosis if disease was severe during immune clearance (158). The HBsAg carrier state can have a number of potential outcomes, including indefinite persistence, resolution of chronic infection (manifested by HBsAg clearance and appearance of anti-HBsAg antibody), or disease reactivation due either to recrudescence of the original infection or the emergence of mutant viruses that fail to express HBeAg (HBeAg⫺ CHB). Approximately 70% of patients remain as inactive carriers indefinitely; however, their course and outcome are not necessarily benign. ALT flares have been observed in 33% of inactive carriers (55). In addition, ongoing inflammation can cause progressive liver disease. Cirrhosis and HCC were observed in 8% and 2% of patients after HBeAg seroconversion (55). Clearance of HBsAg and the emergence of anti-HBsAg antibody has been reported to occur at a rate of 0.5% to 0.8% per year (82, 100). Half of these patients continue to have detectable HBV DNA (82, 100). A small proportion of inactive carriers serorevert to HBeAg-positive status (HBeAg seroreversion). In two Asian cohorts, 3 to 4% of HBeAg-negative patients experienced HBeAg seroreversion during long-term follow-up (55, 165). Higher seroreversion rates (14%) were observed in an Alaskan cohort (100). The fourth phase of chronic infection, HBeAg⫺ CHB, is characterized by lack of detectable HBeAg, the presence of anti-HBeAg antibody, detectable HBV DNA, fluctuating liver enzymes, and active inflammation upon biopsy (Table 2). Progression to this phase occurs spontaneously or in the setting of immune suppression in inactive carriers. Some patients can progress directly from HBeAg⫹ CHB to HBeAg⫺ CHB (55). Replicating viruses have mutations that prevent HBeAg expression, including two sequence changes in the basal core promoter that drives HBeAg transcription (A1762T and VOL. 20, 2007 G1764A) (110) or a stop codon in the second-to-last codon of the precore region (codon 28, nucleotide 1896) (12). These mutations can occur singly or in combination. The stop codon mutation is found only after infection with certain genotypes, due to constraints imposed by secondary structure (46). Nucleotide 1896 is base-paired to nucleotide 1858 within a stem loop structure required for HBV replication. Nucleotide 1858 varies according to genotype (T or U in B, D, E, G and some C strains; C in A, F, and some C strains). The G-to-A stop codon mutation at nucleotide 1896 stabilizes the stem loop in B, D E, G, and some C strains. The opposite effect occurs in A, F, and some C strains, likely leading to a relative lack of replicative fitness. The stop codon mutation is therefore found in infections with genotypes B, D, E, and G (and some C strains). HBeAg⫺ CHB was once thought to be uncommon outside the Mediterranean region, but it is now recognized to be variably prevalent, ranging from 15% of total chronic hepatitis B in the United States, northern Europe, and Asia-Pacific to 33% in the Mediterranean region (38). The prevalent mutation (basal core promoter versus stop codon) varies by geographic locale, with the basal core promoter mutation found in Asia (median, 77%) and the stop codon mutation found in the Mediterranean region (median, 92%), likely due to the genotypes present in each region (38). Occult hepatitis B infection, defined as detectable HBV DNA in peripheral blood or liver by sensitive nucleic acid detection methods in patients without detectable HBsAg, is now widely recognized to occur, although it has not been discussed as a phase of chronic hepatitis B in any recent guidelines. Occult hepatitis B has been observed in patients with unexplained liver disease (hepatocellular carcinoma and cryptogenic cirrhosis), in HBV-seropositive individuals with resolving or recovered chronic infection (spontaneously or after interferon [IFN] therapy), and in HBV-seronegative patients without evidence of liver disease, such as hemodialysis patients and blood donors (11, 20, 44, 101, 102, 139). Although data from properly designed cross-sectional studies are lacking, the available data suggest that the prevalence of occult HBV is correlated to the rate of endemic infection (29). Within a given population, occult HBV appears to be found more frequently in patients with evidence of liver disease than in other subjects. For example, occult hepatitis B was found in 16% of North American patients with HCC (62) versus 2% of hepatitis B core antibody-positive blood donors (50) and 4% of hemodialysis patients (101). Occult HBV is found more frequently in patients with serologic evidence of HBV infection (anti-hepatitis B core antibody positive) than in core antibody-negative individuals (11, 102). Finally, occult HBV is found in a significant proportion of patients with chronic hepatitis due to hepatitis C virus, with HBV DNA detectable in up to 30% of serum samples and 50% of liver biopsies (11). Although viruses with replication deficits could theoretically explain occult HBV, the finding of cccDNA, RNA transcripts, and pregenomic replicative RNA intermediates in a large proportion of patients suggests that most occult infections are due to low-level replication of wild-type virus (97, 98). In addition, the transmissibility of acute HBV via liver transplant or blood product transfusion from donors with occult infection (5, 85, 88) provides evidence against the presence of defective viruses. MOLECULAR TESTING FOR CHRONIC HEPATITIS B 429 Occult HBV may also result from mutations in HBsAg coding or transcription control regions that alter antigenicity or expression levels (19, 54, 61). Such mutant viruses have been reported as the sole circulating strain in up to 40% of patients with occult HBV (2, 15, 54, 101). A higher prevalence of HBsAg mutants (⬃60%) in individuals with occult HBV from an isolated Inuit community has recently been reported and awaits further study (102). Serum virus loads in occult HBV cases are usually below the limits of detection of hybridization assays, and detection usually requires more-sensitive methods, such as nested or realtime PCR. In anti-core antibody-positive individuals with occult HBV, virus loads are usually ⬍10,000 copies/ml, or ⬍2,000 IU/ml with the approximate conversion factor of 5 copies per international unit for PCR methods (50, 68, 102, 108). A single study reported viral loads of ⬎4 million copies/ml in anti-core antibody-positive individuals (145); however, further description of patients with such high-titer viremia was not provided. The consequences of occult HBV infection are not clear. Given the prevalence of HBV DNA in the livers of affected patients (11, 20), it may play a role in the development of cirrhosis and HCC. However, an overall risk of disease progression has not been defined. In hepatitis C virus-infected patients, the data on the effects of occult HBV on fibrosis are conflicting, with some studies documenting more-severe fibrosis (139) and others demonstrating no effect (57). However, a number of studies suggest that occult HBV is associated with an increased risk of HCC in chronic hepatitis C (104, 134, 139). There are currently no consensus guidelines for diagnosis of occult HBV. Testing has been advocated for a variety of settings, including cryptogenic liver disease, impending immunosuppression in patients with HBV risk factors (who may experience flares), and isolated positivity for core antibody, since the presence of HBV DNA may affect solid-organ-donation and HBV vaccination decisions (29, 139). From the perspective of diagnostic yield, liver biopsy samples may be better specimens than serum, as HBV DNA positivity rates have been found to be higher in liver than in serum in studies of paired samples (2, 11, 40, 162). There are currently no available reports on treatment for occult HBV. ANTIVIRAL AGENTS Six drugs have been approved by the U.S. Food and Drug Administration for therapy of chronic hepatitis B, including IFN-␣, pegylated IFN-␣, lamivudine, adefovir dipivoxil, entecavir, and telbivudine (Table 3). Tenofovir disoproxil fumarate is currently not approved for use in chronic hepatitis B but has been shown to be effective in human immunodeficiency virus type 1 (HIV-1)/HBV-coinfected patients (Table 3). IFN-␣ (and pegylated formulations) is the only drug that eliminates cccDNA from hepatocytes and is therefore potentially curative. In comparison, prolonged treatment with other agents is required due to greater relapse rates. The therapeutic gain of this approach is balanced by the potential emergence of antiviral resistance. The incidence of resistance varies among the different drugs (Table 3). The highest rates are observed for lamivudine; reported resistance to other nucleoside and nucleotide reverse transcriptase inhibitors is much lower. 430 VALSAMAKIS CLIN. MICROBIOL. REV. TABLE 3. Antiviral therapeutics for chronic hepatitis B Drug a Mechanism of actionb Efficacyc ⫹ Resistance IFN-␣ Immune-mediated clearance HBeAg CHB: 20% greater VR, 6% greater CR than untreated controls (150) HBeAg⫺ CHB: avg 24% SR-12 vs 0% for untreated controls (91) None Pegylated IFN-␣2a and IFN-␣2b* Immune-mediated clearance HBeAg⫹ CHB: 10% greater BR and VR at 6 mo posttreatment than with lamivudine (13, 79); pegylated IFN-␣2a and IFN-␣2b similarly efficacious (60, 79) HBeAg⫺ CHB: 15% greater BR and VR 6 at mo posttreatment than with lamivudine (95) No added benefit of combined pegylated IFN plus lamivudine for HBeAg⫹ or HBeAg⫺ CHB (60, 79, 95) None Lamivudine NRTI; cytidine analog 12-mo regimen: BR and VR at 12 mo posttreatment 10–15% greater than untreated controls for HBeAg⫹ and HBeAg⫺ CHB (46, 91) Prolonged treatment: HBeAg seroconversion rates progressively increase to 50% after 5 yr in HBeAg⫹ CHB; BR and VR peak after 24–36 mo (⬃50%) and then decline in HBeAg⫺ CHB (91) HBeAg⫹ and HBeAg⫺ CHB: 1 yr, 20%; 2 yr, 40%; 3 yr, 60%; 4–5 yr, 70% (18, 80, 115) Adefovir dipivoxil NRTI; dATP analog (chain terminator) HBeAg⫹ CHB (end of treatment): VR, ⬃20%; BR, ⬃50% (compared to placebo) (94) HBeAg⫺ CHB: VR, 70%; BR, 40% (47) HBeAg⫹ and HBeAg⫺ CHB: 1 yr, 0% (47, 149) HBeAg⫺ CHB: 2 yr, 3%; 3 yr, 6% (47); 5 yr, 29% (48) Entecavir 2⬘-Deoxyguanosine analog; inhibits polymerase priming activity and chain elongation HBeAg⫹ CHB: higher rates of undetectable DNA by PCR at end of treatment than with lamivudine; HBeAg loss, seroconversion, and BR equivalent to that with lamivudine (17) HBeAg⫺ CHB: higher rates of undetectable DNA by PCR at end of treatment than with lamivudine; BR equivalent to that with lamivudine (75) HBeAg⫹ and HBeAg⫺ CHB: 1 yr, 0% (17, 75); lamivudineresistant strains have reduced susceptibility in vitro (155) and in vivo (28) Telbivudine NRTI; dTTP analog Not yet published in peer-reviewed literature High-level cross-resistance to lamivudine in vitro (155) Tenofovir disoproxil fumarate NRTI; dATP analog (chain terminator) HIV-1/HBV coinfection: undetectable DNA by PCR in 30– 75% of patients (8, 73) HBV monoinfection: undetectable DNA by PCR in 80–100% of patients (72, 141) 1 yr, 0% (30) Emtricitabine NRTI; nucleoside analog of cytidine Improved histology, higher rates undetectable DNA by PCR, normalized ALT compared to controls (83); approved for use with HIV-1; may be useful in HIV-1/HBV coinfection (133) 1 yr, 13% (83); 2 yr, 18% (42); high-level cross-resistance to lamivudine in vitro (155) a *, IFN-␣2b is not currently FDA approved for treatment of chronic hepatitis B. NRTI, nucleoside reverse transcriptase inhibitor (causes chain termination). VR, virologic response (decrease in serum HBV DNA level to ⬍1 ⫻ 105 copies/ml and loss of HBeAg in patients with HBeAg⫹ CHB); CR, complete response (combined virologic response, biochemical response, and loss of HBsAg); SR-12, sustained response 12 months after therapy cessation; BR, biochemical response (normalization of ALT). b c MOLECULAR ASSAYS IN THE DIAGNOSIS AND MANAGEMENT OF HBV INFECTION Four types of molecular assays are available for the diagnosis and management of HBV infection: quantitative viral load tests, genotyping assays, drug resistance mutation tests, and core promoter/precore mutation assays. Viral load tests that quantify HBV in peripheral blood (serum or plasma fractions) are currently the most useful and most widely used. The applications of other assays are currently more specialized, and their use is more limited. The utility of these assays and their performance characteristics are reviewed below. Quantitative HBV DNA Assays: Utility Practice guidelines for the management of chronic hepatitis B have been published by a number of professional societies (32, 65, 81, 90). These consensus documents recommend the quantifica- tion of HBV DNA in the initial evaluation of chronic hepatitis B and during management, particularly in the decision to initiate treatment and in therapeutic monitoring. High-sensitivity molecular assays are clearly important for the diagnosis of HBeAg⫺ CHB and occult HBV, where viral loads can be quite low. Detectable HBV DNA in plasma or serum is one of the criteria for chronic hepatitis B in all guidelines. Assessment of HBV DNA in plasma or serum should therefore be performed along with other tests to establish this diagnosis. Several guidelines (65, 90, 91) specify a threshold of ⱖ20,000 IU/ml (100,000 copies/ml) for active viral replication during HBeAg⫹ CHB. This value corresponds to the limit of detection of hybridization-based quantitative HBV DNA assays and was originally proposed in an early consensus statement on HBV management (89) without much supporting evidence. Subsequent studies have demonstrated that ⬃90% of patients with HBeAg⫹ CHB had viral loads of ⬎20,000 IU/ml (24) and 98% VOL. 20, 2007 of inactive HBsAg carriers consistently had levels of ⬍20,000 IU/ml (96), suggesting that this threshold is reasonable. Measurement of HBV DNA is critical for the diagnosis and management of HBeAg⫺ CHB (core promoter, precore stop mutation), as it is the only marker of viral replication that can be monitored. Distinguishing HBeAg⫺ CHB from the inactive carrier state can be difficult due to fluctuating ALT and HBV DNA levels. A cutoff of 2,000 IU/ml was found to optimally differentiate HBeAg⫺ CHB from the inactive carrier state after short-term and long-term follow-up, particularly in patients with normal ALT levels (93, 166). Application of a higher threshold (20,000 IU/ml) was not found to be useful in this setting given the inability to identify 30% of patients with HBeAg⫺ CHB even after multiple sequential tests (24). Despite the observed predictive value of the 2,000-IU/ml threshold, serial testing should be performed in patients with low HBV DNA and normal ALT levels to optimally distinguish the inactive carrier state from HBeAg⫺ CHB (32). Viral loads can fall below the detection limit of low-sensitivity hybridizationbased assays in 40 to 60% of patients with HBeAg⫺ CHB (93); therefore, more-sensitive assay formats should be used in this setting. The detection of HBV DNA in peripheral blood (plasma or serum) is also important to establish the diagnosis of occult HBV (by definition, detectable HBV DNA in peripheral blood or liver in the absence of HBsAg). Testing for occult hepatitis B has been recommended in the following settings: (i) in cryptogenic liver disease, especially if serology is positive for previous exposure to HBV (anti-core antibody-positive individuals); (ii) prior to immunosuppression, due to the potential for hepatitis flares; and (iii) in solid organ transplant donors whose only serological marker of HBV infection is anti-core antibody, due to the potential for transmission (29, 139). There is general agreement between practice guidelines that the decision to initiate therapy should be based on the demonstration of active viral replication (defined as an HBV DNA level of ⱖ20,000 IU/ml in HBeAg⫹ CHB) and moderate to severe disease, as evidenced by persistent ALT elevation (3 to 6 months) and/or histologic demonstration of moderate to severe hepatitis (32, 65, 81, 90). HBV DNA levels are not primary determinants of therapy, because viral loads are not necessarily reflective of disease severity. Median viral loads in inactive chronic hepatitis B are lower than in HBeAg-positive patients; however, there is some overlap in viral load between individuals in these phases (66, 96, 129). Cirrhosis has also been observed in patients with low viral loads (⬍2,000 IU/ml) (160). One recent set of treatment guidelines (65) has applied viral load thresholds more extensively than others. For example, these guidelines suggest that viral loads can be helpful in the decision to obtain a biopsy from HBeAg⫹ CHB or HBeAg⫺ CHB patients with normal ALT levels. Individuals with viral loads greater than threshold (20,000 IU/ml for HBeAg⫹ CHB; 2,000 IU/ml for HBeAg⫺ CHB) may benefit from biopsy to identify histologic evidence of disease requiring initiation of treatment. Additionally, these guidelines recommend treatment for patients with compensated cirrhosis and viral loads greater than 2,000 IU/ml, while those with lower viral loads could either be treated or observed. Of note, the threshold for HBeAg⫹ CHB without cirrhosis (20,000 IU/ml) was greater than that for HBeAg⫺ CHB and for compensated cirrhosis MOLECULAR TESTING FOR CHRONIC HEPATITIS B 431 (2,000 IU/ml), largely reflecting the lower viral loads associated with the latter two states. Once the decision to treat has been made, viral load testing is useful at baseline to predict the response to antivirals and the emergence of antiviral resistance, particularly to lamivudine (16, 74). Low viral loads are associated with response to IFN-␣ (120) and pegylated IFN-␣ (60) in patients with HBeAg⫹ CHB. The influence of pretreatment viral load on IFN responsiveness in HBeAg⫺ CHB has not been reported. The value of baseline viral load as a predictor of response to lamivudine appears to depend on the patient cohort. In HBeAg⫹ CHB, it has not been found to be a significant variable for initial response, defined as HBeAg loss (52, 119), but was found to be associated with relapse (143). In HBeAg⫺ CHB, high viral loads were observed to be associated with lack of response to lamivudine (116). The influence of pretreatment viral load on response and the emergence of resistance to newer nucleoside/ nucleotide antivirals has not yet been reported. Viral load measurement plays a significant role during therapy, as most guidelines propose that suppression of HBV replication is a major therapeutic goal. Measurement of viral load at 3- to 6-month intervals during treatment has been recommended (32, 81). Monitoring intervals can also be guided by specific treatment, with shorter intervals (every 3 months) for lamivudine and longer intervals (at least every 6 months) for other nucleoside/nucleotide reverse transcriptase inhibitors due to the more rapid emergence of resistance during lamivudine therapy (65). The treatment duration for IFN is relatively fixed (4 to 6 months for HBeAg⫹ CHB; 12 to 24 months for HBeAg⫺ CHB). However, viral load measurement is helpful in determining when to end treatment with nucleoside/nucleotide reverse transcriptase inhibitors. One study has suggested that viral load can be used as a primary end point to assess the response to nucleoside antiviral therapy, since a correlation between a decline in viral load, an improved histologic necroinflammatory score, and HBeAg seroconversion was observed (103). However, most guidelines also incorporate serologic testing (HBeAg and anti-HBeAg antibody to assess HBeAg seroconversion) in patients with HBeAg⫹ CHB. In this setting, therapy can be stopped 4 to 12 months after HBeAg seroconversion occurs and HBV DNA levels decrease to less than 20,000 IU/ml (32, 90) or become undetectable by PCR (⬍40 IU/ml) (65, 81). Data documenting the relative utility of high (20,000 IU/ml) versus low (40 IU/ml) thresholds are not available. In HBeAg⫺ CHB, HBV DNA is the only virologic marker that can guide the decision to end treatment; however, no specific stopping point for nucleoside/nucleotide reverse transcriptase inhibitors has been recommended in practice guidelines largely due to the higher relapse rates in these patients (37, 116). The identification of nonresponders through determination of viral load kinetics at early time points posttreatment compared to baseline has become the standard of care in pegylated IFN/ribavirin treatment for chronic hepatitis C. A similar paradigm for chronic hepatitis B therapy has not yet been adopted, largely due to the lack of trials designed to address this question; however, preliminary data are emerging. In one recent study of IFN-␣ treatment in HBeAg⫹ CHB, nonresponders could be predicted accurately at week 8 and week 12 432 VALSAMAKIS CLIN. MICROBIOL. REV. TABLE 4. Commercially available assays and reagents for HBV DNA quantification Test or reagent (manufacturer) Method Analytical sensitivitya Linear rangea Reference(s) IU/ml Copies/ml RealTime HBV PCR (Abbott Molecular) Real-time PCR Hybrid Capture II (Digene) Hybrid capture COBAS Amplicor HBV Monitor (Roche Diagnostics) COBAS TaqMan HBV (Roche Diagnostics) Europe, CE; United States, ASR 5 1.9 ⫻ 105–1.7 ⫻ 109 3.0 ⫻ 106–1.0 ⫻ 109 107 118 United States, RUO 8 ⫻ 103 8.0 ⫻ 103–1.7 ⫻ 109 107 United States, RUO 56 Europe, CE; United States, not available 1.9 ⫻ 10 54–3.6 ⫻ 109 Real-time PCR 2.5 ⫻ 10 –1.0 ⫻ 10 2 9 2 ⫻ 102 Semiautomated PCR 135 2.0 ⫻ 102–2.0 ⫻ 105 1 ⫻ 10 –3 ⫻ 10 3 Real-time PCR 5 8 54–1.1 ⫻ 108‡ 30–1.1 ⫻ 108§ 6–2.1 ⫻ 108¶ 3.3 ⫻ 103 69, 90 Europe, CE; United States, RUO 118 1.7 ⫻ 10 –8.5 ⫻ 10 † 2 35† 12‡ ⬃35§ ⬃2.5¶ VERSANT HBV DNA 3.0 bDNA (Siemens Medical Solutions Diagnostics) Regulatory statusb Copies/ml 9–4 ⫻ 109* 4* Ultrasensitive Hybrid Capture Hybrid capture II (Digene) Artus HBV PCR (QIAGEN Diagnostics) IU/ml 146 Europe, CE; United States, ASR, RUO (with HighPure extraction) 51 124 39 3.3 ⫻ 103–1.0 ⫻ 108 156 Europe, CE; United States, RUO a Symbols: *, data from company website (http://www.international.abbott.molecular.com/HBVPCRkit_1137.asp); †, nucleic acid extraction with HighPure (Roche Diagnostics); ‡, extraction with COBAS AmpliPrep and HBV-specific chemistry (Roche Diagnostics); §, extraction with COBAS AmpliPrep and Total Nucleic Acid chemistry (Roche Diagnostics); ¶, extraction with MagNAPure (Roche Diagnostics). b CE, Conformité Européen (approval according to European In Vitro Diagnostic Directive 98/79/EC); ASR, analyte-specific reagent; RUO, research use only. of treatment, although sustained responders were identified more accurately by measuring the response at week 12 (142). HBV viral load measurement has been widely recommended as part of a panel of follow-up tests (including transaminases and HBeAg/HBeAg antibody, if relevant) to assess the durability of the antiviral response after treatment cessation. Recommended posttreatment monitoring intervals vary from every 1 to 3 months for 12 months and then every 6 to 12 months (32) to monthly for 3 months then once again at 6 months, with continued monitoring only for nonresponders to identify delayed therapeutic responses or the need to reinitiate treatment (81). Molecular Tests for HBV Quantification: Available Assays and Performance Characteristics First-generation assays for HBV DNA quantification in peripheral blood (usually serum or plasma) were based on solution hybridization technology and measured HBV DNA in picograms per milliliter. Solution hybridization was successful due to high viral loads in many patients with chronic hepatitis B. This assay was relatively insensitive (approximately 5.0 log10 copies/ml), and its linearity ranged from 5.0 to 10.0 log10 copies/ml (90). Adaptation of advanced molecular technologies, such as signal and target amplification, led to the development of second-generation assays with enhanced sensitivity (as low as 200 copies/ml) (117). The latest generation HBV quantification assays utilize realtime PCR and have improved analytical performance characteristics, including low limits of detection, broad linear ranges, and excellent precision (Table 4). These advances have been demonstrated to translate into better clinical performance as manifested by better detection of low virus concentrations and elimination of specimen dilution for a large proportion of specimens with high viral loads (43, 56). A WHO international standard was created in response to the need for standardization of quantification (126). This virus preparation (code 97/746) was based on a high-titer genotype A EUROHEP reference preparation (49). Results are reported in international units per milliliter for most currently available assays. In the current WHO HBV standard preparation, 1 IU is equivalent to 5.4 genome equivalents (copies). However, accurate conversion factors are likely to be dependent on the chemistry used for HBV DNA quantification. In support of this, PCR-based quantification assays were demonstrated to have similar conversion factors (Amplicor, 7.3 copies per IU; SuperQuant, 6.2 copies per IU) that were higher than those for bDNA (VERSANT, v2.0, 4.5 copies per IU) and Hybrid Capture II (2.3 copies per IU) (132). Significant variability in quantification among different assays can occur randomly (39, 69) despite the standardization of reporting units and the finding of generally good correlation between different assays (56, 77, 122). Patients should therefore be monitored with a single assay. The HBV genotype is a variable that may influence quantification by different methods. Genotype F-dependent underquantification by conventional PCR (COBAS Amplicor Monitor) has been observed (77, 146). For real-time PCR, no bias in amplification has been observed (51, 146). Quantification by the Hybrid Capture II and Ultrasensitive Hybrid Capture II VOL. 20, 2007 MOLECULAR TESTING FOR CHRONIC HEPATITIS B tests also appears to be free of genotype-dependent bias (107). Data on genotype influence in quantification by VERSANT HBV DNA 3.0 have not been published. Results of specificity studies have been reported for most of the assays described in Table 4. Excellent results (⬎97%) have been obtained in studies using samples from HBV-seronegative subjects (51, 107, 118, 146, 156). Problems with reproducibility at concentrations near the limits of quantification of Hybrid Capture II and Ultrasensitive Hybrid Capture II have been described (25 to 40% of samples originally determined to have 5,000 to 10,000 copies/ml were undetectable after repeat testing), suggesting that implementation of an equivocal zone should be considered for these assays (69). False-positive results (ranging from 1 to 3%) at the lower limit of quantification of VERSANT HBV DNA 3.0 have also been observed (39, 124, 156). Real-time PCR assays have utilized a number of different methods for extraction of HBV DNA. Automated extraction methods with COBAS AmpliPrep (Roche Diagnostics) and MagNAPure (Roche Diagnostics) have been reported for RealArt HBV LC PCR reagents (Artus HBV PCR; QIAGEN Diagnostics) (56, 135) and COBAS TaqMan reagents (39, 51, 124). Manual column methods have also been reported for use with COBAS TaqMan reagents (84, 146). HBV Genotyping: Utility The HBV genotype is a variable that could potentially influence the outcome of chronic hepatitis B and the success of antiviral therapy. The influence of the HBV genotype on chronic infection has been most intensively studied in highprevalence populations in Asia, where genotypes B and C predominate. Outcome data for these genotypes are probably the most valid, since infection duration (due predominantly to perinatal acquisition) is not a confounding variable (36). In general, patients with genotype B infections have more-favorable outcomes than those with genotype C, including younger age at HBeAg seroconversion, lower rates of active liver disease, and slower progression to cirrhosis (87). The influence of genotype on HCC is complex. In most cases, genotype C correlates with higher risk (112, 138, 159); however, the association may also be dependent on other factors, such as host age and geographically dependent virus strain (87). For example, a high rate of HCC has been observed in young patients (63, 106) and in Taiwanese patients with genotype B infection (63). An HBV genotype-dependent response to antiviral therapy has been observed for some drugs but not for others. For IFN-␣ treatment of HBeAg⫹ CHB, greater rates of HBeAg seroconversion have been observed for genotype A than for genotype D (49% versus 26%) (31) and for B than for C (39% versus 17%) (144). Similar results have been found with pegylated IFN-␣2b (33, 60). A trend of higher HBeAg seroconversion rates after pegylated IFN-␣2a treatment of HBeAg⫹ CHB has been observed for genotype A compared to genotypes B, C, and D (79). For nucleoside/nucleotide analogs, potential genotype-dependent predictive parameters include therapeutic response and emergence of resistance. On the whole, there seems to be little evidence for genotype-dependent responses to these agents. For lamivudine treatment of HBeAg⫹ CHB, information is available primarily for genotypes B and C. There are 433 strong data demonstrating no effect of genotype (B versus C) on lamivudine response, including equivalent rates of HBeAg seroconversion, log HBV DNA reduction, and median aminotransferase normalization 12 months after end of therapy (163). Another study, measuring similar parameters at the same times (22), showed a greater sustained response in genotype B than in genotype C patients; however, the ratio of genotype B to C patients in this study was 3:1, introducing a potential for bias in the results. No genotype-dependent differences in HBV DNA responses have been observed for adefovir (148) or tenofovir (8). Development of resistance appears to be equivalent among the different genotypes. Most data pertain to lamivudine, since it has the highest resistance rates of the nucleoside/nucleotide analogs. No difference in rates of resistance has been observed between genotypes B and C (3, 64). More rapid emergence of resistance has been reported for genotype A than for genotype D; however, the difference between the two genotypes was actually quite small (167). Recent data on adefovir resistance identified a potential association with genotype D that bears further investigation (35). The above observations and data suggest that genotype determination in chronic hepatitis B has a more limited role in clinical management than it does in chronic hepatitis C virus infection. Genotyping is probably most important if IFN therapy is a consideration, and genotype determination has been suggested prior to initiation of IFN therapy in one set of practice guidelines (65). None of the current guidelines have advocated a role for genotyping in counseling patients on the outcome of chronic hepatitis B. HBV Genotyping Methods Given its limited utility, HBV genotype testing has not yet been widely adopted in clinical laboratories. A variety of methods have been used, including whole- or partial-genome sequencing, restriction fragment length polymorphism, genotype-specific PCR amplification, PCR plus hybridization, and serology (7). Whole-genome sequencing is the “gold standard,” and it is particularly accurate for detecting recombinant viruses. However, it is cumbersome and time-consuming and has limited ability to detect mixed-genotype infections. Single gene sequencing, most commonly the S gene, is technically less demanding. The most common method of sequence-based HBV genotype determination has been searching of the GenBank database for homologous sequences using BlastN. This approach was noted to be complicated by the paucity of genotype-annotated HBV sequences within GenBank (7); however, the deposition of a growing number of annotated sequences into the database has made this approach more practical. PCR plus hybridization has been adapted into a commercial product (INNO-LiPA; Innogenetics [research use only in Europe and the United States]). The amplification target lies in the major hydrophilic domain of HBsAg and is encoded within the pre-S1 gene, which has been found to be useful for genotype determination by direct sequencing. Amplicons are generated by nested PCR, although hybridization can be performed after the first PCR round if product is visualized by gel electrophoresis. Data on the analytical and clinical performance characteristics of this assay are limited to two reports 434 VALSAMAKIS (58, 113). The reported accuracy of single-genotype identification was 97 to 99.9% compared to direct sequencing (58, 113). This method has several advantages over direct sequencing. It can effectively identify mixed infection, as 65% of mixed infections were verified by clonal analysis (113). In addition, it can be used to identify genotypes in samples that yield poor quality sequence data (113). One disadvantage is the potential for indeterminate results for viruses with single nucleotide polymorphisms or deletions in sequences that are complementary to probes used in the hybridization phase. An indeterminate rate of up to 5% has been observed (113). Reasonable analytical and clinical sensitivity has been achieved with a modified protocol incorporating a number of different improvements to the recommended PCR plus hybridization procedure, including automated extraction (MagNAPure; Roche Diagnostics), single-round PCR (versus the recommended nested PCR), and uracil N-glycosylase (123). A commercial direct sequencing assay for genotype determination (TRUGENE HBV Genotyping Kit; Siemens Medical Solutions Diagnostics) is available as a research-use-only kit. Published assay performance data are limited. Correlation data with other direct sequencing methods or with INNO-LiPA have not been reported. The assay can be used in samples with fairly low HBV concentrations (200 to 900 IU/ml) (41). Antiviral Resistance Testing: Utility and Assays The emergence of drug-resistant HBV should be suspected when a 10-fold increase in viral load compared to nadir is confirmed in a patient with documented therapeutic response. Documenting the mutation that confers drug resistance has not been part of routine clinical practice. This may change with the growing armamentarium of antivirals and reported crossresistance among drugs within the same structural class (for example, cross-resistance observed between lamivudine and other L-nucleosides such as entecavir, emtricitabine, and telbivudine [28, 155]). Resistance can be documented by phenotypic or sequence analysis. Each strategy has advantages and disadvantages. Phenotypic analysis entails assessment of mutant replication in the presence of drug and requires some form of genetic engineering (either site-directed mutagenesis of wild-type sequence or construction of full-length mutant clones expressed in baculovirus models) followed by expression in cell culture systems (130). This approach is the most effective means of ascertaining whether a complex set of mutations confers antiviral resistance. However, it is far too cumbersome for standard clinical molecular laboratories and is usually limited to specialized laboratories with a specific interest in antiviral resistance. Direct sequencing can identify known and potential new resistance mutations. However, this method is not sufficiently sensitive for the detection of emerging, resistant mutants that are present in low concentrations. These minor populations can be identified by large-scale cloning and sequencing protocols; however, this is cumbersome and beyond the capacity of clinical laboratories. In comparison, hybridization-based methods are more sensitive and less labor-intensive. This approach also has a number of disadvantages: (i) only known mutations can be identified, (ii) individual probes are required to detect each mutation, and (iii) single-nucleotide polymorphisms that CLIN. MICROBIOL. REV. have no effect on phenotype can impair probe binding and produce false-negative results (92). While sequence determination is relatively simple to perform compared to phenotypic analysis, interpretation of sequence data is not always straightforward. For example, some mutations confer resistance to multiple drugs (rtA181T is observed after long-term lamivudine therapy and in adefovirresistant viruses) (34, 157). Other sequence changes may not confer resistance when present alone but apparently cause resistance in combination with other mutations. While lamivudine resistance has not been observed with the single mutation rtL180M, it does occur with the double mutant rtL180M/ rtA181T, which apparently alters the position of rtM204, a critical residue in the nucleotide binding pocket (130). Finally, it has been noted that patients who have been treated with multiple drugs will likely have a combination of sequence changes that represent (i) drug resistance mutations, (ii) compensatory mutations that improve the fitness of drug-resistant viruses, and (iii) singlenucleotide polymorphisms that have no phenotype (130). A small number of sequence determination assays are commercially available, including hybridization (biotinylated amplicons hybridized to membrane-bound oligonucleotides specific for each mutation) and direct-sequencing formats. The hybridization-based assay is in its second generation. The first-generation product (for the detection of lamivudine resistance mutations at amino acids 180, 204, and 207) was found to be more sensitive for the detection of emerging variants than sequencing and was able to detect mutations in specimens with low viral loads (approximately 200 IU/ml) (1, 92). The second-generation product (INNO-LiPA DR, version 2.0) has a refined, expanded lamivudine resistance panel (codons 80, 173, 180, and 204) and also detects adefovir resistance mutations (codons 181 and 236). This test demonstrated a high degree of concordance with direct sequencing (⬃95% of codons interrogated) (59, 114). INNO-LiPA DR, version 2.0, detected a greater number of mixed (resistance mutation plus wild-type virus) infections, and lamivudine resistance was detected earlier (mean, 6 to 7months). Dual resistance (lamivudine and adefovir) was also demonstrated (59). Operational disadvantages have been noted, including a large number of reaction lines per strip (34 lines per strip), faint bands, absence of bands (false negatives), and indeterminate results due to lack of amplification by primers (114). There is limited published experience with commercially available direct-sequencing resistance detection assays. The TRUGENE HBV genotyping kit (Siemens Medical Diagnostic Solutions), modified to incorporate automated extraction (versus the recommended manual method), was able to provide sequence data from specimens with low viral loads (⬍900 IU/ ml) (41). In one comparison between TRUGENE and INNOLiPA HBV DR, concordance was high (approximately 80%) with clinical samples from HIV-1/HBV-coinfected patients treated with lamivudine as part of antiretroviral therapy. However, the hybridization-based assay was better able to detect mixed populations (125), as has been reported in other comparisons between PCR plus hybridization and noncommercial direct sequencing protocols. Another commercial platform (Affigene HBV DE/3TC assay; Sangtec Molecular Diagnostics AB) combines hybridization and direct sequencing in a microwell plate format to VOL. 20, 2007 detect lamivudine resistance mutations. PCR amplicons are immobilized by hybridization; codons 180 and 204 are subsequently interrogated by microsequencing. This assay demonstrated concordance with direct sequencing that was similar to INNO-LiPA DR, version 1.0 (111). Indeterminate rates were greater with Affigene than with INNO-LiPA (13 versus 3%). Detection of Core Promoter/Precore Mutations in HBeAgⴚ CHB: Utility and Assays The diagnosis of HBeAg⫺ CHB is made primarily through assessment of a combination of virological markers (HBsAg positive, HBeAg negative, detectable HBV DNA), serology (anti-HBeAg antibody positive), and evidence of hepatic injury (elevated aminotransferases and or histologic evidence of necroinflammation). Assays to detect mutations that abrogate HBeAg expression are commercially available. Test formats for the detection of antiviral resistance mutations have been applied to the identification of precore/core promoter mutations (PCR plus hybridization, INNO-LiPA HBV PreCore; hybridization/direct sequencing, Affigene HBV Mutant VL19 [Sangtec Molecular Diagnostics AB]). The PCR-plus-hybridization format detects three mutations (basal core promoter nucleotides 1762 and 1764 and precore codon 28), while the hybridization/direct-sequencing format detects two of these three mutations (basal core promoter nucleotide 1764 and precore codon 28). Both assays demonstrated high concordance with direct sequencing (approximately 90%); however, they more effectively detected mixed populations (mutant variant plus wild type) (111). Indeterminate results in lowviral-load specimens were observed only with the hybridization/direct sequencing assay, suggesting that PCR plus hybridization may be a more sensitive format (111). Procedural improvements to the PCR-plus-hybridization test (automated extraction, single round of PCR, incorporation of uracil Nglycosylase) have been published (123). FUTURE TRENDS IN MOLECULAR DIAGNOSTIC TESTING FOR CHRONIC HEPATITIS B The adoption of new technologies and the identification of new virological or host markers could potentially provide opportunities for growth and evolution of molecular testing in chronic hepatitis B. Emerging technologies that have not yet penetrated significantly into diagnostic laboratories may become useful in the future. For example, a high-density array that can compete with sequencing for the identification of mutations (polymerase-based resistance mutations and core promoter/precore mutations) and genotypes has been reported (140). This system has a number of advantages, including flexibility and throughput. Microarrays currently pose numerous technical, regulatory, and cost challenges that will have to be overcome in order for these types of platforms to become widely adopted in clinical laboratories. Virtual phenotyping, the prediction of drug resistance from analysis of complex sequence changes, is used routinely in managing HIV-1 resistance to antivirals. This method has great applicability to the management of chronic hepatitis B. A database that can be used to process HBV DNA sequence information to identify genotype and detect mutations that MOLECULAR TESTING FOR CHRONIC HEPATITIS B 435 inhibit HBeAg expression and to predict resistance has been reported (SEQHEPB) (161). Access is provided on a subscription basis. Clinical information can be deposited so that mutations can be correlated with patient presentation, course, and treatment. One particularly interesting feature of this database is that the potential effects of mutations on drug binding can be visualized through a three-dimensional model of drug-bound reverse transcriptase. As the number of drugs to treat chronic hepatitis B expands, such databases may become increasingly useful. New virological markers, such as cccDNA, the minichromosome that serves as the transcriptional template for viral RNAs, could become clinically useful. cccDNA quantification was once cumbersome but is now readily achievable by realtime PCR and other homogeneous amplification/detection chemistries such as cleavase (Invader HBV DNA; Third Wave Technologies). Studies utilizing these techniques are beginning to elucidate the dynamics of HBV replication during the course of infection and treatment. Median total intrahepatic HBV DNA and cccDNA levels have been found to be greater in HBeAg⫹ CHB than in HBeAg⫺ CHB (78, 147, 152, 154). Interestingly, higher levels of cccDNA transcriptional activity, defined as the ratio of pregenomic RNA to cccDNA, were observed in HBeAg⫹ CHB than in HBeAg⫺ CHB (78), providing a potential explanation for the higher levels of viral replication that occur in the former group. In HCC, tumor tissue has been found to contain higher median cccDNA levels and proportions of cccDNA to intrahepatic total HBV DNA than adjacent nontumor tissue (151), supporting the current paradigm that HBV replication is partially or completely downregulated in HCC. Total intrahepatic HBV DNA and cccDNA loads decline after antiviral therapy (137, 147, 154), reflecting response to therapy. Preliminary data on the utility of intrahepatic cccDNA as a predictor of response to antiviral therapy are emerging. Whether cccDNA will be useful to predict a sustained response is not yet resolved. Data from studies with small cohorts are conflicting (137, 147). Initial evidence suggests that baseline intrahepatic cccDNA is useful for predicting HBeAg seroconversion after adefovir monotherapy or combination therapy with pegylated IFN-␣ (147, 154). Whether this applies to other therapies is unknown. While novel intrahepatic markers of chronic HBV infection and response to therapy are promising and of interest, diagnostic targets that can be monitored less invasively in peripheral blood are likely to be more useful and more widely applicable. One potential candidate is cccDNA measurement in serum. This molecule has been detected in a greater proportion of patients with HBeAg⫹ CHB than with HBeAg⫺ CHB, and median levels have been found to be greater in HBeAg⫹ CHB (152). In addition, a statistically significant decrease in serum cccDNA was observed compared to placebo in patients with HBeAg⫹ CHB who responded virologically to a 52-week course of lamivudine (164). These studies indicate that measurement of cccDNA in peripheral blood is possible, but its potential clinical utility awaits further investigation. Overall, the field of molecular testing for the diagnosis and management of HBV infection has shown steady improvement in technology and standardization. Emerging, more-sensitive technologies have facilitated diagnostics and therapeutics, particularly for HBeAg⫺ CHB and occult hepatitis B. They have 436 VALSAMAKIS CLIN. MICROBIOL. REV. also helped to better define the natural history of chronic hepatitis B and how the host responds to therapy. Importantly, these new methods have also raised questions and spurred debate on the most useful virological markers for documenting the response to antivirals, which can only improve the application of molecular methods to this field (4, 86, 103). In the natural transfer of information that occurs between the basic and clinical sciences, new markers are being identified during investigation into chronic hepatitis B pathogenesis. As has happened in the past, the most promising molecules will be adapted for clinical use, propelling the field of clinical diagnostics forward yet again. 16. 17. 18. 19. ACKNOWLEDGMENTS 20. Many thanks to Michael Forman for thoughtful commentary on the manuscript, John Ticehurst for spontaneously fielding numerous questions, and Sherron Wilson-Alexander for assistance with manuscript preparation. This work was supported by the HIV Prevention Trials Network (HPTN), sponsored by NIAID, NIDA, NIMH, and the Office of AIDS Research of the NIH, DHHS (U01-AI-068613). 21. REFERENCES 24. 1. Aberle, S. W., J. Kletzmayr, B. Watschinger, B. Schmied, N. Vetter, and E. Puchhammer-Stockl. 2001. Comparison of sequence analysis and the INNO-LiPA HBV DR line probe assay for detection of lamivudine-resistant hepatitis B virus strains in patients under various clinical conditions. J. Clin. Microbiol. 39:1972–1974. 2. Ahn, S. H., Y. N. Park, J. Y. Park, H. Y. Chang, J. M. Lee, J. E. Shin, K. H. Han, C. Park, Y. M. Moon, and C. Y. Chon. 2005. Long-term clinical and histological outcomes in patients with spontaneous hepatitis B surface antigen seroclearance. J. Hepatol. 42:188–194. 3. Akuta, N., F. Suzuki, M. Kobayashi, A. Tsubota, Y. Suzuki, T. Hosaka, T. Someya, M. Kobayashi, S. Saitoh, Y. Arase, K. Ikeda, and H. Kumada. 2003. The influence of hepatitis B virus genotype on the development of lamivudine resistance during long-term treatment. J. Hepatol. 38:315–321. 4. Alberti, A. 2003. Can serum HBV-DNA be used as a primary end point to assess efficacy of new treatments for chronic hepatitis B? Hepatology 38: 18–20. 5. Allain, J. P. 2004. Occult hepatitis B virus infection: implications in transfusion. Vox Sang. 86:83–91. 6. Arauz-Ruiz, P., H. Norder, B. H. Robertson, and L. O. Magnius. 2002. Genotype H: a new Amerindian genotype of hepatitis B virus revealed in Central America. J. Gen. Virol. 83:2059–2073. 7. Bartholomeusz, A., and S. Schaefer. 2004. Hepatitis B virus genotypes: comparison of genotyping methods. Rev. Med. Virol. 14:3–16. 8. Benhamou, Y., H. Fleury, P. Trimoulet, I. Pellegrin, R. Urbinelli, C. Katlama, W. Rozenbaum, G. Le Teuff, A. Trylesinski, and C. Piketty. 2006. Anti-hepatitis B virus efficacy of tenofovir disoproxil fumarate in HIVinfected patients. Hepatology 43:548–555. 9. Bortolotti, F., P. Cadrobbi, C. Crivellaro, M. Guido, M. Rugge, F. Noventa, R. Calzia, and G. Realdi. 1990. Long-term outcome of chronic type B hepatitis in patients who acquire hepatitis B virus infection in childhood. Gastroenterology 99:805–810. 10. Bortolotti, F., M. Guido, S. Bartolacci, P. Cadrobbi, C. Crivellaro, F. Noventa, G. Morsica, M. Moriondo, and A. Gatta. 2006. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 43:556–562. 11. Brechot, C., V. Thiers, D. Kremsdorf, B. Nalpas, S. Pol, and P. PaterliniBrechot. 2001. Persistent hepatitis B virus infection in subjects without hepatitis B surface antigen: clinically significant or purely “occult”? Hepatology 34:194–203. 12. Carman, W. F., M. R. Jacyna, S. Hadziyannis, P. Karayiannis, M. J. McGarvey, A. Makris, and H. C. Thomas. 1989. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet ii:588–591. 13. Chan, H. L., N. W. Leung, A. Y. Hui, V. W. Wong, C. T. Liew, A. M. Chim, F. K. Chan, L. C. Hung, Y. T. Lee, J. S. Tam, C. W. Lam, and J. J. Sung. 2005. A randomized, controlled trial of combination therapy for chronic hepatitis B: comparing pegylated interferon-alpha2b and lamivudine with lamivudine alone. Ann. Intern. Med. 142:240–250. 14. Chan, H. L., and J. J. Sung. 2006. Hepatocellular carcinoma and hepatitis B virus. Semin. Liver Dis. 26:153–161. 15. Chan, H. L., S. W. Tsang, N. W. Leung, C. H. Tse, Y. Hui, J. S. Tam, F. K. Chan, and J. J. Sung. 2002. Occult HBV infection in cryptogenic liver 22. 23. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. cirrhosis in an area with high prevalence of HBV infection. Am. J. Gastroenterol. 97:1211–1215. Chang, M. L., R. N. Chien, C. T. Yeh, and Y. F. Liaw. 2005. Virus and transaminase levels determine the emergence of drug resistance during long-term lamivudine therapy in chronic hepatitis B. J. Hepatol. 43:72–77. Chang, T. T., R. G. Gish, R. de Man, A. Gadano, J. Sollano, Y. C. Chao, A. S. Lok, K. H. Han, Z. Goodman, J. Zhu, A. Cross, D. DeHertogh, R. Wilber, R. Colonno, and D. Apelian. 2006. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 354: 1001–1010. Chang, T. T., C. L. Lai, R. N. Chien, R. Guan, S. G. Lim, C. M. Lee, K. Y. Ng, G. J. Nicholls, J. C. Dent, and N. W. Leung. 2004. Four years of lamivudine treatment in Chinese patients with chronic hepatitis B. J. Gastroenterol. Hepatol. 19:1276–1282. Chaudhuri, V., R. Tayal, B. Nayak, S. K. Acharya, and S. K. Panda. 2004. Occult hepatitis B virus infection in chronic liver disease: full-length genome and analysis of mutant surface promoter. Gastroenterology 127: 1356–1371. Chemin, I., and C. Trepo. 2005. Clinical impact of occult HBV infections. J. Clin. Virol. 34(Suppl. 1):S15–S21. Chen, M. T., J. N. Billaud, M. Sallberg, L. G. Guidotti, F. V. Chisari, J. Jones, J. Hughes, and D. R. Milich. 2004. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 101:14913–14918. Chien, R. N., C. T. Yeh, S. L. Tsai, C. M. Chu, and Y. F. Liaw. 2003. Determinants for sustained HBeAg response to lamivudine therapy. Hepatology 38:1267–1273. Chisari, F. V. 2000. Rous-Whipple Award lecture. Viruses, immunity, and cancer: lessons from hepatitis B. Am. J. Pathol. 156:1117–1132. Chu, C. J., M. Hussain, and A. S. Lok. 2002. Quantitative serum HBV DNA levels during different stages of chronic hepatitis B infection. Hepatology 36:1408–1415. Chu, C. J., E. B. Keeffe, S. H. Han, R. P. Perrillo, A. D. Min, C. SoldevilaPico, W. Carey, R. S. Brown, Jr., V. A. Luketic, N. Terrault, and A. S. Lok. 2003. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 125:444–451. Chu, C. M. 2000. Natural history of chronic hepatitis B virus infection in adults with emphasis on the occurrence of cirrhosis and hepatocellular carcinoma. J. Gastroenterol. Hepatol. 15(Suppl.):E25–E30. Chu, C. M., S. J. Hung, J. Lin, D. I. Tai, and Y. F. Liaw. 2004. Natural history of hepatitis B e antigen to antibody seroconversion in patients with normal serum aminotransferase levels. Am. J. Med. 116:829–834. Colonno, R. J., R. Rose, C. J. Baldick, S. Levine, K. Pokornowski, C. F. Yu, A. Walsh, J. Fang, M. Hsu, C. Mazzucco, B. Eggers, S. Zhang, M. Plym, K. Klesczewski, and D. J. Tenney. 2006. Entecavir resistance is rare in nucleoside naive patients with hepatitis B. Hepatology 44:1656–1665. Conjeevaram, H. S., and A. S. Lok. 2001. Occult hepatitis B virus infection: a hidden menace? Hepatology 34:204–206. Dore, G. J., D. A. Cooper, A. L. Pozniak, E. DeJesus, L. Zhong, M. D. Miller, B. Lu, and A. K. Cheng. 2004. Efficacy of tenofovir disoproxil fumarate in antiretroviral therapy-naive and -experienced patients coinfected with HIV-1 and hepatitis B virus. J. Infect. Dis. 189:1185–1192. Erhardt, A., D. Blondin, K. Hauck, A. Sagir, T. Kohnle, T. Heintges, and D. Haussinger. 2005. Response to interferon alfa is hepatitis B virus genotype dependent: genotype A is more sensitive to interferon than genotype D. Gut 54:1009–1013. European Association for the Study of the Liver. 2003. Proceedings of the European Association for the Study of the Liver (EASL) International Consensus Conference on Hepatitis B. September 14–16, 2002. Geneva, Switzerland. J. Hepatol. 39(Suppl. 1):S1–S235. Flink, H. J., M. van Zonneveld, B. E. Hansen, R. A. de Man, S. W. Schalm, and H. L. Janssen. 2006. Treatment with Peg-interferon alpha-2b for HBeAg-positive chronic hepatitis B: HBsAg loss is associated with HBV genotype. Am. J. Gastroenterol. 101:297–303. Fung, S. K., P. Andreone, S. H. Han, K. Rajender Reddy, A. Regev, E. B. Keeffe, M. Hussain, C. Cursaro, P. Richtmyer, J. A. Marrero, and A. S. Lok. 2005. Adefovir-resistant hepatitis B can be associated with viral rebound and hepatic decompensation. J. Hepatol. 43:937–943. Fung, S. K., H. B. Chae, R. J. Fontana, H. Conjeevaram, J. Marrero, K. Oberhelman, M. Hussain, and A. S. Lok. 2006. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J. Hepatol. 44:283–290. Fung, S. K., and A. S. Lok. 2004. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 40:790–792. Fung, S. K., F. Wong, M. Hussain, and A. S. Lok. 2004. Sustained response after a 2-year course of lamivudine treatment of hepatitis B e antigennegative chronic hepatitis B. J. Viral Hepat. 11:432–438. Funk, M. L., D. M. Rosenberg, and A. S. Lok. 2002. World-wide epidemiology of HBeAg-negative chronic hepatitis B and associated precore and core promoter variants. J. Viral Hepat. 9:52–61. Germer, J. J., M. O. Qutub, J. N. Mandrekar, P. S. Mitchell, and J. D. Yao. 2006. Quantification of hepatitis B virus (HBV) DNA with a TaqMan HBV VOL. 20, 2007 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. analyte-specific reagent following sample processing with the MagNA Pure LC instrument. J. Clin. Microbiol. 44:1490–1494. Ghisetti, V., A. Marzano, F. Zamboni, A. Barbui, A. Franchello, S. Gaia, G. Marchiaro, M. Salizzoni, and M. Rizzetto. 2004. Occult hepatitis B virus infection in HBsAg negative patients undergoing liver transplantation: clinical significance. Liver Transpl. 10:356–362. Gintowt, A. A., J. J. Germer, P. S. Mitchell, and J. D. Yao. 2005. Evaluation of the MagNA Pure LC used with the TRUGENE HBV genotyping kit. J. Clin. Virol. 34:155–157. Gish, R. G., H. Trinh, N. Leung, F. K. Chan, M. W. Fried, T. L. Wright, C. Wang, J. Anderson, E. Mondou, A. Snow, J. Sorbel, F. Rousseau, and L. Corey. 2005. Safety and antiviral activity of emtricitabine (FTC) for the treatment of chronic hepatitis B infection: a two-year study. J. Hepatol. 43:60–66. Gordillo, R. M., J. Gutierrez, and M. Casal. 2005. Evaluation of the COBAS TaqMan 48 real-time PCR system for quantitation of hepatitis B virus DNA. J. Clin. Microbiol. 43:3504–3507. Gregorek, H., K. Dzierzanowska-Fangrat, M. Woynarowski, P. Jozwiak, E. Witkowska-Vogtt, J. Socha, M. Syczewska, and K. Madalinski. 2005. Persistence of HBV-DNA in children with chronic hepatitis B who seroconverted to anti-HBs antibodies after interferon-alpha therapy: correlation with specific IgG subclass responses to HBsAg. J. Hepatol. 42:486–490. Guidotti, L. G., B. Matzke, C. Pasquinelli, J. M. Shoenberger, C. E. Rogler, and F. V. Chisari. 1996. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. J. Virol. 70:7056–7061. Hadziyannis, S. J., and G. V. Papatheodoridis. 2006. Hepatitis B e antigennegative chronic hepatitis B: natural history and treatment. Semin. Liver Dis. 26:130–141. Hadziyannis, S. J., N. C. Tassopoulos, E. J. Heathcote, T. T. Chang, G. Kitis, M. Rizzetto, P. Marcellin, S. G. Lim, Z. Goodman, J. Ma, S. Arterburn, S. Xiong, G. Currie, and C. L. Brosgart. 2005. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 352:2673–2681. Hadziyannis, S. J., N. C. Tassopoulos, E. J. Heathcote, T. T. Chang, G. Kitis, M. Rizzetto, P. Marcellin, S. G. Lim, Z. Goodman, J. Ma, C. L. Brosgart, K. Borroto-Esoda, S. Arterburn, and S. L. Chuck. 2006. Longterm therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology 131:1743–1751. Heermann, K. H., W. H. Gerlich, M. Chudy, S. Schaefer, R. Thomssen, and the Eurohep Pathobiology Group. 1999. Quantitative detection of hepatitis B virus DNA in two international reference plasma preparations. J. Clin. Microbiol. 37:68–73. Hennig, H., I. Puchta, J. Luhm, P. Schlenke, S. Goerg, and H. Kirchner. 2002. Frequency and load of hepatitis B virus DNA in first-time blood donors with antibodies to hepatitis B core antigen. Blood 100:2637–2641. Hochberger, S., D. Althof, R. Gallegos de Schrott, N. Nachbaur, H. Rock, and H. Leying. 2006. Fully automated quantitation of hepatitis B virus (HBV) DNA in human plasma by the COBAS AmpliPrep/COBAS TaqMan system. J. Clin. Virol. 35:373–380. Hom, X., N. R. Little, S. D. Gardner, and M. M. Jonas. 2004. Predictors of virologic response to lamivudine treatment in children with chronic hepatitis B infection. Pediatr. Infect. Dis. J. 23:441–445. Horvat, R. T., and G. E. Tegtmeier. 2007. Hepatitis B and D viruses, p. 1641–1659. In P. R. Murray, E. J. Baron, J. H. Jorgensen, M. L. Landry, and M. A. Pfaller (ed.), Manual of clinical microbiology, 9th ed., vol. 2. ASM Press, Washington, DC. Hou, J., Z. Wang, J. Cheng, Y. Lin, G. K. Lau, J. Sun, F. Zhou, J. Waters, P. Karayiannis, and K. Luo. 2001. Prevalence of naturally occurring surface gene variants of hepatitis B virus in nonimmunized surface antigen-negative Chinese carriers. Hepatology 34:1027–1034. Hsu, Y. S., R. N. Chien, C. T. Yeh, I. S. Sheen, H. Y. Chiou, C. M. Chu, and Y. F. Liaw. 2002. Long-term outcome after spontaneous HBeAg seroconversion in patients with chronic hepatitis B. Hepatology 35:1522–1527. Hui, C. K., S. Bowden, H. Y. Zhang, A. Wong, S. Lewin, F. Rousseau, H. Mommeja-Marin, N. P. Lee, J. M. Luk, S. Locarnini, N. Leung, N. V. Naoumov, and G. K. Lau. 2006. Comparison of real-time PCR assays for monitoring serum hepatitis B virus DNA levels during antiviral therapy. J. Clin. Microbiol. 44:2983–2987. Hui, C. K., E. Lau, H. Wu, A. Monto, M. Kim, J. M. Luk, G. K. Lau, and T. L. Wright. 2006. Fibrosis progression in chronic hepatitis C patients with occult hepatitis B co-infection. J. Clin. Virol. 35:185–192. Hussain, M., C. J. Chu, E. Sablon, and A. S. Lok. 2003. Rapid and sensitive assays for determination of hepatitis B virus (HBV) genotypes and detection of HBV precore and core promoter variants. J. Clin. Microbiol. 41: 3699–3705. Hussain, M., S. Fung, E. Libbrecht, E. Sablon, C. Cursaro, P. Andreone, and A. S. Lok. 2006. Sensitive line probe assay that simultaneously detects mutations conveying resistance to lamivudine and adefovir. J. Clin. Microbiol. 44:1094–1097. Janssen, H. L., M. van Zonneveld, H. Senturk, S. Zeuzem, U. S. Akarca, Y. Cakaloglu, C. Simon, T. M. So, G. Gerken, R. A. de Man, H. G. Niesters, P. Zondervan, B. Hansen, and S. W. Schalm. 2005. Pegylated interferon MOLECULAR TESTING FOR CHRONIC HEPATITIS B 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 437 alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 365:123–129. Jeantet, D., I. Chemin, B. Mandrand, A. Tran, F. Zoulim, P. Merle, C. Trepo, and A. Kay. 2004. Cloning and expression of surface antigens from occult chronic hepatitis B virus infections and their recognition by commercial detection assays. J. Med. Virol. 73:508–515. Kannangai, R., E. Molmenti, L. Arrazola, A. Klein, M. Choti, D. L. Thomas, and M. Torbenson. 2004. Occult hepatitis B viral DNA in liver carcinomas from a region with a low prevalence of chronic hepatitis B infection. J. Viral Hepat. 11:297–301. Kao, J. H., P. J. Chen, M. Y. Lai, and D. S. Chen. 2000. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 118:554–559. Kao, J. H., C. J. Liu, and D. S. Chen. 2002. Hepatitis B viral genotypes and lamivudine resistance. J. Hepatol. 36:303–304. Keeffe, E. B., D. T. Dieterich, S. H. Han, I. M. Jacobson, P. Martin, E. R. Schiff, H. Tobias, and T. L. Wright. 2006. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: an update. Clin. Gastroenterol. Hepatol. 4:936–962. Kessler, H. H., S. Preininger, E. Stelzl, E. Daghofer, B. I. Santner, E. Marth, H. Lackner, and R. E. Stauber. 2000. Identification of different states of hepatitis B virus infection with a quantitative PCR assay. Clin. Diagn. Lab Immunol. 7:298–300. Kleinman, S. H., and M. P. Busch. 2006. Assessing the impact of HBV NAT on window period reduction and residual risk. J. Clin. Virol. 36(Suppl. 1):S23–S29. Kleinman, S. H., M. C. Kuhns, D. S. Todd, S. A. Glynn, A. McNamara, A. DiMarco, and M. P. Busch. 2003. Frequency of HBV DNA detection in US blood donors testing positive for the presence of anti-HBc: implications for transfusion transmission and donor screening. Transfusion 43:696–704. Konnick, E. Q., M. Erali, E. R. Ashwood, and D. R. Hillyard. 2005. Evaluation of the COBAS Amplicor HBV Monitor assay and comparison with the Ultrasensitive HBV Hybrid Capture 2 assay for quantification of hepatitis B virus DNA. J. Clin. Microbiol. 43:596–603. Koziel, M. J., and S. Siddiqui. 2005. Hepatitis B virus and hepatitis delta virus. In G. L. Mandell, J. E. Bennett, and R. Dolin (ed.), Principles and practice of infectious diseases. Elsevier, Philadelphia, PA. Kuhns, M. C., and M. P. Busch. 2006. New strategies for blood donor screening for hepatitis B virus: nucleic acid testing versus immunoassay methods. Mol. Diagn. Ther. 10:77–91. Kuo, A., J. L. Dienstag, and R. T. Chung. 2004. Tenofovir disoproxil fumarate for the treatment of lamivudine-resistant hepatitis B. Clin. Gastroenterol. Hepatol. 2:266–272. Lacombe, K., J. Gozlan, P. Y. Boelle, L. Serfaty, F. Zoulim, A. J. Valleron, and P. M. Girard. 2005. Long-term hepatitis B virus dynamics in HIVhepatitis B virus-co-infected patients treated with tenofovir disoproxil fumarate. AIDS 19:907–915. Lai, C. L., J. Dienstag, E. Schiff, N. W. Leung, M. Atkins, C. Hunt, N. Brown, M. Woessner, R. Boehme, and L. Condreay. 2003. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin. Infect. Dis. 36:687–696. Lai, C. L., D. Shouval, A. S. Lok, T. T. Chang, H. Cheinquer, Z. Goodman, D. DeHertogh, R. Wilber, R. C. Zink, A. Cross, R. Colonno, and L. Fernandes. 2006. Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 354:1011–1020. Lamberts, C., M. Nassal, I. Velhagen, H. Zentgraf, and C. H. Schroder. 1993. Precore-mediated inhibition of hepatitis B virus progeny DNA synthesis. J. Virol. 67:3756–3762. Laperche, S., V. Thibault, F. Bouchardeau, S. Alain, S. Castelain, M. Gassin, M. Gueudin, P. Halfon, S. Larrat, F. Lunel, M. Martinot-Peignoux, B. Mercier, J. M. Pawlotsky, B. Pozzetto, A. M. Roque-Afonso, F. RoudotThoraval, K. Saune, and J. J. Lefrere. 2006. Expertise of laboratories in viral load quantification, genotyping, and precore mutant determination for hepatitis B virus in a multicenter study. J. Clin. Microbiol. 44:3600–3607. Laras, A., J. Koskinas, E. Dimou, A. Kostamena, and S. J. Hadziyannis. 2006. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 44:694– 702. Lau, G. K., T. Piratvisuth, K. X. Luo, P. Marcellin, S. Thongsawat, G. Cooksley, E. Gane, M. W. Fried, W. C. Chow, S. W. Paik, W. Y. Chang, T. Berg, R. Flisiak, P. McCloud, and N. Pluck. 2005. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 352:2682–2695. Leung, N. W., C. L. Lai, T. T. Chang, R. Guan, C. M. Lee, K. Y. Ng, S. G. Lim, P. C. Wu, J. C. Dent, S. Edmundson, L. D. Condreay, and R. N. Chien. 2001. Extended lamivudine treatment in patients with chronic hepatitis B enhances hepatitis B e antigen seroconversion rates: results after 3 years of therapy. Hepatology 33:1527–1532. Liaw, Y. F., N. Leung, R. Guan, G. K. Lau, I. Merican, G. McCaughan, E. Gane, J. H. Kao, and M. Omata. 2005. Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2005 update. Liver Int. 25: 472–489. 438 VALSAMAKIS 82. Liaw, Y. F., I. S. Sheen, T. J. Chen, C. M. Chu, and C. C. Pao. 1991. Incidence, determinants and significance of delayed clearance of serum HBsAg in chronic hepatitis B virus infection: a prospective study. Hepatology 13:627–631. 83. Lim, S. G., T. M. Ng, N. Kung, Z. Krastev, M. Volfova, P. Husa, S. S. Lee, S. Chan, M. L. Shiffman, M. K. Washington, A. Rigney, J. Anderson, E. Mondou, A. Snow, J. Sorbel, R. Guan, and F. Rousseau. 2006. A doubleblind placebo-controlled study of emtricitabine in chronic hepatitis B. Arch. Intern. Med. 166:49–56. 84. Lindh, M., and C. Hannoun. 2005. Dynamic range and reproducibility of hepatitis B virus (HBV) DNA detection and quantification by COBAS TaqMan HBV, a real-time semiautomated assay. J. Clin. Microbiol. 43: 4251–4254. 85. Liu, C. J., D. S. Chen, and P. J. Chen. 2006. Epidemiology of HBV infection in Asian blood donors: emphasis on occult HBV infection and the role of NAT. J. Clin. Virol. 36(Suppl. 1):S33–S44. 86. Liu, C. J., J. H. Kao, and D. S. Chen. 2003. Molecular assays for hepatitis B virus infection. Hepatology 38:1311. 87. Liu, C. J., J. H. Kao, and D. S. Chen. 2005. Therapeutic implications of hepatitis B virus genotypes. Liver Int. 25:1097–1107. 88. Liu, C. J., S. C. Lo, J. H. Kao, P. T. Tseng, M. Y. Lai, Y. H. Ni, S. H. Yeh, P. J. Chen, and D. S. Chen. 2006. Transmission of occult hepatitis B virus by transfusion to adult and pediatric recipients in Taiwan. J. Hepatol. 44:39–46. 89. Lok, A. S., E. J. Heathcote, and J. H. Hoofnagle. 2001. Management of hepatitis B: 2000—summary of a workshop. Gastroenterology 120:1828– 1853. 90. Lok, A. S., and B. J. McMahon. 2001. Chronic hepatitis B. Hepatology 34:1225–1241. 91. Lok, A. S., and B. J. McMahon. 2004. Chronic hepatitis B: update of recommendations. Hepatology 39:857–861. 92. Lok, A. S., F. Zoulim, S. Locarnini, A. Mangia, G. Niro, H. Decraemer, G. Maertens, F. Hulstaert, K. De Vreese, and E. Sablon. 2002. Monitoring drug resistance in chronic hepatitis B virus (HBV)-infected patients during lamivudine therapy: evaluation of performance of INNO-LiPA HBV DR assay. J. Clin. Microbiol. 40:3729–3734. 93. Manesis, E. K., G. V. Papatheodoridis, V. Sevastianos, E. Cholongitas, C. Papaioannou, and S. J. Hadziyannis. 2003. Significance of hepatitis B viremia levels determined by a quantitative polymerase chain reaction assay in patients with hepatitis B e antigen-negative chronic hepatitis B virus infection. Am. J. Gastroenterol. 98:2261–2267. 94. Marcellin, P., T. T. Chang, S. G. Lim, M. J. Tong, W. Sievert, M. L. Shiffman, L. Jeffers, Z. Goodman, M. S. Wulfsohn, S. Xiong, J. Fry, and C. L. Brosgart. 2003. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 348:808–816. 95. Marcellin, P., G. K. Lau, F. Bonino, P. Farci, S. Hadziyannis, R. Jin, Z. M. Lu, T. Piratvisuth, G. Germanidis, C. Yurdaydin, M. Diago, S. Gurel, M. Y. Lai, P. Button, and N. Pluck. 2004. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 351:1206–1217. 96. Martinot-Peignoux, M., N. Boyer, M. Colombat, R. Akremi, B. N. Pham, S. Ollivier, C. Castelnau, D. Valla, C. Degott, and P. Marcellin. 2002. Serum hepatitis B virus DNA levels and liver histology in inactive HBsAg carriers. J. Hepatol. 36:543–546. 97. Marusawa, H., S. Uemoto, M. Hijikata, Y. Ueda, K. Tanaka, K. Shimotohno, and T. Chiba. 2000. Latent hepatitis B virus infection in healthy individuals with antibodies to hepatitis B core antigen. Hepatology 31:488– 495. 98. Mason, A. L., L. Xu, L. Guo, M. Kuhns, and R. P. Perrillo. 1998. Molecular basis for persistent hepatitis B virus infection in the liver after clearance of serum hepatitis B surface antigen. Hepatology 27:1736–1742. 99. McMahon, B. J., W. L. Alward, D. B. Hall, W. L. Heyward, T. R. Bender, D. P. Francis, and J. E. Maynard. 1985. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J. Infect. Dis. 151:599–603. 100. McMahon, B. J., P. Holck, L. Bulkow, and M. Snowball. 2001. Serologic and clinical outcomes of 1536 Alaska natives chronically infected with hepatitis B virus. Ann. Intern. Med. 135:759–768. 101. Minuk, G. Y., D. F. Sun, R. Greenberg, M. Zhang, K. Hawkins, J. Uhanova, A. Gutkin, K. Bernstein, A. Giulivi, and C. Osiowy. 2004. Occult hepatitis B virus infection in a North American adult hemodialysis patient population. Hepatology 40:1072–1077. 102. Minuk, G. Y., D. F. Sun, J. Uhanova, M. Zhang, S. Caouette, L. E. Nicolle, A. Gutkin, K. Doucette, B. Martin, and A. Giulivi. 2005. Occult hepatitis B virus infection in a North American community-based population. J. Hepatol. 42:480–485. 103. Mommeja-Marin, H., E. Mondou, M. R. Blum, and F. Rousseau. 2003. Serum HBV DNA as a marker of efficacy during therapy for chronic HBV infection: analysis and review of the literature. Hepatology 37:1309–1319. 104. Momosaki, S., Y. Nakashima, M. Kojiro, and E. Tabor. 2005. HBsAgnegative hepatitis B virus infections in hepatitis C virus-associated hepatocellular carcinoma. J. Viral Hepat. 12:325–329. CLIN. MICROBIOL. REV. 105. Naumann, H., S. Schaefer, C. F. Yoshida, A. M. Gaspar, R. Repp, and W. H. Gerlich. 1993. Identification of a new hepatitis B virus (HBV) genotype from Brazil that expresses HBV surface antigen subtype adw4. J. Gen. Virol. 74:1627–1632. 106. Ni, Y. H., M. H. Chang, K. J. Wang, H. Y. Hsu, H. L. Chen, J. H. Kao, S. H. Yeh, Y. M. Jeng, K. S. Tsai, and D. S. Chen. 2004. Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology 127:1733–1738. 107. Niesters, H. G., M. Krajden, L. Cork, M. de Medina, M. Hill, E. Fries, and A. D. Osterhaus. 2000. A multicenter study evaluation of the Digene Hybrid Capture II signal amplification technique for detection of hepatitis B virus DNA in serum samples and testing of EUROHEP standards. J. Clin. Microbiol. 38:2150–2155. 108. Noborg, U., A. Gusdal, P. Horal, and M. Lindh. 2000. Levels of viraemia in subjects with serological markers of past or chronic hepatitis B virus infection. Scand. J. Infect. Dis. 32:249–252. 109. Norder, H., A. M. Courouce, and L. O. Magnius. 1994. Complete genomes, phylogenetic relatedness, and structural proteins of six strains of the hepatitis B virus, four of which represent two new genotypes. Virology 198: 489–503. 110. Okamoto, H., F. Tsuda, H. Sakugawa, R. I. Sastrosoewignjo, M. Imai, Y. Miyakawa, and M. Mayumi. 1988. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J. Gen. Virol. 69:2575–2583. 111. Olivero, A., A. Ciancio, M. L. Abate, S. Gaia, A. Smedile, and M. Rizzetto. 2006. Performance of sequence analysis, INNO-LiPA line probe assays and AFFIGENE assays in the detection of hepatitis B virus polymerase and precore/core promoter mutations. J. Viral Hepat. 13:355–362. 112. Orito, E., T. Ichida, H. Sakugawa, M. Sata, N. Horiike, K. Hino, K. Okita, T. Okanoue, S. Iino, E. Tanaka, K. Suzuki, H. Watanabe, S. Hige, and M. Mizokami. 2001. Geographic distribution of hepatitis B virus (HBV) genotype in patients with chronic HBV infection in Japan. Hepatology 34:590– 594. 113. Osiowy, C., and E. Giles. 2003. Evaluation of the INNO-LiPA HBV Genotyping assay for determination of hepatitis B virus genotype. J. Clin. Microbiol. 41:5473–5477. 114. Osiowy, C., J. P. Villeneuve, E. J. Heathcote, E. Giles, and J. Borlang. 2006. Detection of rtN236T and rtA181V/T mutations associated with resistance to adefovir dipivoxil in samples from patients with chronic hepatitis B virus infection by the INNO-LiPA HBV DR line probe assay (version 2). J. Clin. Microbiol. 44:1994–1997. 115. Papatheodoridis, G. V., E. Dimou, K. Dimakopoulos, S. Manolakopoulos, I. Rapti, G. Kitis, D. Tzourmakliotis, E. Manesis, and S. J. Hadziyannis. 2005. Outcome of hepatitis B e antigen-negative chronic hepatitis B on long-term nucleos(t)ide analog therapy starting with lamivudine. Hepatology 42:121–129. 116. Papatheodoridis, G. V., E. Dimou, A. Laras, V. Papadimitropoulos, and S. J. Hadziyannis. 2002. Course of virologic breakthroughs under long-term lamivudine in HBeAg-negative precore mutant HBV liver disease. Hepatology 36:219–226. 117. Pawlotsky, J. M. 2003. Hepatitis B virus (HBV) DNA assays (methods and practical use) and viral kinetics. J. Hepatol. 39(Suppl. 1):S31–S35. 118. Pawlotsky, J. M., A. Bastie, C. Hezode, I. Lonjon, F. Darthuy, J. Remire, and D. Dhumeaux. 2000. Routine detection and quantification of hepatitis B virus DNA in clinical laboratories: performance of three commercial assays. J. Virol. Methods 85:11–21. 119. Perrillo, R. P., C. L. Lai, Y. F. Liaw, J. L. Dienstag, E. R. Schiff, S. W. Schalm, E. J. Heathcote, N. A. Brown, M. Atkins, M. Woessner, and S. D. Gardner. 2002. Predictors of HBeAg loss after lamivudine treatment for chronic hepatitis B. Hepatology 36:186–194. 120. Perrillo, R. P., E. R. Schiff, G. L. Davis, H. C. Bodenheimer, Jr., K. Lindsay, J. Payne, J. L. Dienstag, C. O’Brien, C. Tamburro, I. M. Jacobson, and the Hepatitis Interventional Therapy Group. 1990. A randomized, controlled trial of interferon alfa-2b alone and after prednisone withdrawal for the treatment of chronic hepatitis B. N. Engl. J. Med. 323:295–301. 121. Perz, J. F., G. L. Armstrong, L. A. Farrington, Y. J. Hutin, and B. P. Bell. 2006. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 45:529–538. 122. Plentz, A., G. Koller, K. M. Weinberger, and W. Jilg. 2004. Precision and stability of hepatitis B virus DNA levels in chronic surface antigen carriers. J. Med. Virol. 73:522–528. 123. Qutub, M. O., J. J. Germer, S. P. Rebers, J. N. Mandrekar, M. G. Beld, and J. D. Yao. 2006. Simplified PCR protocols for INNO-LiPA HBV Genotyping and INNO-LiPA HBV PreCore assays. J. Clin. Virol. 37:218–221. 124. Ronsin, C., A. Pillet, C. Bali, and G. A. Denoyel. 2006. Evaluation of the COBAS AmpliPrep-total nucleic acid isolation-COBAS TaqMan hepatitis B virus (HBV) quantitative test and comparison to the VERSANT HBV DNA 3.0 assay. J. Clin. Microbiol. 44:1390–1399. 125. Roque-Afonso, A. M., M. P. Ferey, V. Mackiewicz, L. Fki, and E. Dussaix. 2003. Monitoring the emergence of hepatitis B virus polymerase gene variants during lamivudine therapy in human immunodeficiency virus coin- VOL. 20, 2007 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146. fected patients: performance of CLIP sequencing and line probe assay. Antivir. Ther. 8:627–634. Saldanha, J., W. Gerlich, N. Lelie, P. Dawson, K. Heermann, and A. Heath. 2001. An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang. 80:63–71. Scaglioni, P. P., M. Melegari, and J. R. Wands. 1997. Posttranscriptional regulation of hepatitis B virus replication by the precore protein. J. Virol. 71:345–353. Schaefer, S. 2005. Hepatitis B virus: significance of genotypes. J. Viral Hepat. 12:111–124. Seo, Y., S. Yoon, B. X. Truong, H. Kato, K. Hamano, M. Kato, Y. Yano, M. Katayama, T. Ninomiya, Y. Hayashi, and M. Kasuga. 2005. Serum hepatitis B virus DNA levels differentiating inactive carriers from patients with chronic hepatitis B. Eur. J. Gastroenterol. Hepatol. 17:753–757. Shaw, T., A. Bartholomeusz, and S. Locarnini. 2006. HBV drug resistance: mechanisms, detection and interpretation. J. Hepatol. 44:593–606. Shepard, C. W., E. P. Simard, L. Finelli, A. E. Fiore, and B. P. Bell. 2006. Hepatitis B virus infection: epidemiology and vaccination. Epidemiol. Rev. 28:112–125. Shyamala, V., P. Arcangel, J. Cottrell, D. Coit, A. Medina-Selby, C. McCoin, D. Madriaga, D. Chien, and B. Phelps. 2004. Assessment of the target-capture PCR hepatitis B virus (HBV) DNA quantitative assay and comparison with commercial HBV DNA quantitative assays. J. Clin. Microbiol. 42:5199–5204. Soriano, V., M. Puoti, M. Bonacini, G. Brook, A. Cargnel, J. Rockstroh, C. Thio, and Y. Benhamou. 2005. Care of patients with chronic hepatitis B and HIV co-infection: recommendations from an HIV-HBV international panel. AIDS 19:221–240. Squadrito, G., T. Pollicino, I. Cacciola, G. Caccamo, D. Villari, T. La Masa, T. Restuccia, E. Cucinotta, C. Scisca, D. Magazzu, and G. Raimondo. 2006. Occult hepatitis B virus infection is associated with the development of hepatocellular carcinoma in chronic hepatitis C patients. Cancer 106:1326– 1330. Stelzl, E., Z. Muller, E. Marth, and H. H. Kessler. 2004. Rapid quantification of hepatitis B virus DNA by automated sample preparation and real-time PCR. J. Clin. Microbiol. 42:2445–2449. Stuyver, L., S. De Gendt, C. Van Geyt, F. Zoulim, M. Fried, R. F. Schinazi, and R. Rossau. 2000. A new genotype of hepatitis B virus: complete genome and phylogenetic relatedness. J. Gen. Virol. 81:67–74. Sung, J. J., M. L. Wong, S. Bowden, C. T. Liew, A. Y. Hui, V. W. Wong, N. W. Leung, S. Locarnini, and H. L. Chan. 2005. Intrahepatic hepatitis B virus covalently closed circular DNA can be a predictor of sustained response to therapy. Gastroenterology 128:1890–1897. Toan, N. L., H. Song le, P. G. Kremsner, D. N. Duy, V. Q. Binh, B. Koeberlein, S. Kaiser, R. Kandolf, J. Torresi, and C. T. Bock. 2006. Impact of the hepatitis B virus genotype and genotype mixtures on the course of liver disease in Vietnam. Hepatology 43:1375–1384. Torbenson, M., and D. L. Thomas. 2002. Occult hepatitis B. Lancet Infect. Dis. 2:479–486. Tran, N., R. Berne, R. Chann, M. Gauthier, D. Martin, M. A. Armand, A. Ollivet, C. G. Teo, S. Ijaz, D. Flichman, M. Brunetto, K. P. Bielawski, C. Pichoud, F. Zoulim, and G. Vernet. 2006. European multicenter evaluation of high-density DNA probe arrays for detection of hepatitis B virus resistance mutations and identification of genotypes. J. Clin. Microbiol. 44: 2792–2800. van Bommel, F., B. Zollner, C. Sarrazin, U. Spengler, D. Huppe, B. Moller, H. H. Feucht, B. Wiedenmann, and T. Berg. 2006. Tenofovir for patients with lamivudine-resistant hepatitis B virus (HBV) infection and high HBV DNA level during adefovir therapy. Hepatology 44:318–325. van der Eijk, A. A., H. G. Niesters, B. E. Hansen, R. A. Heijtink, H. L. Janssen, S. W. Schalm, and R. A. de Man. 2006. Quantitative HBV DNA levels as an early predictor of nonresponse in chronic HBe-antigen positive hepatitis B patients treated with interferon-alpha. J. Viral Hepat. 13:96– 103. van Nunen, A. B., B. E. Hansen, D. J. Suh, H. F. Lohr, L. Chemello, H. Fontaine, J. Heathcote, B. C. Song, H. L. Janssen, R. A. de Man, and S. W. Schalm. 2003. Durability of HBeAg seroconversion following antiviral therapy for chronic hepatitis B: relation to type of therapy and pretreatment serum hepatitis B virus DNA and alanine aminotransferase. Gut 52:420– 424. Wai, C. T., C. J. Chu, M. Hussain, and A. S. Lok. 2002. HBV genotype B is associated with better response to interferon therapy in HBeAg(⫹) chronic hepatitis than genotype C. Hepatology 36:1425–1430. Weber, B., W. Melchior, R. Gehrke, H. W. Doerr, A. Berger, and H. Rabenau. 2001. Hepatitis B virus markers in anti-HBc only positive individuals. J. Med. Virol. 64:312–319. Weiss, J., H. Wu, B. Farrenkopf, T. Schultz, G. Song, S. Shah, and J. Siegel. 2004. Real time TaqMan PCR detection and quantitation of HBV genotypes A-G with the use of an internal quantitation standard. J. Clin. Virol. 30:86–93. MOLECULAR TESTING FOR CHRONIC HEPATITIS B 439 147. Werle-Lapostolle, B., S. Bowden, S. Locarnini, K. Wursthorn, J. Petersen, G. Lau, C. Trepo, P. Marcellin, Z. Goodman, W. E. T. Delaney, S. Xiong, C. L. Brosgart, S. S. Chen, C. S. Gibbs, and F. Zoulim. 2004. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 126:1750–1758. 148. Westland, C., W. T. Delaney, H. Yang, S. S. Chen, P. Marcellin, S. Hadziyannis, R. Gish, J. Fry, C. Brosgart, C. Gibbs, M. Miller, and S. Xiong. 2003. Hepatitis B virus genotypes and virologic response in 694 patients in phase III studies of adefovir dipivoxil. Gastroenterology 125: 107–116. 149. Westland, C. E., H. Yang, W. E. T. Delaney, C. S. Gibbs, M. D. Miller, M. Wulfsohn, J. Fry, C. L. Brosgart, and S. Xiong. 2003. Week 48 resistance surveillance in two phase 3 clinical studies of adefovir dipivoxil for chronic hepatitis B. Hepatology 38:96–103. 150. Wong, D. K., A. M. Cheung, K. O’Rourke, C. D. Naylor, A. S. Detsky, and J. Heathcote. 1993. Effect of alpha-interferon treatment in patients with hepatitis B e antigen-positive chronic hepatitis B. A meta-analysis. Ann. Intern. Med. 119:312–323. 151. Wong, D. K., M. F. Yuen, R. T. Poon, J. C. Yuen, J. Fung, and C. L. Lai. 2006. Quantification of hepatitis B virus covalently closed circular DNA in patients with hepatocellular carcinoma. J. Hepatol. 45:553–559. 152. Wong, D. K., M. F. Yuen, H. Yuan, S. S. Sum, C. K. Hui, J. Hall, and C. L. Lai. 2004. Quantitation of covalently closed circular hepatitis B virus DNA in chronic hepatitis B patients. Hepatology 40:727–737. 153. World Health Organization. 2000. Hepatitis B. (Fact sheet no. 204.) World Health Organization, Geneva, Switzerland. 154. Wursthorn, K., M. Lutgehetmann, M. Dandri, T. Volz, P. Buggisch, B. Zollner, T. Longerich, P. Schirmacher, F. Metzler, M. Zankel, C. Fischer, G. Currie, C. Brosgart, and J. Petersen. 2006. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology 44:675–684. 155. Yang, H., X. Qi, A. Sabogal, M. Miller, S. Xiong, and W. E. T. Delaney. 2005. Cross-resistance testing of next-generation nucleoside and nucleotide analogues against lamivudine-resistant HBV. Antivir. Ther. 10:625–633. 156. Yao, J. D., M. G. Beld, L. L. Oon, C. H. Sherlock, J. Germer, S. Menting, S. Y. Se Thoe, L. Merrick, R. Ziermann, J. Surtihadi, and H. J. Hnatyszyn. 2004. Multicenter evaluation of the VERSANT Hepatitis B Virus DNA 3.0 assay. J. Clin. Microbiol. 42:800–806. 157. Yeh, C. T., R. N. Chien, C. M. Chu, and Y. F. Liaw. 2000. Clearance of the original hepatitis B virus YMDD-motif mutants with emergence of distinct lamivudine-resistant mutants during prolonged lamivudine therapy. Hepatology 31:1318–1326. 158. Yim, H. J., and A. S. Lok. 2006. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 43:S173–S181. 159. Yu, M. W., S. H. Yeh, P. J. Chen, Y. F. Liaw, C. L. Lin, C. J. Liu, W. L. Shih, J. H. Kao, D. S. Chen, and C. J. Chen. 2005. Hepatitis B virus genotype and DNA level and hepatocellular carcinoma: a prospective study in men. J. Natl. Cancer Inst. 97:265–272. 160. Yuan, H. J., M. F. Yuen, D. Ka-Ho Wong, E. Sablon, and C. L. Lai. 2005. The relationship between HBV-DNA levels and cirrhosis-related complications in Chinese with chronic hepatitis B. J. Viral Hepat. 12:373–379. 161. Yuen, L. K., A. Ayres, M. Littlejohn, D. Colledge, A. Edgely, W. J. Maskill, S. A. Locarnini, and A. Bartholomeusz. 22 December 2006, posting date. SEQHEPB: a sequence analysis program and relational database system for chronic hepatitis B. Antiviral Res. doi:10.1016/j.antiviral.2006.11.014. 162. Yuen, M. F., D. K. Wong, E. Sablon, E. Tse, I. O. Ng, H. J. Yuan, C. W. Siu, T. J. Sander, E. J. Bourne, J. G. Hall, L. D. Condreay, and C. L. Lai. 2004. HBsAg seroclearance in chronic hepatitis B in the Chinese: virological, histological, and clinical aspects. Hepatology 39:1694–1701. 163. Yuen, M. F., D. K. Wong, E. Sablon, H. J. Yuan, S. M. Sum, C. K. Hui, A. O. Chan, B. C. Wang, and C. L. Lai. 2003. Hepatitis B virus genotypes B and C do not affect the antiviral response to lamivudine. Antivir. Ther. 8:531– 534. 164. Yuen, M. F., D. K. Wong, S. S. Sum, H. J. Yuan, J. C. Yuen, A. O. Chan, B. C. Wong, and C. L. Lai. 2005. Effect of lamivudine therapy on the serum covalently closed-circular (ccc) DNA of chronic hepatitis B infection. Am. J. Gastroenterol. 100:1099–1103. 165. Yuen, M. F., H. J. Yuan, C. K. Hui, D. K. Wong, W. M. Wong, A. O. Chan, B. C. Wong, and C. L. Lai. 2003. A large population study of spontaneous HBeAg seroconversion and acute exacerbation of chronic hepatitis B infection: implications for antiviral therapy. Gut 52:416–419. 166. Zacharakis, G. H., J. Koskinas, S. Kotsiou, M. Papoutselis, F. Tzara, N. Vafeiadis, A. J. Archimandritis, and K. Papoutselis. 2005. Natural history of chronic HBV infection: a cohort study with up to 12 years follow-up in North Greece (part of the Interreg I-II/EC-project). J. Med. Virol. 77:173– 179. 167. Zollner, B., J. Petersen, E. Puchhammer-Stockl, J. Kletzmayr, M. Sterneck, L. Fischer, M. Schroter, R. Laufs, and H. H. Feucht. 2004. Viral features of lamivudine resistant hepatitis B genotypes A and D. Hepatology 39:42–50. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 440–458 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00001-07 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Carbapenemases: the Versatile -Lactamases Anne Marie Queenan* and Karen Bush Johnson & Johnson Pharmaceutical Research & Development, L.L.C., Raritan, New Jersey 08869 INTRODUCTION .......................................................................................................................................................440 CLASSIFICATION SCHEMES ................................................................................................................................440 MOLECULAR CLASS A CARBAPENEMASES ....................................................................................................441 Chromosomally Encoded Enzymes: SME, NMC, and IMI...............................................................................441 Plasmid-Encoded Enzymes: KPC and GES ........................................................................................................444 CLASS B METALLO--LACTAMASES .................................................................................................................445 Recent Epidemiology ..............................................................................................................................................446 CLASS D SERINE-CARBAPENEMASES: THE OXA -LACTAMASES ..........................................................448 DETECTION OF CARBAPENEMASES .................................................................................................................450 MICs .........................................................................................................................................................................450 Microbiological Tests with Inhibitors ..................................................................................................................450 Biochemical and Molecular Tests ........................................................................................................................452 CARBAPENEMASE ORIGINS AND TRANSMISSION.......................................................................................452 CONCLUDING REMARKS ......................................................................................................................................452 REFERENCES ............................................................................................................................................................453 fication of plasmid-encoded IMP-1, a metallo--lactamase in Pseudomonas aeruginosa (228), ARI-1 (OXA-23), a class D carbapenemase in Acinetobacter baumannii (157, 191), and KPC-1, a class A carbapenemase in Klebsiella pneumoniae (245), has changed the patterns of carbapenemase dissemination. What was once considered to be a problem of clonal spread has now become a global problem of interspecies dispersion. Because of the proliferation of new members of established carbapenemase families, it is even more important to try to understand the properties of these enzymes, with all their strengths and limitations. Several excellent carbapenemase reviews have appeared recently, including detailed compilations of the kinetic characteristics of these enzymes (112, 150, 221, 225). Therefore, this review focuses on updated information on the epidemiological and biochemical characteristics of both metallo- and serine carbapenemases. INTRODUCTION Carbapenemases represent the most versatile family of -lactamases, with a breadth of spectrum unrivaled by other -lactam-hydrolyzing enzymes. Although known as “carbapenemases,” many of these enzymes recognize almost all hydrolyzable -lactams, and most are resilient against inhibition by all commercially viable -lactamase inhibitors (114, 150, 225). Some investigators have preferred the nomenclature “carbapenem-hydrolyzing enzymes” to the term “carbapenemases,” suggesting that carbapenems are but one segment of their substrate spectrum (182). However, the term carbapenemase has become entrenched in the -lactamase literature and is used throughout this review. Carbapenemases belong to two major molecular families, distinguished by the hydrolytic mechanism at the active site. The first carbapenemases described were from gram-positive bacilli. Unlike other -lactamases known at that time, these enzymes were inhibited by EDTA, thereby establishing them as metalloenzymes. Later work has shown that all metallocarbapenemases contain at least one zinc atom at the active site that serves to facilitate hydrolysis of a bicyclic -lactam ring (46). In the mid- to late 1980s, another set of carbapenem-hydrolyzing enzymes emerged among the Enterobacteriaceae (134), but EDTA did not inhibit their activity (183). Subsequent studies showed that these enzymes utilized serine at their active sites and were inactivated by the -lactamase inhibitors clavulanic acid and tazobactam (183, 243). Until the early 1990s, all carbapenemases were described as species-specific, chromosomally encoded -lactamases, each with a well-defined set of characteristics. However, the identi- CLASSIFICATION SCHEMES Classification of -lactamases can be defined according to two properties, functional and molecular. In the early work with -lactamases, before genes were routinely cloned and sequenced, a new -lactamase was analyzed biochemically by isolating the protein and determining its isoelectric point, followed by enzymatic studies to determine substrate hydrolysis and inhibition characteristics (185, 202). The relative rates of hydrolysis for a broad spectrum of -lactam substrates, and inhibitor profiles, allowed for the classification of the new -lactamase. This functional classification process evolved over many years into a widely accepted scheme currently dividing the known -lactamases into four major functional groups (groups 1 to 4), with multiple subgroups under group 2 that are differentiated according to group-specific substrate or inhibitor profiles (22). In this functional classification scheme, carbapenemases are found primarily in groups 2f and 3. Classification based on amino acid homology has resulted in * Corresponding author. Mailing address: Johnson & Johnson Pharmaceutical Research & Development, L.L.C., Raritan, NJ 08869. Phone: (908) 704-5515. Fax: (908) 707-3501. E-mail: aqueenan@prdus .jnj.com. 440 CARBAPENEMASES: THE VERSATILE -LACTAMASES VOL. 20, 2007 441 TABLE 1. Classification of class B metallo--lactamases: subclass designations of representative metallo--lactamases and consensus sequences based on structural similarities to identified Zn2⫹ ligandsa Ligand(s)b -Lactamase Substrate(s) Zn1 116 Subclass B1 (BcII, IMP-1, CcrA, VIM-2, SPM-1) Subclass B2c (CphA, Sfh-1) Subclass B3 (L1, FEZ-1, Gob-1, CAU-1) 118 Zn2 196 120/121 221 263 Broad spectrum His His His Asp/(Arg/Cys) Cys His Preferential hydrolysis of carbapenems Preferential hydrolysis of cephalosporins (Asn) His/Gln (His) His (His) His Asp/(Arg) Asp/Hisd Cys (Ser) His His a Data are from references 46, 50, and 51. Amino acids in parentheses do not appear to be active-site zinc ligands. Zn1 ligand for subclass B2 is inhibitory to enzymatic activity. The amino acids indicated have not been confirmed crystallographically to bind this Zn2⫹ atom. d His121 replaces Cys221 as the second zinc ligand. b c four major classes (5, 76, 84), which correlate well with the functional scheme but lack the detail concerning the enzymatic activity of the enzyme. Molecular classes A, C, and D include the -lactamases with serine at their active site, whereas molecular class B -lactamases are all metalloenzymes with an active-site zinc. Carbapenemases, -lactamases with catalytic efficiencies for carbapenem hydrolysis, resulting in elevated carbapenem MICs, include enzymes from classes A, B, and D. Functional classification schemes that included carbapenemases were first proposed by Bush in 1988 (21). A number of subclassification schemes for the metallo--lactamases have subsequently been proposed over the past 10 years. Rasmussen and Bush in 1997 suggested that the functional group 3 metalloenzymes (22) could be divided into three functional subgroups, based primarily on substrate specificities (182), whereas molecular subclasses have been proposed by Frere and colleagues for a number of years (46, 50, 51). In addition to these proposals, other modifications have been suggested for the classification of the metallo--lactamases (60). These enzymes are currently divided into three subclasses based on a combination of structural features, zinc affinities for the two binding sites, and hydrolysis characteristics. A consensus scheme is shown in Table 1. Subclasses B1 and B3, divided by amino acid homology, bind two zinc atoms for optimal hydrolysis, while enzymes in subclass B2 are inhibited when a second zinc is bound. Subclass B2 also differs in hydrolysis spectrum, as it preferentially hydrolyzes carbapenems, in contrast to the broad hydrolysis spectrum observed for B1 and B3 enzymes (46). MOLECULAR CLASS A CARBAPENEMASES Class A serine carbapenemases of functional group 2f have appeared sporadically in clinical isolates since their first discovery over 20 years ago (134). These -lactamases have been detected in Enterobacter cloacae, Serratia marcescens, and Klebsiella spp. as single isolates or in small outbreaks (134, 149, 243). Bacteria expressing these enzymes are characterized by reduced susceptibility to imipenem, but MICs can range from mildly elevated (e.g., imipenem MICs of ⱕ4 g/ml) to fully resistant. These -lactamases, therefore, may go unrecognized following routine susceptibility testing. Three major families of class A serine carbapenemases include the NMC/IMI, SME, and KPC enzymes. Their hydrolytic mechanism requires an active-site serine at position 70 in the Ambler numbering system for class A -lactamases (6). All have the ability to hydrolyze a broad variety of -lactams, including carbapenems, cephalosporins, penicillins, and aztreonam, and all are inhibited by clavulanate and tazobactam, placing them in the group 2f functional subgroup of -lactamases. A fourth member of this class, the GES -lactamases, was originally identified as an ESBL family, but over time variants were discovered that had low, but measurable, imipenem hydrolysis. This subgroup of GES enzymes is also classified as functional group 2f carbapenemases. Table 2 shows the geographical and chronological detection of the class A carbapenemases. Chromosomally Encoded Enzymes: SME, NMC, and IMI The antibiotic resistance profile of strains expressing the chromosomal group 2f -lactamases is distinctive: carbapenem resistance coupled with susceptibility to extendedspectrum cephalosporins. SME-1 (for “Serratia marcescens enzyme”) was first detected in England from two S. marcescens isolates that were collected in 1982 (146, 243). The SME-1 -lactamase, along with the nearly identical SME-2 and SME-3, has been found sporadically throughout the United States (49, 178, 179, 208). Infections caused by SMEproducing S. marcescens infections were found as single incidents or small clusters of up to 19 isolates. SME-producing S. marcescens isolates from different geographical locations are not identical, as defined by pulsed-field gel electrophoresis (PFGE), although there may be some degree of clonal spread when several isolates are collected from one location (178, 208). The IMI (for “imipenem-hydrolyzing -lactamase”) and NMC-A (for “not metalloenzyme carbapenemase”) enzymes have been detected in rare clinical isolates of E. cloacae in the United States, France, and Argentina (149, 175, 181, 183). NMC-A and IMI-1 have 97% amino acid identity and are related to SME-1, with approximately 70% amino acid identity (146, 183). They all contain the conserved active-site motifs S-X-X-K, S-D-N, and K-T-G of the class A -lactamases. In addition, these carbapenemases have conserved cysteine 442 QUEENAN AND BUSH CLIN. MICROBIOL. REV. TABLE 2. Emergence of class A carbapenemases -Lactamase Yr Location No. of isolates Organism Gene locationa pIb GenBank accession no. Reference(s) SME-1 1982 1985 1999 London, United Kingdom Minnesota Illinois S. marcescens S. marcescens S. marcescens 2 1 2 Chrom Chrom NR 9.7 9.5 8.5 U60295c 146, 199, 243 179, 199 49 SME-2 1992 1994–1999 California Massachusetts S. marcescens S. marcescens 5 19 Chrom Chrom 9.5 9.5 AF275256 179 179 SME-3 2003 Illinois S. marcescens 2 Chrom 9.5 AY584237 178 IMI-1 1984 California E. cloacae 2 Chrom 7.05 U50278 183 IMI-2 2001 Hangzhou, China E. cloacae 1 Plasmid 8.1 AY780889 249 NMC-A 1990 2000 2003 Paris, France Buenos Aires, Argentina Washington E. cloacae E. cloacae E. cloacae 1 1 1 Chrom NR NR 6.9 6.9 6.9 Z21956 AJ536087 143, 149 181 175 KPC-1 1996 North Carolina K. pneumoniae 1 Plasmid 6.7 AF297554 245 KPC-2 1998–1999 Maryland K. pneumoniae 4 Plasmid 6.9 AY034847 138 KPC-2 1998 Maryland 1 Plasmid 6.7 AF481906 137 1998 1997–2001 New York New York 2002–2003 2003–2004 2004 2004 2005 2005 2005 2005 2006 2006 New York New York New York Zhejiang, China Paris, France Medellin, Colombia New York Israel Medellin, Colombia Medellin, Colombia Plasmid NR NR Plasmid NR NR NR NR NR Plasmid Plasmid Plasmid NR Plasmid Plasmid/Chrom Plasmid 6.7 6.7 6.7 6.9 6.7 6.7 NR NR 6.7 6.7 6.8 7.0 NR 6.7 6.8 6.8 246 13 Massachusetts New York 1 18 1 4 1 1 29 95 59 1 1 2 3 4 3 1 AY210887 2001 2001–2003 Salmonella enterica serotype Cubana K. oxytoca K. pneumoniae K. oxytoca Enterobacter spp. E. cloacae E. aerogenes K. pneumoniae K. pneumoniae K. pneumoniae K. pneumoniae K. pneumoniae K. pneumoniae K. pneumoniae E. coli P. aeruginosa C. freundii KPC-3 2000–2001 2003 2004 New York New York New York K. pneumoniae E. cloacae K. pneumoniae 24 1 3 Plasmid NR NR 6.5 6.7 6.7 AF395881 AY522950 233 14 16 KPC-4 2004 Scotland Enterobacter spp. NR NR AY700571 None (unpublished) GES-2 2000 2000 2004 South Africa South Africa South Africa P. aeruginosa P. aeruginosa P. aeruginosa 1 8 51 Plasmid Plasmid NR 5.8 5.8 NR AF326355 174 173 230 GES-4 2002 Japan K. pneumoniae 1 Plasmid 6.9 AB11620 219, 220 GES-5 2004 2004 Athens, Greece Korea E. coli K. pneumoniae 1 6 Plasmid Plasmid 5.8 5.8 AY494717 218 86 GES-6 2004 Athens, Greece K. pneumoniae 1 Plasmid 6.9 AY494718 218 1 74 14 DQ897687 DQ523564 15 17 16 229 145 217 118 147 216 216 a Chrom, chromosome; NR, not reported. NR, not reported. Two sequences differing by a single amino acid, Z28968 and U60295, for the S. marcescens S6 carbapenemase are reported in the GenBank database. Structural analysis and further sequencing confirmed U60295 as the correct sequence for the SME-1 enzyme (179, 199). b c residues at positions 69 and 238 that form a disulfide bridge. The genes for these three -lactamases are all chromosomally located, with no evidence of mobile element association, a fact that may have contributed to their rarity. More re- cently, however, genes encoding IMI-2 -lactamases were found on plasmids in Enterobacter asburiae from United States rivers and on a plasmid from an E. cloacae isolate from China (9, 249). VOL. 20, 2007 Substrate Rel kcat (%) 185 28 92 4.4 90 956 NA 93 125 Km (M) 15 9.3 11 2.7 0.052 0.30 NA 0.053 5.7 kcat/Km 2,000 36 89 10 0.0068 3.4 NA 0.3 51 kcat (s⫺1) 100 1.8 4.5 0.5 0.00034 0.17 NA 0.015 2.6 Rel kcat (%) 1,070 64 170 26 270 190 NA 45 93 Km (M) 1.9 0.56 0.52 0.38 0.000025 0.018 NA 0.0067 0.55 kcat/Km 530 63 31 3.6 0.49 17 12 0.95 66 kcat (s⫺1) 100 12 5.8 0.68 0.092 3.2 2.3 0.18 12 Rel kcat (%) 510 30 90 13 230 100 540 140 420 Km (M) 1.0 2.1 0.34 0.28 0.0021 0.17 0.022 0.0068 0.16 kcat/Km 490 130 0.38 NA 2.5 17 NA 85 NDH kcat (s⫺1) 100 27 0.078 NA 0.51 3.4 NA 17 ND Rel kcat (%) 2,200 160 4.7 NA 1,500 700 NA 810 ND Km (M) 0.22 0.81 0.081 NA 0.0017 0.024 NA 0.11 ND kcat/Km TABLE 3. Steady-state kinetic parameters for representative class A carbapenemasesa kcat (s⫺1) 100 9.2 37 0.43 0.17 10 NA 0.18 25 GES-4 kcat/Km 2,820 260 1,040 12 4.7 286 NA 5.0 707 KPC-2 Km (M) 1.3 1.1 0.51 0.68 ND ND NA ND 0.42 IMI-1 Rel kcat (%) 770 17 202 13 ND ND NA ND 260 NMC-A kcat (s⫺1) 100 1.9 11 0.91 ⬍0.01 ⬍0.10 NA ⬍0.02 11 SME-1 980 19 104 8.9 ⬍0.07 ⬍0.98 NA ⬍0.15 108 References: SME-1, 179; NMC-A, 124; IMI-1, 183; KPC-2, 246; GES-4, 219. Abbreviations: Rel kcat, kcat relative to that of cephaloridine; ND, not determinable; NA, no data available; NDH, no detectable hydrolysis. Cephaloridine Benzylpenicillin Imipenem Meropenem Ceftazidime Cefotaxime Cefepime Cefoxitin Aztreonam a Biochemical characterization of purified SME, NMC, and IMI enzymes revealed a broad hydrolysis spectrum that includes the penicillins, early cephalosporins, aztreonam, and carbapenems (Tables 3 and 4). Imipenem hydrolysis was easily measurable, with kcat values of ⬎30 s⫺1. Meropenem had lower kcat and Km values than did imipenem. Cefoxitin and extended-spectrum cephalosporins were inefficiently hydrolyzed, when hydrolysis could be detected at all, and cefotaxime was hydrolyzed faster than ceftazidime (124, 179, 183). These chromosomal -lactamases can be induced in response to imipenem and cefoxitin. Sequencing of the regions upstream of NMC-A, IMI-1, and SME-1 revealed the presence of a divergently transcribed gene related to the LysR family of DNA-binding transcriptional regulatory genes (143, 146, 183). Deletions within the nmcR gene eliminated the inducibility of NMC-A and reduced carbapenem MICs, defining a role for this protein as a positive regulator of NMC-A expression (143). The ImiR protein is 97% identical to NmcR, but an analysis of its regulatory effects has not been published. SmeR is 69% identical to NmcR but has weaker activity as a positive regulator of the blaSME-1 gene. Two crystal structures of NMC-A, one native and the other complexed with a penicillanic acid inhibitor, and a structure for SME-1 all show that the overall structure and catalytic residues are similar to those of other class A -lactamases (139, 199, 201). For NMC-A, the structure of the enzyme did not change conformation when the inhibitor was bound. The disulfide bond between positions 69 and 238 is located near the active site, a feature common among the class A carbapenemases. This disulfide bond is necessary for hydrolytic activity, not just for imipenem hydrolysis, suggesting that it is required to stabilize the enzyme structurally (123, 199). The two most prominent distinctions between the class A TEM-1 structure and NMC-A are a 1-Å alteration of the position of Asn132, which enlarges the substrate-binding cavity, and a change in conformation of the S3 strand between residues 237 and 240, which increases access to the active site. However, in the crystal structure of SME-1, the position of Asn132 was almost identical to the Asn132 residue position of TEM-1 (199). The major difference in the SME-1 structure compared to TEM and NMC-A was crowding of Ser70 and Glu166 in the active-site cleft, leaving no room for a catalytic water molecule. It is possible that conformational rearrangements upon substrate binding may allow a catalytic water molecule into this position. The structure of SME-1 complexed with a -lactam molecule would test this hypothesis. In structure/function studies using site-directed mutagenesis, the ability of the SME-1 -lactamase to hydrolyze imipenem could not be attributed to a single amino acid residue (122). A substitution of alanine for serine at position 237 reduced imipenem kcat values from 100 s⫺1 to 20 s⫺1; hydrolysis of cephalothin was also reduced fivefold, but the benzylpenicillin kcat values remained unchanged (200). It seems likely that the important conserved residues of the group 2f carbapenemases together contribute to form an active site that can accommodate and hydrolyze the carbapenems. CARBAPENEMASES: THE VERSATILE -LACTAMASES 443 444 QUEENAN AND BUSH CLIN. MICROBIOL. REV. TABLE 4. Substrate and inhibition profiles of the carbapenemases Hydrolysis profilea Molecular class Functional group A 2f B1 D Inhibition profileb Early cephalosporins Extendedspectrum cephalosporins Aztreonam Carbapenems EDTA Clavulanic acid Reference(s) Penicillins NMC IMI SME KPC GES ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫾ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ ⫹ ⫹ ⫹ ⫹ ⫾ ⫺ ⫺ ⫺ ⫺ ⫺ ⫹ ⫹ ⫹ ⫹ ⫹ 124 183 179 4 174, 219 3 IMP VIM GIM SPM ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ ⫺ ⫺ ⫺ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ ⫺ ⫺ ⫺ 224 224 224 224 2d OXA ⫹ ⫹ ⫾ ⫺ ⫾ ⫺ ⫾ 225 Enzyme a Symbols: ⫹, strong hydrolysis (generally, kcat of ⬎2 s⫺1); ⫾, weak hydrolysis (generally, kcat of 0.5 to 2 s⫺1); ⫺, no measurable hydrolysis reported (generally, kcat of ⬍0.5 s⫺1). b Symbols: ⫹, reported inhibition; ⫾, variable inhibition among -lactamase family members; ⫺, no inhibition reported. Plasmid-Encoded Enzymes: KPC and GES Two characteristics separate the KPC (for “Klebsiella pneumoniae carbapenemase”) carbapenemases from the other functional group 2f enzymes. First, the KPC enzymes are found on transferable plasmids; second, their substrate hydrolysis spectrum includes the aminothiazoleoxime cephalosporins, such as cefotaxime. Although the KPC -lactamases are predominantly found in K. pneumoniae, there have been reports of these enzymes in Enterobacter spp. and in Salmonella spp. (14, 74, 137). The first member of the KPC family was discovered through the ICARE surveillance project in a K. pneumoniae clinical isolate from North Carolina in 1996 (245). This isolate was resistant to all -lactams tested, but carbapenem MICs decreased in the presence of clavulanic acid. Carbapenemase activity, first detected with a bioassay, was associated with a large plasmid that encoded the KPC-1 -lactamase. The discovery of KPC-1 was quickly followed by several reports of a single-amino-acid variant, KPC-2, along the east coast of the United States (137, 138, 246). KPC-2 was first identified in 2003 as the result of a point mutation in KPC-1 and appeared in four isolates with imipenem MICs of 2 to 8 g/ml from Baltimore, MD, from 1998 to 1999. The KPC-2producing gene resided on a transferable plasmid, and it was noted that while all isolates exhibited reduced susceptibility to imipenem, none were technically resistant according to approved CLSI (formerly NCCLS) breakpoints (138). KPC-2 was then described in another Maryland site on a plasmid in Salmonella enterica (137). Genetic regions around this KPC-2 gene contained three open reading frames with sequence homology to transposases. Reports of KPC-2 in the New York, NY, area began to appear in 2004, with KPC-expressing K. pneumoniae currently an alarming problem (13, 15, 17). This is especially disturbing because New York has had large outbreaks of ESBL-producing klebsiellae (135, 177) for which carbapenems were considered to be one of the few treatment options (156). When ribotyping was conducted on the KPC-2-producing strains, the majority of the isolates were clonal, even when the surveillance included multiple hospitals in the New York metropolitan area. Notably, several of these reports described inconsistencies in recognizing some of these strains as carbapenem resistant, because carbapenem MICs were less than the approved MIC breakpoints (13, 15, 16, 118). Concurrent with the increasing reports of KPC-2, a singleamino-acid variant of KPC-2, KPC-3, was reported from a 2000 to 2001 Klebsiella pneumoniae outbreak in New York (233). KPC-3 has also been detected in Enterobacter spp. (14), where MICs for imipenem were also not consistently in the resistant range. Kinetic analysis of the KPC-3 enzyme revealed a profile similar to those of KPC-1 and KPC-2, with a slight increase in the hydrolysis of ceftazidime (4). After the rapid expansion of the KPC class of carbapenemases along the east coast of the United States, worldwide reports began to appear. A report from France in 2005 documented KPC-2 in a K. pneumoniae strain from a patient who had been in New York for medical treatment (145). KPC carbapenemases have recently been detected in Scotland (KPC-4 [GenBank accession no. AY700571]), Colombia (217), Israel (147), and China (229). The first detection of KPC-2 on a plasmid in P. aeruginosa has been reported; this represents a disturbing development in the spread of these carbapenemases (216). KPC enzymes have the conserved active-site motifs S-XX-K, S-D-N, and K-T-G of the class A -lactamases and have the closest amino acid identity (⬃45%) to the SME carbapenemases. In addition, these -lactamases have the conserved C69 and C238 residues that form a disulfide bond described for the SME and NMC/IMI enzymes. The structure of the KPC-2 -lactamase, compared to the SME-1 and NMC-A carbapenemases and the TEM-1 and SHV-1 noncarbapenemases, reveals characteristics conserved among the carbapenemases. KPC-2, along with the other carbapenemases, had a decrease in the size of the water pocket and had the catalytic serine in a more shallow position of the active-site cleft (92). The combination of subtle active-site adjustments in the class A car- VOL. 20, 2007 bapenemases is proposed to allow carbapenems access to the catalytic site, resulting in the altered specificity of these enzymes. KPC carbapenemases hydrolyze -lactams of all classes, with the most efficient hydrolysis observed for nitrocefin, cephalothin, cephaloridine, benzylpenicillin, ampicillin, and piperacillin (Tables 3 and 4). Imipenem and meropenem, as well as cefotaxime and aztreonam, were hydrolyzed 10-fold-less efficiently than the penicillins and early cephalosporins. Weak but measurable hydrolysis was observed for cefoxitin and ceftazidime, giving the KPC family a broad hydrolysis spectrum that includes most -lactam antibiotics. Of the functional group 2f carbapenemases, the KPC family has the greatest potential for spread due to its location on plasmids, especially since it is most frequently found in K. pneumoniae, an organism notorious for its ability to accumulate and transfer resistance determinants. In addition, the clonal spread seen in several epidemics points to difficulties with infection control for this organism (13, 15–17, 233). Most worrisome, treatment of infections caused by these organisms is extremely difficult because of their multidrug resistance, which results in high mortality rates (15). The GES/IBC family of -lactamases is an infrequently encountered family that was first described in 2000 with reports of IBC-1 (for “integron-borne cephalosporinase”) from an E. cloacae isolate in Greece (53) and GES-1 (for “Guiana extended spectrum”) in a K. pneumoniae isolate from French Guiana (168). These enzymes differ by only two amino acid substitutions and possess the class A active site-motifs with the cysteine residues at Ambler positions 69 and 238 that have been found in the KPC, SME, and NMC/IMI families. Their amino acid sequences show them to be distantly related to these carbapenemases, with identities of 36% to KPC-2, 35% to SME-1, and 31% to NMC-A (174). The genes encoding the GES family of enzymes were located in integrons on plasmids. Because the enzymes had a broad hydrolysis spectrum that included penicillins and extended-spectrum cephalosporins, they were initially classified as extended-spectrum -lactamases (53). Their hydrolysis spectrum was expanded in 2001 to include imipenem, with the report of GES-2 in a clinical isolate of P. aeruginosa (174). GES-2, from a multidrug-resistant P. aeruginosa isolate from South Africa, had a single amino acid substitution of glycine to asparagine at position 170. Imipenem hydrolysis by GES enzymes was slow, with hydrolytic rates of ⱕ0.004 s⫺1and 0.004 s⫺1 for GES-1 and GES-2, respectively. However, the hydrolytic efficiency for imipenem was 100-fold higher for GES-2, due to a 100-fold decrease in the Km value. The GES-4 -lactamase differed from GES-2 by three amino acids, one of which was a serine at position 170. Carbapenem kcat values for purified GES-4 were higher than those for GES-2, with imipenem hydrolyzed at a rate of 0.38 s⫺1 (219). Nomenclature of the GES/IBC family has undergone several revisions (105). A consensus nomenclature has been reached whereby the IBC names have been converted to the GES nomenclature (83, 105). At least nine GES variants have been described, with GES-9 recently identified in a P. aeruginosa isolate from France (164). Of these closely related enzymes, GES-2, GES-4, GES-5, and GES-6 have substitutions of as- CARBAPENEMASES: THE VERSATILE -LACTAMASES 445 paragine or serine at position 170, associated with imipenem hydrolysis (105, 218). Although rare, GES enzymes have been identified worldwide, with reports from Greece, France, Portugal, South Africa, French Guiana, Brazil, Argentina, Korea, and Japan (25, 33, 42, 86, 90, 155, 164, 168, 173, 186, 218–220). These enzymes have been most frequently associated with single occurrences. However, P. aeruginosa strains expressing GES-2 have caused a small nosocomial outbreak in eight patients (173), and six patients in Korea had infections caused by GES-5-producing K. pneumoniae (86). CLASS B METALLO--LACTAMASES The metallo--lactamases have been thoroughly reviewed recently (221, 224), and so this section serves as a summary and epidemiological update. This class of -lactamases is characterized by the ability to hydrolyze carbapenems and by its resistance to the commercially available -lactamase inhibitors but susceptibility to inhibition by metal ion chelators. The substrate spectrum is quite broad; in addition to the carbapenems, most of these enzymes hydrolyze cephalosporins and penicillins but lack the ability to hydrolyze aztreonam. The mechanism of hydrolysis is dependent on interaction of the -lactams with zinc ions in the active site of the enzyme, resulting in the distinctive trait of their inhibition by EDTA, a chelator of Zn2⫹ and other divalent cations. The first metallo--lactamases detected and studied were chromosomal enzymes present in environmental and opportunistic pathogenic bacteria such as Bacillus cereus (98, 109), Aeromonas spp. (78), and Stenotrophomonas maltophilia (188). These chromosomal enzymes were usually found in bacteria that also expressed at least one serine -lactamase, with both -lactamases inducible after exposure to -lactams. Fortunately, with the exception of S. maltophilia, these bacteria have not been frequently associated with serious nosocomial infections, as they are generally opportunistic pathogens, and the chromosomal metallo--lactamase genes are not easily transferred. Early classification based on functional analyses of purified proteins indicated that these -lactamases were distinctive from the groups of enzymes that had a serine-based hydrolytic mechanism (21). Major distinctive properties included the requirement of Zn2⫹ for the efficient hydrolysis of -lactams and a lack of inhibition by clavulanic acid and tazobactam. A defining aspect of their substrate spectrum was the ability to hydrolyze carbapenems (Table 4). Interestingly, not all of the metallo--lactamases readily hydrolyzed nitrocefin, the popular colorimetric indicator of -lactamase activity (130). The “hidden” carbapenemases of Aeromonas spp. hydrolyzed nitrocefin and other cephalosporins imperceptibly and could be detected only by using a carbapenem bioassay or hydrolysis test (64, 65, 196, 223, 241). Recently, the VIM-2 enzyme was shown to share this property and was detected on IEF gels by an imipenem hydrolysis detection method (171). Since the initial identification of the metallo--lactamases, a considerable amount of sequencing work has demonstrated high variability in primary sequences and in molecular structures. The first metallo--lactamases for which an amino acid sequence was determined was the BCII metallo--lactamase 446 QUEENAN AND BUSH from Bacillus cereus (77), the prototypical metallo--lactamase for many years. Extensive molecular characterization, including biochemical and crystallization studies, has also been completed with the CcrA (CfiA) chromosomal metallo--lactamase that appears in a small percentage of Bacteroides fragilis strains (32, 242). Amino acid sequence identities are as low as 23% across the metallo--lactamases, including enzymes such as CcrA, CphA, and L1 (Table 1) (182, 224). However, all of the enzymes have conserved residues that bind zinc at two sites (see “Classification Schemes,” above, and Table 1) and exhibit a well-conserved active site, as demonstrated through sophisticated modeling analyses (127) and in the crystal structures published to date (31, 32, 34, 44, 52, 141, 214). In contrast to the chromosomal metallo--lactamases, whose presence is directly correlated with the prevalence of the producing species, there has been a dramatic increase in the detection and spread of the acquired or transferable families of these metalloenzymes. The most common metallo--lactamase families include the VIM, IMP, GIM, and SIM enzymes, which are located within a variety of integron structures, where they have been incorporated as gene cassettes. When these integrons become associated with plasmids or transposons, transfer between bacteria is readily facilitated. Transferable imipenem resistance was first detected in Japan, initially in a P. aeruginosa isolate, in 1990 (228), followed by a second report of a transferable carbapenemase in B. fragilis (11). The transferable B. fragilis enzyme was one of the first characterized metallo--lactamases, but this species has not caused widespread clinical outbreaks in Japan. In contrast, IMP-1 (for “active on imipenem”), located on a conjugative plasmid in the P. aeruginosa clinical isolate (228), was found on an integron in S. marcescens and other Enterobacteriaceae in Japan (7, 72, 80, 195). This enzyme hydrolyzed imipenem, penicillins, and extended-spectrum cephalosporins but not aztreonam. The hydrolytic activity was inhibited by EDTA and restored by the addition of Zn2⫹. The first member of the IMP family found in Europe was in an A. baumannii isolate from Italy, which produced a related enzyme, IMP-2, as the first cassette on a class 1 integron (184). Since that time, the IMP family has been found throughout the world, with the most recent spread to the United States and Australia (62, 159). The historical discovery of IMP-type -lactamases is well documented in the recent review by Walsh et al. (224). Currently, the IMP family members number up to 18 in the published literature, with 23 assigned IMP sequences listed on the -lactamase nomenclature website (http://www.lahey.org/Studies/ [updated 17 November 2006]). Another prevalent family of integron-associated metallo-lactamases is composed of the VIM enzymes. VIM-1 (for “Verona integron-encoded metallo--lactamase”) was first isolated in Verona, Italy, in 1997 (101), with the identification of VIM-2 in France in 1996 subsequently reported (171). Both of these enzymes were initially found in P. aeruginosa clinical isolates and resided in class 1 integrons. The VIM family currently consists of 14 members (http://www.lahey.org/Studies/ [updated 17 November 2006]), with occurrences mostly in P. aeruginosa within multiple-integron cassette structures. VIM-2 has the dubious distinction of being the most-reported metallo--lactamase worldwide (224). Identification of the SPM-1 metallo--lactamase defined a CLIN. MICROBIOL. REV. new family with 35.5% amino acid identity to IMP-1 (206). SPM-1 (for “Sao Paulo metallo--lactamase”) was first isolated in a P. aeruginosa strain in Sao Paolo, Brazil. Since the initial report, single clones of SPM-1-containing P. aeruginosa have caused multiple hospital outbreaks with high mortality in Brazil (126, 169, 251). Genetic analysis of regions around the SPM-1 gene revealed that it was not part of an integron but instead was associated with common regions that contain a new type of transposable structure with potential recombinase and promoter sequences (169, 206). GIM-1 (for “German imipenemase”) was isolated in Germany in 2002 (26). GIM had approximately 30% homology to VIM, 43% homology to IMPs, and 29% homology to SPM. GIM-1 has characteristics similar to those of the other acquired metallo--lactamases in that it was found in five clonal P. aeruginosa isolates within a class 1 integron on a plasmid. At this time, it has not been reported elsewhere in the world. The latest family of acquired metallo--lactamases to be described comes from Korea. The enzyme SIM-1 (for “Seoul imipenemase”) has the closest amino acid identity to the IMP family (64 to 69%). SIM-1 was discovered in a large-scale screen of 1,234 imipenem-resistant Pseudomonas sp. and Acinetobacter sp. isolates, of which 211 (17%) were positive for metallo--lactamases. In this screening study, mostly VIM (74%) and IMP (22%) alleles were identified; however, seven putative metallo--lactamase-producing A. baumannii isolates were negative by PCR testing. Investigation with PCR primers designed to amplify entire integrons revealed that they had the novel SIM-1 metallo--lactamase located within a class 1 integron (104). Two clonal groups, of four and three strains, carried the same integron, suggesting independent acquisition events. Since their initial discoveries, SPM, GIM, and SIM metallo-lactamases have not spread beyond their countries of origin. However, VIM and IMP continue to be detected worldwide, with an overall trend of these two metallo--lactamases moving beyond P. aeruginosa and into the Enterobacteriaceae (Table 5). Recent Epidemiology Overall, worldwide susceptibility to carbapenems is 98% among the Enterobacteriaceae, whereas imipenem susceptibility ranges from 60% to 83% for P. aeruginosa and A. baumannii (2004 to 2005 surveys) (93, 209, 210). The current epidemiology of metallo--lactamase production generally follows patterns of increasing occurrences that are country specific. Presumably this is due to multiple factors, including antibiotic usage, dosing regimens, and local hospital practices concerning isolation of patients with multiresistant pathogens. While outside the focus of this review, it should be noted that the loss of the OprD porin in P. aeruginosa, not the acquisition of carbapenemases, is the most common mechanism associated with imipenem resistance in this pathogen (113, 176). In a recent survey from Korea of 15,960 gram-negative clinical isolates (247), metallo--lactamases were found in 36 of 581 imipenem-resistant isolates. In P. aeruginosa, VIM-2-like enzymes were the majority of metallo--lactamases in this sample; however, two strains tested positive by PCR for IMP1-like enzymes. For the Acinetobacter species, 26.5% (136 of CARBAPENEMASES: THE VERSATILE -LACTAMASES VOL. 20, 2007 447 TABLE 5. Worldwide emergence of metallo--lactamases in the Enterobacteriaceae Country Isolation datea Publication date Australia Australia Australia 2002 2004 NA 2005 Brazil China Greece Greece Greece Greece Greece Greece France Italy 2003 NA 2001 2001 2002 2003 2003–2005 2004–2005 2003–2004 2002 2005 2001 2003 2004 2003 2005 2006 2005 2006 2004 Japan 1993 1995 Japan 1994–1995 1996 Japan Japan NA 1991–1996 1998 1998 Japan Japan 1996 1998–2000 2001 2003 Japan 2001–2002 2003 Portugal Singapore South Korea South Korea South Korea NA 1996 2000 2000 2003–2004 2005 1999 2003 2002 2006 Spain 2003 2005 Taiwan Taiwan 1998 1999–2000 2001 2002 Tunisia Turkey 2005 Before 2002 2006 2005 Organism Enzymeb K. pneumoniae E. coli K. pneumoniae E. cloacae C. amalonaticus K. pneumoniae C. youngae E. coli E. coli K. pneumoniae E. cloacae K. pneumoniae K. pneumoniae K. pneumoniae K. pneumoniae E. cloacae S. marcescens IMP-4 IMP-4 IMP-4 IMP-4 IMP-4 IMP-1 IMP-4 VIM-1 VIM-1 VIM-1 VIM-1 VIM-1 VIM-1 VIM-1 VIM-4 VIM-4 IMP-1-like 1 1 2 1 1 1 1 1 4 (clonal) 17 1 5 (clonal) 27 (clonal) 8 (clonal) 1 1 4 Plasmid Plasmid Plasmid Plasmid Plasmid Chromosome Plasmid Plasmid Plasmid Plasmid Chromosome Plasmid Plasmid Plasmid Plasmid Plasmid Multiple plasmids K. pneumoniae S. marcescens S. flexneri S. marcescens C. freundii S. marcescens S. marcescens C. freundii P. vulgaris S. marcescens K. pneumoniae E. coli E. cloacae C. freundii K. oxytoca P. rettgeri M. morganii E. aerogenes K. oxytoca K. pneumoniae E. cloacae S. marcescens K. pneumoniae E. cloacae S. marcescens K. pneumoniae E. coli K. pneumoniae E. cloacae C. freundii K. pneumoniae E. cloacae IMP-1-like IMP-1-like IMP-3 IMP-1-like IMP-1-like IMP-6 IMP-1 IMP-1 IMP-1 IMP-1-like IMP-1-like IMP-1-like IMP-1-like IMP-1-like IMP-1-like IMP-1-like IMP-1-like IMP-1-like VIM-2 IMP-1 VIM-2 VIM-2 VIM-2-like VIM-2-like VIM-2-like VIM-1 VIM-1 IMP-8 IMP-8 VIM-2 VIM-4 VIM-5 1 9 1 13 (some clonal) 1 1 5 1 1 47 23 17 5 3 2 2 1 1 4 (clonal) 1 1 1 2 1 1 1 1 1 36 (mostly clonal) 1 20 (clonal) 1 Not tested Not tested Plasmid Not tested Not tested Plasmid Not tested Not tested Not tested Not tested Not tested Not tested Not tested Not tested Not tested Not tested Not tested Not tested Plasmid Plasmid Chromosome Not tested Not tested Not tested Not tested Plasmid Plasmid Plasmid Plasmid Plasmid Plasmid Plasmid No. of isolates Location Plasmid type Not tested Not tested Conjugative Conjugative Conjugative Conjugative Conjugative Conjugative Conjugative Conjugative Conjugative Nonconjugative Conjugative Conjugative Conjugative and nonconjugative Not tested Not tested Conjugative Conjugative Nonconjugative Conjugative Conjugative Conjugative Conjugative Conjugative Conjugative Nonconjugative Integron type Reference(s) 1 1 1 1 1 1 ? 1 1 1 1 1 1 1 Not tested Not tested Not tested 172 172 43 43 43 110 63 136 193 54 48 117 79 91 120 120 80 3 3 1 Not Not 1 Not Not Not 1 1 1 1 1 1 1 1 1 1 Not 1 1 Not Not Not 1 1 1 Not Not 1 1 195 195 81, 152 72 72 244 235 8 8 197 197 197 197 197 197 197 197 197 30 95 87 250 250 250 250 207 207 238 237 237 97 47 tested tested tested tested tested tested tested tested tested tested tested a NA, not available. b IMP-1-like and VIM-2-like enzymes were detected by PCR or colony hybridization but were not sequenced. 513) of the imipenem-resistant strains carried metallo--lactamases: 64% were VIM-2-like, 29% were IMP-1-like, and 7% were SIM-like. Among the Enterobacteriaceae, resistance to imipenem was 2% for K. pneumoniae and ⬍1% for E. cloacae and S. marcescens, which tested positive by PCR for VIM-2. IMP metallo--lactamases continue to persist as the major metalloenzymes found in Japan, both at a local (148) and a country-wide (94) level. However, VIM-2-like genes have also been detected in P. aeruginosa. IMP-1 and IMP-2-like enzymes were found in Acinetobacter spp., as well as S. marcescens, P. rettgeri, C. freundii, E. cloacae, and M. morganii. Among almost 20,000 clinical gram-negative isolates from 13 clinical laboratories, the overall rate of metallo--lactamase detection was 0.5% in Japan. In these reports, there was as much as 2.6% metallo--lactamase detection in imipenem-resistant P. aeruginosa; S. marcescens had the highest rate of IMP gene detection in Japan, at 3%. In a SENTRY surveillance study conducted in Japan, 1.1% of the P. aeruginosa strains expressed metallo-- lactamases (88); among these, IMP alleles were the only metallo--lactamases detected. The first metallo--lactamase reported from China was IMP-4, which was found on a plasmid in C. youngae and reported in a 2001 publication (63), followed by a report of IMP-1 in a P. aeruginosa isolate (226). Recently, IMP-9 has emerged in this country, as reported in a surveillance study that included 11 hospitals (234). The IMP-9 genes were all on identical integrons, and only two strains that came from the same ward were related, based on random amplified polymorphic DNA typing. A very large plasmid (⬃450 kb) was shown to carry the IMP-9 gene by conjugation experiments, suggesting that the spread of this IMP-containing integron was through horizontal transfer. The VIM-2 metallo--lactamase was first reported in China only in 2006 (227). This single P. aeruginosa isolate occurred in the same hospital as the IMP-1 isolate (226). At this point, the route for emergence of these enzymes is unknown, but further 448 QUEENAN AND BUSH epidemiological analysis, sequencing of the integrons, and typing of the strains could answer questions about whether the strains imported or acquired metallo--lactamases independently. In Taiwan, IMP-1 was reported in two A. baumannii isolates that were unrelated by PFGE. In these isolates, the IMP-1 gene was on two different plasmids within class 1 integrons (111). VIM-2 and VIM-3 have also been detected within integrons in multidrug-resistant P. aeruginosa (236). In southern Europe, particularly in Italy, there has been almost continuous detection of VIM and IMP metallo--lactamases throughout many countries over the past decade. In Italy, a recent SENTRY study for the years 2001 and 2002 found that 25 of 383 (6.5%) P. aeruginosa isolates from three medical centers carried metallo--lactamases (88). The majority of metallo--lactamases identified were VIM-1, but IMP-13 was also detected. Some of the VIM-1-expressing isolates from Rome and Catania carried the integron described in the original VIM-1 from Verona; differences in ribotypes demonstrated the mobility of this integron. Several unique integron cassette arrangements were reported, including some strains possessing as many as four integrons, thus leaving open the possibility for more integron gene rearrangements (100). In another study reported by Pagani et al., IMP-13 was responsible for a large clonal outbreak of at least 86 metallo--lactamase-producing P. aeruginosa strains in southern Italy (154). The integron, which was the same as the IMP-13 found in the SENTRY study, was localized to the chromosome and not transferable to P. aeruginosa or Escherichia coli by conjugation. Another surveillance study of 506 P. aeruginosa isolates from a single center in Varese showed 82% IPM susceptibility, but with only four strains carrying metallo--lactamases, (VIM-1 and VIM-2), demonstrating the regional variation in resistance patterns due to these enzymes (121). More frequent reports of metallo--lactamases are now being published from other European countries, documenting further detections of acquired carbapenem resistance. P. aeruginosa isolates with integron-associated VIM alleles have been reported in Germany (66), Turkey (10), Croatia (190), Hungary (107, 108), and Poland (45). Metallo--lactamase outbreaks in South America are dominated by the SPM and VIM families. In a recent SENTRY study of 183 P. aeruginosa isolates, 44.8% were imipenem resistant; of these, 36 isolates were positive for metallo--lactamases. The majority of the isolates were positive for SPM-1like genes by PCR, (55.6%), followed by VIM-2-type (30.6%) and IMP-1-like genes in three isolates (8.3%) (88, 187). While SPM-1 has been confined to Brazil, other countries in South America that have reported metallo--lactamases are Argentina, Chile, and Venezuela (88), as well as Colombia, where both VIM-2 and VIM-8 were found in P. aeruginosa (36, 217). Emergence of metallo--lactamase-mediated carbapenem resistance has spread to the United States and Canada, with reports of both VIM and IMP metallo--lactamases in P. aeruginosa. VIM-7 was isolated in Texas in 2001 from a single strain that carried the integron on a conjugative plasmid (204). More recently, VIM-2 has also been detected in Texas (1). VIM-2 was also found in Chicago, IL, in an outbreak setting of clonal P. aeruginosa involving 17 patients admitted from 2002 to 2004 (115), where the metallo--lactamase was present on CLIN. MICROBIOL. REV. an integron. Metallo--lactamases have also emerged as outbreaks of VIM-2- and IMP-7-producing P. aeruginosa in Canada (55, 163) and in a single P. aeruginosa isolate from the southwestern United States that produced IMP-18 (62). Australia has also reported detection of metallo--lactamases. The first metalloenzyme that emerged on the Australian continent was IMP-4, reported in 2004 in a single P. aeruginosa clinical isolate (159) and in single K. pneumoniae and E. coli isolates (172), followed by an outbreak of IMP-4-producing gram-negative pathogens (158). Recently, an Aeromonas junii isolate from Australia was found to have two carbapenemases: a class D OXA-58 and an IMP-4 metallo--lactamase (160). Other areas of the world that have recently reported metallo-lactamases include the Middle East, where VIM-2 was the first reported metallo--lactamase. It was detected by PCR in an imipenem-resistant P. aeruginosa isolate from Saudi Arabia (58). Yong et al. recently reported the detection of metallo-lactamases from India by using an EDTA disk test (248), where 13 of 200 (7.5%) Pseudomonas sp. and Acinetobacter sp. isolates were positive for the production of metallo--lactamases. Although further analysis of these enzymes is not yet available, awareness and early detection may prevent uncontrolled outbreaks due to these pathogens (59). CLASS D SERINE-CARBAPENEMASES: THE OXA -LACTAMASES OXA (for “oxacillin-hydrolyzing”) -lactamases represented one of the most prevalent plasmid-encoded -lactamase families in the late 1970s and early 1980s (129, 132, 198). When the molecular class D OXA -lactamases were placed in a separate molecular class from the other serine -lactamases (76), they had been identified mainly in the Enterobacteriaceae and P. aeruginosa (23, 144) and were functionally described as penicillinases capable of hydrolyzing oxacillin and cloxacillin (21). They were in general poorly inhibited by clavulanic acid and EDTA and known to have a large amount of variability in amino acid sequences (22). OXA-11, the first extended-spectrum variant of OXA-10 (previously known as PSE-2), was described in 1993 (61). The extended-spectrum variants OXA11, OXA-15, OXA-18, and OXA-45 had hydrolysis rates for ceftazidime that varied from 1% to 1,150% relative to the hydrolysis rate of penicillin, but imipenem hydrolysis was not detected (38, 61, 162, 205). Currently there have been 102 unique OXA sequences identified (http://www.lahey.org /Studies/), of which 9 are extended spectrum -lactamases and at least 37 are considered to be carbapenemases (Table 6) (225). The first OXA -lactamase with carbapenemase activity was described by Paton et al. in 1993. The enzyme was purified from a multidrug-resistant A. baumannii strain that was isolated in 1985 from a patient in Edinburgh, Scotland (157). Biochemical characterization revealed a -lactamase with a pI value of 6.65 that was poorly inhibited by clavulanic acid and EDTA (Table 4). Imipenem hydrolysis could not be measured spectrophotometrically but was readily detected with a microbiological assay plate. The enzyme was designated ARI-1 (for “Acinetobacter resistant to imipenem”) and was later demonstrated to reside on a large plasmid (191). Sequencing of the ARI-1 enzyme revealed that it belonged to the OXA class D CARBAPENEMASES: THE VERSATILE -LACTAMASES VOL. 20, 2007 TABLE 6. Carbapenemase subgroups of the OXA family of -lactamases Cluster 1 Enzyme subfamily 2 OXA-23 (ARI-1) OXA-24 3 OXA-51 4 5 6 7 8 9 OXA-58 OXA-55 OXA-48 OXA-50 OXA-60 OXA-62 Additional OXA member(s) Reference OXA-27, OXA-49 225 OXA-25, OXA-26, OXA-40, OXA-72 OXA-64 to OXA-71, OXA-75 to OXA-78, OXA-83, OXA-84, OXA-86 to OXA-89, OXA-91, OXA-92, OXA-94, OXA-95 None OXA-SHE OXA-54, OXA-SAR2 OXA-50a to OXA-50d, PoxB OXA-60a to OXA-60d None 225 213, 225 225 225 225 225 225 192 family of -lactamases, and the enzyme was later renamed OXA-23 (41). OXA-23 represented a new subset of the OXA family, with the highest amino acid identity to OXA-5 and OXA-10 at 36%. A recent review of the OXA -lactamases includes many additional details of these enzymes (225). The vast majority of OXA carbapenemases have been discovered in the opportunistic gram-negative pathogen Acinetobacter baumannii. By 1998, carbapenem-hydrolyzing -lactamases had been identified in Acinetobacter species clinical isolates throughout the world (2). For example, OXA-23 has been identified in outbreaks of carbapenem-resistant Acinetobacter in Brazil, the United Kingdom, Korea, and Tahiti (37, 85, 142, 211). OXA-24 and OXA-40, which differ by two amino acids, were found in clonal Acinetobacter outbreaks in hospitals from Spain and Portugal (12, 40, 119). OXA-40 was also the first carbapenem-hydrolyzing oxacillinase reported in the United States (116). Acinetobacter strains with OXA-23 and OXA-58 carbapenemases caused multiple infections in military and civilian personnel serving in Iraq and Afghanistan from 2003 to 2005 (75). At the latest count, there are nine major subgroups of OXA carbapenemases, based on amino acid homologies, as summarized in Table 6 (18, 167, 225). Subgroups 1, 2, and 3 are based on the sequences of OXA-23, OXA-24, and OXA-51, respectively (213). OXA-51-like enzymes have been found in all A. baumannii strains tested and may be a natural component of the chromosome in a subpopulation of that species (69). OXA58, less that 50% identical to other members of the OXA family, stands alone in subgroup 4 and has been found in Acinetobacter spp. from France, Greece, Italy, Romania, Turkey, Argentina, and Kuwait (28, 68, 125, 170, 215). OXA-55 and OXA-SHE, both from Shewanella algae, form the fifth group (71). The OXA-48 enzyme forms the sixth subgroup, along with OXA-54 and additional oxacillinases found in the environmental bacteria Shewanella spp. (165–167). In contrast to the sharp increase in worldwide reports of OXA-expressing Acinetobacter strains, OXA-48 was discovered in a clinical K. pneumoniae isolate from Turkey (167). This OXA variant was plasmid encoded and had less than 50% amino acid identity to the other OXA members. This enzyme also had the highest reported imipenem kcat value, 2 s⫺1, which represents the 449 highest hydrolysis rate of all of the published kinetic parameters for the OXA enzymes. The OXA-50-like enzymes in P. aeruginosa form the seventh group and include a set of enzymes that have been referred to as the poxB enzymes. These PoxB oxacillinases have been reported to be commonly present on the chromosomes of many strains of P. aeruginosa (40 of 70 strains tested) (96) and are thought to be part of the natural component of -lactamases in that species, but they may not be expressed in all strains and do not cause carbapenem resistance (imipenem MIC, ⱕ1 g/ml) (56, 96). Species-specific OXA enzymes include the subgroup 8 OXA-60 family, considered to be a natural component of the genome of Ralstonia pickettii (57), and the subgroup 9 OXA-62, identified as a species-specific oxacillinase in Pandoraea pnomenusa (192). The OXA -lactamases display a wide variety of amino acid sequences. Among those with carbapenem-hydrolyzing activity, there is 40% to 70% amino acid identity between groups. Within a group the identity is greater than or equal to 92.5% (225). If OXA -lactamases without carbapenem-hydrolyzing activity are considered, amino acid identities can be as low as 18%, for example, when OXA-1 is compared with OXA-58 (170). The molecular structures of the OXA -lactamases have been analyzed with the DBL numbering system (35), a classification based on the similarity of motifs around the active sites of the class A enzymes (6). The catalytic serine residue lies in the S-T-F-K tetrad at positions 70 to 73 (102), where the serine and lysine are conserved in both class A and class D enzymes, as well as in penicillin-binding proteins. The Y-G-N motif at positions 144 to 146 and the K-T-G at DBL 216 to 218 are highly conserved among the serine-based -lactamases. The carbapenem-hydrolyzing OXA subgroups 1 and 2 share a substitution of F for Y in the Y-G-N motif, but this is not necessary for imipenem hydrolysis, as the subgroup 3 enzymes and OXA-58 retain the Y-G-N at this position. The first crystal structure of the class D carbapenemases to appear in the literature was that of OXA-24, which was compared to the noncarbapenemase OXA-10 (189). Two amino acids, Tyr-112 and Met-223, make access to the active site smaller and more hydrophobic, allowing favorable interactions with carbapenems. These residues are found in other subgroups of OXA carbapenemases, suggesting that the altered active-site access is an important contributing factor to the carbapenem-hydrolyzing activity among these enzymes. The catalytic mechanism of the OXA -lactamases shares features with other serine carbapenemases. Substrate and enzyme form a covalent acyl intermediate at the catalytic serine, which is subsequently deacylated to yield the inactivated antibiotic hydrolyzed at the C-N bond of the -lactam ring. In addition, CO2 may influence the kinetics of some OXA enzymes, due to carboxylation at lysine 70 (Lys 73 in the DBL scheme) in these class D enzymes. This carbamate activates the catalytic serine side chain so that the acyl intermediate can form (131, 153). To ensure that this modification occurs in studies with isolated enzymes, some researchers add 10 mM NaHCO3 to their reactions (57, 69, 192). OXA enzymes are difficult to purify due to low yield (3) and difficult to characterize biochemically due to low hydrolysis 450 QUEENAN AND BUSH rates and biphasic kinetics for some substrates. The OXA carbapenemases that have been characterized biochemically have measurable hydrolytic activity against the penicillins, some cephalosporins, and imipenem (225). In general, imipenem hydrolysis, which is faster than meropenem hydrolysis, is slow, with the highest kcat values for the group 6 OXA-54 and OXA-48 enzymes at 1 s⫺1 and 2 s⫺1, respectively. The Km values for imipenem are also low, ranging from 2 to 20 M, indicating that the OXA enzymes have very high affinity for these substrates. In contrast, extended-spectrum cephalosporins are not measurably hydrolyzed by the OXA carbapenemases or are hydrolyzed very poorly (225). The hydrolysis of carbapenems by the class D oxacillinase family is weak, but Heritier et al. (70) demonstrated that plasmid-encoded OXA-23 and OXA-58 enzymes contributed to carbapenem resistance in A. baumannii after transformation of the OXA plasmids into carbapenem-susceptible A. baumannii strains. In addition, when a chromosomally located OXA-40 gene was inactivated, susceptibility to carbapenems was observed. Efflux by an overexpressed AdeABC pump was also shown to contribute to carbapenem resistance. DETECTION OF CARBAPENEMASES MICs Detection of carbapenemase activity in a clinical isolate can be challenging for a clinical microbiology laboratory. The first cause for suspicion that a carbapenemase is involved in a clinical infection is an elevated carbapenem MIC. Among P. aeruginosa strains with VIM, IMP, GIM, SIM, and SPM metallo--lactamases, imipenem MICs have been reported in the range of 8 to ⬎128 g/ml (26, 101, 104, 126, 171, 204, 228). However, when the genes for these enzymes were transferred into E. coli, the observed imipenem MIC was usually much lower, sometimes as low as 0.5 g/ml. This demonstrates the presence of other mechanisms contributing to the carbapenem resistance in P. aeruginosa, such as intrinsic impermeability, possibly coupled with an efflux mechanism. This effect of lowlevel transferable resistance has also been observed in K. pneumoniae and A. baumannii with metallo--lactamases (39, 184), as well as with OXA carbapenemases (12, 170, 192). Many OXA carbapenemases have been found in A. baumannii, where MICs for imipenem are usually higher than 8 g/ml, but E. coli cells expressing these enzymes have imipenem MICs of ⱕ2 g/ml (225). Elevated carbapenem MICs are generally predictive of carbapenemase production in the Enterobacteriaceae, but full clinical resistance is not always seen. In a set of 19 K. pneumoniae isolates with imipenem MICs in the susceptible range of 1 to 4 g/ml, a metallo--lactamase was suggested on the basis of disk testing with imipenem in the presence and absence of EDTA and was then confirmed by PCR as VIM-1 (161). A collection of five related K. pneumoniae strains with the VIM-1 gene demonstrated imipenem MICs ranging from 2 to 64 g/ml (susceptible to high-level resistance) (117). Decreased permeability due to the absence of the outer membrane porin OmpK36 was a contributing factor in the most resistant isolate, with an imipenem MIC of 64 g/ml. The KPC serine carbapenemases also have been reported to CLIN. MICROBIOL. REV. be difficult to detect (14, 16, 138). They are often associated with imipenem MICs as low as 2 g/ml (138), and a low inoculum has resulted in susceptible MICs by broth microdilution (15). To document inconsistencies in the detection of KPC-producing K. pneumoniae according to the testing method, Tenover et al. tested 15 characterized imipenem- and meropenem-nonsusceptible KPC-producing isolates for imipenem and meropenem resistance using CLSI broth microdilution, Etest, MicroScan WalkAway, BD Phoenix Sensititre Autoreader, VITEK, and VITEK2. The automated systems reported carbapenem susceptibility in this collection of isolates ranging from 6.7% to 87%, depending on the system used (203). Day-to-day variation was also noted. In addition to the variable results with the automated systems, Etest results were inconsistent due to colonies present in the zones of inhibition. The failure of automated systems to consistently detect KPC-producing isolates indicates the need for improved methodology. One possibility is to screen isolates for resistance to ertapenem, which had the highest sensitivity for detecting KPC-expressing isolates (16). However, the specificity may be reduced due to resistance from other mechanisms, such as AmpC or ESBL production coupled with porin loss (N. Woodford, J. Dallow, R. Hill, M. F. Ralepou, R. Pike, M. Ward, M. Warner, and D. Livermore, presented at the 46th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, 27 to 30 September 2006). The NMC and IMI serine carbapenemases in E. cloacae are associated with imipenem MICs of 16 to 32 g/ml, with ceftazidime MICs often 2 g/ml (149, 183). In S. marcescens, the SME family of -lactamases should be suspected if an isolate displays this pattern of high-level imipenem resistance associated with ceftazidime susceptibility. Microbiological Tests with Inhibitors The disk approximation test with EDTA or 2-mercaptoproionic acid is often used as a screen for metallo--lactamase producers (8, 248). In this test, the zone of inhibition around a -lactam disk is altered by the action of the inhibitor on the metallo--lactamase in the test organism. Imipenem, ceftazidime, and cefepime have been used for this test. The sensitivity with the imipenem-EDTA disk method was 100% for Pseudomonas spp. and 95.7% for Acinetobacter spp. (248). In one study that compared different combinations of antibiotics and inhibitors, imipenem-EDTA combinations were the most sensitive for the detection of metallo--lactamase-producing Pseudomonas and A. baumannii, while ceftazidime-clavulanate with EDTA was the most accurate for K. pneumoniae and cefepime-clavulanate with EDTA was the most accurate for E. cloacae and C. freundii, with an overall sensitivity for this method of 86.7% (239). Etest strips for metallo--lactamase testing are also available as imipenem and imipenem-EDTA combinations (AB BIODISK, Solna, Sweden). A positive test for a metallo-lactamase is interpreted as a threefold-or-greater decrease in the imipenem MIC in the presence of EDTA. This test strip produced a sensitivity of 94% and a specificity of 95% when examined with a set of 138 characterized metallo--lactamase producers (222). However, false-negative results have been reported for the Etest when an isolate had an imipenem MIC CARBAPENEMASES: THE VERSATILE -LACTAMASES VOL. 20, 2007 451 TABLE 7. PCR primers for the detection of -lactamasesa Enzyme family Class A carbapenemases NMC SME IMI KPC GES Class D oxacillinases Subgroup 1 (OXA-23) Subgroup 2 (OXA-24) Subgroup 3 (OXA-69) Subgroup 4 (OXA-58) Subgroup 5 (Shewanella OXA-55) Subgroup 6 (OXA-48) Subgroup 7 (OXA-50) Subgroup 8 (OXA-60) Multiplex PCR for OXAs in A. baumannii Primer sequence (5⬘–3⬘) NMC1* NMC4 IRS-5 IRS-6 IMI-A IMI-B KPC forward KPC reverse GES-C GES-D GCATTGATATACCTTTAGCAGAGA CGGTGATAAAATCACACTGAGCATA AGATAGTAAATTTTATAG CTCTAACGCTAATAG ATAGCCATCCTTGTTTAGCTC TCTGCGATTACTTTATCCTC ATGTCACTGTATCGCCGTCT TTTTCAGAGCCTTACTGCCC GTTTTGCAATGTGCTCAACG TGCCATAGCAATAGGCGTAG Z21956 P5 P6 Forward Reverse OXA-69A AAGCATGATGAGCGCAAAG AAAAGGCCCATTTATCTCAAA GTACTAATCAAAGTTGTGAA TTCCCCTAACATGAATTTGT CTAATAATTGATCTACTCAAG AJ132105 OXA-69B Pre-OXA58prom⫹ PreOXA-58B OXA-55/1 CCAGTGGATGGATGGATAGATTATC TTATCAAAATCCAATCGGC OXA-55/2 OXA-48A OXA-48B S AS OXA-60 A OXA-60 B AGCTGTTCCTGCTTGAGCAC TTGGTGGCATCGATTATCGG GAGCACTTCTTTTGTGATGGC AATCCGGCGCTCATCCATC GGTCGGCGACTGAGGCGG AAAGGAGTTGTCTCATGCTGTCTCG AACCTACAGGCGCGCGTCTCAC GGTG OXA-51-like TAATGCTTTGATCGGCCTTG OXA-23-like OXA-24-like OXA-58-like Class B metalloenzymes IMP-1 IMP-2 VIM-1 VIM-2 SPM-1 GIM-1 SIM-1 Integron PCR GenBank accession no. Primerb Z28968 U50278 Fragment size (bp) Entire coding regionc Reference 68–191 2225–2201 5–22 1142–1128 1291–1310 2108–2089 131–150 1023–1004 176–195 527–546 2,158 Yes 181 1,138 Yes 179 No 9 785–803 1850–1830 Nucleotide positions (in GenBank) 818 893 Yes* 13 371 No 230 1,066 Yes 41 AY859527 1023 Primers external to GenBank sequence 975 Yes Yes 69 AY570763 72–90 934 Yes 68 Yes 71 744 No 167 869 Yes 56 AF297554 AF326355 3 TAACCTCAAACTTCTAATTC CATCTACCTTTAAAATTCCC AY343493 AY236073 1005–986 Primers external to GenBank sequence 969–917 2218–2237 2961–2941 AE004091 AF525303 2757–2782 3605–3579 353 TGGATTGCACTTCATCTTGG GATCGGATTGGAGAACCAGA ATTTCTGACCGCATTTCCAT GGTTAGTTGGCCCCCTTAAA AGTTGAGCGAAAAGGGGATT AAGTATTGGGGCTTGTGCTG CCCCTCTGCGCTCTACATAC Forward Reverse Forward Reverse Forward Reverse Forward Reverse SPM-1F SPM-1R GIM-1F GIM-1R SIM1-F SIM1-R TGAGCAAGTTATCTGTATTC TTAGTTGCTTGGTTTTGATG GGCAGTCGCCCTAAAACAAA TAGTTACTTGGCTGTGATGG TTATGGAGCAGCAACCGATGT CAAAAGTCCCGCTCCAACGA AAAGTTATGCCGCACTCACC TGCAACTTCATGTTATGCCG CCTACAATCTAACGGCGACC TCGCCGTGTCCAGGTATAAC AGAACCTTGACCGAACGCAG ACTCATGACTCCTCACGAGG TACAAGGGATTCGGCATCG TAATGGCCTGTTCCCATGTG 5⬘ CS 3⬘ CS GGCATCCAAGCAGCAAG AAGCAGACTTGACCTGA 57 Yes 232 501 246 599 AJ492820 AJ620678 AY887066 M73819 514–533 1163–1143 837–856 1584–1565 620–638 1190–1171 1190–1206 740 Yes* 240 737 Yes 240 920 Yes 240 865 Yes 240 650 No 26 748 No 26 571 No 104 Variable 106 Some primers are outside the coding regions and amplify the entire -lactamase gene, as indicated; others are internal fragments for diagnostic purposes. *, NMC primers also amplify NMC-R. Where primers are designated “forward” and “reverse,” they were not given names in the referenced study. “Yes” indicates that entire coding region is amplified; “No” indicates that only a part of the coding region is amplified. *, primers cover the extreme ends of the protein. a b c of ⬍4 g/ml (222, 239). It has also been observed that EDTA alone has inhibitory action against some bacteria due to permeabilization of the outer membrane and can lead to falsepositive results (27). Etest metallo--lactamase detection tests have also yielded false-positive results with OXA-23-producing A. baumannii (194). Carbapenem inactivation assays can be a fast, sensitive method for initial characterization of carbapenem-resistant isolates. The cloverleaf test is a microbiological assay of carbapenemase activity where suspensions of whole cells or and/or an extract of the suspect isolate are tested against imipenem on an agar plate (73). Altered growth of an indicator strain around an imipenem disk is a positive result. One advantage of this test is that enzymes that have very weak car- 452 QUEENAN AND BUSH bapenemase activity, such as OXA-23 (73) or GES-5 and GES-6 (218), can be detected by this method. Biochemical and Molecular Tests Isoelectric focusing (IEF) separates proteins by charge, and detection of -lactamases is accomplished with the chromogenic cephalosporin nitrocefin (130). Overlay of the gel with EDTA, clavulanic acid, or aztreonam can detect sensitivity of the enzymes to these potential inhibitors, indicating class B, A, or C -lactamases, respectively. Although IEF results cannot identify a specific -lactamase, information about isoelectric point and inhibition characteristics can be obtained by this method. IEF is especially valuable for the detection of multiple -lactamases present in an isolate. IEF can also be combined with a bioassay to detect the presence of carbapenemases by using an overlay of agar with imipenem and a second overlay with a susceptible indicator organism (103, 192, 241). Growth over an enzyme band indicates a potential carbapenemase. This procedure is especially useful when working with carbapenemases that have poor hydrolysis rates with nitrocefin, such as the metallo--lactamases from Aeromonas spp. (241). Imipenem hydrolysis can most reliably be detected with a spectrophotometric measurement using crude cell extracts or purified -lactamases (101). If the carbapenemase is a metalloenzyme, a brief incubation with EDTA prior to initiation of the reaction will result in a lower hydrolysis rate. Very weak carbapenemases cannot be detected by this method unless large amounts of extracts are used. When the presence of a carbapenemase is suspected, PCR is the fastest way to determine which family of -lactamase is present. Table 7 lists a selection of published primers that have been used with standard PCR technology to detect all of the families and subgroups of carbapenemases known at this time. Some laboratories have used colony blot hybridizations to efficiently screen large numbers of clinical isolates for carbapenemase genes (121, 240). Hybridization techniques are also used with a Southern blot to determine whether the carbapenemase gene resides on a plasmid or is chromosomal (116). Ultimately, the identification of the -lactamase gene requires sequencing of the entire coding region. Cloning of the region around the -lactamase is usually accomplished with a “shotgun” approach, but a clever degenerate PCR method has also been used successfully to amplify 5⬘ and 3⬘ areas surrounding OXA-51 (19). Characterization of a new -lactamase is not complete until both a molecular sequence is obtained and a functional analysis of the hydrolysis and inhibition profiles is performed with purified protein. CLIN. MICROBIOL. REV. capable of degrading these -lactams would be produced by environmental organisms such as Bacillus cereus and Bacillus anthracis, bacteria with well-characterized metallo--lactamases that would provide a selective advantage for growth of these environmental species (98, 128). These chromosomal carbapenemases may have evolved initially as a mechanism for bacteria to protect themselves from external threats to their cell wall (20), but in addition these -lactamases may also play a role in the regulation of cell wall synthesis (151). As described in this review and its many supporting references, the problem of carbapenemase-mediated resistance intensified once genes for these enzymes became associated with acquired genetic determinants. Transmission of carbapenemase genes may occur readily when the gene is located within mobile elements such as plasmids and integrons (82, 133). Investigators looking for prospective sources of carbapenemase genes have been able to find several in environmental species. The class A carbapenemase SFC-1 was described in an environmental isolate of Serratia fonticola (67). Several of the OXA carbapenemase genes such as OXA-50 and its variants in P. aeruginosa (56, 96), OXA-51-like enzymes in A. baumannii (69, 213), OXA-62 in Pandoraea pnomenusa (192), and OXA-54 and OXA-55 in Shewanella spp. (71, 165) appear to be natural components of their respective bacterial chromosomes. In the case of the OXA genes in A. baumannii, insertion sequences of the ISAba1 type, carrying strong promoters, have been detected upstream of the chromosomal oxacillinase, resulting in increased expression and concomitant carbapenemase resistance (212). Transmission of carbapenemase genes is accelerated when the gene is located within mobile elements such as plasmids and integrons. In addition to the finding of novel carbapenemases in environmental isolates, enzymes first detected in the clinic are now being found in environmental bacteria. The VIM-2 carbapenemase was found in a Pseudomonas pseudoalcaligenes strain from a hospital wastewater system (180); further examination found two P. aeruginosa strains with this gene. In another interesting study, bacteriophages carrying -lactamase genes for OXA-type -lactamases were isolated from sewage, suggesting another vector for transfer of these genes between organisms (140). While it is not hard to imagine carbapenemase-producing strains finding their way into the sewage system, the discovery of IMI-2 carbapenemases on plasmids in rare E. asburiae isolates from U.S. rivers is harder to explain, although the location of sampling in relation to population centers was not reported (9). It is likely that the circulation of carbapenemase genes proceeds in two directions: environmental sources may provide genetic material as a source of these enzymes, and clinical strains may disperse this information both within the hospital setting and into the surrounding environment. CARBAPENEMASE ORIGINS AND TRANSMISSION Carbapenem antibiotic design was inspired by the natural product thienamycin, produced by the soil organism Streptomyces cattleya (89). In fact, carbapenems and olivanic acids were some of the most potent naturally occurring -lactams to be identified from diverse sources in early natural product screening programs (24). Because of the prevalence of these molecules in the soil, it is only logical to expect that enzymes CONCLUDING REMARKS Carbapenemase-producing pathogens cause infections that are difficult to treat and have high mortality rates, due to their appearance in multidrug-resistant pathogens such as K. pneumoniae, P. aeruginosa, and Acinetobacter spp. (15, 29, 126). The first descriptions of these enzymes as species-specific chromosomal carbapenemases have more recently been followed by CARBAPENEMASES: THE VERSATILE -LACTAMASES VOL. 20, 2007 the appearance of carbapenemase genes that are easily transferred on mobile elements among species. While considered by some to be relatively rare, reports of their occurrence in outbreak settings have steadily increased. Awareness of their entry into a hospital environment is the first step that clinical microbiologists can take to address this problem. Care in detection is needed, because high carbapenem MICs are not always evident. Evaluation of effective antibiotic options and rigorous infection control measures will help in the fight against carbapenemase-producing organisms. REFERENCES 1. Aboufaycal, H., H. S. Sader, K. Rolston, L. M. Deshpande, M. Toleman, G. Bodey, I. Raad, and R. N. Jones. 2007. blaVIM-2 and blaVIM-7 carbapenemase-producing Pseudomonas aeruginosa isolates detected in a tertiary care medical center in the United States: report from the MYSTIC program. J. Clin. Microbiol. 45:614–615. 2. Afzal-Shah, M., and D. M. Livermore. 1998. Worldwide emergence of carbapenem-resistant Acinetobacter spp. J. Antimicrob. Chemother. 41: 576–577. 3. Afzal-Shah, M., N. Woodford, and D. M. Livermore. 2001. Characterization of OXA-25, OXA-26, and OXA-27, molecular class D -lactamases associated with carbapenem resistance in clinical isolates of Acinetobacter baumannii. Antimicrob. Agents Chemother. 45:583–588. 4. Alba, J., Y. Ishii, K. Thomson, S. Moland Ellen, and K. Yamaguchi. 2005. Kinetics study of KPC-3, a plasmid-encoded class A carbapenem-hydrolyzing -lactamase. Antimicrob. Agents Chemother. 49:4760–4762. 5. Ambler, R. P. 1980. The structure of -lactamases. Philos. Trans. R. Soc. Lond. B 289:321–331. 6. Ambler, R. P., A. F. W. Coulson, J. M. Frere, J. M. Ghuysen, B. Joris, M. Forsman, R. C. Levesque, G. Tiraby, and S. G. Waley. 1991. A standard numbering scheme for the class A -lactamases. Biochem. J. 276:269–270. 7. Arakawa, Y., M. Murakami, K. Suzuki, H. Ito, R. Wacharotayankun, S. Ohsuka, N. Kato, and M. Ohta. 1995. A novel integron-like element carrying the metallo--lactamase gene blaIMP. Antimicrob. Agents Chemother. 39:1612–1615. 8. Arakawa, Y., N. Shibata, K. Shibayama, H. Kurokawa, T. Yagi, H. Fujiwara, and M. Goto. 2000. Convenient test for screening metallo--lactamase-producing gram-negative bacteria by using thiol compounds. J. Clin. Microbiol. 38: 40–43. 9. Aubron, C., L. Poirel, J. Ash Ronald, and P. Nordmann. 2005. Carbapenemase-producing Enterobacteriaceae, U.S. rivers. Emerg. Infect. Dis. 11: 260–264. 10. Bahar, G., A. Mazzariol, R. Koncan, A. Mert, R. Fontana, G. M. Rossolini, and G. Cornaglia. 2004. Detection of VIM-5 metallo--lactamase in a Pseudomonas aeruginosa clinical isolate from Turkey. J. Antimicrob. Chemother. 54:282–283. 11. Bandoh, K., K. Watanabe, Y. Muto, Y. Tanaka, N. Kato, and K. Ueno. 1992. Conjugal transfer of imipenem resistance in Bacteroides fragilis. J. Antibiot. (Tokyo) 45:542–547. 12. Bou, G., A. Oliver, and J. Martinez-Beltran. 2000. OXA-24, a novel class D -lactamase with carbapenemase activity in an Acinetobacter baumannii clinical strain. Antimicrob. Agents Chemother. 44:1556–1561. 13. Bradford, P. A., S. Bratu, C. Urban, M. Visalli, N. Mariano, D. Landman, J. J. Rahal, S. Brooks, S. Cebular, and J. Quale. 2004. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenemhydrolyzing KPC-2 and inhibitor-resistant TEM-30 -lactamases in New York City. Clin. Infect. Dis. 39:55–60. 14. Bratu, S., D. Landman, M. Alam, E. Tolentino, and J. Quale. 2005. Detection of KPC carbapenem-hydrolyzing enzymes in Enterobacter spp. from Brooklyn, New York. Antimicrob. Agents Chemother. 49:776–778. 15. Bratu, S., D. Landman, R. Haag, R. Recco, A. Eramo, M. Alam, and J. Quale. 2005. Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: a new threat to our antibiotic armamentarium. Arch. Intern. Med. 165:1430–1435. 16. Bratu, S., M. Mooty, S. Nichani, D. Landman, C. Gullans, B. Pettinato, U. Karumudi, P. Tolaney, and J. Quale. 2005. Emergence of KPC-possessing Klebsiella pneumoniae in Brooklyn, New York: Epidemiology and recommendations for detection. Antimicrob. Agents Chemother. 49:3018–3020. 17. Bratu, S., P. Tolaney, U. Karumudi, J. Quale, M. Mooty, S. Nichani, and D. Landman. 2005. Carbapenemase-producing Klebsiella pneumoniae in Brooklyn, N.Y.: molecular epidemiology and in vitro activity of polymyxin B and other agents. J. Antimicrob. Chemother. 56:128–132. 18. Brown, S., and S. Amyes. 2006. OXA -lactamases in Acinetobacter: the story so far. J. Antimicrob. Chemother. 57:1–3. 19. Brown, S., H. K. Young, and S. G. B. Amyes. 2005. Characterisation of OXA-51, a novel class D carbapenemase found in genetically unrelated 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 453 clinical strains of Acinetobacter baumannii from Argentina. Clin. Microbiol. Infect. 11:15–23. Bush, K. 1997. The evoution of -lactamases, p. 152–166. In D. J. Chadwick and J. Goode (ed.), Antibiotic resistance: origins, evolution, selection and spread, vol. 207. John Wiley & Sons, Chichester, United Kingdom. Bush, K. 1988. Recent developments in -lactamase research and their implications for the future. Rev. Infect. Dis. 10:681–690; 739–743. Bush, K., G. A. Jacoby, and A. A. Medeiros. 1995. A functional classification scheme for -lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 39:1211–1233. Bush, K., and R. B. Sykes. 1987. Characterization and epidemiology of -lactamases. Elsevier Science Publishers BV, Philadelphia, PA. Butterworth, D., M. Cole, G. Hanscomb, and G. N. Rolinson. 1979. Olivanic acids, a family of -lactam antibiotics with -lactamase inhibitory properties produced by Streptomyces species. I. Detection, properties and fermentation studies. J. Antibiot. 32:287–294. Castanheira, M., R. E. Mendes, T. R. Walsh, A. C. Gales, and R. N. Jones. 2004. Emergence of the extended-spectrum -lactamase GES-1 in a Pseudomonas aeruginosa strain from Brazil: report from the SENTRY antimicrobial surveillance program. Antimicrob. Agents Chemother. 48: 2344–2345. Castanheira, M., M. A. Toleman, R. N. Jones, F. J. Schmidt, and T. R. Walsh. 2004. Molecular characterization of a -lactamase gene, blaGIM-1, encoding a new subclass of metallo--lactamase. Antimicrob. Agents Chemother. 48:4654–4661. Chu, Y. W., T. K. M. Cheung, J. Y. W. Ngan, and K. M. Kam. 2005. EDTA susceptibility leading to false detection of metallo--lactamase in Pseudomonas aeruginosa by Etest and an imipenem-EDTA disk method. Int. J. Antimicrob. Agents 26:340–341. Coelho, J., N. Woodford, M. Afzal-Shah, and D. Livermore. 2006. Occurrence of OXA-58-like carbapenemases in Acinetobacter spp. collected over 10 years in three continents. Antimicrob. Agents Chemother. 50:756–758. Coelho, J. M., J. F. Turton, M. E. Kaufmann, J. Glover, N. Woodford, M. Warner, M.-F. Palepou, R. Pike, T. L. Pitt, B. C. Patel, and D. M. Livermore. 2006. Occurrence of carbapenem-resistant Acinetobacter baumannii clones at multiple hospitals in London and southeast England. J. Clin. Microbiol. 44:3623–3627. Conceicao, T., A. Brizio, A. Duarte, and R. Barros. 2005. First isolation of blaVIM-2 in Klebsiella oxytoca clinical isolates from Portugal. Antimicrob. Agents Chemother. 49:476. Concha, N. O., C. A. Janson, P. Rowling, S. Pearson, C. A. Cheever, B. P. Clarke, C. Lewis, M. Galleni, J. M. Frere, D. J. Payne, J. H. Bateson, and S. S. Abdel-Meguid. 2000. Crystal structure of the IMP-1 metallo -lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298. Concha, N. O., B. A. Rasmussen, K. Bush, and O. Herzberg. 1996. Crystal structure of the wide-spectrum binuclear zinc -lactamase from Bacteroides fragilis. Structure 4:823–836. Correia, M., F. Boavida, F. Grosso, M. J. Salgado, L. M. Lito, J. M. Cristino, S. Mendo, and A. Duarte. 2003. Molecular characterization of a new class 3 integron in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 47:2838–2843. Costello, A. L., N. P. Sharma, K.-W. Yang, M. W. Crowder, and D. L. Tierney. 2006. X-ray absorption spectroscopy of the zinc-binding sites in the class B2 metallo--lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry 45:13650–13658. Couture, F., J. Lachapelle, and R. C. Levesque. 1992. Phylogeny of LCR-1 and OXA-5 with class A and class D -lactamases. Mol. Microbiol. 6:1693– 1705. Crespo, M. P., N. Woodford, A. Sinclair, M. E. Kaufmann, J. Turton, J. Glover, J. D. Velez, C. R. Castaneda, M. Recalde, and D. M. Livermore. 2004. Outbreak of carbapenem-resistant Pseudomonas aeruginosa producing VIM-8, a novel metallo--lactamase, in a tertiary care center in Cali, Colombia. J. Clin. Microbiol. 42:5094–5101. Dalla-Costa, L. M., J. M. Coelho, H. A. P. H. M. Souza, M. E. S. Castro, C. J. N. Stier, K. L. Bragagnolo, A. Rea-Neto, S. R. Penteado-Filho, D. M. Livermore, and N. Woodford. 2003. Outbreak of carbapenem-resistant Acinetobacter baumannii producing the OXA-23 enzyme in Curitiba, Brazil. J. Clin. Microbiol. 41:3403–3406. Danel, F., L. M. C. Hall, D. Gur, and D. M. Livermore. 1997. OXA-15, an extended-spectrum variant of OXA-2 -lactamase, isolated from a Pseudomonas aeruginosa strain. Antimicrob. Agents Chemother. 41:785–790. Da Silva, G. J., M. Correia, C. Vital, G. Ribeiro, J. C. Sousa, R. Leitao, L. Peixe, and A. Duarte. 2002. Molecular characterization of blaIMP-5, a new integron-borne metallo--lactamase gene from an Acinetobacter baumannii nosocomial isolate in Portugal. FEMS Microbiol. Lett. 215:33–39. Da Silva, G. J., S. Quinteira, E. Bertolo, J. C. Sousa, L. Gallego, A. Duarte, and L. Peixe. 2004. Long-term dissemination of an OXA-40 carbapenemase-producing Acinetobacter baumannii clone in the Iberian Peninsula. J. Antimicrob. Chemother. 54:255–258. Donald, H. M., W. Scaife, S. G. Amyes, and H. K. Young. 2000. Sequence 454 42. 43. 44. 45. 46. 47. 48. 49. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. analysis of ARI-1, a novel OXA -lactamase, responsible for imipenem resistance in Acinetobacter baumannii 6B92. Antimicrob. Agents Chemother. 44:196–199. Duarte, A., F. Boavida, F. Grosso, M. Correia, L. M. Lito, J. M. Cristino, and M. J. Salgado. 2003. Outbreak of GES-1 -lactamase-producing multidrug-resistant Klebsiella pneumoniae in a university hospital in Lisbon, Portugal. Antimicrob. Agents Chemother. 47:1481–1482. Espedido, B., J. Iredell, L. Thomas, and A. Zelynski. 2005. Wide dissemination of a carbapenemase plasmid among gram-negative bacteria: implications of the variable phenotype. J. Clin. Microbiol. 43:4918–4919. Fabiane, S. M., M. K. Sohi, T. Wan, D. J. Payne, J. H. Bateson, T. Mitchell, and B. J. Sutton. 1998. Crystal structure of the zinc-dependent -lactamase from Bacillus cereus at 1.9 Å resolution: binuclear active site with features of a mononuclear enzyme. Biochemistry 37:12404–12411. Fiett, J., A. Baraniak, A. Mrowka, M. Fleischer, Z. Drulis-Kawa, L. Naumiuk, A. Samet, W. Hryniewicz, and M. Gniadkowski. 2006. Molecular epidemiology of acquired-metallo--lactamase-producing bacteria in Poland. Antimicrob. Agents Chemother. 50:880–886. Frere, J. M., M. Galleni, K. Bush, and O. Dideberg. 2005. Is it necessary to change the classification of -lactamases? J. Antimicrob. Chemother. 55: 1051–1053. Gacar, G. G., K. Midilli, F. Kolayli, K. Ergen, S. Gundes, S. Hosoglu, A. Karadenizli, and H. Vahaboglu. 2005. Genetic and enzymatic properties of metallo--lactamase VIM-5 from a clinical isolate of Enterobacter cloacae. Antimicrob. Agents Chemother. 49:4400–4403. Galani, I., M. Souli, Z. Chryssouli, K. Orlandou, and H. Giamarellou. 2005. Characterization of a new integron containing blaVIM-1 and aac(6⬘)-IIc in an Enterobacter cloacae clinical isolate from Greece. J. Antimicrob. Chemother. 55:634–638. Gales, A. C., D. J. Biedenbach, P. Winokur, D. M. Hacek, M. A. Pfaller, and R. N. Jones. 2001. Carbapenem-resistant Serratia marcescens isolates producing Bush group 2f -lactamase (SME-1) in the United States: results from the MYSTIC Programme. Diagn. Microbiol. Infect. Dis. 39:125–127. Galleni, M., J. Lamotte-Brasseur, G. M. Rossolini, J. Spencer, O. Dideberg, and J. M. Frere. 2001. Standard numbering scheme for class B -lactamases. Antimicrob. Agents Chemother. 45:660–663. Garau, G., I. Garcia-Saez, C. Bebrone, C. Anne, P. Mercuri, M. Galleni, J.-M. Frere, and O. Dideberg. 2004. Update of the standard numbering scheme for class B -lactamases. Antimicrob. Agents Chemother. 48:2347– 2349. Garcia-Saez, I., J. Hopkins, C. Papamicael, N. Franceschini, G. Amicosante, G. M. Rossolini, M. Galleni, J.-M. Frere, and O. Dideberg. 2003. The 1.5-Å structure of Chryseobacterium meningosepticum zinc -lactamase in complex with the inhibitor, D-captopril. J. Biol. Chem. 278:23868–23873. Giakkoupi, P., L. S. Tzouvelekis, A. Tsakris, V. Loukova, D. Sofianou, and E. Tzelepi. 2000. IBC-1, a novel integron-associated class A -lactamase with extended-spectrum properties produced by an Enterobacter cloacae clinical strain. Antimicrob. Agents Chemother. 44:2247–2253. Giakkoupi, P., A. Xanthaki, M. Kanelopoulou, A. Vlahaki, V. Miriagou, S. Kontou, E. Papafraggas, H. Malamou-Lada, L. S. Tzouvelekis, N. J. Legakis, and A. C. Vatopoulos. 2003. VIM-1 metallo--lactamase-producing Klebsiella pneumoniae strains in Greek hospitals. J. Clin. Microbiol. 41:3893–3896. Gibb, A. P., C. Tribuddharat, R. A. Moore, T. J. Louie, W. Krulicki, D. M. Livermore, M.-F. I. Palepou, and N. Woodford. 2002. Nosocomial outbreak of carbapenem-resistant Pseudomonas aeruginosa with a new blaIMP allele, blaIMP-7. Antimicrob. Agents Chemother. 46:255–258. Girlich, D., T. Naas, and P. Nordmann. 2004. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48:2043–2048. Girlich, D., T. Naas, and P. Nordmann. 2004. OXA-60, a chromosomal, inducible, and imipenem-hydrolyzing class D -lactamase from Ralstonia pickettii. Antimicrob. Agents Chemother. 48:4217–4225. Guerin, F., C. Henegar, G. Spiridon, O. Launay, D. Salmon-Ceron, and C. Poyart. 2005. Bacterial prostatitis due to Pseudomonas aeruginosa harboring the blaVIM-2 metallo--lactamase gene from Saudi Arabia. J. Antimicrob. Chemother. 56:601–602. Gupta, V., P. Datta, and J. Chander. 2006. Prevalence of metallo--lactamase (MBL) producing Pseudomonas spp. and Acinetobacter spp. in a tertiary care hospital in India. J. Infect. 52:311–314. Hall, B. G., and M. Barlow. 2005. Revised Ambler classification of -lactamases. J. Antimicrob. Chemother. 55:1050–1051. Hall, L. M. C., D. M. Livermore, D. Gur, M. Akova, and H. E. Akalin. 1993. OXA-11, an extended-spectrum variant of OXA-10 (PSE-2) -lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 37:1637– 1644. Hanson, N. D., A. Hossain, L. Buck, S. Moland Ellen, and S. Thomson Kenneth. 2006. First occurrence of a Pseudomonas aeruginosa isolate in the United States producing an IMP metallo--lactamase, IMP-18. Antimicrob. Agents Chemother. 50:2272–2273. Hawkey, P. M., J. Xiong, H. Ye, H. Li, and F. H. M’Zali. 2001. Occurrence of a new metallo--lactamase IMP-4 carried on a conjugative plasmid in CLIN. MICROBIOL. REV. 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. Citrobacter youngae from the People’s Republic of China. FEMS Microbiol. Lett. 194:53–57. Hayes, M. V., C. J. Thomson, and S. G. B. Amyes. 1996. The “hidden” carbapenemase of Aeromonas hydrophila. J. Antimicrob. Chemother. 37: 33–44. Hayes, M. V., C. J. Thomson, and S. G. B. Amyes. 1994. Three -lactamases isolated from Aeromonas salmonicida, including a carbapenemase not detectable by conventional methods. Eur. J. Clin. Microbiol. Infect. Dis. 13:805–811. Henrichfreise, B., I. Wiegand, K. J. Sherwood, and B. Wiedemann. 2005. Detection of VIM-2 metallo--lactamase in Pseudomonas aeruginosa from Germany. Antimicrob. Agents Chemother. 49:1668–1669. Henriques, I., A. Moura, A. Alves, M. J. Saavedra, and A. Correia. 2004. Molecular characterization of a carbapenem-hydrolyzing class A -lactamase, SFC-1, from Serratia fonticola UTAD54. Antimicrob. Agents Chemother. 48:2321–2324. Heritier, C., A. Dubouix, L. Poirel, N. Marty, and P. Nordmann. 2005. A nosocomial outbreak of Acinetobacter baumannii isolates expressing the carbapenem-hydrolysing oxacillinase OXA-58. J. Antimicrob. Chemother. 55:115–118. Heritier, C., L. Poirel, P.-E. Fournier, J.-M. Claverie, D. Raoult, and P. Nordmann. 2005. Characterization of the naturally occurring oxacillinase of Acinetobacter baumannii. Antimicrob. Agents Chemother. 49:4174–4179. Heritier, C., L. Poirel, T. Lambert, and P. Nordmann. 2005. Contribution of acquired carbapenem-hydrolyzing oxacillinases to carbapenem resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother. 49: 3198–3202. Heritier, C., L. Poirel, and P. Nordmann. 2004. Genetic and biochemical characterization of a chromosome-encoded carbapenem-hydrolyzing Ambler class D -lactamase from Shewanella algae. Antimicrob. Agents Chemother. 48:1670–1675. Hirakata, Y., K. Izumikawa, T. Yamaguchi, H. Takemura, H. Tanaka, R. Yoshida, J. Matsuda, M. Nakano, K. Tomono, S. Maesaki, M. Kaku, Y. Yamada, S. Kamihira, and S. Kohno. 1998. Rapid detection and evaluation of clinical characteristics of emerging multiple-drug-resistant gram-negative rods carrying the metallo--lactamase gene blaIMP. Antimicrob. Agents Chemother. 42:2006–2011. Hornstein, M., C. Sautjeau-Rostoker, J. Peduzzi, A. Vessieres, L. T. Hong, M. Barthelemy, M. Scavizzi, and R. Labia. 1997. Oxacillin-hydrolyzing -lactamase involved in resistance to imipenem in Acinetobacter baumannii. FEMS Microbiol. Lett. 153:333–339. Hossain, A., M. J. Ferraro, R. M. Pino, R. B. Dew III, E. S. Moland, T. J. Lockhart, K. S. Thomson, R. V. Goering, and N. D. Hanson. 2004. Plasmidmediated carbapenem-hydrolyzing enzyme KPC-2 in an Enterobacter sp. Antimicrob. Agents Chemother. 48:4438–4440. Hujer, K. M., A. M. Hujer, E. A. Hulten, S. Bajaksouzian, J. M. Adams, C. J. Donskey, D. J. Ecker, C. Massire, M. W. Eshoo, R. Sampath, J. M. Thomson, P. N. Rather, D. W. Craft, J. T. Fishbain, A. J. Ewell, M. R. Jacobs, D. L. Paterson, and R. A. Bonomo. 2006. Analysis of antibiotic resistance genes in multidrug-resistant Acinetobacter sp. isolates from military and civilian patients treated at the Walter Reed Army Medical Center. Antimicrob. Agents Chemother. 50:4114–4123. Huovinen, P., S. Huovinen, and G. A. Jacoby. 1988. Sequence of PSE-2 -lactamase. Antimicrob. Agents Chemother. 32:134–136. Hussain, M., A. Carlino, M. J. Madonna, and J. O. Lampen. 1985. Cloning and sequencing of the metallothioprotein -lactamase II gene of Bacillus cereus 569/H in Escherichia coli. J. Bacteriol. 164:223–229. Iaconis, J. P., and C. C. Sanders. 1990. Purification and characterization of inducible -lactamases in Aeromonas spp. Antimicrob. Agents Chemother. 34:44–51. Ikonomidis, A., D. Tokatlidou, I. Kristo, D. Sofianou, A. Tsakris, P. Mantzana, S. Pournaras, and A. N. Maniatis. 2005. Outbreaks in distinct regions due to a single Klebsiella pneumoniae clone carrying a blaVIM-1 metallo--lactamase gene. J. Clin. Microbiol. 43:5344–5347. Ito, H., Y. Arakawa, S. Ohsuka, R. Wacharotayankun, N. Kato, and M. Ohta. 1995. Plasmid-mediated dissemination of the metallo--lactamase gene blaIMP among clinically isolated strains of Serratia marcescens. Antimicrob. Agents Chemother. 39:824–829. Iyobe, S., H. Kusadokoro, J. Ozaki, N. Matsumura, S. Minami, S. Haruta, T. Sawai, and K. O’Hara. 2000. Amino acid substitutions in a variant of IMP-1 metallo--lactamase. Antimicrob. Agents Chemother. 44:2023– 2027. Jacoby, G., and K. Bush. 2005. -Lactam resistance in the 21st century, p. 53–65. In D. G. White, M. N. Alekshun, and P. F. McDermott (ed.), Frontiers in antimicrobial resistance: a tribute to Stuart B. Levy. ASM Press, Washington, DC. Jacoby, G. A. 2006. -Lactamase nomenclature. Antimicrob. Agents Chemother. 50:1123–1129. Jaurin, B., and T. Grundstrom. 1981. ampC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of -lactamases of the penicillinase type. Proc. Natl. Acad. Sci. USA 78:4897–4901. Jeon, B.-C., S. H. Jeong, I. K. Bae, S. B. Kwon, K. Lee, D. Young, J. H. Lee, cmr.asm.orgm at UNIV OF ARKANSAS on July 30, 2007 50. QUEENAN AND BUSH VOL. 20, 2007 86. 87. 88. 89. 90. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. 125. 126. 127. 128. 455 integrons reveals several novel combinations of resistance genes. Antimicrob. Agents Chemother. 39:185–191. Libisch, B., M. Gacs, K. Csiszar, M. Muzslay, L. Rokusz, and M. Fuezi. 2004. Isolation of an integron-borne blaVIM-4 type metallo--lactamase gene from a carbapenem-resistant Pseudomonas aeruginosa clinical isolate in Hungary. Antimicrob. Agents Chemother. 48:3576–3578. Libisch, B., M. Muzslay, M. Gacs, J. Minarovits, M. Knausz, J. Watine, G. Ternak, E. Kenez, I. Kustos, L. Rokusz, K. Szeles, B. Balogh, and M. Fuzi. 2006. Molecular epidemiology of VIM-4 metallo--lactamase-producing Pseudomonas sp. isolates in Hungary. Antimicrob. Agents Chemother. 50: 4220–4223. Lim, H. M., J. J. Pene, and R. W. Shaw. 1988. Cloning, nucleotide sequence, and expression of the Bacillus cereus 5/B/6 -lactamase II structural gene. J. Bacteriol. 170:2873–2878. Lincopan, N., J. A. McCulloch, C. Reinert, V. C. Cassettari, A. C. Gales, and E. M. Mamizuka. 2005. First isolation of metallo--lactamase-producing multiresistant Klebsiella pneumoniae from a patient in Brazil. J. Clin. Microbiol. 43:516–519. Liu, S. Y., J. Y. Lin, C. Chu, L. H. Su, T. Y. Lin, and C. H. Chiu. 2006. Integron-associated imipenem resistance in Acinetobacter baumannii isolated from a regional hospital in Taiwan. Int. J. Antimicrob. Agents. 27: 81–84. Livermore, D. M. 2002. The impact of carbapenemases on antimicrobial development and therapy. Curr. Opin. Investig. Drugs 3:218–224. Livermore, D. M. 2001. Of Pseudomonas, porins, pumps and carbapenems. J. Antimicrob. Chemother. 47:247–250. Livermore, D. M., and N. Woodford. 2006. The -lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 14: 413–420. Lolans, K., A. M. Queenan, K. Bush, A. Sahud, and J. P. Quinn. 2005. First nosocomial outbreak of Pseudomonas aeruginosa producing an integronborne metallo--lactamase (VIM-2) in the United States. Antimicrob. Agents Chemother. 49:3538–3540. Lolans, K., T. W. Rice, L. S. Munoz-Price, and J. P. Quinn. 2006. Multicity outbreak of carbapenem-resistant Acinetobacter baumannii isolates producing the carbapenemase OXA-40. Antimicrob. Agents Chemother. 50:2941– 2945. Loli, A., L. S. Tzouvelekis, E. Tzelepi, A. Carattoli, A. C. Vatopoulos, P. T. Tassios, and V. Miriagou. 2006. Sources of diversity of carbapenem resistance levels in Klebsiella pneumoniae carrying blaVIM-1. J. Antimicrob. Chemother. 58:669–672. Lomaestro, B. M., E. H. Tobin, W. Shang, and T. Gootz. 2006. The spread of Klebsiella pneumoniae carbapenemase-producing K. pneumoniae to upstate New York. Clin. Infect. Dis. 43:e26–28. Lopez-Otsoa, F., L. Gallego, K. J. Towner, L. Tysall, N. Woodford, and D. M. Livermore. 2002. Endemic carbapenem resistance associated with OXA-40 carbapenemase among Acinetobacter baumannii isolates from a hospital in northern Spain. J. Clin. Microbiol. 40:4741–4743. Luzzaro, F., J.-D. Docquier, C. Colinon, A. Endimiani, G. Lombardi, G. Amicosante, G. M. Rossolini, and A. Toniolo. 2004. Emergence in Klebsiella pneumoniae and Enterobacter cloacae clinical isolates of the VIM-4 metallo-lactamase encoded by a conjugative plasmid. Antimicrob. Agents Chemother. 48:648–650. Luzzaro, F., A. Endimiani, J.-D. Docquier, C. Mugnaioli, M. Bonsignori, G. Amicosante, G. M. Rossolini, and A. Toniolo. 2004. Prevalence and characterization of metallo--lactamases in clinical isolates of Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 48:131–135. Majiduddin, F. K., and T. Palzkill. 2005. Amino acid residues that contribute to substrate specificity of class A -lactamase SME-1. Antimicrob. Agents Chemother. 49:3421–3427. Majiduddin, F. K., and T. Palzkill. 2003. Amino acid sequence requirements at residues 69 and 238 for the SME-1 -lactamase to confer resistance to -lactam antibiotics. Antimicrob. Agents Chemother. 47:1062– 1067. Mariotte-Boyer, S., M. H. Nicolas-Chanoine, and R. Labia. 1996. A kinetic study of NMC-A -lactamase, an Ambler class A carbapenemase also hydrolyzing cephamycins. FEMS Microbiol. Lett. 143:29–33. Marque, S., L. Poirel, C. Heritier, S. Brisse, M. D. Blasco, R. Filip, G. Coman, T. Naas, and P. Nordmann. 2005. Regional occurrence of plasmidmediated carbapenem-hydrolyzing oxacillinase OXA-58 in Acinetobacter spp. in Europe. J. Clin. Microbiol. 43:4885–4888. Marra, A. R., C. A. P. Pereira, A. C. Gales, L. C. Menezes, R. G. R. Cal, J. M. A. de Souza, M. B. Edmond, C. Faro, and S. B. Wey. 2006. Bloodstream infections with metallo--lactamase-producing Pseudomonas aeruginosa: epidemiology, microbiology, and clinical outcomes. Antimicrob. Agents Chemother. 50:388–390. Massova, I., and S. Mobashery. 1998. Kinship and diversification of bacterial penicillin-binding proteins and -lactamases. Antimicrob. Agents Chemother. 42:1–17. Materon, I. C., A. M. Queenan, T. M. Koehler, K. Bush, and T. Palzkill. 2003. Biochemical characterization of -lactamases Bla1 and Bla2 from Bacillus anthracis. Antimicrob. Agents Chemother. 47:2040–2042. Downloaded from cmr.asm.org at UNIV OF ARKANSAS on July 30, 2007 91. J. S. Song, and S. H. Lee. 2005. Investigation of a nosocomial outbreak of imipenem-resistant Acinetobacter baumannii producing the OXA-23 -lactamase in Korea. J. Clin. Microbiol. 43:2241–2245. Jeong, S. H., I. K. Bae, D. Kim, S. G. Hong, J. S. Song, J. H. Lee, and S. H. Lee. 2005. First outbreak of Klebsiella pneumoniae clinical isolates producing GES-5 and SHV-12 extended-spectrum -lactamases in Korea. Antimicrob. Agents Chemother. 49:4809–4810. Jeong, S. H., K. Lee, Y. Chong, J. H. Yum, S. H. Lee, H. J. Choi, J. M. Kim, K. H. Park, B. H. Han, S. W. Lee, and T. S. Jeong. 2003. Characterization of a new integron containing VIM-2, a metallo--lactamase gene cassette, in a clinical isolate of Enterobacter cloacae. J. Antimicrob. Chemother. 51:397–400. Jones, R. N., D. J. Biedenbach, H. S. Sader, T. R. Fritsche, M. A. Toleman, and T. R. Walsh. 2005. Emerging epidemic of metallo--lactamase-mediated resistances. Diagn. Microbiol. Infect. Dis. 51:77–84. Kahan, J. S., F. M. Kahan, R. Goegelman, S. A. Currie, M. Jackson, E. O. Stapley, T. W. Miller, A. K. Miller, D. Hendlin, S. Mochales, S. Hernandez, H. B. Woodruff, and J. Birnbaum. 1979. Thienamycin, a new -lactam antibiotic. I. Discovery, taxonomy, isolation and physical properties. J. Antibiot. 32:1–12. Kartali, G., E. Tzelepi, S. Pournaras, C. Kontopoulou, F. Kontos, D. Sofianou, A. N. Maniatis, and A. Tsakris. 2002. Outbreak of infections caused by Enterobacter cloacae producing the integron-associated -lactamase IBC-1 in a neonatal intensive care unit of a Greek hospital. Antimicrob. Agents Chemother. 46:1577–1580. Kassis-Chikhani, N., D. Decre, V. Gautier, B. Burghoffer, F. Saliba, D. Mathieu, D. Samuel, D. Castaing, J.-C. Petit, E. Dussaix, and G. Arlet. 2006. First outbreak of multidrug-resistant Klebsiella pneumoniae carrying blaVIM-1 and blaSHV-5 in a French university hospital. J. Antimicrob. Chemother. 57:142–145. Ke, W., C. R. Bethel, J. M. Thomson, R. A. Bonomo, and F. van den Akker. 2007. Crystal structure of KPC-2: insights into carbapenemase activity in class A -lactamases. Biochemistry 46:5732–5740. Kiffer, C. R. V., C. Mendes, J. L. Kuti, and D. P. Nicolau. 2004. Pharmacodynamic comparisons of antimicrobials against nosocomial isolates of Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii and Pseudomonas aeruginosa from the MYSTIC surveillance program: the OPTAMA Program, South America 2002. Diagn. Microbiol. Infect. Dis. 49:109–116. Kimura, S., J. Alba, K. Shiroto, R. Sano, Y. Niki, S. Maesaki, K. Akizawa, M. Kaku, Y. Watanuki, Y. Ishii, and K. Yamaguchi. 2005. Clonal diversity of metallo--lactamase-possessing Pseudomonas aeruginosa in geographically diverse regions of Japan. J. Clin. Microbiol. 43:458–461. Koh, T. H., G. S. Babini, N. Woodford, L. H. Sng, L. M. Hall, and D. M. Livermore. 1999. Carbapenem-hydrolysing IMP-1 -lactamase in Klebsiella pneumoniae from Singapore. Lancet 353:2162. Kong, K.-F., S. R. Jayawardena, A. Del Puerto, L. Wiehlmann, U. Laabs, B. Tummler, and K. Mathee. 2005. Characterization of poxB, a chromosomalencoded Pseudomonas aeruginosa oxacillinase. Gene 358:82–92. Ktari, S., G. Arlet, B. Mnif, V. Gautier, F. Mahjoubi, M. Ben Jmeaa, M. Bouaziz, and A. Hammami. 2006. Emergence of multidrug-resistant Klebsiella pneumoniae isolates producing VIM-4 metallo--lactamase, CTXM-15 extended-spectrum -lactamase, and CMY-4 AmpC -lactamase in a Tunisian university hospital. Antimicrob. Agents Chemother. 50:4198– 4201. Kuwabara, S., and E. P. Abraham. 1967. Some properties of two extracellular -lactamases from Bacillus cereus 569/H. Biochem. J. 103:27C–30C. Reference deleted. Lagatolla, C., E. A. Tonin, C. Monti-Bragadin, L. Dolzani, F. Gombac, C. Bearzi, E. Edalucci, F. Gionechetti, and G. M. Rossolini. 2004. Endemic carbapenem-resistant Pseudomonas aeruginosa with acquired metallo-lactamase determinants in European hospital. Emerg. Infect. Dis. 10:535– 538. Lauretti, L., M. L. Riccio, A. Mazzariol, G. Cornaglia, G. Amicosante, R. Fontana, and G. M. Rossolini. 1999. Cloning and characterization of blaVIM, a new integron-borne metallo--lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 43:1584– 1590. Ledent, P., X. Raquet, B. Joris, J. Van Beeumen, and J. M. Frere. 1993. A comparative study of class-D -lactamases. Biochem. J. 292:555–562. Lee, K., J. B. Lim, J. H. Yum, D. Yong, Y. Chong, J. M. Kim, and D. M. Livermore. 2002. blaVIM-2 cassette-containing novel integrons in metallo-lactamase-producing Pseudomonas aeruginosa and Pseudomonas putida isolates disseminated in a Korean hospital. Antimicrob. Agents Chemother. 46:1053–1058. Lee, K., J. H. Yum, D. Yong, H. M. Lee, H. D. Kim, J.-D. Docquier, G. M. Rossolini, and Y. Chong. 2005. Novel acquired metallo--lactamase gene, blaSIM-1, in a class 1 integron from Acinetobacter baumannii clinical isolates from Korea. Antimicrob. Agents Chemother. 49:4485–4491. Lee, S. H., and S. H. Jeong. 2005. Nomenclature of GES-type extendedspectrum -lactamases. Antimicrob. Agents Chemother. 49:2148. Levesque, C., L. Piche, C. Larose, and P. H. Roy. 1995. PCR mapping of CARBAPENEMASES: THE VERSATILE -LACTAMASES 456 QUEENAN AND BUSH 154. 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. 171. 172. 173. 174. Strynadka. 2000. Crystal structure of the class D -lactamase OXA-10. Nat. Struct. Biol. 7:918–925. Pagani, L., C. Colinon, R. Migliavacca, M. Labonia, J.-D. Docquier, E. Nucleo, M. Spalla, M. Li Bergoli, and G. M. Rossolini. 2005. Nosocomial outbreak caused by multidrug-resistant Pseudomonas aeruginosa producing IMP-13 metallo--lactamase. J. Clin. Microbiol. 43:3824–3828. Pasteran, F., D. Faccone, A. Petroni, M. Rapoport, M. Galas, M. Vazquez, and A. Procopio. 2005. Novel variant (blaVIM-11) of the metallo--lactamase blaVIM family in a GES-1 extended-spectrum--lactamase-producing Pseudomonas aeruginosa clinical isolate in Argentina. Antimicrob. Agents Chemother. 49:474–475. Paterson, D. L., and R. A. Bonomo. 2005. Extended-spectrum -lactamases: a clinical update. Clin. Microbiol. Rev. 18:657–686. Paton, R., R. S. Miles, J. Hood, and S. G. B. Amyes. 1993. ARI 1: -lactamase-mediated imipenem resistance in Acinetobacter baumannii. Int. J. Antimicrob. Agents 2:81–87. Peleg, A. Y., C. Frankiin, J. M. Bell, and D. W. Spelman. 2005. Dissemination of the metallo--lactamase gene blaIMP-4 among Gram-negative pathogens in a clinical setting in Australia. Clin. Infect. Dis. 41:1549–1556. Peleg, A. Y., C. Franklin, J. Bell, and D. W. Spelman. 2004. Emergence of IMP-4 metallo--lactamase in a clinical isolate from Australia. J. Antimicrob. Chemother. 54:699–700. Peleg, A. Y., C. Franklin, L. J. Walters, J. M. Bell, and D. W. Spelman. 2006. OXA-58 and IMP-4 carbapenem-hydrolyzing -lactamases in an Acinetobacter junii blood culture isolate from Australia. Antimicrob. Agents Chemother. 50:399–400. Petropoulou, D., K. Tzanetou, V. P. Syriopoulou, G. L. Daikos, G. Ganteris, and E. Malamou-Lada. 2006. Evaluation of imipenem/imipenem ⫹ EDTA disk method for detection of metallo--lactamase-producing Klebsiella pneumoniae isolated from blood cultures. Microb. Drug Resist. 12:39–43. Philippon, L. N., T. Naas, A.-T. Bouthors, V. Barakett, and P. Nordmann. 1997. OXA-18, a class D clavulanic acid-inhibited extended-spectrum -lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 41: 2188–2195. Pitout, J. D. D., B. L. Chow, D. B. Gregson, K. B. Laupland, S. Elsayed, and D. L. Church. 2007. Molecular epidemiology of metallo--lactamase-producing Pseudomonas aeruginosa in the Calgary Health Region: emergence of VIM-2-producing isolates. J. Clin. Microbiol. 45:294–298. Poirel, L., L. Brinas, N. Fortineau, and P. Nordmann. 2005. Integronencoded GES-type extended-spectrum -lactamase with increased activity toward aztreonam in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:3593–3597. Poirel, L., C. Heritier, and P. Nordmann. 2004. Chromosome-encoded Ambler class D -lactamase of Shewanella oneidensis as a progenitor of carbapenem-hydrolyzing oxacillinase. Antimicrob. Agents Chemother. 48: 348–351. Poirel, L., C. Heritier, and P. Nordmann. 2005. Genetic and biochemical characterization of the chromosome-encoded class B -lactamases from Shewanella livingstonensis (SLB-1) and Shewanella frigidimarina (SFB-1). J. Antimicrob. Chemother. 55:680–685. Poirel, L., C. Heritier, V. Toluen, and P. Nordmann. 2004. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 48:15–22. Poirel, L., I. Le Thomas, T. Naas, A. Karim, and P. Nordmann. 2000. Biochemical sequence analyses of GES-1, a novel class A extended-spectrum -lactamase, and the class 1 integron In52 from Klebsiella pneumoniae. Antimicrob. Agents Chemother. 44:622–632. Poirel, L., M. Magalhaes, M. Lopes, and P. Nordmann. 2004. Molecular analysis of metallo--lactamase gene blaSPM-1-surrounding sequences from disseminated Pseudomonas aeruginosa isolates in Recife, Brazil. Antimicrob. Agents Chemother. 48:1406–1409. Poirel, L., S. Marque, C. Heritier, C. Segonds, G. Chabanon, and P. Nordmann. 2005. OXA-58, a novel class D -lactamase involved in resistance to carbapenems in Acinetobacter baumannii. Antimicrob. Agents Chemother. 49:202–208. Poirel, L., T. Naas, D. Nicolas, L. Collet, S. Bellais, J.-D. Cavallo, and P. Nordmann. 2000. Characterization of VIM-2, a carbapenem-hydrolyzing metallo--lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob. Agents Chemother. 44:891–897. Poirel, L., J. N. Pham, L. Cabanne, B. J. Gatus, S. M. Bell, and P. Nordmann. 2004. Carbapenem-hydrolysing metallo--lactamases from Klebsiella pneumoniae and Escherichia coli isolated in Australia. Pathology (Philadelphia) 36:366–367. Poirel, L., G. F. Weldhagen, C. De Champs, and P. Nordmann. 2002. A nosocomial outbreak of Pseudomonas aeruginosa isolates expressing the extended-spectrum -lactamase GES-2 in South Africa. J. Antimicrob. Chemother. 49:561–565. Poirel, L., G. F. Weldhagen, T. Naas, C. De Champs, M. G. Dove, and P. Nordmann. 2001. GES-2, a class A -lactamase from Pseudomonas aeruginosa with increased hydrolysis of imipenem. Antimicrob. Agents Chemother. 45:2598–2603. Downloaded from cmr.asm.org at UNIV OF ARKANSAS on July 30, 2007 129. Matthew, M. 1979. Plasmid mediated -lactamases of gram-negative bacteria: properties and distribution. J. Antimicrob. Chemother. 5:349–358. 130. Matthew, M., A. M. Harris, M. J. Marshall, and G. W. Ross. 1975. The use of analytical isoelectric focusing for detection and identification of -lactamases. J. Gen. Microbiol. 88:169–178. 131. Maveyraud, L., D. Golemi-Kotra, A. Ishiwata, O. Meroueh, S. Mobashery, and J.-P. Samama. 2002. High-resolution X-ray structure of an acyl-enzyme species for the class D OXA-10 -lactamase. J. Am. Chem. Soc. 124:2461– 2465. 132. Medeiros, A. A. 1984. -Lactamases. Br. Med. Bull. 40:18–27. 133. Medeiros, A. A. 1997. Evolution and dissemination of -lactamases accelerated by generations of -lactam antibiotics. Clinic. Infect. Dis. 24:S19– S45. 134. Medeiros, A. A., and R. S. Hare. 1986. -Lactamase-mediated resistance to penems and carbapenems amongst Enterobacteriaceae, abstr. 116. 26th Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC. 135. Meyer, K. S., C. Urban, J. A. Eagan, B. J. Berger, and J. J. Rahal. 1993. Nosocomial outbreak of Klebsiella infection resistant to late-generation cephalosporins. Ann. Intern. Med. 119:353–358. 136. Miriagou, V., E. Tzelepi, D. Gianneli, and L. S. Tzouvelekis. 2003. Escherichia coli with a self-transferable, multiresistant plasmid coding for metallo-lactamase VIM-1. Antimicrob. Agents Chemother. 47:395–397. 137. Miriagou, V., L. S. Tzouvelekis, S. Rossiter, E. Tzelepi, F. J. Angulo, and J. M. Whichard. 2003. Imipenem resistance in a Salmonella clinical strain due to plasmid-mediated class A carbapenemase KPC-2. Antimicrob. Agents Chemother. 47:1297–1300. 138. Moland, E. S., N. D. Hanson, V. L. Herrera, J. A. Black, T. J. Lockhart, A. Hossain, J. A. Johnson, R. V. Goering, and K. S. Thomson. 2003. Plasmidmediated, carbapenem-hydrolysing -lactamase, KPC-2, in Klebsiella pneumoniae isolates. J. Antimicrob. Chemother. 51:711–714. 139. Mourey, L., K. Miyashita, P. Swaren, A. Bulychev, J.-P. Samama, and S. Mobashery. 1998. Inhibition of the NMC-A -lactamase by a penicillanic acid derivative and the structural bases for the increase in substrate profile of this antibiotic resistance enzyme. J. Am. Chem. Soc. 120:9382–9383. 140. Muniesa, M., A. Garcia, E. Miro, B. Mirelis, G. Prats, J. Jofre, and F. Navarro. 2004. Bacteriophages and diffusion of -lactamase genes. Emerg. Infect. Dis. 10:1134–1137. 141. Murphy, T. A., L. E. Catto, S. E. Halford, A. T. Hadfield, W. Minor, T. R. Walsh, and J. Spencer. 2006. Crystal structure of Pseudomonas aeruginosa SPM-1 provides insights into variable zinc affinity of metallo--lactamases. J. Mol. Biol. 357:890–903. 142. Naas, T., M. Levy, C. Hirschauer, H. Marchandin, and P. Nordmann. 2005. Outbreak of carbapenem-resistant Acinetobacter baumannii producing the carbapenemase OXA-23 in a tertiary care hospital of Papeete, French Polynesia. J. Clin. Microbiol. 43:4826–4829. 143. Naas, T., and P. Nordmann. 1994. Analysis of a carbapenem-hydrolyzing class A -lactamase from Enterobacter cloacae and of its LysR-type regulatory protein. Proc. Natl. Acad. Sci. USA 91:7693–7697. 144. Naas, T., and P. Nordmann. 1999. OXA-type -lactamases. Curr. Pharm. Des. 5:865–879. 145. Naas, T., P. Nordmann, G. Vedel, and C. Poyart. 2005. Plasmid-mediated carbapenem-hydrolyzing -lactamase KPC in a Klebsiella pneumoniae isolate from France. Antimicrob. Agents Chemother. 49:4423–4424. 146. Naas, T., L. Vandel, W. Sougakoff, D. M. Livermore, and P. Nordmann. 1994. Cloning and sequence analysis of the gene for a carbapenem-hydrolyzing class A -lactamase, Sme-1, from Serratia marcescens S6. Antimicrob. Agents Chemother. 38:1262–1270. 147. Navon-Venezia, S., I. Chmelnitsky, A. Leavitt, M. J. Schwaber, D. Schwartz, and Y. Carmeli. 2006. Plasmid-mediated imipenem-hydrolyzing enzyme KPC-2 among multiple carbapenem-resistant Escherichia coli clones in Israel. Antimicrob. Agents Chemother. 50:3098–3101. 148. Nishio, H., M. Komatsu, N. Shibata, K. Shimakawa, N. Sueyoshi, T. Ura, K. Satoh, M. Toyokawa, T. Nakamura, Y. Wada, T. Orita, T. Kofuku, K. Yamasaki, M. Sakamoto, S. Kinoshita, M. Aihara, and Y. Arakawa. 2004. Metallo--lactamase-producing gram-negative bacilli: laboratory-based surveillance in cooperation with 13 clinical laboratories in the Kinki region of Japan. J. Clin. Microbiol. 42:5256–5263. 149. Nordmann, P., S. Mariotte, T. Naas, R. Labia, and M.-H. Nicolas. 1993. Biochemical properties of a carbapenem-hydrolyzing -lactamase for Enterobacter cloacae and cloning of the gene into Escherichia coli. Antimicrob. Agents Chemother. 37:939–946. 150. Nordmann, P., and L. Poirel. 2002. Emerging carbapenemases in Gramnegative aerobes. Clin. Microbiol. Infect. 8:321–331. 151. Normark, S. 1995. -Lactamase induction in gram-negative bacteria is intimately linked to peptidoglycan recycling. Microb. Drug Resist. Mech. Epidemiol. Dis. 1:111–114. 152. O’Hara, K., S. Haruta, T. Sawai, M. Tsunoda, and S. Iyobe. 1998. Novel metallo -lactamase mediated by a Shigella flexneri plasmid. FEMS Microbiol. Lett. 162:201–206. 153. Paetzel, M., F. Danel, L. de Castro, S. C. Mosimann, M. G. Page, and N. C. CLIN. MICROBIOL. REV. VOL. 20, 2007 198. 199. 200. 201. 202. 203. 204. 205. 206. 207. 208. 209. 210. 211. 212. 213. 214. 215. 216. 217. 457 for metallo--lactamases and integrases carried by gram-negative bacteria isolated in Japan, with focus on the class 3 integron. J. Clin. Microbiol. 41:5407–5413. Simpson, I. N., P. B. Harper, and C. H. O’Callaghan. 1980. Principal -lactamases responsible for resistance to -lactam antibiotics in urinary tract infections. Antimicrob. Agents Chemother. 17:929–936. Sougakoff, W., G. L’Hermite, L. Pernot, T. Naas, V. Guillet, P. Nordmann, V. Jarlier, and J. Delettre. 2002. Structure of the imipenem-hydrolyzing class A -lactamase SME-1 from Serratia marcescens. Acta Crystallogr. D 58:267–274. Sougakoff, W., T. Naas, P. Nordmann, E. Collatz, and V. Jarlier. 1999. Role of Ser-237 in the substrate specificity of the carbapenem-hydrolyzing class A -lactamase Sme-1. Biochim. Biophys. Acta 1433:153–158. Swaren, P., L. Maveyraud, X. Raquet, S. Cabantous, C. Duez, J.-D. Pedelacq, S. Mariotte-Boyer, L. Mourey, R. Labia, M.-H. Nicolas-Chanoine, P. Nordmann, J.-M. Frere, and J.-P. Samama. 1998. X-ray analysis of the NMC-A -lactamase at 1.64-Å resolution, a class A carbapenemase with broad substrate specificity. J. Biol. Chem. 273:26714–26721. Sykes, R. B., and M. Matthew. 1976. The -lactamases of Gram-negative bacteria and their role in resistance to -lactam antibiotics. J. Antimicrob. Chemother. 2:115–157. Tenover, F. C., R. K. Kalsi, P. P. Williams, R. B. Carey, S. Stocker, D. Lonsway, J. K. Rasheed, J. W. Biddle, J. E. McGowan, Jr., and B. Hanna. 2006. Carbapenem resistance in Klebsiella pneumoniae not detected by automated susceptibility testing. Emerg. Infect. Dis. 12:1209–1213. Toleman, M. A., K. Rolston, R. N. Jones, and T. R. Walsh. 2004. blaVIM-7, an evolutionarily distinct metallo--lactamase gene in a Pseudomonas aeruginosa isolate from the United States. Antimicrob. Agents Chemother. 48:329–332. Toleman, M. A., K. Rolston, R. N. Jones, and T. R. Walsh. 2003. Molecular and biochemical characterization of OXA-45, an extended-spectrum class 2d⬘ -lactamase in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 47:2859–2863. Toleman, M. A., A. M. Simm, T. A. Murphy, A. C. Gales, D. J. Biedenbach, R. N. Jones, and T. R. Walsh. 2002. Molecular characterization of SPM-1, a novel metallo--lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J. Antimicrob. Chemother. 50:673–679. Tortola, M. T., S. Lavilla, E. Miro, J. J. Gonzalez, N. Larrosa, M. Sabate, F. Navarro, and G. Prats. 2005. First detection of a carbapenem-hydrolyzing metalloenzyme in two Enterobacteriaceae isolates in Spain. Antimicrob. Agents Chemother. 49:3492–3494. Troillet, N., Y. Carmeli, L. Venkataraman, P. DeGirolami, and M. H. Samore. 1999. Epidemiological analysis of imipenem-resistant Serratia marcescens in hospitalized patients. J. Hosp. Infect. 42:37–43. Turner, P. J. 2004. Susceptibility of meropenem and comparators tested against 30,634 Enterobacteriaceae isolated in the MYSTIC Programme (1997–2003). Diagn. Microbiol. Infect. Dis. 50:291–293. Turner, P. J. 2005. Trends in antimicrobial susceptibilities among bacterial pathogens isolated from patients hospitalized in European medical centers: 6-year report of the MYSTIC Surveillance Study (1997–2002). Diagn. Microbiol. Infect. Dis. 51:281–289. Turton, J. F., M. E. Kaufmann, J. Glover, J. M. Coelho, M. Warner, R. Pike, and T. L. Pitt. 2005. Detection and typing of integrons in epidemic strains of Acinetobacter baumannii found in the United Kingdom. J. Clin. Microbiol. 43:3074–3082. Turton, J. F., M. E. Ward, N. Woodford, M. E. Kaufmann, R. Pike, D. M. Livermore, and T. L. Pitt. 2006. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol. Lett. 258:72–77. Turton, J. F., N. Woodford, J. Glover, S. Yarde, M. E. Kaufmann, and T. L. Pitt. 2006. Identification of Acinetobacter baumannii by detection of the blaOXA-51-like carbapenemase gene intrinsic to this species. J. Clin. Microbiol. 44:2974–2976. Ullah, J. H., T. R. Walsh, I. A. Taylor, D. C. Emery, C. S. Verma, S. J. Gamblin, and J. Spencer. 1998. The crystal structure of the L1 metallo-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J. Mol. Biol. 284:125–136. Vahaboglu, H., F. Budak, M. Kasap, G. Gacar, S. Torol, A. Karadenizli, F. Kolayli, and C. Eroglu. 2006. High prevalence of OXA-51-type class D -lactamases among ceftazidime-resistant clinical isolates of Acinetobacter spp.: co-existence with OXA-58 in multiple centres. J. Antimicrob. Chemother. 58:537–542. Villegas, M. V., K. Lolans, A. Correa, J. N. Kattan, J. A. Lopez, J. P. Quinn, and the Colombian Nosocomial Resistance Study Group. 2007. First identification of Pseudomonas aeruginosa isolates producing a KPC-type carbapenem-hydrolyzing -lactamase. Antimicrob. Agents Chemother. 51:1553– 1555. Villegas, M. V., K. Lolans, O. M. del Rosario, C. J. Suarez, A. Correa, A. M. Queenan, and J. P. Quinn. 2006. First detection of metallo--lactamase VIM-2 in Pseudomonas aeruginosa isolates from Colombia. Antimicrob. Agents Chemother. 50:226–229. Downloaded from cmr.asm.org at UNIV OF ARKANSAS on July 30, 2007 175. Pottumarthy, S., E. S. Moland, S. Jeretschko, S. R. Swanzy, K. S. Thomson, and T. R. Fritsche. 2003. NmcA carbapenem-hydrolyzing enzyme in Enterobacter cloacae in North America. Emerg. Infect. Dis. 9:999–1002. 176. Quale, J., S. Bratu, J. Gupta, and D. Landman. 2006. Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 50: 1633–1641. 177. Quale, J. M., D. Landman, P. A. Bradford, M. Visalli, J. Ravishankar, C. Flores, D. Mayorga, K. Vangala, and A. Adedeji. 2002. Molecular epidemiology of a citywide outbreak of extended-spectrum beta-lactamase-producing Klebsiella pneumoniae infection. Clin. Infect. Dis. 35:834–841. 178. Queenan, A. M., W. Shang, P. Schreckenberger, K. Lolans, K. Bush, and J. Quinn. 2006. SME-3, a novel member of the Serratia marcescens SME family of carbapenem-hydrolyzing -lactamases. Antimicrob. Agents Chemother. 50:3485–3487. 179. Queenan, A. M., C. Torres-Viera, H. S. Gold, Y. Carmeli, G. M. Eliopoulos, R. C. Moellering, Jr., J. P. Quinn, J. Hindler, A. A. Medeiros, and K. Bush. 2000. SME-type carbapenem-hydrolyzing class A -lactamases from geographically diverse Serratia marcescens strains. Antimicrob. Agents Chemother. 44:3035–3039. 180. Quinteira, S., and H. F. Luisa-Peixe. 2005. First isolation of blaVIM-2 in an environmental isolate of Pseudomonas pseudoalcaligenes. Antimicrob. Agents Chemother. 49:2140–2141. 181. Radice, M., P. Power, G. Gutkind, K. Fernandez, C. Vay, A. Famiglietti, N. Ricover, and J. Ayala. 2004. First class A carbapenemase isolated from Enterobacteriaceae in Argentina. Antimicrob. Agents Chemother. 48:1068– 1069. 182. Rasmussen, B. A., and K. Bush. 1997. Carbapenem-hydrolyzing -lactamases. Antimicrob. Agents Chemother. 41:223–232. 183. Rasmussen, B. A., K. Bush, D. Keeney, Y. Yang, R. Hare, C. O’Gara, and A. A. Medeiros. 1996. Characterization of IMI-1 -lactamase, a class A carbapenem-hydrolyzing enzyme from Enterobacter cloacae. Antimicrob. Agents Chemother. 40:2080–2086. 184. Riccio, M. L., N. Franceschini, L. Boschi, B. Caravelli, G. Cornaglia, R. Fontana, G. Amicosante, and G. M. Rossolini. 2000. Characterization of the metallo--lactamase determinant of Acinetobacter baumannii AC-54/97 reveals the existence of blaIMP allelic variants carried by gene cassettes of different phylogeny. Antimicrob. Agents Chemother. 44:1229–1235. 185. Richmond, M. H., and R. B. Sykes. 1973. The -lactamases of Gramnegative bacteria and their possible physiological role, p. 31–88. In A. H. Rose and D. W. Tempest (ed.), Advances in microbial physiology, vol. 9. Academic Press, New York, NY. 186. Ryoo, N. H., E.-C. Kim, S. G. Hong, Y. J. Park, K. Lee, I. K. Bae, E. H. Song, and S. H. Jeong. 2005. Dissemination of SHV-12 and CTX-M-type extendedspectrum -lactamases among clinical isolates of Escherichia coli and Klebsiella pneumoniae and emergence of GES-3 in Korea. J. Antimicrob. Chemother. 56:698–702. 187. Sader, H. S., A. O. Reis, S. Silbert, and A. C. Gales. 2005. IMPs, VIMs and SPMs: the diversity of metallo--lactamases produced by carbapenem-resistant Pseudomonas aeruginosa in a Brazilian hospital. Clin. Microbiol. Infect. 11:73–76. 188. Saino, Y., F. Kobayashi, M. Inoue, and S. Mitsuhashi. 1982. Purification and properties of inducible penicillin -lactamase isolated from Pseudomonas maltophilia. Antimicrob. Agents Chemother. 22:564–570. 189. Santillana, E., A. Beceiro, G. Bou, and A. Romero. 2007. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc. Natl. Acad. Sci. USA 104:5354–5359. 190. Sardelic, S., L. Pallecchi, V. Punda-Polic, and G. M. Rossolini. 2003. Carbapenem-resistant Pseudomonas aeruginosa-carrying VIM-2 metallo--lactamase determinants, Croatia. Emerg. Infect. Dis. 9:1022–1023. 191. Scaife, W., H.-K. Young, R. H. Paton, and S. G. B. Amyes. 1995. Transferable imipenem-resistance in Acinetobacter species from a clinical source. J. Antimicrob. Chemother. 36:585–586. 192. Schneider, I., A. M. Queenan, and A. Bauernfeind. 2006. Novel carbapenem-hydrolyzing oxacillinase OXA-62 from Pandoraea pnomenusa. Antimicrob. Agents Chemother. 50:1330–1335. 193. Scoulica, E. V., I. K. Neonakis, A. I. Gikas, and Y. J. Tselentis. 2004. Spread of blaVIM-1-producing E. coli in a university hospital in Greece. Genetic analysis of the integron carrying the blaVIM-1 metallo--lactamase gene. Diagn. Microbiol. Infect. Dis. 48:167–172. 194. Segal, H., and B. G. Elisha. 2005. Use of Etest MBL strips for the detection of carbapenemases in Acinetobacter baumannii. J. Antimicrob. Chemother. 56:598. 195. Senda, K., Y. Arakawa, S. Ichiyama, K. Nakashima, H. Ito, S. Ohsuka, K. Shimokata, N. Kato, and M. Ohta. 1996. PCR detection of metallo-lactamase gene (blaIMP) in gram-negative rods resistant to broad-spectrum -lactams. J. Clin. Microbiol. 34:2909–2913. 196. Shannon, K., A. King, and I. Phillips. 1986. -Lactamases with high activity against imipenem and Sch 34343 from Aeromonas hydrophila. J. Antimicrob. Chemother. 17:45–50. 197. Shibata, N., Y. Doi, K. Yamane, T. Yagi, H. Kurokawa, K. Shibayama, H. Kato, K. Kai, and Y. Arakawa. 2003. PCR typing of genetic determinants CARBAPENEMASES: THE VERSATILE -LACTAMASES 458 QUEENAN AND BUSH 218. Vourli, S., P. Giakkoupi, V. Miriagou, E. Tzelepi, A. C. Vatopoulos, and L. S. Tzouvelekis. 2004. Novel GES/IBC extended-spectrum -lactamase variants with carbapenemase activity in clinical Enterobacteria. FEMS Microbiol. Lett. 234:209–213. 219. Wachino, J.-I., Y. Doi, K. Yamane, N. Shibata, T. Yagi, T. Kubota, and Y. Arakawa. 2004. Molecular characterization of a cephamycin-hydrolyzing and inhibitor-resistant class A -lactamase, GES-4, possessing a single G170S substitution in the omega-loop. Antimicrob. Agents Chemother. 48:2905–2910. 220. Wachino, J.-I., Y. Doi, K. Yamane, N. Shibata, T. Yagi, T. Kubota, H. Ito, and Y. Arakawa. 2004. Nosocomial spread of ceftazidime-resistant Klebsiella pneumoniae strains producing a novel class A -lactamase, GES-3, in a neonatal intensive care unit in Japan. Antimicrob. Agents Chemother. 48:1960–1967. 221. Walsh, T. R. 2005. The emergence and implications of metallo--lactamases in Gram-negative bacteria. Clin. Microbiol. Infect. 11:2–9. 222. Walsh, T. R., A. Bolmstrom, A. Qwarnstrom, and A. Gales. 2002. Evaluation of a new Etest for detecting metallo--lactamases in routine clinical testing. J. Clin. Microbiol. 40:2755–2759. 223. Walsh, T. R., D. J. Payne, A. P. MacGowan, and P. M. Bennett. 1995. A clinical isolate of Aeromonas sobria with three chromosomally mediated inducible -lactamases: a cephalosporinase, a penicillinase and a third enzyme displaying carbapenemase activity. J. Antimicrob. Chemother. 37: 423–431. 224. Walsh, T. R., M. A. Toleman, L. Poirel, and P. Nordmann. 2005. Metallo-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 18:306–325. 225. Walther-Rasmussen, J., and N. Hoiby. 2006. OXA-type carbapenemases. J. Antimicrob. Chemother. 57:373–383. 226. Wang, C.-X., and Z.-H. Mi. 2004. IMP-1 metallo--lactamase-producing Pseudomonas aeruginosa in a university hospital in the People’s Republic of China. J. Antimicrob. Chemother. 54:1159–1160. 227. Wang, C., J. Wang, and Z. Mi. 2006. Pseudomonas aeruginosa producing VIM-2 metallo--lactamases and carrying two aminoglycoside-modifying enzymes in China. J. Hosp. Infect. 62:522–524. 228. Watanabe, M., S. Iyobe, M. Inoue, and S. Mitsuhashi. 1991. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 35:147–151. 229. Wei, Z.-Q., X.-X. Du, Y.-S. Yu, P. Shen, Y.-G. Chen, and L.-J. Li. 2007. Plasmid-mediated KPC-2 in a Klebsiella pneumoniae isolate from China. Antimicrob. Agents Chemother. 51:763–765. 230. Weldhagen, G. F., and A. Prinsloo. 2004. Molecular detection of GES-2 extended spectrum -lactamase producing Pseudomonas aeruginosa in Pretoria, South Africa. Int. J. Antimicrob. Agents 24:35–38. 231. Reference deleted. 232. Woodford, N., M. J. Ellington, J. M. Coelho, J. F. Turton, M. E. Ward, S. Brown, S. G. B. Amyes, and D. M. Livermore. 2006. Multiplex PCR for genes encoding prevalent OXA carbapenemases in Acinetobacter spp. Int. J. Antimicrob. Agents 27:351–353. 233. Woodford, N., P. M. Tierno, Jr., K. Young, L. Tysall, M.-F. I. Palepou, E. Ward, R. E. Painter, D. F. Suber, D. Shungu, L. L. Silver, K. Inglima, J. Kornblum, and D. M. Livermore. 2004. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A -lactamase, KPC-3, in a New York medical center. Antimicrob. Agents Chemother. 48:4793–4799. 234. Xiong, J., M. F. Hynes, H. Ye, H. Chen, Y. Yang, F. M’Zali, and P. M. Hawkey. 2006. blaIMP-9 and its association with large plasmids carried by Pseudomonas aeruginosa isolates from the People’s Republic of China. Antimicrob. Agents Chemother. 50:355–358. 235. Yamasaki, K., M. Komatsu, T. Yamashita, K. Shimakawa, T. Ura, H. Nishio, K. Satoh, R. Washidu, S. Kinoshita, and M. Aihara. 2003. Production of CTX-M-3 extended-spectrum -lactamase and IMP-1 metallo -lactamase by five Gram-negative bacilli: survey of clinical isolates from seven laboratories collected in 1998 and 2000, in the Kinki region of Japan. J. Antimicrob. Chemother. 51:631–638. CLIN. MICROBIOL. REV. 236. Yan, J.-J., P.-R. Hsueh, J.-J. Lu, F.-Y. Chang, W.-C. Ko, and J.-J. Wu. 2006. Characterization of acquired -lactamases and their genetic support in multidrug-resistant Pseudomonas aeruginosa isolates in Taiwan: the prevalence of unusual integrons. J. Antimicrob. Chemother. 58:530–536. 237. Yan, J.-J., W.-C. Ko, C.-L. Chuang, and J.-J. Wu. 2002. Metallo--lactamase-producing Enterobacteriaceae isolates in a university hospital in Taiwan: prevalence of IMP-8 in Enterobacter cloacae and first identification of VIM-2 in Citrobacter freundii. J. Antimicrob. Chemother. 50:503–511. 238. Yan, J.-J., W.-C. Ko, and J.-J. Wu. 2001. Identification of a plasmid encoding SHV-12, TEM-1, and a variant of IMP-2 metallo--lactamase, IMP-8, from a clinical isolate of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45:2368–2371. 239. Yan, J.-J., J.-J. Wu, S.-H. Tsai, and C.-L. Chuang. 2004. Comparison of the double-disk, combined disk, and Etest methods for detecting metallo-lactamases in gram-negative bacilli. Diagn. Microbiol. Infect. Dis. 49:5–11. 240. Yan, J. J., P. R. Hsueh, W. C. Ko, K. T. Luh, S. H. Tsai, H. M. Wu, and J. J. Wu. 2001. Metallo--lactamases in clinical Pseudomonas isolates in Taiwan and identification of VIM-3, a novel variant of the VIM-2 enzyme. Antimicrob. Agents Chemother. 45:2224–2228. 241. Yang, Y., and K. Bush. 1996. Biochemical characterization of the carbapenem-hydrolyzing -lactamase AsbM1 from Aeromonas sobria AER 14M: a member of a novel subgroup of metallo--lactamases. FEMS Microbiol. Lett. 137:193–200. 242. Yang, Y., B. A. Rasmussen, and K. Bush. 1992. Biochemical characterization of the metallo--lactamase CcrA from Bacteroides fragilis TAL3636. Antimicrob. Agents Chemother. 36:1155–1157. 243. Yang, Y., P. Wu, and D. M. Livermore. 1990. Biochemical characterization of a -lactamase that hydrolyzes penems and carbapenems from two Serratia marcescens isolates. Antimicrob. Agents Chemother. 34:755–758. 244. Yano, H., A. Kuga, R. Okamoto, H. Kitasato, T. Kobayashi, and M. Inoue. 2001. Plasmid-encoded metallo--lactamase (IMP-6) conferring resistance to carbapenems, especially meropenem. Antimicrob. Agents Chemother. 45:1343–1348. 245. Yigit, H., A. M. Queenan, G. J. Anderson, A. Domenech-Sanchez, J. W. Biddle, C. D. Steward, S. Alberti, K. Bush, and F. C. Tenover. 2001. Novel carbapenem-hydrolyzing -lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45:1151– 1161. 246. Yigit, H., A. M. Queenan, K. Rasheed, J. W. Biddle, A. Domenech-Sanchez, S. Alberti, K. Bush, and F. C. Tenover. 2003. Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzing -lactamase KPC-2. Antimicrob. Agents Chemother. 47:3881–3889. 247. Yong, D., S. Choi Yeoung, H. Roh Kyoung, K. Kim Chang, H. Park Youn, H. Yum Jong, K. Lee, and Y. Chong. 2006. Increasing prevalence and diversity of metallo--lactamases in Pseudomonas spp., Acinetobacter spp., and Enterobacteriaceae from Korea. Antimicrob. Agents Chemother. 50: 1884–1886. 248. Yong, D., K. Lee, J. H. Yum, H. B. Shin, G. M. Rossolini, and Y. Chong. 2002. Imipenem-EDTA disk method for differentiation of metallo--lactamase-producing clinical isolates of Pseudomonas spp. and Acinetobacter spp. J. Clin. Microbiol. 40:3798–3801. 249. Yu, Y.-S., X.-X. Du, Z.-H. Zhou, Y.-G. Chen, and L.-J. Li. 2006. First isolation of blaIMI-2 in an Enterobacter cloacae clinical isolate from China. Antimicrob. Agents Chemother. 50:1610–1611. 250. Yum, J. H., D. Yong, K. Lee, H.-S. Kim, and Y. Chong. 2002. A new integron carrying VIM-2 metallo--lactamase gene cassette in a Serratia marcescens isolate. Diagn. Microbiol. Infect. Dis. 42:217–219. 251. Zavascki, A. P., P. B. Gaspareto, A. F. Martins, A. L. Goncalves, and A. L. Barth. 2005. Outbreak of carbapenem-resistant Pseudomonas aeruginosa producing SPM-1 metallo--lactamase in a teaching hospital in southern Brazil. J. Antimicrob. Chemother. 56:1148–1151. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 459–477 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00039-06 Vol. 20, No. 3 Atmospheric Movement of Microorganisms in Clouds of Desert Dust and Implications for Human Health Dale W. Griffin* U.S. Geological Survey, St. Petersburg, Florida 33701 INTRODUCTION .......................................................................................................................................................459 OCCURRENCE...........................................................................................................................................................461 Bacteria ....................................................................................................................................................................461 Fungi.........................................................................................................................................................................465 Viruses ......................................................................................................................................................................465 PATHOGENS ..............................................................................................................................................................466 Bacteria ....................................................................................................................................................................466 Fungi.........................................................................................................................................................................466 Viruses ......................................................................................................................................................................467 Unknown Agents .....................................................................................................................................................469 DETECTION ...............................................................................................................................................................470 Collection .................................................................................................................................................................470 Identification............................................................................................................................................................471 CONCLUSIONS AND PERSPECTIVES.................................................................................................................472 ACKNOWLEDGMENT..............................................................................................................................................472 REFERENCES ............................................................................................................................................................472 The larger deserts on the planet, which include the Sahara and Sahel regions of North Africa and the Gobi, Takla Makan, and Badain Jaran deserts of Asia, are the primary sources of mobilized desert top soils that move great distances through the atmosphere each year (Fig. 1). The current estimate for the annual quantity of desert dust that makes regional or global airborne migrations is 0.5 to 5.0 billion tons (184). While it is believed that the Sahara and Sahel regions of North Africa have been and are the dominant sources of dust in the atmosphere (50 to 75% of the current estimate), there has been increased Asian dust activity over the last 20 years that has been attributed to climate change and desertification (73, 166, 191, 265). Between 1975 and 1987, the desertification rate in China was ⬃2,100 km2 year⫺1 (266). Other regions of known dust storm activity include the arid regions of the continental United States (the Great Basin), Central America, South America (Salar de Uyuni), Central Australia, South Africa (Etosha and Mkgadikgadi basins), and the Middle East (244). In general, high-energy wind conditions in arid regions can result in the mobilization of significant quantities of soils into the atmosphere, and large dust storm events are capable of continent-wide, transoceanic, and global dispersion (68, 193). Dust emanates from North Africa year-round and at times throughout the year impacts air quality in Africa, the Middle East, Europe, Asia, the Caribbean, and the Americas (Fig. 2). Dust source areas in the Sahara and Sahel, primary latitudinal transport routes, and the influence of climate and climate systems (North Atlantic Oscillation and El Niño, etc.) on year- * Present address: U.S. Geological Survey, 2010 Levy Ave., Tallahassee, FL 32310. Phone: (850) 942-9500, ext. 3062. Fax: (850) 9429521. E-mail: [email protected]. 459 c to-year dust flux have been previously reported (73, 74, 156, 166, 189, 191, 192, 212, 224, 235). Dust storm activity in the deserts of Asia is seasonal, with the majority of atmospheric transport occurring during the spring (February to May) (258). Although Asian dust generation is seasonal, significant quantities are generated and can be dispersed universally in the Northern Hemisphere. A large Asian dust event in 1990 moved across the Pacific, the North American continent, and the Atlantic Ocean and was later identified in the French Alps via isotopic analysis of deposited particulate matter (84). A large dust event impacting the west coast of North America in 1998 reduced solar radiation levels by 30 to 40% and left a chemical fingerprint of deposited dust extending inland to the state of Minnesota (100). Asian dust storms can take from 7 to 9 days to cross the Pacific Ocean. Dust storms moving off the west coast of Africa can take from 3 to 5 days to reach the Caribbean and Americas. As Fig. 1 illustrates, continuous transmission off the North African deserts can result in prolonged exposure of individuals living at considerable distances (the Caribbean and Americas) from the source of particulates. Using a handheld laser particle counter, I recorded a background particulate load of 2.6 ⫻ 106 airborne particles m⫺3 at a location south of Tampa Bay, Florida, on 15 July 2005. During an African dust event that occurred from 25 to 28 July 2005, the particle count in the same region south of Tampa Bay on 25 July was 26.1 ⫻ 106 particles m⫺3. Ninety-nine percent of the particles were within a size range of ⬎0.3 m to ⬍1.0 m, the sub-2.5-m fraction that can penetrate deep into the lung environment (Fig. 1C is an image of that dust event period). These data demonstrate the ability of dust storms to impact air quality at significant distances from their sources (Tampa Bay is ⬎6,500 km west of the coast of North Africa). A higher risk to human health would obviously be associated with populations closer to dust INTRODUCTION 460 GRIFFIN CLIN. MICROBIOL. REV. FIG. 1. African and Asian dust storms. Stars identify dust cloud source regions, and arrows identify dust clouds and the general direction of movement. (A) NASA image, via the moderate-resolution imaging spectroradiometer (MODIS) aboard the Terra satellite, of a dust storm blowing over the Sea of Japan on 1 April 2002. (Image courtesy of Jacques Descloitres, MODIS Land Rapid Response Team, NASA/Goddard Space Flight Center.) (B) NASA image, via MODIS, of a dust storm blowing out of Africa over the Mediterranean Sea in the direction of Turkey. The black spot in the tongue of dust is the Troödos mountain range of Cyprus, which protrudes through the top of the dust cloud. The image was taken on 25 February 2006. (Courtesy of Jeff Schmaltz, MODIS Land Rapid Response Team, NASA/Goddard Space Flight Center.) (C) NASA SeaViewing Wide Field-of-View Sensor (SeaWiFS) image of a large dust cloud blowing across the Atlantic. The image was taken on 19 July 2005 and is courtesy of the SeaWiFS Project, NASA/Goddard Space Flight Center, and ORBIMAGE. This dust cloud impacted the air quality in Florida. Airborne particle measurements taken by the author with a handheld laser particle counter south of Tampa Bay, FL, went from 2.6 ⫻ 106 m⫺3 on 15 July 2005 (normal clear atmosphere) to 26.1 ⫻ 106 m⫺3 on 25 July 2005 (dust conditions). Over 90% of the particles ranged from ⬎0.3 to 0.5 m in size. source regions, as this excerpt regarding the American Dust Bowl period illustrates: It is stated by some authorities that man breathes, on an average, approximately 25 pounds of air daily. Nelson’s “Loose Leaf Living Medicine” says that a normal individual inhales 30.5 cubic inches of air at each breath and breathes 17 times per minute. Then with an average of 0.0368 g of dust per cubic foot of air over a period of ten hours (the average duration of the dust storms) the straining apparatus of the respiratory system is confronted with a very large task. From above figures it can be computed that an individual breathes 6.6240 g of dust during an average dust storm (18). Every human breath taken is laden with particulate matter, and human evolution produced the most obvious and familiar front line of defense, nose hair. Less obvious are the mucus glands that line our airways. These glands function to trap and aid in the expulsion of particulates via secretion, ciliated transport, and ingestion or cough. Of particular concern are particles of ⬍10 m in size that can penetrate into the lungs and those of ⬍2.5 m that may penetrate into deep lung tissue and the subepithelial environment. These very small particles cause adverse health effects via oxidative stress (47, 52, 263). The deposition rate of ultrafine particles (⬍100 nm) in the lungs has been shown to increase as particle size decreases and to increase with exercise versus resting (46). Health studies conducted in urban and suburban environments have demonstrated mortality risk with exposure to particulate matter and have attributed this risk to anthropogenic particulates generated through automotive and industrial combustion versus those of crustal origin (130, 210). Several studies conducted to investigate the role of dust storms that consist of concentrated crustal particulates have shown an associated allergic, asthma, and silicosis/pulmonary fibrosis risk (36, 127, 173, 180, 202, 259). Areas impacted by desert dust storms, such as communities in the Middle East and the Caribbean, are known to have some of the highest incidences of asthma on the planet (8, 16, 96). On the Caribbean island of Barbados, the incidence of asthma increased 17-fold between 1973 and 1996, a period that coincided with increased flux of African dust to the area of the island (97, 188). Gyan et al. recently demonstrated a link between African dust and pediatric respiratory stress on the southern Caribbean island of Trinidad (85). Allergens commonly associated with dust storms include fungal spores, plant and grass pollens, anthropogenic emissions, and organic detritus (62, 103, 125). Desert dust cloud toxicity may be influenced by anthropogenic material as a result of particulate/pollutant aerosoliza- VOL. 20, 2007 DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 461 FIG. 2. Primary sources of desert dust and their atmospheric pathways. (1) During summer in the Northern Hemisphere (approximately June through October), African desert dust is transported across the Atlantic to the northern Caribbean and North America. (2) During winter in the Northern Hemisphere (approximately November through May), African desert dust is transported across the Atlantic to the southern Caribbean and South America. (3) The Asian dust season typically lasts from late February to late April. (4) Large Asian dust events can travel significant distances in the Northern Hemisphere. Yellow lines show Asian desert dust atmospheric routes, orange lines show African dust routes, brown lines show routes of other desert dust sources, and broken black lines depict wind patterns. (Base map image courtesy of NASA’s Geospatial Interoperability Office, GSFC [http://viewer.digitalearth.gov/].) tion during cloud formation or cloud capture during downwind transport (adsorption of pesticides, herbicides, and industrial emissions, etc.). Inherent toxicity is due to differences in native soil elemental composition (metals and naturally occurring or synthetic radioisotopes, etc.), atmospheric chemical alteration, size fractionation, and extreme particulate load (5, 44, 88, 131, 174, 176, 263). Toxic metals such as arsenic and mercury have been shown to occur in airborne desert dust in downwind environments at concentrations higher than regional crustal concentrations (93). In addition to the presence of these toxic metals, dust can indirectly impact human health by spurring toxic algal blooms in coastal environments (i.e., red tides, in which marine organisms utilize dust components such as iron as a nutrient in nutrient-depleted waters) (93, 138, 243). Dust clouds may contain high concentrations of organics composed of plant detritus and microorganisms (80, 108) and may pick up additional biological loads (fungal spores, bacteria, viruses, and pollen, etc.) as the clouds move through and sandblast downwind terrestrial environments and/or over aquatic environments through the adhesion of microbe-laden fine aquatic sprays to dust particles. All of these potential dust cloud constituents may negatively influence human health in regional and downwind environments, with the greatest risk factors being frequency of exposure, concentration of and composition of particulates, and immunological status. Dust-borne microorganisms in particular can directly impact human health via pathogenesis, exposure of sensitive individuals to cellular components (pollen and fungal allergens and lipopolysaccharide [LPS], etc.), and the development of sensitivities (i.e., asthma) through prolonged exposure. Dust-borne dispersion of microorganisms may also play a significant role in the biogeographical distribution of both pathogenic and nonpathogenic species, as long-range atmospheric transport routes and concentrations shift through time due to climatic and geologic change (148, 163). OCCURRENCE Reflecting on the whole of this, I conclude that the germs float through the atmosphere in groups or clouds, and that now and then a cloud specifically different from the prevalent ones is wafted through the air. The touching of a nutritive fluid by a Bacteria cloud would naturally have a different effect from the touching of it by the sterile air between two clouds. But, as in the case of a mottled sky, the various portions of the landscape are successively visited by shade, so, in the long run, are the various tubes of our tray touched by the Bacterial clouds, the final fertilization or infection of them all being the consequence (236). Bacteria Desert topsoils are laden with viable and diverse prokaryote communities (29, 32, 53, 124, 177). Globally, a gram of topsoil contains ⬃107 (forest) to 109 (arid and other soil types) prokaryotes (247). These populations are believed to consist of ⬃10,000 bacterial types with ⬃0.1% of the total population composing ⬃99.9% of the diversity (67, 233, 234). Studies that have examined the number of culturable prokaryotes in desert soils have reported concentrations ranging from 0 to ⬃107 gram⫺1 (126, 144, 169). Dominant phyla found in soils, as determined by their prevalence in sequence libraries, include the Proteobacteria, Acidobacteria, and Actinobacteria (110). Although this community 462 GRIFFIN CLIN. MICROBIOL. REV. TABLE 1. Genera of bacteria and fungi found in dust storm samples where identified to at least the genus levela Bacterial genus/genera Fungal genera Method of identification (bacterial/fungal) Location (reference) Dust source region Arthrobacter, Bacillus, Cryptococcus, Flavimonas, Kurthia, Neisseria, Paenibacillus, Pseudomonas, Ralstonia, and Staphylococcus Alternaria, Cryptococcus, Mortierella, Penicillium, Phoma, Rhodotorula, and Stemphylium Fatty acid methyl esters and 16S rRNA gene sequencing/26S rRNA gene sequencing Kuwait (141) Middle East Bacillus, Pseudomonas, and Staphylococcus Alternaria, Aspergillus, Botrytis, Cladosporium, Mortierella, Mucor, Penicillium, Pythium Ulocladium, and Verticillium Microscope/microscope Saudi Arabia (126) Saudi Arabia Arthrobacter, Corynebacterium, Microbacterium, Nocardioides, Planococcus, Saccharothrix, and Streptomyces Alternaria, Cladosporium, Microsporum, and Penicillium 16S rRNA gene sequencing/ microscope and 18S rRNA gene sequencing Turkey (82) Sahara Arthrobacter, Agrococcus, Bacillus, Curtobacterium, Duganella, Kocuria, Massilia, and Microbacterium Acremonium, Alternaria, Cladosporium, Fusarium, Microsporum, Penicillium, and Trichophyton 16S rRNA gene sequencing/ microscope and 18S rRNA gene sequencing Turkey (82) Middle East ND Alternaria, Aspergillus, Cladosporium, Coleophoma, Fusarium, Libertella, Lophiostoma, Penicillium, Phoma, and Zygosporium NA/microscope Israel (207) Sahara Acinetobacter, Agrococcus, Arthrobacter, Aureobacterium, Bacillus, Corynebacterium, Deinococcus, Dietzia, Gordonia, Kocuria, Microbacterium, Micrococcus, Paenibacillus, Paracoccus, Planococcus, Rhodococcus, Saccharococcus, Staphylococcus, Streptomyces, and Zoogloea Cladosporium, Alternaria, and Aspergillus 16S rRNA gene sequencing/ 18S rRNA gene sequencing Mali, Africa (117) Sahara/Sahel Actinomyces, Bacillus, Brevibacterium, Cellulomonas, Frigoribacterium, Gordonia, Kocuria, Lechevalieria, Leifsonia, Lentzea, Novosphingobium, Pseudomonas, and Staphylococcus Alternaria, Cladosporium, Dendryphion, Lojkania, Lithothelium, Massaria, Myriangium, Neotestudina, Penicillium, Phoma, Setosphaeria, Stachybotrys, Trichophyton, and Ulocladium 16S rRNA gene sequencing/ 18S rRNA gene sequencing Tropical mid-Atlantic ridge (83) Sahara/Sahel Actinosynnema, Afipia, Agrococcus, Ancylobacter, Arthrobacter, Bacillus, Bosea, Curtobacterium, Frankiaceae, Fulvimaria, Hymenobacter, Kocuria, Kineococcus, Mesorhizobium, Nocardioides, Paracoccus, Propionibacterium, Pseudomonas, Rhizobium, Saccharothrix, Sphingomonas, Sinorhizobium, Streptomyces, and Taxeobacter Acremonium, Aspergillus, Aureobasidium, Bipolaris, Chromelosporium, Chrysosporium, Cladosporium, Coccodinium, Cochliobolus, Geotrichum, Gibberella, Microsporum, Monocillium, Nigrospora, Oidiodendron, Paecilomyces, Penicillium, Pleospora, Rhizomucor, Scytalidium, and Trichophyton 16S rRNA gene sequencing/ 18S rRNA gene sequencing U.S. Virgin Islands (78, 79) Sahara/Sahel Bacillus ND Microscope/NA Sweden (20) North of the Black Sea Bacillus Mycelia sterilia (unidentified imperfect fungi with no known spore stage, 48% of isolates), Alternaria, Arthrinium, Aspergillus, Cladosporium, Curvularia, Neurospora, Penicillium, and Periconium Microscope/microscope Barbados (190) Sahara/Sahel ND Fusarium, Aspergillus, Penicillium, and Basipetospora NA/microscope Korea (261) Gobi/Takla Makan ND Penicillium, Aspergillus, Nigrospora, Arthrinium, Curvularia, Stemphylium Cercospora, and Pithomyces NA/microscope Korea (256) Gobi/Takla Makan Continued on facing page VOL. 20, 2007 DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 463 TABLE 1—Continued Bacterial genus/genera ND a Fungal genera Alternaria, Arthrinium, Aspergillus, Botrytis, Cercospora, Cladosporium, Curvularia, Drechslera, Fusarium, Ganoderma, Nigrospora, Papulara, Penicillium, Periconia, Pithomyces, Stemphylium, and Ulocladium Method of identification (bacterial/fungal) Location (reference) Dust source region NA/microscope Taiwan (92) Gobi/Takla Makan All studies listed were culture based. ND, no data; NA, not applicable. composition is determined by a number of factors, including pH, temperature, elemental composition, and nutrient and moisture content, members of other phyla also occur less frequently (67, 110). Pigments produced by desert soil taxa such as Nostoc sp. and Scytonema sp. are believed to provide UV shielding compared to less-pigmented community members (21). Novel or rare actinomycetes (Citricoccus alkalitolerans, Jiangella gansuensis, and species of Nocardiopsis and Saccharothrix) have been isolated from a number of desert soil samples collected in the deserts of both Africa and Asia (216, 267). Similar to the potential hazards of life in the soil, atmospheric sources of stress to airborne microorganisms include UV damage, desiccation (drying by wind), temperature (both low and high temperatures depending on the tolerance range of an organism), and atmospheric chemistry (humidity levels dependent on tolerance and oxygen radicals, etc.) (54, 76, 158). Dust-borne transport of microorganisms, particularly over aquatic environments, should be enhanced due to tolerable humidity levels and attenuation of UV by the particle load of the various dust clouds (78). NASA research has shown that the particle load of large dust clouds can attenuate UV by more than 50% (91). Table 1 lists the genera identified in dust storm microbiology studies as described below. While it would be interesting to compare and contrast the diversity of isolates identified in each study, it should be noted that some relied on microscopy for identification while others employed molecular methods. It should also be emphasized that all were culture-based studies and few utilized like assays (i.e., collection and culture media, etc.), which restricts the comparison of data. Desert dust research in Kuwait, which focused on “military personnel health protection,” identified 147 bacterial CFU, which included representatives from 10 genera, by gas chromatographic analysis of fatty acid methyl esters and 16S rRNA gene sequencing in settled dust (141). Seven genera were isolated from the atmosphere over Erdemli, Turkey, during Saharan dust events in March 2002, with the most prevalent being species of Streptomyces (82). During a dust event of Middle East origin that impacted the same location in October 2002, species from eight genera were recovered, with the most prevalent belonging to the genus Kocuria (82). In Bamako, Mali, Africa, Kellogg et al. identified a small subset of the observed (n ⫽ 94 [by 16S rRNA gene sequencing]) airborne bacterial isolates collected during four dust events and one nondust event. These isolates were composed of 20 genera, and Bacillus sp. represented 38% of the isolates, followed by Kocuria sp. (12.8%), Planococcus sp.(8.5%), Saccharococcus sp. (7.4%), and Micrococcus sp. (6.4%) (117). Twenty-five bacterial iso- lates collected from the atmosphere over the mid-Atlantic ridge (⬃15°N, 45°W) during periods of elevated African desert dust concentrations consisted of 13 genera, with those dominant being Bacillus (32%), Gordonia (12%), and Staphylococcus (8%) (83). When African dust was visibly present in the northern Caribbean air, a subset of isolates from various samples was identified by 16S rRNA gene sequences as consisting of 25 genera (78, 79). The most dominant genera detected in these samples were Microbacterium (14.4%), Sphingomonas (7.2%), Bacillus (6.5%), and Streptomyces (4.0%). Eleven of the isolates (closest GenBank neighbor) had previously been identified in marine environments, demonstrating that aerosolized marine spray may adhere to dust particles as the clouds move over marine environments (78, 79). The few other dust storm-related studies devoted to the prevalence of bacteria in atmospheric samples were based on the presence of spores (in aerobic culture; i.e., species of Bacillus were identified) or other cellular morphologies (19, 126, 190) or on only reported CFU (39). Concentrations of bacteria observed during dust storms are noted in Table 2. Between April and November 1934, 30 flights were conducted at altitudes of 0 to 7,772 m in the vicinity of Boston, MA, to collect air samples for microbial analysis (187). Flight 4 recovered samples at altitudes of 457 to 7,772 m at a time when visibility was hampered by dust transported within air masses from over the tropical Atlantic (area of the Sargasso Sea). These samples resulted in the growth of 16 to 266 bacteria (overall average of the five samples, 144) and 11 to 43 fungi (overall average of five samples, 23) m⫺3 of air (187). These CFU counts were by far the highest combined (bacterial and fungal) counts for all of the flights (for the other 29 flights, there were 137 samples yielding 0 to 138 bacterial CFU, average 14, and 0 to 267 fungal CFU, average 11). Flight 4 was the only flight of the 30 to be affected by Transatlantic dust (187). In Kansas, in 1935, numbers of bacteria on nutrient agar settle plates exposed to dust storms ranged from 2,880 to 42,735 m⫺2 min⫺1 (23). Images of a clear-day petri dish and a dust storm petri dish (both plates exposed to the atmosphere for 1.5 min.) showed fewer than 10 colonies isolated on the clear day and overgrowth on the dust storm plate (23). Atmospheric samples collected by plane from three altitudes (365, 1,280, and 2,500 m) over Junction, TX, demonstrated that the highest concentrations of bacterial and fungal isolates occurred when dust generated by frontal activity was present (66). Graph-based data showed ⬎1,544 combined CFU m⫺3 at the 365-m altitude versus ⬍450 combined CFU m⫺3 at each of the three altitudes during normal/no-dust conditions. The lowest CFU collections (⬍100 at each altitude) followed frontal precipitation events 464 GRIFFIN CLIN. MICROBIOL. REV. TABLE 2. Concentrations of culturable bacteria and fungi and fungal spores in dust storms Sample site (reference) Dust storm source Sampling methoda Concnb (bacterial CFU m⫺3) (avg) Background Boston, MA (187) Air mass from over the Sargasso Sea Kansas (23) Kansas Junction, TX (66) Texas Saudi Arabia (126) Saudi Arabia Mali (117) Sahara/Sahel Israel (207) Sahara Turkey (82) Sahara/region Tropical midAtlantic (15°N, 45°W) (83) U.S. Virgin Islands (78) U.S. Virgin Islands (79) Barbados (190) Sahara/Sahel Sahara/Sahel Korea (39) Taiwan (92) Taiwan (256) Gobi/Takla Mahan Gobi/Takla Mahan Gobi/Takla Mahan Sahara/Sahel Sahara/Sahel Oil-saturated lens paper from a plane; altitudes, 0–7,620 m For background, 1.5 min; for dust storms, 5- to 15-s exposures of nutrient agar settle plates Exposure of nutrient agar plates from a plane; altitudes, 365–2,500 m Dust plate trap followed by a dilution method (also 6-h nutrient agar settle plate counts) ⬍1 h, low-flow rate; MF 25 min, 28.3 liters min⫺1; Andersen sampler ⬍1 h, low-flow rate; MF ⬍1–12 h, low-flow rate; MF ⬍1 h, low-flow rate; MF ⬍1 h, low-flow rate; MF ⬃24 h, low-flow rate; MF Andersen sampler Spore trap Spore trap 0–138 (14) Dust 16–266 (144) Photograph of one plate with less than 10 colonies ⬍450 (avg) combined bacterial and fungal CFU; NS 2,880–42,735 ⬎1,544 (avg) combined bacterial and fungal CFU; NS 1,892 ⫾ 325 (100⫻ increase over background) ND 200–1,100 (537) 720–15,700 (6,194) Concnc (fungal CFU m⫺3) (avg) Background 0–267 (11) Dust 11–43 (23) ND ND See bacterial concn See bacterial concn ND 869 ⫾ 75 (40⫻ increase over background) 0–130 (62) 80–370 (195) 31–115 (73) 205–226 (215) 79–108 (93) 694–995 (844) 0–41* (3) 0–59† (6) 0–291* (25) NA 0–24 (1)‡ NA 0–100 (12) 90–350 (203) 0–60 (15) 30–60 (48) 1–100 (13) 0–353 (80) 0–57 (13) 0–90 (31) 0–(⬃10) (NS) 0–(⬃20) (NS) 0–(⬃4) (NS) 0–(⬃16) (NS) 225–8,212 (1,642) ND ND 100–8,510 (1,702) 4,839 28,683 336–6,992 (1,398) 6,078 29,038 105–1,930 (386) ND ND 0–703† (66) 0–27 (3)‡ Low-flow rate, ⬍20 liters min⫺1; MF, membrane filtration. Values for the Kansas study are bacterial cells per square meter per min. NA, not applicable due to some level of dust always being present; ND, no data; NS, not specified (graph-based data); *, dust concentration below 12 g m⫺3; †, dust concentration at or above 12 g m⫺3; ‡, dust concentration above 6 g m⫺3 at 10 m above sea level. c Values for the Taiwan studies are mean numbers of spores per cubic meter. NA, not applicable due to some level of dust always being present; ND, no data; NS, not specified (graph-based data); *, dust concentration below 12 g m⫺3; †, dust concentration at or above 12 g m⫺3; ‡, dust concentration above 6 g m⫺3 at 10 m above sea level. a b (66). In Saudi Arabia, an increase of 100% over background levels was observed during dust storms with a settle plate technique. Kellogg et al. (117) reported background levels of 200 to 1,100 bacterial CFU m⫺3 in the atmosphere over Bamako, Mali, Africa, and dust condition levels ranging from 720 to 15,700 bacterial CFU m⫺3. During two Saharan dust events impacting Israel’s atmosphere (January and April 2004), airborne bacteria averaged 844 CFU m⫺3, versus a 2-day background average of 93 CFU m⫺3, and ratios of total CFU (bacteria and fungi) for dust days versus non-dust days were similar to those reported by Griffin et al. in the Caribbean (207). At Erdemli, Turkey, dust event bacterial concentrations ranged from 0 to 59 CFU m⫺3 (average, 6), versus background concentrations of 0 to 41 CFU m⫺3 (average, 3) (82). In the northern Caribbean (⬃18°N), normal background concentrations of bacteria in air samples collected over the islands and open water averaged 13.6 CFU m⫺3 (28 samples) (79). The same study noted an increase in airborne bacterial CFU (average, 80.1 CFU m⫺3 [15 samples]) when African desert dust was visibly present in the atmosphere, with the higher concen- trations (average, 143.1 CFU m⫺3; n, 8) occurring during late July and early August (79). Dust-borne microbial concentrations in the early summer (May, June, and early July) were not distinguishable above normal background levels in this Caribbean research site (79). To determine if bacteria and fungi being transported to the Caribbean in the early summer months of May and June could be detected in air samples at a site closer to the continent of Africa, a study was conducted over the mid-Atlantic ridge at 15°N over a 6-week period (22 May to 30 June 2003). Results showed that bacterial CFU ranged from 0.1 CFU to 23.7 CFU m⫺3 when culturable bacteria were recovered from the air samples (12 of 85 samples) (83). The U.S. Navy’s Naval Aerosol Analysis and Prediction System, an atmospheric model that can determine airborne aerosol concentrations at various altitudes on a global scale (94, 114, 196, 209), demonstrated that most of the microorganisms recovered in the mid-Atlantic study were recovered during periods of elevated aerosol (dust) concentrations (83). On the island of Barbados, at ⬃13.1°N in the southern Caribbean, African desert dust-borne bacterial CFU (morphological iden- VOL. 20, 2007 DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 465 tifications were via spore staining, and almost all were identified as Bacillus sp.) ranged from 0.0 to approximately 20.0 CFU m⫺3, with the highest concentrations occurring primarily during the summer (190). During Asian dust events impacting air quality in Taejon, Korea, the bacterial CFU concentration increased on average 4.3 times over that observed under normal atmospheric conditions (39). (78, 79, 82, 117). Concentrations of fungal CFU and spores observed during dust storms at various geographical locations are noted in Table 2. In all cases, with the exception of the data reported by Choi et al. (39) (Table 2), the presence of desert dust in the atmosphere resulted in an increase in the concentration of culturable fungi or fungal spores relative to background or clear atmospheric conditions. Fungi Viruses The total number of fungal species has been estimated at 1.5 ⫻ 106, with approximately 7.4 ⫻ 104 to 1.2 ⫻ 105 having been identified (89). The number of fungi typically found in a gram of topsoil is approximately 106 (225). One of the genetic advantages that fungi have over many other microorganisms is that they are capable of producing spores. Spores enhance survival during transport and periods of prolonged environmental stress. Fungal spores are egg-like vesicles covered with a thin layer of hydrophobic proteins that provide protection from physical stresses such as UV exposure and desiccation (56). Viable fungi recovered from extreme altitudes in the atmosphere have demonstrated the ability to survive harsh conditions (77, 142, 143, 153, 200, 242). Mycological studies conducted in desert environments have demonstrated that diverse communities exist in desert topsoils worldwide (1, 2, 9, 165, 218). An early review of outdoor airborne concentrations of fungal spores reported that the highest concentrations typically occur in temperate and tropical regions (106 spores m⫺3 of air) and the lowest in desert environments (400 spores m⫺3 of air) (129). In a survey of airborne fungi in the eastern and western deserts of Egypt, 44 genera and 102 species (Aspergillus was the dominant genus) were recovered, with the areas of highest fungal concentrations and diversity corresponding to increases in vegetative cover and anthropogenic activity (107). The most prevalent fungal genera in airborne dust samples collected from the atmosphere of Taif, Saudi Arabia, were (31 genera and 70 species) Aspergillus, Drechslera, Fusarium, Mucor, Penicillium, Phoma, and Stachybotrys, and it was noted that genus prevalence was dependent on the nutrient medium used (3). A similar study conducted in Egypt identified 27 genera (64 species), and again, prevalence was determined by the nutrient medium employed (4). While most studies have shown that desert environments harbor diverse mycological communities, fungi such as Coccidioides immitis and Coccidioides posadasii are known to be restricted geographically (the Americas in this case) (90). In contrast to microbial studies of soil or air environments, far fewer studies have been devoted to dust-borne transport of microorganisms. Table 1 lists those fungal genera found in dust storm studies where dust-associated isolates were identified to at least the genus level. Several studies, while identifying bacterial and or fungal isolates, did not distinguish those recovered during clear versus dust conditions and thus are not included in Table 1 (66, 187). As can be seen from Table 1, the dust-associated fungal community is diverse, and the true extent of diversity is probably much greater given that some of the more recent studies, which used molecular methods for identification, identified only a small fraction of the cultured isolates due to budget constraints Viral abundance in soil has been shown to occur at a concentration of ⬃108 per gram (198, 252). Certain desert soil characteristics (high temperatures, elemental composition [the ability of viruses to adsorb to soil particulates], soil chemistry [pH], and moisture content) are known to impact viral survival and thus community composition and concentration (260). Poliovirus and bacteriophage MS2 survival in seeded desert soil samples was limited in the summer months when temperatures of ⬃33.0°C and a decrease in soil moisture content (from 40% to ⬍5%) were recorded (in comparison to winter survival) (222). Low pH, low moisture, and high concentrations of clay, cations, and total dissolved solids have been shown to enhance virus survival by increasing adsorption of viruses to other soil constituents (99, 215, 260). As in soil, milder temperatures and moderate to high humidity (50 to 80%, depending on the virus type) favor viral survival in aerosols, with atmospheric transport over open bodies of water (higher humidity versus that of overland environments) favoring long-range dispersion and infection (70, 104, 217). Bacteriophages that integrate their genome into the host genome (prophage) may be more apt to survive long-range atmospheric transport than are virulent phages (phages that directly replicate and lyse host cells following infection and therefore do not benefit from the shielding potential of a host cell). The scientific community has yet to address this topic of research. Since prophages are persistent in environmental bacterial populations and are important vectors of bacterial virulence factors (and other genomic material), these organisms may play an unforeseen role in the global dispersion of prokaryotic genomic information through long-range atmospheric dispersion (30, 146). Viral transmission in aerosols (influenza viruses and rhinoviruses, etc.) has been reviewed, and most of the documented cases have been limited to short-range laboratory tests (animal and human hosts) or indoor studies in hospitals (206). Data referencing long-range transmission of infectious viruses have been obtained only with aerosol models (no field detection of the aerosolized virus) (206). Several papers have hypothesized transoceanic movement of viruses through the atmosphere based on favorable atmospheric conditions (i.e., wind patterns, mild temperatures) and incidence of disease (81, 87). One research project that used a direct-count assay (use of a nucleic acid stain to count microorganisms via epifluorescence microscopy) to tabulate the number of virus-like particles in U.S. Virgin Island atmospheric samples reported a background concentration of 1.8 ⫻ 104 m⫺3 and an African dust event concentration of 2.13 ⫻ 105 m⫺3 (78). In summary, little research has been conducted to address the naturally occurring populations of desert soil viral communities, those viruses introduced by humans and other an- 466 GRIFFIN CLIN. MICROBIOL. REV. imals, or occurrence and survival issues associated with airborne desert soils. PATHOGENS Bacteria Culture-based dust-borne microbiological studies have established that a diverse bacterial community can move through the atmosphere in clouds of desert dust, but no study has demonstrated the movement of a prokaryote pathogen and linked it to the occurrence of disease. The closest known associations of dust storms and human disease of microbial origin are the outbreaks of meningitis (primarily due to Neisseria meningitidis infection) that occur within the “meningitis belt” of North Africa (223). These outbreaks occur frequently in the Sahel region of North Africa between (and within) the months of February and May and affect as many as 200,000 individuals annually (160, 223). The conditions during this period are characterized as dry with frequent dust storms. Outbreaks usually cease with the onset of the wet season (160, 161). Dust storms are believed to promote infection via dust particles causing abrasions of the nasopharyngeal mucosa upon inhalation, allowing Neisseria meningitidis cells residing in the mucosa access to underlying tissue and blood, thus initiating infection (161). An additional and complementary hypothesis argues that cells of Neisseria meningitidis associated with the inhaled dust particulates are an alternate route of infection (80). Five viable isolates of Neisseria meningitidis recently identified in settled-dust samples from Kuwait demonstrate that dust can serve as a carrier for the pathogen (141). Other pathogens were also identified in this project (141), including Staphylococcus aureus (wide range of infections), Bacillus circulans, (opportunistic), Bacillus licheniformis (opportunistic, peritonitis), Pantoea agglomerans (opportunistic, peritonitis), Ralstonia paucula, (opportunistic, septicemia, peritonitis, abscess, and tenosynovitis), and Cryptococcus albidus (opportunistic, disseminated) (13, 122, 136, 140, 159, 179). Given the current state of affairs in the Middle East, many of these opportunistic dust-borne pathogens may play a significant role in human health with regard to combat-related injuries, treatment, and recovery. Bacterial species that are known human pathogens have been collected during dust events in Bamako, Mali (117). The species include Acinetobacter calcoaceticus (nosocomial respiratory tract infections), Corynebacterium aquaticum (urinary tract infections), Gordonia terrae (nervous system, skin), a Kocuria sp. found in advanced noma lesions in Nigerian children, and Kocuria rosea (bacteremia) (10, 28, 55, 117, 147, 181, 226). Additionally, Acinetobacter calcoaceticus has been linked to mad cow disease, and another isolate, Staphylococcus xylosus, was previously identified as the causative agent of septicemia in a loggerhead turtle in the Canary Islands (117, 229, 232). Of the 95 bacteria identified in this study, Kellogg et al. reported that approximately 10% were potential animal pathogens, 5% were potential plant pathogens, and 25% were opportunistic human pathogens (117). In the African dust corridor over the mid-Atlantic ridge, two of the human pathogens identified in the Mali dust study (83, 117), Kocuria rosea and Gordonia terrae, were also recovered when elevated concentra- tions of African dust were present in the atmosphere (83). Furthermore, atmospheric dust-borne bacteria isolated in this mid-Atlantic study that are recognized as pathogens included Brevibacterium casei (opportunistic, sepsis) and Staphylococcus epidermidis (opportunistic, endocarditis, urinary tract infections) (22, 254). In the atmosphere over the U.S. Virgin Islands, the opportunistic pathogen Pseudomonas aeruginosa, which can cause fatal infections in burn patients, was isolated when African dust was present (79). Additionally, identified microorganisms found in a Nigerian noma lesion study were also isolated (GenBank closest neighbor similarities: Kocuria sp., 97%; Microbacterium arborescens, 98%; Sphingomonas sp., 96%) (79). At Erdemli, Turkey, Kocuria rosea, Corynebacterium aquaticum (opportunistic, urinary tract infections, sepsis), and Massilia timonae (which has been isolated from the blood of an immunocompromised patient) were isolated during dust events (82, 133, 162, 226). Other desert dust studies included isolates of Bacillus, although no specific species-level pathogenic identifications were made (20, 190). The bacterial endotoxin LPS can also directly impact human health via inhalation (205). LPS is a cell wall component of gram-negative bacteria and is commonly found in organic dusts (240). Human and animal inhalation trials have shown increases in neutrophils and lymphocytes with a reduction in alveolar macrophage phagocytosis (205). Short-term exposure can result in fever and reduced airflow. Long-term exposure can result in the development of lung diseases such as asthma, bronchitis, and irreversible airflow obstruction (240). Since gram-negative bacteria are dominant in marine waters, aerosolized marine bacteria that adhere to traversing dust particles may enhance dust cloud toxicity and affect human health in coastal environments that are typically impacted by significant quantities of desert dust. African dust reaches coastal communities in Europe, the Middle East, the Caribbean, and the Americas. Asian dust reaches Korea, Taiwan, Okinawa, Japan, regional islands, the Artic, and North America. Approximately half of the bacteria found in African dust studies conducted in the Caribbean were gram-negative bacteria. Given the volume of annual African dust exposure, LPS may contribute to the high incidence of asthma in the region (79, 96). Sea spray generation in near-shore environments may also produce elevated humidity levels in near-surface atmospheric layers, which can both concentrate and “rain out” dust-borne pathogens and toxins, thus increasing exposure and risk. It is obvious from the few dust-borne microbiology studies that have identified pathogenic bacteria that there is some degree of human health risk associated with exposure to airborne desert dust. While most of the pathogens identified to date are opportunistic in nature, these are but a small number of culture-based studies, and as in most outbreaks of disease it is often the young, old, and immunocompromised who are within the higher levels of risk. These few studies provide a glimpse of risk evidence and highlight the need for non-culture-based assays to advance this field of study. Fungi In 1941 several air fields were established in the southern San Joaquin Valley. Coccidioidal infection was recognized to be a hazard, but it was preferred to VOL. 20, 2007 the hazards of mountains and fog in alternative locations. During the first year clouds of dust billowed over the fields, and a quarter of the susceptible personnel became infected. C. E. Smith, appointed consultant to the Secretary of War, proposed rigid dust control measures. Roads were paved, air strips were hard-surfaced, and swimming pools were substituted for dusty athletic fields. Lawns were planted by the acre, and military personnel were ordered, at risk of court martial, to avoid unprotected areas as much as possible (64). 467 Acremonium, Alternaria, Arthrinium, Aspergillus, Cladosporium, Curvularia, Emericella, Fusarium, Nigrospora, Paecilomyces, Pithomyces, Phoma, Penicillium, Torula, Trichophyton, and Ulocladium). Outside of human disease, an outbreak of aspergillosis was documented in a captive locust population (⬃40% of the population) in Bikaner, India, following a dust storm (239). Sahara/Sahel dust storms have also been identified as longrange transport mechanisms for the pathogen responsible for the Caribbean region-wide outbreak of Aspergillus infections (A. sydowii) in seafans (213, 245). The long-range atmospheric dispersal of fungal crop pathogens has been well documented. Brown and Hovmøller present a review of those data (24). Viruses Transport of infectious human viruses in dust storms has not been investigated. Hammond et al. hypothesized that longrange transport of human influenza virus from Asia to the Americas could occur in the winter months given the prevailing wind patterns over the Pacific and the low dose of virus required for infection (87). With the annual peak of the Asian dust season occurring in March and April, the attachment of infectious viruses to dust particles moving across the Pacific may serve to enhance long-range host-to-host transport, as virus survival studies (water and air based) have shown a relationship between particle association/attachment and enhanced survival (41, 45, 128, 194). Obviously, short-range transmission (person to person) of infectious viruses such as the influenza viruses and rhinoviruses is an evolved means of movement. Short-range atmospheric transmission via dried aerosolized rodent excreta (dust) is the primary route of hantavirus infection, and elevated risk has been documented in the arid American southwest during droughts that follow El Niño events (59). Evidence of longer-range transmission (building to building) of the severe acute respiratory syndrome virus has been documented (262), but what about even longer-range transmission? Is there a range limit with various human viral pathogens that are transmitted through the air? How does the limit vary between viral pathogens, and what is the evidence that long-range transmission occurs? In livestock populations, dust storms have been identified as a possible source of foot-and-mouth disease virus outbreaks that occurred in Korea and Japan. Some of the outbreaks followed Gobi/Takla Makan dust events (115, 175, 203). Although two studies (115, 175) screened dust samples for the presence of foot-and-mouth disease virus by reverse transcriptase PCR (⬎400 samples) with no positive results, the authors suggested additional testing before ruling out the transmission route. Griffin et al. hypothesized that foot-and-mouth disease virus outbreaks in Europe could result from long-range transmission in African dust based on the similarity of endemic (Africa) and outbreak serotypes, dust events and outbreak occurrences, and a favorable atmospheric route (over water) (81). Evidence of regional (European) airborne transmission of foot-and-mouth disease virus to downwind locations within the United Kingdom, from France across the English Channel to the United Kingdom, and from Germany over the Baltic Sea to Denmark, has been well documented (51, 69, 71, 72, 157, 217). Using an atmospheric model and inputting optimal climate and topography, Sorensen et al. estimated that 1,000 pigs V The most well-known human pathogen associated with desert dust storms is the fungus Coccidioides immitis (64). This fungus has been found only in the Americas. Annual outbreaks following dust storms (the primary means of exposure) or dust clouds generated from natural disasters or anthropogenic activity (e.g., earthquakes and construction, etc.) are well documented (90, 113, 178, 208). An outbreak at a Naval Air Station in California following a large dust storm event resulted in a coccidioidomycosis incidence rate of 36 per 100,000 for personnel classified as white and 254 per 100,000 for those classified as nonwhite, indicating differences in racial susceptibility (250). In Arizona, an increase in annual cases from 33 per 100,000 in 1998 to 43 per 100,000 in 2001 was attributed to climate change (increased wind speed and drought), with peak periods occurring in the winter and the age group at highest risk being those ⱖ65 years old (33, 178). Of 128 studied cases of coccidioidomycosis that occurred between 1 September and 31 December 1991 in Tulare County, CA, males, Asians and blacks, and those over 20 years old were identified as the highest risk groups (57). In addition to wind-generated dust storms, an outbreak of coccidioidomycosis resulting in 203 cases and three fatalities followed exposure from earthquakegenerated dust clouds (208). Between 1991 and 1993, medical expenses associated with outbreaks were estimated at $66 million (113). While no other fungal outbreaks in human populations have been attributed to dust storm exposure, the cited fungal prevalence studies in desert soils and dust storms have demonstrated diverse communities composed of both nonpathogenic and pathogenic genera and species. In most of these studies, only a fraction of the observed culturable community was identified, or the methods employed for identification, such as morphological or spore identification, limited the ability to identify pathogenic species. As a result, research has produced only an indication of the true community structure in any of the dust source regions. While they have yet to be identified in dust storm studies, fungal pathogens such as Histoplasma capsulatum, the causative agent of histoplasmosis, often associated with exposure to dust originating from dried bird or bat feces, are certainly a potential risk factor (31, 164). Table 3 lists the dust storm studies in which fungal CFU or spores have been identified, the genera known to contain pathogenic species, and, where information was available, the pathogenic species. Information available on these genera indicates that most are cosmopolitan in nature. These pathogenic or opportunistic pathogens are known to cause a wide range of human diseases (mild to fatal disseminating infections). Some of the fungi in Table 3 are known to be mild or potent allergens (species of DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 468 GRIFFIN CLIN. MICROBIOL. REV. TABLE 3. Fungi capable of affecting humansa Organism Location(s) (reference关s兴)b Dust source region(s) Miscellaneous informationc Disease(s)d Mycetoma (colonization of tissue/bone), onychomycosis (colonization of nail), mycotic keratitis, a number of different allergen-related disease types in the immunocompromised Cutaneous phaeohyphomycosis (colonization of skin), potent allergen (common cause of extrinsic asthma)-related disease, deep tissue infections in the immunocompromised Phaeohyphomycosis Acremonium USVI, Turkey (82, 83) Sahara/Sahel, Middle East Common in soil, on plants, and indoors Alternaria Mali, Africa, mid-Atlantic, Barbados, Taiwan, Saudi Arabia, Israel, Turkey (82, 83, 92, 117, 126, 190, 207, 256) Sahara/Sahel, Gobi/ Takla Makan, Middle East Common in soil, on plants, and indoors Alternaria infectoria Turkey (82) Sahara Arthrinium Barbados (190) Sahara/Sahel Aspergillus Mali, Africa, USVI, Barbados, Taiwan, Saudi Arabia, Israel (79, 83, 92, 117, 126, 207) Sahara/Sahel, Gobi/ Takla Makan, Saudi Arabia Common environmental isolate Common environmental isolate Common in soil, organic detritus, and indoors Aspergillus clavatus Barbados (190) Sahara/Sahel Aspergillus flavus Barbados, Israel (190, 207) Sahara/Sahel Aspergillus fumigatus Aspergillus niger Barbados, Israel (190, 207) Sahara/Sahel Sahara/Sahel Aspergillus terreus Mali, Africa, Barbados, Israel (117, 190, 207) Barbados (190) Sahara/Sahel Aspergillus ustus Israel (207) Saharan Aspergillus versicolor Aureobasidium Mali, Africa, Israel (117, 207) Mid-Atlantic, USVI (79, 83) Sahara/Sahel Bipolaris USVI (79) Sahara/Sahel Cladosporium Mali, Africa, mid-Atlantic, USVI, Barbados, Taiwan, Saudi Arabia, Turkey (78, 79, 82, 83, 92, 117, 126, 256) Sahara/Sahel, Gobi/ Takla Makan, Middle East Most commonly isolated fungus in outdoor studies Cladosporium cladosporioides Cladosporium sphaerospermum Chrysosporium Mali, Africa, USVI, Israel (78, 117, 207) Israel (207) Sahara/Sahel Sahara USVI (79) Sahara/Sahel Curvularia Taiwan, Barbados (92) Gobi/Takla Makan Common isolate Common isolate Common isolate Common isolate Emericella nidulans Mid-Atlantic (83) Sahara/Sahel Fusarium Taiwan, Turkey (82, 92, 256) Gobi/Takla Makan, Middle East Common in tropical and subtropical environments Common soil and indoor isolate Microsporum USVI, Turkey (79, 82) Sahara/Sahel, Middle East Some species are geographically restricted Sahara/Sahel Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Distributed in temperate areas; common on plant tissue and indoors Common plant and indoor isolate environmental environmental environmental environmental Allergen-related disease Aspergillosis (pulmonary 关allergic and colonizing兴, disseminated, central nervous system, cutaneous, nasalorbital, and iatrogenic), a number of different allergenrelated disease types in the immunocompromised Invasive aspergillosis Invasive aspergillosis Invasive aspergillosis Invasive aspergillosis Invasive aspergillosis Rare, invasive aspergillosis Invasive aspergillosis Cutaneous phaeohyphomycosis, invasive disease in the immunocompromised Pansinusitis, meningoencephalitis, chronic pulmonary disease Cutaneous phaeohyphomycosis, chromoblastomycosis (subcutaneous skin infections), mycotic keratitis, potent allergen-related disease Cutaneous phaeohyphomycosis, chromoblastomycosis Cutaneous phaeohyphomycosis, chromoblastomycosis Opportunistic, infecting brain, nasal and skin tissue Allergen-related disease, opportunistic, pneumonia, disseminated Allergic alveolitis Invasive cutaneous (erythematous lesions and nodules), systemic granulomatous disease, allergen-related disease Dermatophytosis (i.e., ringworm) Continued on facomg page VOL. 20, 2007 DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 469 TABLE 3—Continued Organism b Location(s) (reference关s兴) Dust source region(s) Mortierella Saudi Arabia (126) Saudi Arabia Mucor Saudi Arabia (126) Saudi Arabia Neotestudina rosatii Mid-Atlantic (83) Sahara/Sahel Nigrospora USVI, Taiwan (79, 92, 256) Paecilomyces USVI (79) Sahara/Sahel, Gobi/ Takla Makan Sahara/Sahel Pithomyces Taiwan (92, 256) Gobi/Takla Makan Phoma Pythium Penicillium Mid-Atlantic (83) Saudi Arabia (126) Mid-Atlantic, USVI, Barbados, Taiwan, Saudi Arabia, Turkey (79, 82, 83, 92, 126, 190, 256) Turkey (82) Sahara/Sahel Saudi Arabia Sahara/Sahel, Gobi/ Takla Makan, Middle East Israel (207) Sahara Penicillium brevicompactum Penicillium chrysogenum Penicillium spinulosum Phoma cava Israel (207) Sahara Israel (207) Sahara Rhizomucor USVI (79) Sahara/Sahel Stachybotrys Mid-Atlantic (83) Sahara/Sahel Stemphylium Torula Taiwan (256) Taiwan (92, 256) Gobi/Takla Makan Gobi/Takla Makan Trichophyton Mid-Atlantic, USVI, Turkey (79, 82, 83) Taiwan, Saudi Arabia (92, 126) Sahara/Sahel, Middle East Gobi/Takla Makan, Saudi Arabia Mid-Atlantic (83) Sahara/Sahel Ulocladium Ulocladium botrytis Sahara Miscellaneous informationc Common environmental isolate Common environmental isolate Common environmental isolate; Africa, Australia, India Common in soil, organic detritus, and indoors Common in soil, organic detritus, and indoors Typically found on dead plant detritus Common plant pathogens Common plant pathogen Very common in temperate regions, common in soil and indoors Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Common environmental isolate Commonly found in soil and decaying plants Common plant pathogens Common environmental isolate Commonly found in soils and indoors Found in soil, on plants, and in high-moisture environments Found in soil, on plants, and in high-moisture environments Disease(s)d Rare, cutaneous Rare, opportunistic, pulmonary, disseminating, cutaneous Mycetoma Allergen-related disease Mycotic keratitis paecilomycosis, pneumonia, allergen-related disease Allergen-related disease Allergen-related disease Pythiosis Bronchopulmonary penicilliosis, potent allergen-related disease (hypersensitivity and allergic alveolitis) Rare, necrotic lung ball Rare, endocarditis, necrotizing pneumonia Allergen-related disease Subcutaneous phaeohyphomycosis Rare opportunistic, pulmonary, disseminating, cutaneous zygomycosis Rare, toxin producer, pulmonary Phaeohyphomycosis Allergen-related disease Dermatophytosis, allergenrelated disease Phaeohyphomycosis Allergic alveolitis a The fungi were cultured and identified—to the species level, where the information was available—in atmospheric desert dust samples. Where no species is given, disease information is relevant to pathogenic strains. b USVI, U.S. Virgin Islands. c See references 199 and 221. d See references 199 and 221 and http://www.doctorfungus.org. Unknown Agents knock two people down, for the static electricity from the dusters was so strong. Ike Osteen’s life spans the flu epidemic of 1918, the worst depression in American history, and a world war that ripped apart the globe. Nothing compares to the black dusters of the 1930s, he says, a time when the simplest thing in life—taking a breath—was a threat (58). Cattle went blind and suffocated. When farmers cut them open, they found stomachs stuffed with fine sand. Horses ran madly against the storms. Children coughed and gagged, dying of something the doctors called “dust pneumonia.” In desperation, some families gave away their children. The instinctive act of hugging a loved one or shaking someone’s hand could Incidence of pneumonia of unknown etiology in populations exposed to airborne desert dust storms has been reported throughout the course of time (18, 23, 121, 174, 248). In the American Dust Bowl era of the 1930s, the numbers of cases of pneumonia were 50 to 100% higher during dust periods than during previous low-dust years (23). In dust storm-impacted regions around the Aral Sea, pneumonia rates are the highest excreting foot-and-mouth disease virus could infect cattle located 300 km downwind (217). Modeling was also used to demonstrate the 1988 atmospheric spread of pseudorabies virus between swine herds over an area of 150 km2 in Decatur County, IN (40, 75). 470 GRIFFIN CLIN. MICROBIOL. REV. in the former USSR, and 50% of all childhood illnesses are respiratory (105, 237). Pneumonia from dust storm exposure has also been reported in the Middle East, especially those cases pertaining to deployed military personnel (14, 34, 121, 214). Of 15,459 surveyed military personnel who were deployed in Afghanistan or Iraq during 2003 and 2004, 69.1% reported some form of respiratory illness (204). Pneumonia acquired from exposure to inorganic and organic material in dust storms has been termed Al Eskan disease, Persian Gulf syndrome, Persian Gulf War syndrome, Gulf War syndrome, and desert dust pneumonitis (50, 109, 120, 121). In addition to pneumonia, adverse health conditions reported by military personnel who were deployed in the area include fatigue, fever, respiratory stress, arthromyoneuropathy, and neurological impairment, and epidemiological studies demonstrated higher prevalence in exposed/deployed groups than in controls (14, 50, 65, 86, 108, 116). In contrast, a number of studies designed to identify the incidence of disease in deployed versus nondeployed groups noted no identifiable differences between the groups (101, 106, 118, 134, 201). In China and the Himalayas, studies have shown that indigenous populations in dust areas have higher incidences of silicosis than do populations residing in nondust or low-dust areas (173, 202, 259). Cell culture studies have demonstrated that dust storm constituents can inhibit defense functions of alveolar macrophages and increase reactive oxygen species, causing DNA base damage (61, 186). Mouse- and rat-based inhalation studies have shown that desert dust causes increases in the levels of neutrophils, lymphocytes, eosinophils, interleukin-2 and -6, and tumor necrosis factor alpha and that responses are dose dependent (102, 137). DETECTION One of the most striking characteristics of aerobiological studies, and more specifically of the few on dust storm microbiology cited in this article, is the different techniques used to collect and analyze air samples, as previously reviewed (27, 255). The only instances of dust storm microbiology research that employed “like protocols” are those papers published by the same research groups. Due to the drawbacks of various collection and evaluation assays, comparisons of results among studies that utilized different techniques can answer only the most basic of questions: that, yes, a diverse community of culturable bacteria and fungi is capable of surviving long-range atmospheric transport in clouds of desert dust. The limited number of dust-borne projects, in addition to the lack of a recognized or standardized protocol, has and will impede progression in this field of study. Moreover, there is a clear need for culture-independent analysis of dust-borne microbial communities (16S and 18S rRNA gene sequencing), without which we will never be able to see the “big picture” (98). For a comprehensive review of aeromicrobiology methods, see Buttner et al. (27). Collection Protocols that have been used to collect microorganisms in the atmosphere include impaction, impingement, centrifugation, filtration, and gravity deposition (27, 54, 219). Gravity deposition is one of the simplest methods of collecting air- borne microorganisms and involves exposing nutrient agar to an open-air environment for some period of time. The benefits of this protocol are that it is inexpensive and cell death from impaction is insignificant, and yet it has been shown to be an unacceptable method in comparison to volumetric assays (26, 76). Impaction of microorganisms onto surfaces such as tape, nutrient agar, or hard surfaces coated with an adhesive fluid such as oil is commonly utilized for the collection of airborne microorganisms. Exposure of a petri dish containing nutrient agar or a small metal surface coated with glycerol to an air stream has been used to collect airborne microorganisms from aircraft for morphological identification of airborne biological constituents or culture-based studies (77, 152, 154, 155). While a reliable estimate can be made of the volume of air sampled based on the collector surface area, air speed, and time exposed, particle capture on the surfaces can be negatively influenced by wind speed, impactor shape, and presentation angle (collector surface angle to wind/flight direction) (76). Impaction of spores or pollen onto adhesive tape via an air pumpdriven system such as a Burkard spore trap (Burkard Manufacturing Co. Ltd., Rickmansworth, United Kingdom) is an efficient means of obtaining total counts of fungal spores (and identifications based on spore morphology), as demonstrated by Ho et al. and Wu et al. in their Taiwan-based Asian dust studies (92, 256). The primary limitations of spore traps are restricted use in the analysis of other microbial types (bacteria and viruses) and the fact that morphology-based identifications are limited to genus level classification. Another method of impaction is the use of an air pump to move air over the surface of a petri dish (cassettes and strips are also used) containing nutrient agar. Airflow over nutrient agar is controlled by slits or holes that are arranged to distribute the airflow evenly over the agar surface and in some cases to control for particle size ranges. Numerous companies manufacture air pump devices, including the portable handheld Millipore M Air T Air Tester (Millipore Corporation, Bedford, MA) and the popular Andersen six-stage sampler (Andersen 6-STG; Graseby Andersen, Smyrna, GA) (12; K. R. Lentine, N. Le, J. Lemonnier, A. Entzmann, and M. Pickett, presented at the 99th General Meeting of the American Society for Microbiology, Chicago, IL, 30 May to 3 June 1999). The benefits of these types of samplers are ease of use, portability, cost, assessment of culturable populations of bacteria and fungi per volume of air, and, as in the case of the Andersen 6-STG-type sampler, the ability to determine the numbers of microorganisms associated with various size ranges of airborne particulates. Drawbacks to the use of these impactors are loss of viability due to impact stress, loss of recovery efficiency due to microorganisms not adhering to agar surfaces, low sample volumes due to low flow rates, and low recovery of ultrafine biological matter in the size range of viruses (220). Centrifugation has been used to collect microorganisms on container walls, wet or dry slides, or nutrient agar (petri dishes and strips, etc.) (249, 251, 257). The benefits of centrifugal concentration are the use of high flow rates (⬎100 liters min⫺1) and efficient capture (251). The drawback of this method is the loss of viability due to impaction stress (257). Impingement of microorganisms into a liquid matrix (via bubbling) is another method of capture used in aeromicrobiology (7, 27, 227). A widely used liquid impinger is the AGI-30 VOL. 20, 2007 DESERT DUST MICROBIOLOGY AND HUMAN HEALTH (Ace Glass Inc., Vineland, NJ), which utilizes a low flow rate to bubble air through a liquid matrix. Research has shown that the AGI-30 is a low-cost and efficient method of collecting aerosolized microorganisms for culture- and non-culturebased analyses (227). However, this impinger is constructed of glass and can be easily broken in the rigors of field studies. High-flow-rate liquid impinger units have been developed to detect low concentrations of microorganisms in the atmosphere and have also been utilized for culture- and non-culture-based analyses for the presence of bacteria, fungi, and viruses (17). The benefit of using liquid impingers for aeromicrobiology studies is that the liquid matrix can be split for various analyses to include media cultures, direct counts, molecular assays, and cell cultures. The drawbacks of liquid impingement include the low capture rate of some low-flow-rate impingers (the use of large-bore air lines and, thus, large bubble production in the liquid matrix), high cost (high-flow-rate impingers), loss of collection fluid to evaporation and violent bubbling (some of the high-flow-rate impingers limit this loss), low capture rate of virus-sized particles, and loss of viability (i.e., viral inactivation due to bubbling) (6). Membrane filtration in which air is pumped through a porous membrane is also utilized in aeromicrobiology and has been used by a number of researchers in desert dust microbiology (27, 78, 79, 83, 117, 190). This method can be employed for both culture- and non-culture-based studies, is inexpensive, can be portable, and is highly efficient at trapping microorganisms larger than the pore size on the filter surface (111, 145). Filters commonly utilized for collection include cellulosebased, nylon, and glass fibers with pore sizes down to 0.02 m (for viral analysis) (78). The drawbacks of filtration include desiccation of the microorganisms on the filter surface due to filtration rate and time (higher flow rates and longer filtration times), the preferential concentration of spore-forming microbes over other community members as non-spore formers are desiccated during rapid or long filtration times (culturebased studies), filter size (37 mm versus 47 mm; i.e., larger diameters limit stacking of cells in a high particle load environment), and filter type (a thicker filter can wick nutrients to cells, thereby limiting close-contact nutrient shock of stressed cells) (27, 83, 111, 150, 231). In regard to these currently used methods of recovery, use of a high-volume liquid impinger for aeromicrobiology studies in general is the most versatile and arguably one of the most efficient capture methods available. While the capture efficiency of virus-range particles is a concern with today’s machines, virus-based analyses are still possible. Furthermore, advances in technology should enhance the capture of all microbial size fractions and at the same time allow what no other isolation assay currently does: rapid large-volume sampling for the full suite of microbial types and the ability to split a single sample for multiple analyses. Identification Microorganisms collected in samples (soil, aquatic, air, and clinical, etc.) are characterized by culture-based assays (for CFU counts and identification via substrate utilization) and/or non-culture-based assays (morphology-based identification; cell, spore, physiological, and nucleic acid stains; identification 471 via nucleic acid amplification and/or hybridization; immunological identification; direct counts; and fatty-acid analysis). Usually, in any environment, less than 1.0% of the bacterial community is culturable under the most favorable (low-stress) conditions, and thus any data obtained from this approach are limited in regard to community composition (11). For culturebased studies of airborne microorganisms, particularly in the case of long-range transport studies in which a majority of the community may be stressed by desiccation, temperature, and UV exposure, selection of a nutrient source and incubation temperature that will maximize recovery is critical. It is known that both nutrient and temperature shock can negatively impact the ability to culture stressed bacteria (197). Numerous studies comparing low-nutrient broths or agars (diluted highnutrient recipes or low-nutrient recipes such as Reasoner’s 2 agar [R2A]) have shown superior recovery rates of culturable bacteria versus rates with high-nutrient agars in different settings (water, air, soil, and serum, etc.) (15, 95, 112, 149, 195, 238). The incubation temperature can significantly impact culturability (in general, a temperature close to the environmental temperature from which the sample was taken gives better recoveries), and most dust-borne culture-based studies have used moderate temperatures for growth (⬃23.0°C [room temperature]) (43, 79, 83, 117, 170, 185, 190). Several nutrientbased identification assays, such as the Analytab System (Analytab Products, Syosset, NY) and BioLog plates (Hayward, CA), are utilized in environmental studies. Although these are low-cost assays, they are subject to variation (i.e., reproducibility issues due to variation in cell densities and physiological state, etc.) and false-positive results (previously unclassified organisms producing similar or like carbon utilization patterns) (25, 119, 135). Recent advances in culture technology, such as the employment of single-cell encapsulation into microdroplets containing naturally occurring nutrients, have demonstrated improved recovery rates and hold promise for future studies (264). Microscopy is one of the oldest tools used for the study of bacteria and fungi. Identifications or descriptions are based on staining characteristics, cellular morphology, spore shape, the presence or absence of spores, and pigmentation. At the microscopic level, many organisms cannot be identified, due to similarities among species and genera and to significant differences in the education and experience of the investigator. Species- and strain-specific identifications are possible with the use of immunological (antigen-specific antibodies tagged with a reporter) and genetic (i.e., fluorescence in situ hybridization [FISH]) tools, as well as direct-count assays and physiological studies using nucleic acid or cellular stains, respectively (37, 48, 172, 183, 228, 241). Whereas stains that utilize direct-count assays are invaluable in assessing total cell counts in a sample, certain cells are known to resist staining, and similar-sized particles that are not cells may take up the stain, resulting in false-positive counts (27). Another limitation is that more than 104 cells per sample are required for useful data (27). The result is that microscopic methods can be prone to error. Such methods also require expertise and can be time-consuming (27). Although sequence-based identification assays, such as those using oligonucleotides (dot blot assays and FISH, etc.), have served microbial ecologists well, the invention of nucleic acid 472 GRIFFIN amplification protocols such as PCR and nucleic acid sequence-based amplification has enabled researchers to assess community composition at the strain level efficiently and to detect organisms in the community that occur in very low numbers (42, 167, 182). Nucleic acid amplification methods target regions of conserved genes, such as those found in the ribosomal genes, for use in identification (42, 167). Once the target gene is amplified, the community amplicon can be evaluated with ecology-based gene chips or through the classical approach of clone library production and analysis (35, 253). Several other widely used types of postamplification analysis include denaturing gradient gel electrophoresis and temperature gradient gel electrophoresis (168). These assays produce a microbial community fingerprint that can be analyzed with specific group probes for individual band identification, or individual bands can be excised and sequenced directly. The primary benefits of PCR-based assays are ease of use as DNA and RNA extraction kits, PCR amplification kits, cloning kits, the availability of commercial sequencing facilities, and information on the community (culturable and nonculturable) are obtained. The primary drawbacks are the cost of cloning kits, the required expertise in sequence interpretation, debate among mycologists on the appropriateness of relying on DNA sequences alone to identify fungi, and the lack of a universal gene for virus ecology (211). Phospholipid ester-linked fatty acid and intact phospholipid profiling can also be utilized to identify bacteria and fungi in both culture- and non-culture-based studies via unique lipid structures (63, 132, 246). The benefits of these assays are that they address viable biomass due to rapid phospholipid turnover (63, 246). The drawbacks are costs of analysis, overlapping profiles, biomass to cell count conversion, and compositional shift from variance in growth conditions (63). With the rapid progression of technology, new and emerging tools, such as optical tweezers, phylogenic gene chips, chip use in cross-hybridization studies, cellular circuits, mass spectrometry, and the miniaturization of existing equipment, should greatly enhance our understanding of dust-borne microbial ecology through availability, affordability, adaptation, and use (38, 60, 123, 151, 171, 230, 253). CONCLUSIONS AND PERSPECTIVES Two common misconceptions are that desert soils are too inhospitable to host a diverse microbial community and that those microorganisms that are present cannot withstand the physical stresses (UV, desiccation, temperature) of atmospheric transport. Research in the twentieth and twenty-first centuries has shown that microorganisms mobilized into the atmosphere along with desert soils are capable of surviving long-range transport on a global scale. As with the betterknown pathogens that use aerosolization to move from host to host (Bacillus anthracis, Yersinia pestis, Mycobacterium tuberculosis, Legionella pneumophila, Histoplasma capsulatum, influenza viruses, and rhinoviruses, etc.), the microbiological research conducted to date has identified a wide range of dust-borne pathogenic microorganisms that move great distances through the atmosphere. In light of the few dust-oriented studies outlined in Table 2, which are based only on culture or spore-counting techniques, it is clear that we have CLIN. MICROBIOL. REV. only begun to grasp the true numbers of microorganisms capable of using the atmosphere as an infectious route or as a means of extending the limit of their dispersion. The prime question, of course, is related to risk: do microorganisms in dust clouds pose a risk to human health? The evidence of outbreaks of coccidiomycosis following dust storm exposure in the Americas demonstrates risk. Outside of this example, the risk from other pathogenic microorganisms is unknown, due to the limited number of studies and study design (all to date have been culture based, and none have been risk oriented). While some of the dust storm microbiology studies cited in this review have identified pathogenic microorganisms, information relative to concentrations over a realistic period of exposure does not exist, nor do we know the full extent of fate and survival issues as they relate to geographical variance and seasonal influences. The adaptation of existing and emerging molecular techniques will give us a clearer understanding in future studies. We need non-culture-based studies to advance this field in regard to issues in microbial ecology, biogeography, pathogen prevalence (fungal, bacterial, and viral), epidemiology, bacteria as carriers of potentially harmful genetic elements (i.e., phage related), microbial fate and survival, and risk. Integrated and global-scale collaborations need to be advocated and supported to allow multiple purchases of appropriate equipment (i.e., high-volume liquid impingers) for “true” matched studies in source and downwind environments. We also do not know what roles the synergistic influences of the many different constituents of dust play in human health. How does exposure to dust storms differ between source regions? When individuals who have never been exposed to desert environments (or dust storms) visit them (or are deployed in them), how is their health impacted in both the short and long terms? With the advent of the industrial age and the resulting widespread use and release of anthropogenic chemicals and emissions, are modern dust clouds more of a health threat than those that blew around the planet before the influence of humanity? How will climate change influence dust emissions and their associated microbial communities? The questions are many. As if this point has not been made enough, I close this review with a quotation from a paper published by Fred C. Meier and Charles A. Lindbergh in 1935: While it is generally known that bacteria, spores of higher fungi and pollen grains are present among dust particles in the atmosphere near the earth’s surface, much detailed information of practical value remains to be revealed by further research (154). ACKNOWLEDGMENT The use of trade names is for descriptive purposes only and does not imply endorsement by the U.S. Government. REFERENCES 1. Abdel-Hafez, S. I. I. 1982. Cellulose-decomposing fungi of desert soils in Saudi Arabia. Mycopathologia 78:73–78. 2. Abdel-Hafez, S. I. I. 1982. Survey of the mycoflora of desert soils in Saudi Arabia. Mycopathologia 80:3–8. 3. Abdel-Hafez, S. I. I., and A. A. M. Shoreit. 1985. Mycotoxins producing fungi and mycoflora of air-dust from Taif, Saudi Arabia. Mycopathologia 92:65–71. 4. Abdel-Hafez, S. I. I., A. A. M. Shoreit, A. I. I. Abdel-Hafez, and M. O. E. Maghraby. 1986. Mycoflora and mycotoxin-producing fungi of air-dust particles from Egypt. Mycopathologia 93:25–32. VOL. 20, 2007 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 473 PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol. Ecol. 6:475–482. Chang, C. C., I. M. Lee, S. S. Tsai, and C. Y. Yang. 2006. Correlation of Asian dust storm events with daily clinic visits for allergic rhinitis in Taipei, Taiwan. J. Toxicol. Environ. Health A 69:229–235. Chen, F., J.-R. Lu, B. J. Binder, Y.-C. Liu, and R. E. Hodson. 2001. Application of digital image analysis and flow cytometry to enumerate marine viruses stained with SYBR gold. Appl. Environ. Microbiol. 67:539– 545. Cho, J. C., and J. M. Tiedje. 2000. Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl. Environ. Microbiol. 66: 5448–5456. Choi, D. S., Y. K. Park, S. K. Oh, H. J. Yoon, J. C. Kim, W. J. Seo, and S. H. Cha. 1997. Distribution of airborne microorganisms in yellow sands of Korea. J. Microbiol. 35:1–9. Christensen, L. S., S. Mortensen, A. Botner, B. S. Strandbygaard, L. Ronsholt, C. A. Henricksen, and J. B. Anderson. 1993. Further evidence of long distance airborne transmission of Aujeszky’s disease (pseudorabies) virus. Vet. Rec. 132:317–321. Chung, H., and M. D. Sobsey. 1993. Comparative survival of indicator viruses and enteric viruses in seawater and sediment. Water Sci. Technol. 27:425–428. Compton, J. 1991. Nucleic acid sequence-based amplification. Nature 350: 91–92. Connon, S. A., and S. J. Giovannoni. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 68:3878–3885. Cook, A. G., P. Weinstein, and J. A. Centeno. 2005. Health effects of natural dust—role of trace elements and compounds. Biol. Trace Element Res. 103:1–15. Cox, C. 1995. Stability of airborne microbes and allergens, p. 77–99. In C. S. Cox and C. M. Wathes (ed.), Bioaerosols handbook. Lewis Publishers, London, United Kingdom. Daigle, C. C., D. C. Chalupa, F. R. Gibb, P. E. Morrow, G. Oberdorster, M. J. Utell, and M. W. Frampton. 2003. Ultrafine particle deposition in humans during rest and exercise. Inhal. Toxicol. 15:539–552. Delfino, R. J., C. Sioutas, and S. Malik. 2005. Potential role of ultrafine particles in associations between airborne particle mass and cardiovascular health. Environ. Health Perspect. 113:934–946. DeLong, E. F., L. T. Taylor, T. L. Marsh, and C. M. Preston. 1999. Visualization and enumeration of marine planktonic archaea and bacteria by using polyribonucleotide probes and fluorescent in situ hybridization. Appl. Environ. Microbiol. 65:5554–5563. Reference deleted. Doebbeling, B. N., W. R. Clarke, D. Watson, J. C. Torner, R. F. Woolson, M. D. Voelker, D. H. Barrett, and D. A. Schwartz. 2000. Is there a Persian Gulf War syndrome? Evidence from a large population-based survey of veterans and nondeployed controls. Am. J. Med. 108:695–704. Donaldson, A. I., and S. Alexandersen. 2002. Predicting the spread of foot and mouth disease by airborne virus. Rev. Sci. Technique Off. Int. Epizoot. 21:569–575. Donaldson, K., V. Stone, A. Seaton, and W. MacNee. 2001. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ. Health Perspect. 109:523–527. Dose, K., A. Bieger-Dose, B. Ernst, U. Feister, B. Gomez-Silva, A. Klein, S. Risi, and C. Stridde. 2001. Survival of microorganisms under the extreme conditions of the Atacama Desert. Orig. Life Evol. Biosph. 31:287–303. Dowd, S. E., and R. M. Maier. 2000. Aeromicrobiology. Academic Press, San Diego, CA. Drancourt, M., J. Pelletier, A. A. Cherif, and D. Raoult. 1997. Gordona terrae central nervous system infection in an immunocompetent patient. J. Clin. Microbiol. 35:379–382. Dufrene, Y. F. 2000. Direct characterization of the physicochemical properties of fungal spores using functionalized AFM probes. Biophys. J. 78: 3286–3291. Durry, E., D. Pappagianis, S. B. Werner, L. Hutwagner, R. K. Sun, M. Maurer, M. M. McNeil, and R. W. Pinner. 1997. Coccidioidomycosis in Tulare County, California, 1991: reemergence of an endemic disease. Med. Mycol. 35:321–326. Egan, T. 2006. The worst hard time. Houghton Mifflin Company, New York, NY. Engelthaler, D. M., D. G. Mosley, J. E. Cheek, C. E. Levy, K. K. Komatsu, P. Ettestad, T. Davis, D. T. Tanda, L. Miller, J. W. Frampton, R. Porter, and R. T. Bryan. 1999. Climatic and environmental patterns associated with hantavirus pulmonary syndrome, Four Corners Region, United States. Emerg. Infect. Dis. 5:87–94. Ericsson, M., D. Hanstorp, P. Hagberg, J. Enger, and T. Nystrom. 2000. Sorting out bacterial viability with optical tweezers. J. Bacteriol. 182:5551– 5555. Ezeamuzie, C. I., M. U. Beg, and D. Al-Ajmi. 1998. Responses of alveolar macrophages to post-Gulf War airborne dust from Kuwait. Environ. Int. 24:213–220. 7 5. Abrahams, P. W. 2002. Soils: their implications to human health. Sci. Total Environ. 291:1–32. 6. Agranovski, I. E., A. S. Safatov, A. I. Borodulin, O. V. Pyankov, V. A. Petrishchenko, A. N. Sergeev, A. P. Agafonov, G. M. Ignatiev, A. A. Sergeev, and V. Agranovski. 2004. Inactivation of viruses in bubbling processes utilized for personal bioaerosol monitoring. Appl. Environ. Microbiol. 70: 6963–6967. 7. Agranovski, I. E., A. S. Safatov, O. V. Pyankov, A. A. Sergeev, A. N. Sergeev, and S. A. Grinshpun. 2005. Long-term sampling of viable airborne viruses. Aerosol Sci. Technol. 39:912–918. 8. Al Frayh, A. R., Z. Shakoor, M. O. G. E. Rab, and S. M. Hasnain. 2001. Increased prevalence of asthma in Saudi Arabia. Ann. Allergy Asthma Immunol. 86:292–296. 9. al-Musallam, A. A. 1989. Distribution of keratinophilic fungi in desert soil of Kuwait. Mycoses 32:296–302. 10. Altuntas, F., O. Yildiz, B. Eser, K. Gundogan, B. Sumerkan, and M. Cetin. 2004. Catheter-related bacteremia due to Kocuria rosea in a patient undergoing peripheral blood stem cell transplantation. BMC Infect. Dis. 4:62. 11. Amann, R. I., W. Ludwig, and K. H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169. 12. Andersen, A. A. 1958. New sampler for the collection, sizing, and enumeration of viable airborne particles. J. Bacteriol. 76:471–484. 13. Archer, G. L. 1998. Staphylococcus aureus: a well-armed pathogen. Clin. Infect. Dis. 26:1179–1181. 14. Aronson, N. E., J. W. Sanders, and K. A. Moran. 2006. In harm’s way: infection in deployed American military forces. Clin. Infect. Dis. 43:1045– 1051. 15. Azevedo, N. F., A. P. Pacheco, C. W. Keevil, and M. J. Vieira. 2004. Nutrient shock and incubation atmosphere influence recovery of culturable Helicobacter pylori from water. Appl. Environ. Microbiol. 70:490–493. 16. Bener, A., Y. M. Abdulrazzaq, J. Al-Mutawwa, and P. Debuse. 1996. Genetic and environmental factors associated with asthma. Hum. Biol. 68:405– 414. 17. Bergman, W., J. Shinn, R. Lochner, S. Sawyer, F. Milanovich, and R. Milanovich, Jr. 2005. High volume, low pressure drop, bioaerosol collector using a multi-slit virtual impactor. J. Aerosol Sci. 36:619–638. 18. Blue, J. A. 1938. Dust—its effect on man from a medical standpoint with special reference to the dust bowl. Southern Med. J. 31:1101–1106. 19. Bovallius, A., B. Bucht, R. Roffey, and P. Anas. 1978. Long-range air transmission of bacteria. Appl. Environ. Microbiol. 35:1231–1232. 20. Bovallius, A., R. Roffey, and E. Henningson. 1980. Long range transmission of bacteria. Ann. N. Y. Acad. Sci. 353:186–200. 21. Bowker, M. A., S. C. Reed, J. Belnap, and S. L. Phillips. 2002. Temporal variation in community composition, pigmentation, and Fv/Fm of desert cyanobacterial soil crusts. Microb. Ecol. 43:13–25. 22. Brazzola, P., R. Zbinden, C. Rudin, U. B. Schaad, and U. Heininger. 2000. Brevibacterium casei sepsis in an 18-year-old female with AIDS. J. Clin. Microbiol. 38:3513–3514. 23. Brown, E. G., S. Gottlieb, and R. L. Laybourn. 1935. Dust storms and their possible effect on health. Public Health Rep. 50:1369–1383. 24. Brown, J. K. M., and M. S. Hovmoeller. 2002. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science 297:537–541. 25. Butler, D. A., C. M. Lobregat, and T. L. Gavan. 1975. Reproducibility of the Analytab (API 20E) system. J. Clin. Microbiol. 2:322–326. 26. Buttner, M. P., and L. D. Stetzenbach. 1993. Monitoring airborne fungal spores in an experimental indoor environment to evaluate sampling methods and the effects of human activity on air sampling. Appl. Environ. Microbiol. 59:219–226. 27. Buttner, M. P., K. Willeke, and S. A. Grinshpun. 1997. Sampling and analysis of airborne microorganisms, p. 629–640. In C. J. Hurst, G. R. Knudsen, M. J. McInerney, L. D. Stetzenbach, and M. V. Walter (ed.), Manual of environmental microbiology. ASM Press, Washington, DC. 28. Buxton, A. E., R. L. Anderson, D. Werdegar, and E. Atlas. 1978. Nosocomial respiratory tract infection and colonization with Acinetobacter calcoaceticus. Epidemiologic characteristics. Am. J. Med. 65:507–513. 29. Caimi, P., and A. Eisenstark. 1986. Sensitivity of Deinococcus radiodurans to near-ultraviolet radiation. Mutat. Res. 162:145–151. 30. Canchaya, C., C. Proux, G. Fournous, A. Bruttin, and H. Brussow. 2003. Prophage genomics. Microbiol. Mol. Biol. Rev. 67:238–276. 31. Cano, M. V., and R. A. Hajjeh. 2001. The epidemiology of histoplasmosis: a review. Semin. Respir. Infect. 16:109–118. 32. Carlton, C. A., F. Westall, and R. T. Schelble. 2001. Importance of a Martian hematite site for astrobiology. Astrobiology 1:111–123. 33. Centers for Disease Control and Prevention. 2003. Increase in coccidioidomycosis—Arizona, 1998–2001. Morbid. Mortal. Wkly. Rep. 52:109–112. 34. Centers for Disease Control and Prevention. 2003. Severe acute pneumonitis among deployed U.S. military personnel—southwest Asia, March– August 2003. Morbid. Mortal. Wkly. Rep. 290:1845–1846. 35. Chandler, D. P., J. K. Fredrickson, and F. J. Brockman. 1997. Effect of DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 474 GRIFFIN 62. Ezeamuzie, C. I., M. S. Thomson, S. Al-Ali, A. Dowaisan, M. Khan, and Z. Hijazi. 2000. Asthma in the desert: spectrum of the sensitizing aeroallergens. Allergy 55:157–162. 63. Fang, J., M. J. Barcelona, and P. J. J. Alvarez. 2000. A direct comparison between fatty acid analysis and intact phospholipid profiling for microbial identification. Org. Geochem. 31:881–887. 64. Fiese, M. J. 1958. Coccidioidomycosis. Charles C. Thomas, Springfield, MA. 65. Fukuda, K., R. Nisenbaum, B. Stewart, W. W. Thompson, L. Robin, R. M. Washko, D. L. Noah, D. H. Barrett, B. Randall, B. L. Herwaldt, A. C. Mawle, and W. C. Reeves. 1998. Chronic multisymptom illness affecting Air Force veterans of the Gulf War. JAMA 280:981–988. 66. Fulton, J. D. 1966. Microorganisms of the upper atmosphere. V. Relationship between frontal activity and the micropopulation at altitude. Appl. Environ. Microbiol. 14:245–250. 67. Gans, J., M. Wolinsky, and J. Dunbar. 2005. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390. 68. Gillies, J. A., W. G. Nickling, and G. H. McTainsh. 1996. Dust concentrations and particle-size characteristics of an intense dust haze event: Inland Delta region, Mali, West Africa. Atmosph. Environ. 30:1081–1090. 69. Gloster, J. 1982. Risk of airborne spread of foot-and-mouth disease from the continent to England. Vet. Rec. 111:290–295. 70. Gloster, J., and S. Alexandersen. 2004. New directions: airborne transmission of foot-and-mouth disease virus. Atmosph. Environ. 38:503–505. 71. Gloster, J., R. M. Blackall, R. F. Sellers, and A. I. Donaldson. 1981. Forecasting the airborne spread of foot-and-mouth disease. Vet. Rec. 108: 370–374. 72. Gloster, J., R. F. Sellers, and A. I. Donaldson. 1982. Long-distance transport of foot-and-mouth-disease virus over the sea. Vet. Rec. 110:47–52. 73. Goudie, A. S., and N. J. Middleton. 2001. Saharan dust storms: nature and consequences. Earth Sci. Rev. 56:179–204. 74. Graham, W. F., and R. A. Duce. 1979. Atmospheric pathways of the phosphorus cycle. Geochim. Cosmochim. Acta 43:1195–1208. 75. Grant, R. H., A. B. Scheidt, and L. R. Rueff. 1994. Aerosol transmission of a viable virus affecting swine: explanation of an epizootic of pseudorabies. Int. J. Biometeorol. 38:33–39. 76. Gregory, P. H. 1961. The microbiology of the atmosphere. Leonard Hill Books Ltd., London, United Kingdom. 77. Griffin, D. W. 2004. Terrestrial microorganisms at an altitude of 20,000 m in Earth’s atmosphere. Aerobiologia 20:135–140. 78. Griffin, D. W., V. H. Garrison, J. R. Herman, and E. A. Shinn. 2001. African desert dust in the Caribbean atmosphere: microbiology and public health. Aerobiologia 17:203–213. 79. Griffin, D. W., C. A. Kellogg, V. H. Garrison, J. T. Lisle, T. C. Borden, and E. A. Shinn. 2003. African dust in the Caribbean atmosphere. Aerobiologia 19:143–157. 80. Griffin, D. W., C. A. Kellogg, V. H. Garrison, and E. A. Shinn. 2002. The global transport of dust. Am. Sci. 90:228–235. 81. Griffin, D. W., C. A. Kellogg, and E. A. Shinn. 2001. Dust in the wind: long-range transport of dust in the atmosphere and its implications for global public and ecosystem health. Global Change Hum. Health 2:20–33. 82. Griffin, D. W., N. Kubilay, M. Kocak, M. A. Gray, T. C. Borden, C. A. Kellogg, and E. A. Shinn. 2007. Airborne desert dust and aeromicrobiology over the Turkish Mediterranean coastline. Atmosph. Environ. 41:4050– 4062 83. Griffin, D. W., D. L. Westphal, and M. A. Gray. 2006. Airborne microorganisms in the African desert dust corridor over the mid-Atlantic ridge, Ocean Drilling Program, leg 209. Aerobiologia 22:211–226. 84. Grousset, F. E., P. Ginoux, A. Bory, and P. E. Biscaye. 2003. Case study of a Chinese dust plume reaching the French Alps. Geophys. Res. Lett. 30: 1277. 85. Gyan, K., W. Henry, S. Lacaille, A. Laloo, C. Lamesee-Eubanks, S. McKay, R. M. Antoine, and M. A. Monteil. 2005. African dust clouds are associated with increased paediatric asthma accident and emergency admissions on the Caribbean island of Trinidad. Int. J. Biometeorol. 49:371–376. 86. Haley, R. W., T. L. Kurt, and J. Hom. 1997. Is there a Gulf War Syndrome? Searching for syndromes by factor analysis of symptoms. JAMA 277:215– 222. 87. Hammond, G. W., R. L. Raddatz, and D. E. Gelskey. 1989. Impact of atmospheric dispersion and transport of viral aerosols on the epidemiology of influenza. Rev. Infect. Dis. 11:494–497. 88. Harrison, R. M., and J. X. Yin. 2000. Particulate matter in the atmosphere: which particle properties are important for its effects on health? Sci. Total Environ. 249:85–101. 89. Hawksworth, D. L. 2001. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 105:1422–1432. 90. Hector, R. F., and R. Laniado-Laborin. 2005. Coccidioidomycosis—a fungal disease of the Americas. PLoS Med. 2:e2. 91. Herman, J. R., N. Krotkov, E. Celarier, D. Larko, and G. Labow. 1999. The distribution of UV radiation at the earth’s surface from TOMS measured UV-backscattered radiances. Geophys. Res. 104:12059–12076. 92. Ho, H. M., C. Y. Rao, H. H. Hsu, Y. H. Chiu, C. M. Liu, and H. J. Chao. CLIN. MICROBIOL. REV. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 2005. Characteristics and determinants of ambient fungal spores in Hualien, Taiwan. Atmosph. Environ. 39:5839–5850. Holmes, C. W., and R. Miller. 2004. Atmospherically transported metals and deposition in the southeastern United States: local or transoceanic? Appl. Geochem. 19:1189–1200. Honrath, R. E., R. C. Owen, M. V. Martin, J. S. Reid, K. Lapina, P. Fialho, M. P. Dziobak, J. Kleissl, and D. L. Westphal. 2004. Regional and hemispheric impacts of anthropogenic and biomass burning emissions on summertime CO2 and O3 in the North Atlantic lower free troposphere. J. Geophys. Res. doi:10.1029/2004JD005147. Horgan, S. E., M. M. Matheson, L. McLoughlin-Borlace, and J. K. Dart. 1999. Use of a low nutrient culture medium for the identification of bacteria causing severe ocular infection. J. Med. Microbiol. 48:701–703. Howitt, M. E. 2000. Asthma management in the Caribbean—an update. Postgrad. Doctor Caribb. 16:86–104. Howitt, M. E., R. Naibu, and T. C. Roach. 1998. The prevalence of childhood asthma and allergy in Barbados. The Barbados National Asthma and Allergy Study. Am. J. Respir. Crit. Care Med. 157:A624. Hugenholtz, P., B. M. Goebel, and N. R. Pace. 1998. Impact of cultureindependent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180:4765–4774. Hurst, C. J., C. P. Gerba, and I. Cech. 1980. Effects of environmental variables and soil characteristics on virus survival in soil. Appl. Environ. Microbiol. 40:1067–1079. Husar, R. B., D. M. Tratt, B. A. Schichtel, S. R. Falke, F. Li, D. Jaffe, S. Gasso, T. Gill, N. S. Laulainen, F. Lu, M. C. Reheis, Y. Chun, D. Westphal, B. N. Holben, C. Gueymard, I. McKendry, N. Kuring, G. C. Feldman, C. McClain, R. J. Frouin, J. Merrill, D. DuBois, F. Vignola, T. Murayama, S. Nickovic, W. W. Wilson, K. Sassen, N. Sugimoto, and W. C. Malm. 2001. Asian dust events of April 1998. J. Geophys. Res. Atmosph. 106:18317– 18330. Hyams, K. C., J. Riddle, D. H. Trump, and J. T. Graham. 2001. Endemic infectious diseases and biological warfare during the Gulf War: a decade of analysis and final concerns. Am. J. Trop. Med. Hyg. 65:664–670. Ichinose, T., M. Nishikawa, H. Takano, N. Sera, K. Sadakane, K. Mori, R. Yanagisawa, T. Oda, H. Tamura, K. Hiyoshi, H. Quan, S. Tomura, and T. Shibamoto. 2005. Pulmonary toxicity induced by intratracheal instillation of Asian yellow dust (Kosa) in mice. Environ. Toxicol. Pharmacol. 20:48–56. Ichinose, T., K. Sadakane, H. Takano, R. Yanagisawa, M. Nishikawa, I. Mori, H. Kawazato, A. Yasuda, K. Hiyoshi, and T. Shibamoto. 2006. Enhancement of mite allergen-induced eosinophil infiltration in the murine airway and local cytokine/chemokine expression by Asian sand dust. J. Toxicol. Environ. Health A 69:1571–1585. Ijaz, M. K., Y. G. Karim, S. A. Sattar, and C. M. Johnson-Lussenburg. 1987. Development of methods to study the survival of airborne viruses. J. Virol. Methods 18:87–106. Ismaelov, S., and S. Akhmedov. 1989. Prevalence and clinico-functional diagnosis of chronic non specific diseases of the lungs among the rural population of Uzbekistan. Prob. Tuberk. 6:19–21. (In Russian.) Ismail, K., B. Everitt, N. Blatchley, L. Hull, C. Unwin, A. David, and S. Wessely. 1999. Is there a Gulf War syndrome? Lancet 353:179–182. Ismail, M. A., S. I. I. Abdel-Hafez, and A. M. Moharram. 2002. Aeromycobiota of western desert of Egypt. Afr. J. Sci. Technol. 3:1–9. Jaenicke, R. 2005. Abundance of cellular material and proteins in the atmosphere. Science 308:73. Jamal, G. A., S. Hansen, F. Apartopoulos, and A. Peden. 1996. The “Gulf War syndrome.” Is there evidence of dysfunction in the nervous system? J. Neurol. Neurosurg. Psych. 60:449–451. Janssen, P. H. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72:1719– 1728. Jensen, P. A., B. Lighthart, A. J. Mohr, and B. T. Shaffer. 1994. Instrumentation used with microbial bioaerosol, p. 226–284. In B. Lighthart and A. J. Mohr (ed.), Atmospheric microbial aerosols: theory and applications. Chapman and Hall, New York, NY. Jensen, P. R., C. A. Kauffman, and W. Fenical. 1996. High recovery of culturable bacteria from the surfaces of marine algae. Mar. Biol. 126:1–7. Jinadu, B. A. 1995. Valley Fever Task Force report on the control of Coccidioides immitis. Kern County Health Department, Kern County, CA. Johnson, K. S., V. A. Elrod, S. E. Fitzwater, J. N. Plant, F. P. Chavez, S. J. Tanner, R. M. Gordon, D. L. Westphal, K. D. Perry, J. Wu, and D. M. Karl. 2003. Surface ocean-lower atmosphere interactions in the Northeast Pacific Ocean Gyre: aerosols, iron and the ecosystem response. Global Biogeochem. Cycles 17:1063. Joo, Y. S., S. H. An, O. K. Kim, J. Lubroth, and J. H. Sur. 2002. Foot-andmouth disease eradication efforts in the Republic of Korea. Can. J. Vet. Res. 66:122–124. Kang, H. K., C. M. Mahan, K. Y. Lee, F. M. Murphy, S. J. Simmens, H. A. Young, and P. H. Levine. 2002. Evidence for a deployment-related Gulf War syndrome by factor analysis. Arch. Environ. Health 57:61–68. Kellogg, C. A., D. W. Griffin, V. H. Garrison, K. K. Peak, N. Royall, R. R. VOL. 20, 2007 118. 119. 120. 121. 122. 123. 124. 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. Smith, and E. A. Shinn. 2004. Characterization of aerosolized bacteria and fungi from desert dust events in Mali, West Africa. Aerobiologia 20:99–110. Knoke, J. D., T. C. Smith, G. C. Gray, K. S. Kaiser, and A. W. Hawksworth. 2000. Factor analysis of self-reported symptoms: does it identify a Gulf War syndrome? Am. J. Epidemiol. 152:379–388. Konopka, A., L. Oliver, and R. F. Turko, Jr. 1998. The use of carbon substrate utilization patterns in environmental and ecological microbiology. Microb. Ecol. 35:103–115. Korenyi-Both, A. L., A. L. Korenyi-Both, and D. J. Juncer. 1997. Al Eskan disease: Persian Gulf syndrome. Military Med. 62:1–13. Korenyi-Both, A. L., A. L. Kornyi-Both, A. C. Molnar, and R. Fidelus-Gort. 1992. Al Eskan disease: Desert Storm pneumonitis. Military Med. 157:452– 462. Krause, A., F. K. Gould, and J. Forty. 1999. Prosthetic heart valve endocarditis caused by Bacillus circulans. J. Infect. 39:160–162. Kuske, C. R. 2006. Current and emerging technologies for the study of bacteria in the outdoor air. Curr. Opin. Biotechnol. 17:291–296. Kuske, C. R., S. M. Barns, and J. D. Busch. 1997. Diverse uncultivated bacterial groups from soils of the arid southwestern United States that are present in many geographic regions. Appl. Environ. Microbiol. 63:3614– 3621. Kwaasi, A. A. 2003. Date palm and sandstorm-borne allergens. Clin. Exp. Allergy 33:419–426. Kwaasi, A. A., R. S. Parhar, F. A. Al-Mohanna, H. A. Harfi, K. S. Collison, and S. T. Al-Sedairy. 1998. Aeroallergens and viable microbes in sandstorm dust. Potential triggers of allergic and nonallergic respiratory ailments. Allergy 53:255–265. Kwon, H. J., S. H. Cho, Y. Chun, F. Lagarde, and G. Pershagen. 2002. Effects of the Asian dust events on daily mortality in Seoul, Korea. Environ. Res. A 90:1–5. Labelle, R., and C. P. Gerba. 1981. Investigations into the protective effect of estuarine sediment on virus survival. Water Res. 16:469–478. Lacey, J. 1981. Aerobiology of conidial fungi, vol. 1. Academic Press Inc., New York, NY. Laden, F., L. M. Neas, D. W. Dockery, and J. Schwarz. 2000. Association of fine particulate matter from different sources with daily mortality in six U.S. cities. Environ. Health Perspect. 108:941–947. Lancaster, N., R. Bamford, and S. Metzger. 1995. Field studies of the potential for wind transport of plutonium—contaminated soils at sites in areas 6 and 11, Nevada Test Site, p. 1–77. U.S. Department of Energy, DOE/NV/10845-60. Larsen, J., P. A. Olsson, and I. Jakobsen. 1998. The use of fatty acid signatures to study mycelial interactions between the arbuscular mycorrhizal fungus Glomus intraradices and the saprotrophic fungus Fusarium culmorum in root-free soil. Mycol. Res. 102:1491–1496. La Scola, B., R. J. Birtles, M. N. Mallet, and D. Raoult. 1998. Massilia timonae gen. nov., sp. nov., isolated from blood of an immunocompromised patient with cerebellar lesions. J. Clin. Microbiol. 36:2847–2852. Lee, H. A., R. Gabriel, J. P. G. Bolton, A. J. Bale, and M. Jackson. 2002. Health status and clinical diagnoses of 3000 UK Gulf War veterans. J. R. Soc. Med. 95:491–497. Lee, H. B., and N. Magan. 1999. Environmental factors and nutritional utilization patterns affect niche overlap indices between Aspergillus ochraceus and spoilage fungi. Lett. Appl. Microbiol. 28:300–304. Lee, Y. A., H. J. Kim, T. W. Lee, M. J. Kim, M. H. Lee, J. H. Lee, and C. G. Ihm. 2004. First report of Cryptococcus albidus-induced disseminated cryptococcosis in a renal transplant recipient. Korean J. Intern. Med. 19:53–57. Lei, Y. C., C. C. Chan, P. Y. Wang, C. T. Lee, and T. J. Cheng. 2004. Effects of Asian dust event particles on inflammation markers in peripheral blood and bronchoalveolar lavage in pulmonary hypertensive rats. Environ. Res. 95:71–76. Lenes, J. M., B. P. Darrow, C. Cattrall, C. A. Heil, M. Callahan, G. A. Vargo, R. H. Byrne, J. M. Prospero, D. E. Bates, K. A. Fanning, and J. J. Walsh. 2001. Iron fertilization and the Trichodesmium response on the West Florida shelf. Limnol. Oceanogr. 46:1261–1277. Reference deleted. Lim, P. S., S. L. Chen, C. Y. Tsai, and M. A. Pai. 2006. Pantoea peritonitis in a patient receiving chronic ambulatory peritoneal dialysis. Nephrology 11:97–99. Lyles, M. B., H. L. Fredrickson, A. J. Bednar, H. B. Fannin, and T. M. Sobecki. 2005. The chemical, biological, and mechanical characterization of airborne micro-particulates from Kuwait. Abstr. 8th Ann. Force Health Protect. Conf., session 2586, Louisville, KY. Lysenko, S. 1980. Resistance of microorganisms of upper layers of the atmosphere to ultraviolet radiation and a high vacuum. Mikrobiologiia 49:175–177. Lysenko, S., and N. S. Demina. 1992. Drying as one of the extreme factors for the microflora of the atmosphere. J. Br. Interplanetary Soc. 45:39–41. Maier, R. M., K. P. Drees, J. W. Neilson, D. A. Henderson, J. Quade, and J. L. Betancourt. 2004. Microbial life in the Atacama Desert. Science 306:1289–1290. Makino, S. I., H. I. Cheun, M. Watarai, I. Uchida, and K. Takeshi. 2001. DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. 171. 475 Detection of anthrax spores from the air by real-time PCR. Lett. Appl. Microbiol. 33:237–240. Marsh, P., and E. M. H. Wellington. 1994. Phage-host interactions in soil. FEMS Microbiol. Ecol. 15:99–108. Martin, T., D. J. Hogan, F. Murphy, I. Natyshak, and E. P. Ewan. 1991. Rhodococcus infection of the skin with lymphadenitis in a nonimmunocompromised girl. J. Am. Acad. Dermatol. 24:328–332. Martiny, J. B., B. J. Bohannon, J. H. Brown, R. K. Colwell, J. A. Fuhrman, J. L. Green, M. C. Horner-Devine, M. Kane, J. A. Krumins, C. R. Kuske, P. J. Morin, S. Naeem, L. Ovreas, A. L. Reysenbach, V. H. Smith, and J. T. Staley. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102–112. Massa, S., M. Caruso, F. Trovatelli, and M. Tosques. 1998. Comparison of plate count agar and R2A medium for enumeration of heterotrophic bacteria in natural mineral water. World J. Microbiol. Biotechnol. 14:727–730. McFeters, G. A., S. C. Cameron, and M. W. LeChevallier. 1982. Influence of diluents, media, and membrane filter on detection of injured waterborne coliform bacteria. Appl. Environ. Microbiol. 43:97–103. Medoro, G., N. Manaresi, A. Leonardi, L. Altomare, M. Tartagni, and R. Guerrieri. 2003. A lab-on-a-chip for cell detection and manipulation. Sensors J. 3:317–325. Meier, F. C. 1936. Collecting microorganisms from winds above the Caribbean Sea. Phytopathology 26:102. Meier, F. C. 1936. Effects of conditions in the stratosphere on spores of fungi. Natl. Geogr. Soc. Stratosph. Ser. 2:152–153. Meier, F. C., and E. Artschwager. 1938. Airplane collections of sugar-beet pollen. Science 88:507–508. Meier, F. C., and C. A. Lindbergh. 1935. Collecting micro-organisms from the arctic atmosphere. Sci. Monthly 40:5–20. Middleton, N. J., and A. S. Goudie. 2001. Saharan dust: sources and trajectories. Trans. Inst. Br. Geogr. 26:165–181. Mikkelsen, T., S. Alexandersen, P. Astrup, H. J. Champion, A. I. Donaldson, F. N. Dunkerley, J. Gloster, J. H. Sorensen, and S. Thykier-Nielsen. 2003. Investigation of airborne foot-and-mouth disease virus transmission during low-wind conditions in the early phase of the UK 2001 epidemic. Atmosph. Chem. Phys. 3:2101–2110. Mohr, A. J. 1997. Fate and transport of microorganisms in air. ASM Press, Washington, DC. Moissenet, D., P. Bidet, A. Garbarg-Chenon, G. Arlet, and H. Vu-Thien. 2001. Ralstonia paucula (formerly CDC group IV c-2): unsuccessful strain differentiation with PCR-based methods, study of the 16S-23S spacer of the rRNA operon, and comparison with other Ralstonia species (R. eutropha, R. pickettii, R. gilardii, and R. solanacearum). J. Clin. Microbiol. 39:381–384. Molesworth, A. M., L. E. Cuevas, S. J. Conner, A. P. Morse, and M. C. Thomson. 2003. Environmental risk and meningitis epidemics in Africa. Emerg. Infect. Dis. 9:1287–1293. Molesworth, A. M., L. E. Cuevas, A. P. Morse, J. R. Herman, and M. C. Thomson. 2002. Dust clouds and spread of infection. Lancet 359:81–82. Moore, C., and R. Norton. 1995. Corynebacterium aquaticum septicaemia in a neutropenic patient. J. Clin. Pathol. 48:971–972. Moreno, A., J. Targarona, J. Henderiks, M. Canals, T. Freudenthal, and H. Meggers. 2001. Orbital forcing of dust supply to the North Canary Basin over the last 250 kyr. Quaternary Sci. Rev. 20:1327–1339. Morgan, J., M. V. Cano, D. R. Feikin, M. Phelan, O. V. Monroy, P. K. Morales, J. Carpenter, A. Weltman, P. G. Spitzer, H. H. Liu, S. A. Mirza, D. E. Bronstein, D. J. Morgan, L. A. Kirkman, M. E. Brandt, N. Iqbal, M. D. Lindsley, D. W. Warnock, R. A. Hajjeh, and the Acapulco Histoplasmosis Working Group. 2003. A large outbreak of histoplasmosis among American travelers associated with a hotel in Acapulco, Mexico, spring 2001. Am. J. Trop. Med. Hyg. 69:663–669. Moubasher, A. H. 1993. Soil fungi in Qatar and other Arab countries. Centre for Scientific and Applied Research, University of Qatar. Moulin, C., C. E. Lambert, F. Dulac, and U. Dayan. 1997. Control of atmospheric export of dust from North Africa by the North Atlantic Oscillation. Nature 387:691–694. Mullis, K., F. Faloona, S. Scharf, R. Saiki, G. Horn, and H. Erlich. 1986. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp. Quant. Biol. 1:263–273. Muyzer, G., and K. Smalla. 1998. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Leeuwenhoek 73:127–141. Navarro-Gonzalez, R., F. A. Rainey, P. Molina, D. R. Bagaley, B. J. Hollen, J. de la Rosa, A. M. Small, R. C. Quinn, F. J. Grunthaner, L. Caceres, B. Gomez-Silva, and C. P. McKay. 2003. Mars-like soils in the Atacama Desert, Chile, and the dry limit of microbial life. Science 302:1018–1021. Nichols, C. M., J. P. Bowman, and J. Geuzennec. 2005. Effects of incubation temperature on growth and production of exopolysaccharides by an Antarctic sea ice bacterium grown in batch culture. Appl. Environ. Microbiol. 71:3519–3523. Nivens, D. E., T. E. McKnight, S. A. Moser, S. J. Osbourn, M. L. Simpson, and G. S. Sayler. 2004. Bioluminescent bioreporter integrated circuits: 476 172. 173. 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. 187. 188. 189. 190. 191. 192. 193. 194. 195. 196. 197. GRIFFIN potentially small, rugged and inexpensive whole-cell biosensors for remote environmental monitoring. J. Appl. Microbiol. 96:33–46. Noble, R. T., and J. A. Furman. 1998. Use of SYBR green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat. Microbiol. Ecol. 14:113–118. Norboo, T., P. T. Angchuk, M. Yahya, S. R. Kamat, F. D. Pooley, B. Corrin, I. H. Kerr, N. Bruce, and K. P. Ball. 1991. Silicosis in a Himalayan village population: role of environmental dust. Thorax 46:861–863. O’Hara, S. L., G. F. S. Wiggs, B. Mamedov, G. Davidson, and R. B. Hubbard. 2000. Exposure to airborne dust contaminated with pesticide in the Aral Sea region. Lancet 355:627. Ozawa, Y., B. L. Ong, and S. H. An. 2001. Traceback systems used during recent epizootics in Asia. Rev. Sci. Technique Off. Int. Epizoot. 20:605–613. Papastefanou, C., M. Manolopoulou, S. Stoulos, A. Ioannidou, and E. Gerasopoulos. 2001. Coloured rain dust from Sahara Desert is still radioactive. J. Environ. Radioact. 55:109–112. Papova, N. A., I. A. Nikolaev, T. P. Turova, A. M. Lysenko, G. A. Osipov, N. V. Verkhovtseva, and N. S. Panikov. 2002. Geobacillus uralicus, a new species of thermophilic bacteria. Mikrobiologiia 71:391–398. Park, B. J., K. Sigel, V. Vaz, K. Komatsu, C. McRill, M. Phelan, T. Colman, A. C. Comrie, D. W. Warnock, J. N. Galgiani, and R. A. Hajjeh. 2005. An epidemic of coccidioidomycosis in Arizona associated with climatic changes, 1998–2001. J. Infect. Dis. 191:1981–1987. Park, D. J., J. C. Yun, J. E. Baek, E. Y. Jung, D. W. Lee, M. A. Kim, and S. H. Chang. 2006. Relapsing Bacillus licheniformis peritonitis in a continuous ambulatory peritoneal dialysis patient. Nephrology 11:21–22. Park, J. W., Y. H. Lim, S. Y. Kyung, C. H. An, S. P. Lee, S. H. Jeong, and Y. S. Ju. 2005. Effects of ambient particulate matter on peak expiratory flow rates and respiratory symptoms of asthmatics during Asian dust periods in Korea. Respirology 10:470–476. Paster, B. J., W. A. J. Falkler, C. O. Enwonwu, E. O. Idigbe, K. O. Savage, V. A. Levanos, M. A. Tamer, R. L. Ericson, C. N. Lau, and F. E. Dewhirst. 2002. Prevalent bacterial species and novel phylotypes in advanced noma lesions. J. Clin. Microbiol. 40:2187–2191. Patterson, S. S., E. T. Casper, L. Garcia-Rubio, M. C. Smith, and J. H. Paul. 2005. Increased precision of microbial RNA quantification using NASBA with an internal control (IC-NASBA). J. Microbiol. Methods 60: 342–352. Paul, J. H. 1982. Use of Hoechst dyes 33258 and 3342 for enumeration of attached and planktonic bacteria. Appl. Environ. Microbiol. 43:939–944. Perkins, S. 2001. Dust, the thermostat. Sci. News 160:200–201. Pettersson, M., and E. Baath. 2003. Temperature-dependent changes in the soil bacterial community in limed and unlimed soil. FEMS Microbiol. Ecol. 45:13–21. Prahalad, A. K., J. Inmon, L. A. Dailey, M. C. Madden, A. J. Ghio, and J. E. Gallagher. 2001. Air pollution particles mediated oxidative DNA base damage in a cell free system and in human airway epithelial cells in relation to particulate metal content and bioreactivity. Chem. Res. Toxicol. 14:879– 887. Proctor, B. E. 1935. The microbiology of the upper air. J. Bacteriol. 30: 363–375. Prospero, J. M. 1999. Long-range transport of mineral dust in the global atmosphere: impact of African dust on the environment of the southeastern United States. Proc. Natl. Acad. Sci. USA 96:3396–3403. Prospero, J. M. 1999. Long-term measurements of the transport of African mineral dust to the southeastern United States: implications for regional air quality. J. Geophys. Res. 104:15917–15927. Prospero, J. M., E. Blades, G. Mathison, and R. Naidu. 2005. Interhemispheric transport of viable fungi and bacteria from Africa to the Caribbean with soil dust. Aerobiologia 21:1–19. Prospero, J. M., and P. J. Lamb. 2003. African droughts and dust transport to the Caribbean: climate change implications. Science 302:1024–1027. Prospero, J. M., and R. T. Nees. 1986. Impact of the North African drought and El Nino on mineral dust in the Barbados trade winds. Nature 320:735– 738. Qian, W., L. Quan, and S. Shi. 2002. Variations of the dust storm in China and its climatic control. J. Climate 15:1216–1229. Rao, V. C., K. M. Seidel, S. M. Goyal, T. G. Metcalf, and J. L. Melnick. 1984. Isolation of enteroviruses from water, suspended solids, and sediments from Galveston Bay: survival of poliovirus and rotavirus adsorbed to sediments. Appl. Environ. Microbiol. 48:404–409. Reasoner, D. J., and E. E. Geldreich. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49:1–7. Reid, J. S., E. M. Prins, D. L. Westphal, C. C. Schmidt, K. A. Richardson, S. A. Christopher, T. F. Eck, E. A. Reid, C. A. Curtis, and J. P. Hoffman. 2004. Real-time monitoring of South American smoke particle emissions and transport using a coupled remote sensing/box-model approach. Geophys. Res. Lett. doi:10.1029/2203GL018845. Reynolds, D. T., and C. R. Fricker. 1999. Application of laser scanning for the rapid and automated detection of bacteria in water samples. J. Appl. Microbiol. 86:785–795. CLIN. MICROBIOL. REV. 198. Reynolds, K. A., and I. L. Pepper. 2000. Microorganisms in the environment, p. 585. In R. M. Maier, I. L. Pepper, and C. P. Gerba (ed.), Environmental microbiology. Academic Press, San Diego, CA. 199. Rippon, J. W. 1988. Medical mycology: the pathogenic fungi and the pathogenic actinomycetes, 3rd ed. W. B. Saunders Company, Philadelphia, PA. 200. Rogers, L. A., and F. C. Meier. 1936. The collection of microorganisms above 36,000 feet. Natl. Geogr. Soc. Stratosph. Ser. 2:146–151. 201. Rook, G. A., and A. Zumla. 1997. Gulf War syndrome: is it due to a systemic shift in cytokine balance towards a Th2 profile? Lancet 349:1831–1833. 202. Saiyed, H. N., Y. K. Sharma, H. G. Sadhu, T. Norboo, P. D. Patel, T. S. Patel, K. Venkaiah, and S. K. Kashyap. 1991. Non-occupational pneumoconiosis at high altitude villages in central Ladakh. Br. J. Indian Med. 48:825–829. 203. Sakamoto, K., and K. Yoshida. 2002. Recent outbreaks of foot and mouth disease in countries of East Asia. Rev. Sci. Technique Off. Int. Epizoot. 21:459–463. 204. Sanders, J. W., S. D. Putnam, C. Frankart, R. W. Frenck, M. R. Monteville, M. S. Riddle, D. M. Rockabrand, T. W. Sharp, and D. R. Tribble. 2005. Impact of illness and non-combat injury during operations Iraqi Freedom and Enduring Freedom (Afghanistan). Am. J. Trop. Med. Hyg. 73:713–719. 205. Sandstrom, T., L. Bjermer, and R. Rylander. 1992. Lipopolysaccharide (LPS) inhalation in health subjects increases neutrophils, lymphocytes and fibronectin levels in bronchoalveolar lavage fluid. Eur. Respir. J. 5:992–996. 206. Sattar, S. A., and M. K. Ijaz. 1997. Airborne viruses, p. 682–692. In C. J. Hurst, G. R. Knudsen, M. J. McInerney, L. D. Stetzenbach, and M. V. Walter (ed.), Manual of environmental microbiology. ASM Press, Washington, DC. 207. Schlesinger, P., Y. Mamane, and I. Grishkan. 2006. Transport of microorganisms to Israel during Saharan dust events. Aerobiologia 22:259–273. 208. Schneider, E., R. A. Hajjeh, R. A. Spiegel, R. W. Jibson, E. L. Harp, G. A. Marshall, R. A. Gunn, M. M. McNeil, R. W. Pinner, R. C. Baron, R. C. Burger, L. C. Hutwagner, C. Crump, L. Kaufman, S. E. Reef, G. M. Feldman, D. Pappagianis, and B. Werner. 1997. A coccidioidomycosis outbreak following the Northridge, California earthquake. JAMA 277:904– 908. 209. Schollaert, S. E., J. A. Yoder, D. L. Westphal, and J. E. O’Reilly. 2003. The influence of dust and sulfate aerosols on ocean color spectra and chlorophyll-a concentrations derived for SeaWiFS of the U.S. coast. J. Geophys. Res. 108:3191. 210. Schwartz, J., G. Norris, T. Larson, L. Sheppard, C. Claiborne, and J. Koenig. 1999. Episodes of high coarse particle concentrations are not associated with increased mortality. Environ. Health Perspect. 107:339–342. 211. Seifert, K. A., B. D. Wingfield, and M. J. Wingfield. 1995. A critique of DNA sequence analysis in the taxonomy of filamentous ascomycetes and ascomycetous anamorphs. Can. J. Botany 73:S760–S767. 212. Shinn, E. A., D. W. Griffin, and D. B. Seba. 2003. Atmospheric transport of mold spores in clouds of desert dust. Arch. Environ. Health 58:498–504. 213. Shinn, E. A., G. W. Smith, J. M. Prospero, P. Betzer, M. L. Hayes, V. Garrison, and R. T. Barber. 2000. African dust and the demise of Caribbean coral reefs. Geol. Res. Lett. 27:3029–3032. 214. Shoor, A. F., S. L. Scoville, S. B. Cersovsky, D. Shanks, C. F. Ockenhouse, B. L. Smoak, W. W. Carr, and B. P. Petruccelli. 2004. Acute eosinophilic pneumonia among US military personnel deployed in or near Iraq. JAMA 292:2997–3005. 215. Sobsey, M. D., C. H. Dean, M. E. Knuckles, and R. A. Wagner. 1980. Interactions and survival of enteric viruses in soil materials. Appl. Environ. Microbiol. 40:92–101. 216. Song, L., W. J. Li, Q. L. Wang, G. Z. Chen, Y. S. Zhang, and L. H. Xu. 2005. Jiangella gansuensis gen. nov., sp. nov., a novel actinomycete from a desert soil in north-west China. Int. J. Syst. Evol. Microbiol. 55:881–884. 217. Sorensen, J. H., D. K. J. Mackay, C. O. Jensen, and A. I. Donaldson. 2000. An integrated model to predict the atmospheric spread of foot-and-mouth disease virus. Epidemiol. Infect. 124:577–590. 218. States, J. S., and M. Christensen. 2001. Fungi associated with biological soil crusts in desert grasslands of Utah and Wyoming. Mycologia 93:432–439. 219. Stetzenbach, L. D., M. P. Buttner, and P. Cruz. 2004. Detection and enumeration of airborne biocontaminants. Curr. Opin. Biotechnol. 15:170– 174. 220. Stewart, S. L., S. A. Grinshpun, K. Willeke, S. Terzieva, V. Ulevicius, and J. Donnelly. 1995. Effect of impact stress on microbial recovery on an agar surface. Appl. Environ. Microbiol. 61:1232–1239. 221. St. Germain, G., and R. Summerbell. 1996. Identifying filamentous fungi. Star Publishing Company, Belmont, CA. 222. Straub, T. M., I. L. Pepper, and C. P. Gerba. 1993. Virus survival in sewage sludge amended desert soil. Water Sci. Technol. 27:421–424. 223. Sultan, B., K. Labadi, J. F. Guegan, and S. Janicot. 2005. Climate drives the meningitis epidemics onset in West Africa. PLoS Med. 2:e6. 224. Swap, R., M. Garstang, S. Greco, R. Talbot, and P. Kallberg. 1992. Saharan dust in the Amazon Basin. Tellus 44:133–149. 225. Tate, R. L., III. 2000. Soil microbiology, 2nd ed. John Wiley and Sons, Inc., New York, NY. 226. Tendler, C., and E. J. Bottone. 1989. Corynebacterium aquaticum urinary VOL. 20, 2007 227. 228. 229. 230. 231. 232. 233. 234. 235. 236. 237. 238. 239. 240. 241. 242. 243. 244. 245. 246. tract infection in a neonate, and concepts regarding the role of the organism as a neonatal pathogen. J. Clin. Microbiol. 27:343–345. Terzieva, S., J. Donnelly, V. Ulevicius, S. A. Grinshpun, K. Willeke, G. N. Stelma, and K. P. Brenner. 1996. Comparison of methods for detection and enumeration of airborne microorganisms collected by liquid impingement. Appl. Environ. Microbiol. 62:2264–2272. Thrane, C., S. Olsson, T. H. Nielsen, and J. Sorensen. 1999. Vital fluorescent stains for detection of stress in Pythium ultimum and Rhizoctonia solani. FEMS Microbiol. Ecol. 30:11–23. Tiwana, H., C. Wilson, J. Pirt, W. Cartmell, and A. Ebringer. 1999. Autoantibodies to brain components and antibodies to Acinetobacter calcoaceticus are present in bovine spongiform encephalopathy. Infect. Immun. 67:6591–6595. Tobias, H. J., M. P. Schafer, M. Pitesky, D. P. Fergenson, J. Horn, M. Frank, and E. E. Gard. 2005. Bioaerosol mass spectrometry for rapid detection of individual airborne Mycobacterium tuberculosis H37Ra particles. Appl. Environ. Microbiol. 71:6086–6095. Tobin, R. S., P. Lomax, and D. J. Kushner. 1980. Comparison of nine brands of membrane filter and the most-probable-number methods for total coliform enumeration in sewage-contaminated drinking water. Appl. Environ. Microbiol. 40:186–191. Torrent, A., S. Deniz, P. Calabuig, and J. Oros. 2000. Septicemia due to Staphylococcus xylosis in a loggerhead sea turtle (Caretta caretta). www.vet .uga.edu/esp/IVCVM/Oros1/. Torsvik, V., J. Goksoyr, and F. L. Daae. 1990. High diversity in DNA of soil bacteria. Appl. Environ. Microbiol. 56:782–787. Torsvik, V., L. Ovreas, and T. F. Thingstad. 2002. Prokaryotic diversity— magnitude, dynamics, and controlling factors. Science 296:1064–1066. Tucker, C. J., and S. E. Nicholson. 1999. Variations in the size of the Sahara Desert from 1980 to 1997. Ambio 28:587–591. Tyndall, J. 1882. Essays on the floating-matter of the air in relation to putrefaction and infection. Johnson Reprint Corporation (1966), New York, NY. United Nations Development Program. 1997. Turkmenistan: human development report 1996. United Nations Development Program, Ashgabat, Turkmenistan. van der Linde, K., B. T. Lim, J. M. M. Rondeel, L. P. M. T. Antonissen, and G. M. T. de Jong. 1999. Improved bacteriological surveillance of haemodialysis fluids: a comparison between Tryptic soy agar and Reasoner’s 2A media. Nephrol. Dialysis Transplant. 14:2433–2437. Venkatesh, M. V., K. R. Joshi, S. C. Harjai, and I. N. Ramdeo. 1975. Aspergillosis in desert locust (Schistocerka gregaria Forsk). Mycopathologia 57:135–138. Vernooy, J. H. J., M. A. Dentener, R. J. van Suylen, W. A. Buurman, and E. F. M. Wouters. 2002. Long-term intratracheal lipopolysaccharide exposure in mice results in chronic lung inflammation and persistent pathology. Am. J. Respir. Cell Mol. Biol. 26:152–159. Vesey, G., J. Narai, N. Ashbolt, K. W. Williams, and D. Veal. 1994. Detection of specific microorganisms in environmental samples using flow cytometry. Methods Cell Biol. Flow Cytom. 42:490–519. Wainwright, M., N. C. Wickramasinghe, J. V. Narlikar, and P. Rajaratnam. 2003. Microorganisms cultured from stratospheric air samples obtained at 41 km. FEMS Microbiol. Lett. 218:161–165. Walsh, J. J., and K. A. Steidinger. 2001. Saharan dust and Florida red tides: the cyanophyte connection. J. Geophys. Res. 106:11597–11612. Washington, R., M. Todd, N. J. Middleton, and A. S. Goudie. 2003. Duststorm source areas determined by the total ozone monitoring spectrometer and surface observations. Ann. Assoc. Am. Geogr. 93:297–313. Weir-Brush, J. R., V. H. Garrison, G. W. Smith, and E. A. Shinn. 2004. The relationship between gorgonian coral (Cnidaria: Gorganacea) diseases and African dust storms. Aerobiologia 20:119–126. White, D. C., H. C. Pinkart, and D. B. Ringelberg. 1997. Biomass measurements: biochemical approaches, p. 91–101. In C. J. Hurst, G. R. Knudsen, M. J. McInerney, L. D. Stetzenback, and M. V. Walter (ed.), Manual of environmental microbiology. ASM Press, Washington, DC. DESERT DUST MICROBIOLOGY AND HUMAN HEALTH 477 247. Whitman, W. B., D. C. Coleman, and W. J. Wiebe. 1998. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA 95:6578–6583. 248. Wiggs, G. F. S., S. L. O’hara, J. Wegerdt, J. V. D. Meer, I. Small, and R. Hubbard. 2003. The dynamics and characteristics of aeolian dust in dryland Central Asia: possible impacts on human exposure and respiratory health in the Aral Sea basin. Geogr. J. 169:142–157. 249. Willeke, K., X. Lin, and S. A. Grinshpun. 1998. Improved aerosol collection by combined impaction and centrifugal motion. Aerosol Sci. Technol. 28: 439–456. 250. Williams, P. L., D. L. Sable, P. Mendez, and L. T. Smyth. 1979. Symptomatic coccidioidomycosis following a severe natural dust storm. An outbreak at the Naval Air Station, Lemoore, California. Chest 76:566–570. 251. Williams, R. H., E. Ward, and H. A. McCartney. 2001. Methods for integrated air sampling and DNA analysis for detection of airborne fungal spores. Appl. Environ. Microbiol. 67:2453–2459. 252. Williamson, K. E., K. E. Wommack, and M. Radosevich. 2003. Sampling natural viral communities from soil for culture-independent analyses. Appl. Environ. Microbiol. 69:6628–6633. 253. Wilson, K. H., W. J. Wilson, J. L. Radosevich, T. Z. DeSantis, V. S. Viswanathan, T. A. Kuczmarski, and G. L. Andersen. 2002. High-density microarray of small-subunit ribosomal DNA probes. Appl. Environ. Microbiol. 68:2535–2541. 254. Wisplinghoff, H., A. E. Rosato, M. C. Enright, M. Noto, W. Craig, and G. L. Archer. 2003. Related clones containing SCCmec type IV predominate among clinically significant Staphylococcus epidermidis isolates. Antimicrob. Agents Chemother. 47:3574–3579. 255. Wolf, F. T. 1943. The microbiology of the upper air. Bull. Torrey Botanical Club 70:1–14. 256. Wu, P. C., J. C. Tsai, F. C. Li, S. C. Lung, and H. J. Su. 2004. Increased levels of ambient fungal spores in Taiwan are associated with dust events from China. Atmosph. Environ. 38:4879–4886. 257. Wust, G., F. Friedl, D. Haas, M. Kock, F. Pichler-Semmelrock, F. F. Reinthaler, R. Schlacher, and E. Marth. 2003. A comparison between Andersen (ACFM) and Reuter Centrifugal Sampler (RCS-plus) for indoor sampling of airborne molds. Aerobiologia 19:125–128. 258. Xiao, C., S. C. Kang, D. Qin, T. D. Yao, and J. W. Ren. 2002. Transport of atmospheric impurities over the Qinghai-Xizang (Tibetan) Plateau as shown by snow chemistry. J. Asian Earth Sci. 20:231–239. 259. Xu, X. Z., X. G. Cai, X. S. Men, P. Y. Yang, J. F. Yang, S. L. Jing, J. H. He, and W. Y. Si. 1993. A study of siliceous pneumoconiosis in the desert area of Sunan County, Gansu Province, China. Biomed. Environ. Sci. 6:217–222. 260. Yates, M. V., and S. R. Yates. 1988. Modeling microbial fate in the subsurface environment. CRC Crit. Rev. Environ. Control 17:307–344. 261. Yeo, H. G., and J. H. Kim. 2002. SPM and fungal spores in the ambient air of west Korea during the Asian dust (yellow sand) period. Atmosph. Environ. 36:5437–5442. 262. Yu, I. T. S., Y. Li, T. W. Wong, W. Tam, M. Phil, A. T. Chan, J. H. W. Lee, D. Y. C. Leung, and T. Ho. 2004. Evidence of airborne transmission of the severe acute respiratory syndrome virus. N. Engl. J. Med. 350:1731–1739. 263. Zanobetti, A., and J. Schwartz. 2005. The effect of particulate air pollution on emergency admissions for myocardial infarction: a multicity case-crossover analysis. Environ. Health Perspect. 113:978–982. 264. Zengler, K., G. Toledo, M. Rappe, J. Elkins, E. J. Mathur, J. M. Short, and M. Keller. 2002. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 99:15681–15686. 265. Zhang, X. Y., S. L. Gong, T. L. Zhao, R. Arimoto, Y. Q. Wang, and Z. J. Zhou. 2003. Sources of Asian dust and role of climate change versus desertification in Asian dust emission. Geophys. Res. Lett. 30:2272. 266. Zhenda, Z., and W. Tao. 1993. The trends of desertification and its rehabilitation in China. Desertification Control Bull. 22:27–29. 267. Zitouni, A., H. Boudjella, L. Lamari, B. Badji, F. Mathieu, A. Lebrihi, and N. Sabaou. 2005. Nocardiopsis and Saccharothrix genera in Saharan soils in Algeria: isolation, biological activities and partial characterization of antibiotics. Res. Microbiol. 156:984–993. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 478–488 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00006-07 Vol. 20, No. 3 Epidemiology of Human Immunodeficiency Virus in the United States Susan Hariri and Matthew T. McKenna* HIV Incidence and Case Surveillance Branch, Division of HIV/AIDS Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD and TB Prevention (Proposed), Centers for Disease Control and Prevention, Atlanta, Georgia INTRODUCTION .......................................................................................................................................................478 BACKGROUND AND EARLY HISTORY ..............................................................................................................478 DATA SOURCES AND METHODS ........................................................................................................................479 HIV/AIDS National Surveillance ..........................................................................................................................479 Population-Based Serosurveys (NHANES) .........................................................................................................479 Other Methods ........................................................................................................................................................479 CURRENT PICTURE ................................................................................................................................................480 Mortality Patterns ..................................................................................................................................................480 AIDS .........................................................................................................................................................................481 HIV Infection (Including AIDS) ...........................................................................................................................482 FUTURE DIRECTIONS ............................................................................................................................................485 Clinical and Epidemiologic Effects of HAART ...................................................................................................485 HIV Incidence..........................................................................................................................................................486 Monitoring HIV Risk Behavior.............................................................................................................................486 CONCLUSION............................................................................................................................................................487 ACKNOWLEDGMENTS ...........................................................................................................................................487 REFERENCES ............................................................................................................................................................487 Columbia have mandated some form of reporting for individuals diagnosed with HIV infection, even in the absence of AIDS, and efforts are under way to standardize methods to comply with CDC-recommended confidential name-based reporting and to integrate data into a national HIV/AIDS surveillance system (17). An essential role of such a surveillance system is to safeguard against duplication of case reports among different states to provide the most accurate and complete picture of the HIV epidemic across the United States. The purpose of this paper is to describe the current state of HIV infection, using national HIV/AIDS surveillance data. INTRODUCTION Advances in the treatment of human immunodeficiency virus (HIV) infection have resulted in a fundamental shift in its epidemiology, from a highly lethal infectious disease to a potentially chronic and manageable condition (46). Thus, overall rates of the most severe clinical presentations of the infection, i.e., AIDS, have declined compared to those in the early 1990s (35). The result is that more people are living with HIV infection, and deaths from AIDS remain low relative to historical levels (33). Unfortunately, successes in preventing new infections appear to have stalled during the late 1990s (33). Therefore, achieving a clear understanding of the sociobehavioral determinants of HIV infection will be essential for successfully controlling the disease. Moreover, racial and ethnic disparities clearly exist both in access to effective treatments for HIV and in rates of new infections (15). Given that the success of prevention and treatment efforts depends on accurate and complete identification of at-risk populations, collection, analysis, and interpretation of surveillance data remain critical for keeping abreast of the evolving nature of the epidemic (12, 50). In particular, the increase in the time from diagnosis of HIV to the onset of AIDS resulting from the use of effective antiretrovirals underscores the need for ongoing enumeration and evaluation of the extents and patterns of diagnosed HIV infections as well as AIDS cases (25). In 1999, the Centers for Disease Control and Prevention (CDC) recommended national implementation of name-based HIV reporting, which had previously been implemented in only 34 areas (11). To date, all states and the District of BACKGROUND AND EARLY HISTORY The first decade of the HIV epidemic was characterized by steady and sharp increases in the incidence of AIDS cases, punctuated by diagnostic and therapeutic milestones as well as changes in the methods used to monitor the clinical manifestations of the infection. These changes were precipitated by increases in the number of new infections and advances in the understanding of the biology of the virus. By the peak of the epidemic in 1993, AIDS had become the leading cause of death in men and women aged 25 to 44 years and was the eighth leading cause of death overall, accounting for 2% of all deaths in the United States (10). Age-adjusted death rates attributed to HIV increased in a linear fashion from 6 deaths per 100,000 in 1987 to 17 deaths per 100,000 in 1995, when deaths due to the disease peaked. Between 1995 and 1998, however, the numbers of reported AIDS cases and deaths in the United States declined precipitously, at rates of 28%, 45%, and 18% each year, after which they remained relatively stable, at approximately 40,000 each year through 2004 (7). Widespread use of improved antiretroviral therapy leading to longer incubation periods and survival after infection is believed to * Corresponding author. Mailing address: CDC, NCCDPHP, 1600 Clifton Road, MS E 47, Atlanta, GA 30333. Phone: (404) 639-2050. Fax: (404) 639-2980. E-mail: [email protected]. 478 VOL. 20, 2007 EPIDEMIOLOGY OF HIV IN THE UNITED STATES explain most of the rapid reduction in mortality in the mid1990s (21). However, other factors, such as increased use of prophylaxis for opportunistic infections and primary prevention, are also likely to have contributed to the observed decline (28). Models that indirectly measured the incidence of HIV infection, using AIDS case reports and knowledge about the distribution of HIV incubation periods, indicated that the transmission incidence peaked around the mid-1980s, approximately a decade earlier than did cases of AIDS (33). In the early years of the epidemic, HIV/AIDS, defined as all HIV infections, with and without AIDS, was characterized as a disease of white men who have sex with men (MSM), the group in whom the disease was first recognized (8). Injecting drug users (IDUs) represented approximately 20% of cases, while heterosexuals and females from all transmission categories accounted for about 5% and 8% of all reported cases, respectively (10). Because direct socioeconomic data were not routinely collected as part of the AIDS national surveillance system, little to no information is available at the national level to describe the socioeconomic characteristics of the early patients. DATA SOURCES AND METHODS HIV/AIDS National Surveillance The national HIV/AIDS surveillance system, supported by the CDC, is the primary national source of information used to track the changing characteristics and patterns of the epidemic. With ongoing technical assistance from the CDC, state and local health departments collect surveillance data for analysis at the local level (41). Local surveillance data are routinely reported to the CDC, where they are analyzed in the aggregate to provide national population-based estimates aimed at elucidating the epidemic. Surveillance data include data on all persons diagnosed with HIV in the entire U.S. population, including institutionalized individuals and noncivilian (i.e., military) persons. Prior to the widespread use of highly active antiretroviral therapy (HAART) in 1996, HIV prevalence was estimated indirectly through mathematical models designed to “backcalculate” numbers of HIV diagnoses (1, 4). The validity of these estimates depended not only on the number of reported AIDS cases but also on knowledge of the HIV incubation period distribution needed to estimate the probability of developing AIDS for each year following HIV infection (23). HAART can differentially affect the incubation period distribution for users, depending on access, adherence, and effectiveness. This situation renders the back-calculation method based on the incidence of AIDS inaccurate for deriving the infection incidence for HIV after the advent of HAART. Since 1994, the number of states using an integrated HIV/AIDS surveillance system has increased (23). Currently, data are available from 33 states with surveillance systems that integrate ascertainment of new HIV diagnoses with monitoring of AIDS cases, using confidential name-based reporting. Sixty-three percent of all new U.S. AIDS cases each year are diagnosed in these states (23). It is important, however, that current surveillance data are limited to persons with a diagnosed HIV infec- 479 tion and do not directly represent the number of infections because some HIV-infected individuals may not get tested. Population-Based Serosurveys (NHANES) The National Health and Nutrition Examination Surveys (NHANES) are a series of population-based cross-sectional surveys designed to provide nationally representative estimates of the health and nutritional status of the civilian noninstitutionalized U.S. population (9). NHANES data are collected through personal interviews and physical examinations of survey participants, with the latter including biologic samples used for anonymous HIV antibody testing from 1988 to 1994 and for confidential name-based HIV antibody testing from 1999 to 2002 (42). As such, NHANES is the only nationally representative survey from which HIV seroprevalence can be estimated. Moreover, data from the 1999–2002 NHANES can be used to examine associations between HIV serostatus and various demographic and risk characteristics for a more thorough investigation of the impact of these factors on the disease (39). Other Methods Mortality data collected through the HIV/AIDS surveillance system are sensitive to changes in the HIV and AIDS case definitions because surveillance reporting is limited to cases that meet the definition in effect at the time of a person’s death. For example, the annual numbers of deaths among persons with HIV infection or AIDS reported to CDC’s HIV/ AIDS surveillance program by state and local health departments may be affected by expansion of the CDC case definition for AIDS and by expansion of the policies of the health departments for reporting of non-AIDS HIV infection cases. Reporting may be less complete for AIDS cases meeting the expanded criteria of the current AIDS case definition but not meeting the narrower criteria of earlier case definitions, particularly for cases diagnosed years before the case definition was expanded in 1993. Further complicating the interpretation of mortality for cases reported to CDC’s HIV/AIDS surveillance program is the fact that no distinction is made between deaths due to HIV/AIDS and deaths due to other causes. In contrast, death certificate data on deaths attributed to HIV disease (regardless of whether it is AIDS or non-AIDS) as the underlying cause are less likely to have been affected by changes in case reporting policies and practices since 1987 and are more likely to reflect true long-term trends in mortality caused by HIV. Therefore, estimates of trends and distributions of deaths due to HIV in the United States are based on data compiled by the National Center for Health Statistics (NCHS) from death certificates of U.S. residents in the 50 states and the District of Columbia. The underlying cause of each death is selected from the conditions reported by physicians, medical examiners, and coroners in the cause-of-death section of the death certificate. The mortality data from NCHS are the sole source of information on all causes of death in the national population, allowing comparison of deaths due to HIV disease and deaths due to other causes (7, 29). Smaller serosurveys lack either power (if population-based) or representativeness (if targeted to special high-risk populations) and thus cannot be generalized to the U.S. population. 480 HARIRI AND MCKENNA FIG. 1. Trends in annual age-adjusted (to 2000 U.S. population) rate of death due to HIV disease in the United States by sex for the period 1987–2002. For comparison with data for 1999 and later years, data for 1987–1988 were modified to account for ICD-10 rules instead of ICD-9 rules. (Reprinted from the CDC website [http://www.cdc.gov /hiv/graphics/mortalit.htm].) Also, in the case of the latter design, the size and characteristics of the population from which samples were drawn (denominator) are often unknown. Longitudinal cohort studies to determine the incidence of HIV infection are not practical due to the associated costs and loss to follow-up and may also lack representativeness. CURRENT PICTURE The most recent estimates indicate that 40 million people are currently infected with HIV (with and without AIDS) worldwide, 1.2 million of whom are in the United States and thus represent 2.5% of the global disease burden (24, 48). Mortality Patterns After years of steady increase followed by precipitous declines, deaths of persons with HIV/AIDS reported to the national HIV/AIDS surveillance system and U.S. Vital Statistics began to stabilize to approximately 5 per 100,000 in 1998 (7, 29). Between 2000 and 2004, in contrast to the steep annual decreases of up to 45% in the latter part of the 1990s, deaths of persons with HIV/AIDS declined only a modest 8%. The lower rates of decline in HIV-related mortality in recent years are largely attributed to the emergence of resistance to currently available HAART regimens and to differential access to treatment. When these medications are appropriately prescribed and the regimens are followed, the progression of the infection can be arrested effectively and substantial immune reconstitution can ensue. However, such treatment cannot eradicate or cure HIV infection. Therefore, ongoing therapy is necessary to arrest disease progression. Late diagnosis, erratic adherence, and adverse side effects of the medications result in poorer outcomes and the emergence of drug resistance. All of these health service challenges are compounded by the fact that HIV is increasingly impacting communities that lack access to treatment, including racial and ethnic minorities and the socioeconomically disadvantaged (3, 15). Although death rates have followed similar patterns across most demographic and behavioral strata, including gender, age, geographic distribution, and race/ethnicity, substantial variation exists in the percentages of decline among different subgroups. For example, although the number of deaths attrib- CLIN. MICROBIOL. REV. FIG. 2. Trends in age-adjusted (to 2000 U.S. population) annual rates of death due to HIV disease in the United States by race/ethnicity for the period 1990–2002. For comparison with data for 1999 and later years, data for 1987–1988 were modified to account for ICD-10 rules instead of ICD-9 rules. (Reprinted from the CDC website [http://www .cdc.gov/hiv/graphics/mortalit.htm].) uted to HIV has declined in both men and women, it has always been and continues to be higher among males (Fig. 1). This reflects the higher incidence rate of the infection in males. However, over time, the ratio of male to female deaths fell from 10:1 in 1987 to 3:1 in 1998 (7, 29). Similarly, although HIV-related deaths declined rapidly in the 1990s in all racial groups, the percent decline was significantly lower for nonHispanic blacks than for any other race/ethnic group, suggesting race/ethnic disparities (Fig. 2). Further stratification of race/ethnicity by gender illustrated similar lower percent decreases in nonwhite men and women. Black women had the smallest percent decline of any category. Moreover, between 2001 and 2004, while the number of deaths continued to decrease steadily, if less dramatically, for non-Hispanic whites, they either stayed the same or increased slightly from year to year among the other race/ethnic groups. Percent declines also differed by geographic region and by poverty level. Geographic comparison of mortality patterns across the U.S. Census Bureau-designated regions of the country (Northeast, South, West, and Midwest) showed the highest death rates in the Northeast and South, with coastal states and large cities carrying the heaviest burden of deaths due to HIV (Fig. 3). Racial and ethnic disparities in death rates were observed in all regions, with non-Hispanic blacks showing the lowest percent FIG. 3. Trends in age-adjusted (to 2000 U.S. population) annual rates of death due to HIV disease in the United States by geographic region for the period 1987–2002. For comparison with data for 1999 and later years, data for 1987–1988 were modified to account for ICD-10 rules instead of ICD-9 rules. The age distribution of the U.S. population was used as the standard for age adjustment. (Reprinted from the CDC website [http://www.cdc.gov/hiv /graphics/mortalit.htm].) VOL. 20, 2007 EPIDEMIOLOGY OF HIV IN THE UNITED STATES 481 are also evident in the prevalence and diagnosis of HIV/AIDS, as presented below. AIDS FIG. 4. Trends in age-adjusted (to 2000 U.S. population) annual rates of death due to HIV disease in the United States by age group for the period 1987–2002. For comparison with data for 1999 and later years, data for 1987–1988 were modified to account for ICD-10 rules instead of ICD-9 rules. (Reprinted from the CDC website [http://www .cdc.gov/hiv/graphics/mortalit.htm].) decline compared to other race/ethnic groups in all four regions of the country. Another factor associated with death rates was poverty, with the poorest areas (defined by the percentage of residents living below the poverty level in 1990) experiencing the lowest percent decreases in mortality. Finally, mortality data suggest an important shift in deaths toward older age groups, such that the median age at death due to HIV infection increased from 36 years in 1987 to the older median age of 43 years in 2002 (Fig. 4). Only 61 deaths due to AIDS were reported for children of ⬍13 years in 2004 (12). The age-related trends are reflective of changes in survival after diagnosis due to HAART as well as of a decreasing incidence of infection from perinatal exposure and transfusions with contaminated blood products for general medical indications as well as for hemophiliacs. Intergroup differences As increasing use of HAART leads to increased survival times and decreasing numbers of HIV-related deaths, the total number of persons living with the infection has increased (Fig. 5) (35). Moreover, as long as the number of new infections decreases or remains the same, the biggest increase in persons living with HIV likely will be among those who would otherwise have died. Results of the surveillance data indicate that despite rapid declines in AIDS diagnoses between 1993 and 2001, estimated AIDS diagnoses increased modestly each year from 2001 through 2004, supporting the assumptions described above. In 2004, the annual incidence of AIDS was estimated to be 14.1 per 100,000 nationally (12). Almost all of the reported AIDS cases in 2004 were in adults (Table 1). Children under the age of 13 years accounted for only 48 of the newly diagnosed cases, representing a very small fraction of the total cases of AIDS and, more importantly, reflecting a substantial decrease of 61% in pediatric AIDS since 2000. Among the mostly adult AIDS diagnoses reported in 2004, males represented almost three times as many cases (⬃31,000) as females (⬃11,000) and were diagnosed with AIDS at higher rates (25.6 per 100,000) than females (9.0 per 100,000) between 2000 and 2004. However, females had the largest increase in absolute numbers of AIDS (10%) compared to their male counterparts (7%) in the same time period. Also, during 2000–2004, the number of AIDS cases increased slightly in all racial/ethnic categories. However, almost double the number of AIDS cases (⬃21,000) occurred among non-Hispanic blacks compared to non-Hispanic whites (⬃12,000), with almost three times as many as those among Hispanics (⬃8,700). In 2004, non-Hispanic blacks also represented the highest rates of AIDS (56.4 FIG. 5. Estimated numbers of AIDS cases, deaths, and persons living with AIDS in the United States for the period 1985–2004. Data have been adjusted for reporting delays. (Reprinted from the CDC website [http://www.cdc.gov/hiv/topics/surveillance/resources/slides/trends/index.htm].) 482 HARIRI AND MCKENNA CLIN. MICROBIOL. REV. TABLE 1. Estimated numbers of AIDS cases, by year of diagnosis and selected characteristics, in the United States in 2000–2004a No. of cases in yr of diagnosisb Characteristic 2000 2001 2002 2003 2004 Sex Male Female 28,974 10,415 28,743 10,348 29,730 10,429 30,578 11,184 31,024 11,442 Age at diagnosis (yr) ⬍13 13–19 20–29 30–39 40–49 50–59 ⱖ60 124 351 4,761 15,427 12,730 4,535 1585 115 353 4,582 14,907 12,903 4,766 1579 109 383 4,746 14,726 13,481 5,154 1668 69 359 4,940 14,766 14,393 5,526 1777 48 386 5,364 13,817 14,992 6,011 1897 Race/ethnicity White, not Hispanic Black, not Hispanic Hispanic Asian/Pacific Islander American Indian/Alaska Native 11,378 19,510 7,957 350 175 11,052 19,473 7,974 381 169 11,604 19,934 7,907 440 186 11,657 20,685 8,632 478 189 12,013 20,965 8,672 488 193 Transmission category Male-to-male sexual contact IDU Male-to-male sexual contact and IDU Heterosexual contact Perinatal Otherc 15,374 10,429 2,102 10,947 122 537 15,510 9,622 2,056 11,370 113 533 16,442 9,255 1,982 11,952 105 528 17,139 9,281 1,996 12,826 68 520 17,691 9,152 1,920 13,128 47 577 Region of residence Northeast Midwest South West U.S. dependency 12,105 3,968 15,841 6,443 1,156 11,212 3,949 16,598 6,258 1,190 10,395 4,303 17,751 6,745 1,073 11,149 4,495 18,612 6,474 1,100 11,158 4,498 19,792 6,083 982 Totald 39,513 39,206 40,267 41,831 42,514 a Data from reference 12. These numbers do not represent reported case counts. Rather, these numbers are point estimates, which result from adjustments of reported case counts. The reported case counts are adjusted for reporting delays and for redistribution of cases for persons initially reported without an identified risk factor. The estimates do not include adjustment for incomplete reporting. c Includes hemophilia, blood transfusion, perinatal transmission, and risk factor not reported or not identified. d Includes persons of unknown race or multiple races and persons of unknown sex. The cumulative total includes 2,308 persons of unknown race or multiple races and 2 persons of unknown sex. Because column totals were calculated independently of the values for the subpopulations, the values in each column may not sum to the column total. b per 100,000), followed by Hispanics (18.6 per 100,000), American Indians/Alaska Natives (7.9 per 100,000), whites (6.0 per 100,000), and Asians/Pacific Islanders (3.7 per 100,000). Although the estimated annual number of AIDS diagnoses increased among persons exposed through heterosexual contact as well as among MSM between 2000 and 2004, the number decreased among IDUs and MSM who were also IDUs in the same time period. The largest number of AIDS cases in 2004 was attributed to male-male sex, followed by heterosexual sex and then IDU. Regionally, the number of reported AIDS cases increased 25% in the South and 13% in the Midwest while decreasing 8% in the Northeast, 6% in the West, and 15% in the U.S. dependencies, possessions, and associated nations (12). Results from the 1999–2002 NHANES indicate racial disparities in treatment that may be partially responsible for the proportional disparities in AIDS deaths and prevalence among non-Hispanic blacks (39). First, by comparing CD4 counts among seropositive respondents who reported HAART use to those among seropositive respondents who did not, NHANES analysis confirmed previous results of HAART effectiveness in slowing disease progression in a population-based sample. Next, the analysis indicated that only 17% of seropositive black respondents reported using HAART medication, in contrast to 78% of respondents in all other categories. Furthermore, among seropositive respondents who were aware of their HIV status, only 67% of blacks reported HAART use, in contrast to a full 100% among all other race/ethnic groups (39). HIV Infection (Including AIDS) Of the estimated 1.2 million people in the United States living with HIV infection, approximately 34% have progressed to AIDS, 42% are classified as having HIV only (not AIDS), and 24% remain undiagnosed and therefore may be at any VOL. 20, 2007 EPIDEMIOLOGY OF HIV IN THE UNITED STATES TABLE 2. Estimated numbers and proportions of persons with HIV infection diagnosis by selected characteristics in 33 states with confidential name-based HIV infection reporting in 2004a Characteristic No. (%) with HIV diagnosisb Sex Male ...................................................................................28,143 (72.7) Female................................................................................10,410 (26.9) Age at diagnosis (yr) ⬍13 ..................................................................................... 174 (0.4) 13–19 .................................................................................. 1,121 (2.9) 20–29 .................................................................................. 8,366 (21.6) 30–39 ..................................................................................12,281 (31.7) 40–49 ..................................................................................10,880 (28.1) 50–59 .................................................................................. 4,370 (11.3) ⱖ60 ..................................................................................... 1,538 (4.0) Race/ethnicity White, not Hispanic .........................................................11,806 (30.5) Black, not Hispanic ..........................................................19,206 (49.6) Hispanic ............................................................................. 6,970 (18.0) Asian/Pacific Islander....................................................... 394 (1.0) American Indian/Alaska Native...................................... 208 (0.5) Transmission category Male-to-male sexual contact ...........................................18,203 (47.1) IDU .................................................................................... 5,962 (15.4) Male-to-male sexual contact and IDU .......................... 1,372 (3.5) Heterosexual contact........................................................12,683 (32.8) Perinatal............................................................................. 145 (0.4) Otherc ................................................................................. 335 (0.9) Total .......................................................................................38,685 (100) a Data from reference 12. Since 2000, the following 33 states have had laws or regulations requiring confidential name-based HIV infection reporting: Alabama, Alaska, Arizona, Arkansas, Colorado, Florida, Idaho, Indiana, Iowa, Kansas, Louisiana, Michigan, Minnesota, Mississippi, Missouri, Nebraska, Nevada, New Jersey, New Mexico, New York, North Carolina, North Dakota, Ohio, Oklahoma, South Carolina, South Dakota, Tennessee, Texas, Utah, Virginia, West Virginia, Wisconsin, and Wyoming. Since July 1997, Florida has had confidential name-based HIV infection reporting only for new diagnoses. b These numbers do not represent reported case counts. Rather, these numbers are point estimates, which result from adjustments of reported case counts. The reported case counts are adjusted for reporting delays and for redistribution of cases for persons initially reported without an identified risk factor. The estimates do not include adjustment for incomplete reporting. Data include persons with a diagnosis of HIV infection. This includes persons with a diagnosis of HIV infection only, a diagnosis of HIV infection and a later AIDS diagnosis, and concurrent diagnoses of HIV infection and AIDS. c Includes hemophilia, blood transfusion, perinatal transmission, and risk factor not reported or not identified. stage along the spectrum of the disease. Table 2 summarizes the demographic and transmission characteristics of persons newly diagnosed with HIV in 2004. The data suggest that HIV-infected individuals who are diagnosed can be characterized as predominantly male (73%) and nonwhite (69%) (24). Non-Hispanic blacks comprise the greatest percentage of HIV infections (50%), while Hispanics represent 18% of all cases. Male-male sex is the most frequently reported transmission category (47%), followed by heterosexual contact (33%) and IDU (5%). Similarly, among non-Hispanic blacks, the most frequently reported transmission route is male-male sex (45%), followed by heterosexual contact (27%) and IDU (22%) (24). Health care personnel are potentially at risk of occupationally acquired HIV infection. However, many health care work- 483 ers also have behavioral risk factors for HIV that make it difficult to differentiate whether the source of infection was attributable specifically to workplace exposures. In general, few HIV infections have been rigorously documented as occurring due to exposures to infectious fluids or tissues from patients. Data from the National Surveillance for Occupationally Acquired HIV Infection and national AIDS surveillance systems identified 57 cases of documented occupationally acquired infection between 1981 and 2001. Specific regimens have been recommended to provide postexposure prophylaxis (PEP) for workers with significant exposures, and among the three cases that occurred after 1996, recommended PEP failed in the only person who received treatment with a two-drug regimen; the other two refused PEP (19, 44). Geographically, HIV infection remains predominantly an urban phenomenon, and southern and northeastern regions of the country contain a disproportionate burden of the disease (24). While the overall trends in new HIV diagnoses suggested no significant change in annual rates between 2001 and 2004 (Fig. 6), subgroup analyses revealed statistically significant increases in annual rates for both males and females in the Asian race/ethnic category (7.8% and 15.1% per year, respectively) (Fig. 7). Annual rates declined significantly in the IDU and heterosexual transmission categories (⫺9.1% and ⫺3.9% per year, respectively) as well as in non-Hispanic black males and females (Fig. 8 and 9) and Hispanic females (⫺4.4%, ⫺6.8%, and ⫺13.0% per year, respectively) during the same time period (12, 22). Although the reductions in rates of HIV infection in certain subgroups are encouraging, important intergroup rate differences exist and must also be considered. In particular, members of nonwhite race/ethnic groups, especially females in these groups, are disproportionately affected by the epidemic. Between 2001 and 2004, 51% of all new HIV diagnoses were in non-Hispanic blacks, who accounted for only 13% of the population from which the data were collected (15). In the same time period, annual HIV diagnosis rates were highest for non-Hispanic black males, followed by non-Hispanic black females, who had higher rates of infection than males of all other race/ethnic categories (12). The 1999–2002 NHANES results are consistent with surveillance data and indicate a high prevalence of HIV in nonHispanic blacks aged 40 to 49 years old (3.58%; 95% confidence interval, 1.88% to 6.71%), with 40- to 49-year-old non-Hispanic black males having the highest prevalence (4.54%; 95% confidence interval, 2.24% to 8.97%) (39). Additionally, surveillance results indicate racial/ethnic differences in transmission characteristics. Analyses of case surveillance data show that male-male sex was the most commonly reported transmission category for all race/ethnic groups (12). However, the proportion of black men reportedly infected through heterosexual contact was over four times greater than that for white men (27% versus 6%). Also, a higher proportion of black men (18%) reported IDU, compared to 10% of white men (12). Infection attributed to heterosexual exposure was reported with greatest frequency among females in all race/ethnic categories. However, infections among white women were almost twice as likely to be attributed to IDU (30%) as those among black women (17%). Therefore, a higher proportion of black women are reportedly infected through heterosexual contact (80%) than are white women (67%) (CDC, unpublished data). 484 HARIRI AND MCKENNA CLIN. MICROBIOL. REV. FIG. 6. Estimated numbers and rates of HIV/AIDS diagnoses in 33 states for the period 2001–2004. EAPC, estimated annual percent change. Stratification of the estimated prevalence of cases of diagnosed and undiagnosed infections by stage of disease (HIV [not AIDS] or AIDS) also reveals some interesting differences. First, females represent a higher proportion of all cases in the HIV (not AIDS) category than in the AIDS category (29% versus 23%). Likewise, non-Hispanic blacks comprise a higher percentage of those with HIV (not AIDS) than those in the AIDS category (47% versus 43%). Finally, 29% of HIV (not AIDS) cases are reported for heterosexuals versus 23% of AIDS cases. In contrast, whites, MSM, and IDUs represent a larger proportion of persons living with AIDS than of those in the HIV (not AIDS) group (24). These proportions, although not statistically significantly different, are suggestive of more new infections in women, nonwhites, and heterosexuals due to a higher percentage of them representing an earlier stage of the disease. However, these differences may be an artifact of differential testing patterns for the various subgroups and must be investigated further using methods to measure HIV incidence. FIG. 7. Estimated annual rates of HIV/AIDS diagnoses in 33 states by sex and race/ethnicity for the period 2001–2004. EAPC, estimated annual percent change; A/PI, Asian/Pacific Islander; AI/AN, American Indian/Alaska Native. Statistically significant percent changes are indicated with asterisks. While people of color currently experience a disproportionate burden of HIV infection, the numbers of children aged ⬍13 years with HIV and of new perinatal infections have declined dramatically. Between 1992 and 2004, the estimated annual number pediatric AIDS cases in the United States decreased steadily each year from 945 to 57, a 94% decrease in just over a decade (12). The reduction of pediatric and perinatal HIV infection has resulted from successful public health interventions targeted to pregnant women. Specifically, since antiretroviral therapy was demonstrated to be effective in reducing mother-to-child transmission of HIV, most professional groups and the CDC have recommended that pregnant women in the United States be provided with universal counseling, voluntary testing, and appropriate treatment to prevent such transmission. If testing and/or treatment is refused or proves ineffective in blocking transmission to a newborn, the child is placed on treatment early to increase survival and decrease the risk of morbidity and mortality due to HIV. These measures have led to the significant reductions in HIV-related infant and child mortality observed in recent years (13). FIG. 8. Estimated annual rates of HIV/AIDS diagnoses among males in 33 states by race/ethnicity for the period 2001–2004. EAPC, estimated annual percent change. Percentages of change were not statistically significant. VOL. 20, 2007 FIG. 9. Estimated annual rates of HIV/AIDS diagnoses among females in 33 states by race/ethnicity for the period 2001–2004. EAPC, estimated annual percent change. Statistically significant percent changes are indicated with asterisks. Studies of sexual transmission of HIV suggest that persons who are unaware of their HIV infection are 3.5 times as likely to transmit the virus to a partner as individuals who are aware of their HIV status (37). Therefore, providing opportunities for widespread testing and early detection would greatly benefit HIV prevention efforts. However, because of the stigma traditionally associated with HIV and AIDS, recommendations and policies surrounding the use of HIV testing have usually included requirements for extensive counseling about the disease, with the solicitation of explicit consent before obtaining the test. This approach is known as the “opt-in” method. However, in the context of screening pregnant women for HIV, the imperatives for optimizing early detection in order to guide therapy and prevent transmission to the newborn have resulted in most recommendations from professional organizations, including the CDC, encouraging an “optout” approach to testing of pregnant women. This approach allows for testing unless the mother explicitly objects. Currently, five states (Delaware, Florida, Indiana, Oregon, and Texas) authorize an “opt-out” approach to HIV testing in pregnant women, and two states (New York and Connecticut) mandate HIV testing for all newborns (26). Recently, the CDC extended the recommendation to testing all persons being evaluated in clinical settings. Persons being tested for HIV can be informed either verbally without extensive counseling or by soliciting written consent (3). Taken together, results from national surveillance and population-based survey data reveal some important trends, underscoring successes while serving as reminders of significant challenges that remain in the context of social, economic, and health care access disparities. Therapeutic advances have resulted in reduced mortality, longer survival, and improved life quality among HIV-infected individuals. Public health campaigns to provide education, testing, and treatment to pregnant women with HIV have led to declines in the number of infants suffering from the disease. Finally, transfusion-transmitted and hemophilia-associated HIV infections have been virtually eliminated in the United States. On the other hand, surveillance data indicate that racial/ethnic, gender, and possibly socioeconomic disparities in HIV infection persist. However, current prevalence estimates from surveillance data cannot discriminate between new and long-standing infections and therefore EPIDEMIOLOGY OF HIV IN THE UNITED STATES 485 FIG. 10. Annual distribution of AIDS cases in the United States by case definition category for the period 1981–2003. do not adequately explain the impact of these disparities on the future of the epidemic. FUTURE DIRECTIONS Clinical and Epidemiologic Effects of HAART During the past decade, the introduction of HAART has been responsible for increased length and quality of life among many HIV-infected individuals and has contributed to significant reductions in HIV-related morbidity and mortality (27, 43). As illustrated in Fig. 10, following the dramatic proportional increase in the number of AIDS cases without opportunistic infections (OIs) relative to those with OIs that resulted from the change in the AIDS case definition in 1993 (which was expanded to include all HIV-infected persons with a CD4⫹ T-lymphocyte count of ⬍200 cells/l or a CD4⫹ Tlymphocyte percentage of total lymphocytes of ⬍14%), the percentage of AIDS cases without OIs continued to increase coincident with the widespread use of HAART in 1996. The absolute number of AIDS cases without OIs also began to increase after 1996, such that 71% of all reported AIDS cases in 2003 were classified on the basis of low CD4 counts without OI, compared to only 35% in 1993. Despite obvious successes, however, widespread use of HAART has also led to new challenges in clinical management of HIV disease due to its failure to completely suppress viral replication, thus resulting in the emergence of HIV drug resistance (5). Most cases of drug resistance are thought to arise as a result of random mutations in the presence of drugselective pressures in treated individuals. However, although the use of HAART has been shown to decrease HIV infectivity by suppressing the viral load (2), transmission of viruses resistant to all classes of antiretroviral drugs has also been demonstrated clearly for all transmission categories. Moreover, transmission of drug-resistant virus appears to be increasing (47). Given that 25% of patients reportedly discontinue treatment within 1 year due to drug toxicities and inconsistent adherence, the long-term success of HAART must be evaluated in the context of this important and rising phenomenon of drug resistance. The drug-associated mutations in most transmitted quasi-species appear to persist as the dominant variant over 486 HARIRI AND MCKENNA time, unlike the situation where mutations in a wild-type variant are selected by drug pressure (4). In the latter situation, there is generally rapid reemergence and dominance of archived wild-type quasi-species when medications are discontinued. Although strains with drug resistance-associated mutations persist in the setting of transmitted resistance, such mutations usually result in reduced viral fitness, as measured by replicative capacity (19). Nevertheless, despite compromised viral fitness and a probable reduced transmissibility of HIV type 1 (HIV-1) drug-resistant variants relative to that of the wild type, primary transmission to drug-naı̈ve individuals can and does occur, and resistant variants persist for years even in the absence of treatment. Persons with acquired or transmitted drug resistance do not respond as well to treatment and take 2 to 8 weeks longer to achieve viral suppression than their drug-susceptible counterparts (18). The prevalence of resistance mutations in drug-naı̈ve individuals newly diagnosed with HIV-1 infection has been shown to vary with place and time. Resistance to all classes of HAART drugs has been documented worldwide, as has resistance to multiple drugs. Various studies have estimated the prevalence of at least one resistance mutation to range from 8% to 28% in the United States (47). Analysis of surveillance data representing a large population-based sample of persons in the United States revealed a prevalence of at least one mutation in 10% of the overall sample. The prevalence of resistance to all classes of drugs was detected and varied by state from 6% to 13% (49). In addition, more sensitive assays are now available to detect minority mutations that were not detectable using bulk sequence detection methods (32). Nevertheless, it is difficult to predict the future trends in transmitted HIV drug resistance. At present, transmitted resistance accounts for only a small fraction of the drug resistance population, and there is evidence of continuing benefits of antiretroviral therapy even among persons with persistent drugresistant variants (18). HAART medications are also associated with substantial side effects that can result in minor as well as substantial metabolic, dermatologic, and neurologic dysfunctions (40). It is beyond the scope of this paper to review the impact of specific HAART medications. However, up to 25% of patients reportedly discontinue treatment within the first 8 months from HAART initiation, in part due to toxic effects of the drugs. Adverse effects of HAART therapy range from mild nuisances, such as fatigue, headaches, and skin rashes, to lifethreatening effects, such as metabolic disruption and hepatotoxicity (40). Given the potential morbidity and mortality associated with HAART therapy and the impact of poor adherence on the development of drug resistance, management of HAART-related adverse outcomes is imperative. On the population level, increased use of HAART must also be considered in the context of HIV incidence and changes in risky behaviors. The impacts of HAART use on the epidemic are decreased mortality and incidence rates (43). However, potential increases in risky behaviors due to treatment availability and complacence among high-risk groups have been shown to negatively impact the amount by which these parameters are reduced (2, 20). Therefore, the potential tradeoff between treatment benefits and negative consequences must be closely monitored. Specifically, continued prevention efforts CLIN. MICROBIOL. REV. targeted to high-risk populations combined with identification and monitoring of viral resistance are critical needs in the next phase of the epidemic. The CDC is currently supporting several surveillance sites to monitor the prevalence of HIV-1 subtypes and primary drug resistance patterns among new cases of HIV infection. As part of the Variant, Atypical, and Resistant Strains of HIV Surveillance System, four cities and 18 state heath departments are currently funded to conduct drug resistance surveillance on remnant sera collected from persons newly diagnosed with HIV infection (1). HIV Incidence According to back-calculations, 40,000 people are newly infected with HIV in the United States annually. However, the accuracy of the statistical models used to derive this estimate is unclear, given their sensitivity to the effects of the change in the AIDS case definition in 1993 and the introduction of HAART in 1996. Yet no alternative was available since, historically, systematic monitoring of new cases of HIV infection was not a part of the national surveillance infrastructure, primarily due to unavailability of a marker able to distinguish newly infected (incident) cases from infections of longer duration. As recommended by the Institute of Medicine in 2001, assessment of HIV incidence trends is a priority for evaluation of current prevention efforts as well as for future resource allocation (30). Incidence estimates are also necessary to evaluate the goal of reducing the estimated annual 40,000 new HIV infections in the United States to 20,000 per year, as outlined in the CDC HIV Prevention Strategic Plan (6). Toward this end, the CDC developed and has begun to implement a National HIV Incidence Surveillance System, which consists of two equally important components (36). The first is an innovative serologic testing strategy called the Serologic Testing Algorithm for Recent HIV Seroconversion (STARHS), which was developed and validated for ascertaining seroincidence (31). This method provided the breakthrough necessary to differentiate between recent and longstanding infections by using a tandem sensitive/less sensitive testing method. The less sensitive assay was subsequently replaced by an enzyme immunoassay that is able to accurately capture immunoglobulin G from infections with HIV subtypes B, E, and D. This test is known as the BED assay and was developed specifically for detecting recent HIV infection (45). However, due to various factors, including widespread use of HAART, that can affect antibody levels measured by these tests, the STARHS test results must be interpreted in the context of supplementary testing history information on each individual tested in order to obtain valid incidence estimates. Collection of the testing history information is the second component of the new surveillance system, which is designed to be fully integrated within the existing case surveillance system. Currently, 34 areas that cover approximately 85% of the epidemic in the United States are implementing this system. Monitoring HIV Risk Behavior In addition to enumerating new HIV infections, ongoing assessment of risk correlates, especially for subgroups at increased risk of infection, is an integral part of the goal to VOL. 20, 2007 EPIDEMIOLOGY OF HIV IN THE UNITED STATES reduce the burden of disease. The CDC is coordinating the National HIV Behavioral Surveillance System (NHBS) to examine HIV-related behaviors in three high-risk groups, including MSM, IDUs, and heterosexual adults, in areas with high rates of HIV by using population-based surveys (14, 22). Data collection began in 2003 among MSM aged ⱖ18 years, the group with the highest rates of HIV infection. As of 2005, surveys have been conducted among eligible MSM in 25 metropolitan statistical areas. Results of surveys conducted between November 2003 and April 2005 indicate that the majority of MSM participants had previously been tested (⬎90%) for HIV infection, with 77% percent reporting testing during the preceding 12 months and 80% having received free condoms during the preceding 12 months (14). In contrast, 58% of respondents reported unprotected anal intercourse with a main partner, while 34% reported this same behavior with a casual partner. Moreover, noninjection drug use was reported by 42% of respondents, and few individual-level (15%) or group-level (8%) HIV prevention programs were utilized by respondents. These data suggest that although HIV testing is common practice among this group, MSM continue to be at increased risk of HIV through sexual and drug-related behaviors (14). Interestingly, data collected between 1990 and 1999 in New York City indicated a 15% decline in HIV seroprevalence among male IDUs who have sex with other men, despite their engaging in these high-risk behaviors. The same study also showed HIV seroprevalence to have decreased in male IDUs who did not report sex with other men (38). Another risk group for whom national HIV-related behavior data have been collected is adolescents. Data from the national Youth Risk Behavior Survey indicate a decline in the percentage of high school students engaging in high-risk sexual behavior during 1991–2005 (16). Overall, self-reported sexual experience decreased 13%, from 54% to 47%; multiple sex partners (defined as four or more in the preceding 3 months) decreased 24%, from 19% to 14%; current sexual activity decreased 9%, from 37% to 34%; and condom use increased 36%, from 46% to 63%. Despite overall declines, however, disparities that must be addressed further remained among subgroups. For example, black students were more likely than white and Hispanic students to report high-risk sexual behaviors, while no decrease was observed in the prevalence of sexual experience, having had multiple sex partners, or current sexual activity among Hispanic students. Moreover, the percentage of high school students reporting injection drug use remained stable, at 4%, during the same time period. The second group assessed by the NHBS was IDUs (34). The next phase of NHBS focuses on high-risk heterosexuals and is under way. Data generated from NHBS and other national and local surveys will provide more information on HIVrelated behaviors and help us to move toward more effective and targeted prevention strategies to reduce the burden of HIV disease. CONCLUSION The emergence of the HIV epidemic in the United States has been characterized by many transitions, initially presenting as a rapidly lethal disease primarily affecting white MSM in a limited number of urban areas. Systematic public health sur- 487 veillance data have served as the foundation for extensive clinical, epidemiologic, viral, behavioral, and pharmacologic research. The result of these efforts was the rapid identification of a novel retroviral infection with a long incubation period as the cause of a severe, cellular immunodeficiency. The development of diagnostic tests, effective behavioral preventive interventions for adults, and the implementation of treatment protocols that decrease perinatal transmission were followed, within a decade, by the introduction of pharmacologic therapies that arrest disease progression and allow for immune restoration. Despite the generally encouraging evolution of these events, the epidemic presents persistent challenges. HIV now exacts enormous morbidity and mortality in minority communities, particularly among African-American men and women. The infection has also disseminated into rural areas. Finally, although treatable, HIV infections are incurable. The result has been the emergence of drug resistance among treated persons as well as drug-naı̈ve, newly diagnosed persons. Surveillance efforts must continue to adapt to the changing characteristics of the epidemic to accurately capture the molecular, clinical, and behavioral dimensions of the epidemic so that activities focused on prevention, treatment, and care can continue to progress. ACKNOWLEDGMENTS We are grateful to Richard Selik for his input and assistance in summarizing the HIV mortality data. The findings and conclusions in this paper are those of the authors and do not necessarily reflect the views of the Centers for Disease Control and Prevention. REFERENCES 1. Bennett, D. 2005. HIV [corrected] genetic diversity surveillance in the United States. J. Infect. Dis. 192:4–9. 2. Blower, S., E. Bodine, J. Kahn, and W. McFarland. 2005. The antiretroviral rollout and drug-resistant HIV in Africa: insights from empirical data and theoretical models. AIDS 19:1–14. 3. Branson, B. M., H. H. Handsfield, M. A. Lampe, R. S. Janssen, A. W. Taylor, S. B. Lyss, and J. E. Clark. 2006. Revised recommendations for HIV testing of adults, adolescents, and pregnant women in health-care settings. Morb. Mortal. Wkly. Rep. Recomm. Rep. 55:1–17. 4. Brookmeyer, R. 2003. Temporal factors in epidemics: the role of the incubation period, p. 127–145. In R. Brookmeyer and D. F. Stroup (ed.), Monitoring the health of populations: statistical principles and methods for public health surveillance. Oxford University Press, Oxford, United Kingdom. 5. Cane, P. A. 2005. Stability of transmitted drug-resistant HIV-1 species. Curr. Opin. Infect. Dis. 18:537–542. 6. CDC. August 2003, posting date. HIV prevention strategic plan through 2005. CDC, Atlanta, GA. http://www.cdc.gov/hiv/partners/PSP/Goal4-Objective1-2-3 .htm#obj1. 7. CDC. 2002, posting date. HIV/AIDS mortality surveillance slides. CDC, Atlanta, GA. http://www.cdc.gov/hiv/graphics/mortalit.htm. 8. CDC. 1981. Kaposi’s sarcoma and Pneumocystis pneumonia among homosexual men—New York City and California. Morb. Mortal. Wkly. Rep. 30:305–308. 9. CDC. 1994. Plan and operation of the Third National Health and Nutrition Examination Survey, 1988–94. Series 1: programs and collection procedures. Vital Health Stat. 1:1–407. 10. CDC. 1996. Update: mortality attributable to HIV infection among persons aged 25–44 years—United States, 1994. Morb. Mortal. Wkly. Rep. 45:121– 125. 11. CDC. December 2000, posting date. HIV/AIDS surveillance report, 1999, vol. 11. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA. http://www.cdc.gov/hiv/stats /hasr1102.pdf. 12. CDC. December 2005, posting date. HIV/AIDS surveillance report, 2004, vol. 17. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA. http://www.cdc.gov/hiv/topics /surveillance/resources/reports/2004report/pdf/2004SurveillanceReport.pdf. 13. CDC. 2006. Achievements in public health. Reduction in perinatal transmission of HIV infection—United States, 1985–2005. Morb. Mortal. Wkly. Rep. 55:592–597. 488 HARIRI AND MCKENNA 14. CDC. 2006. Human immunodeficiency virus (HIV) risk, prevention, and testing behaviors—United States, National HIV Behavioral Surveillance System: men who have sex with men, November 2003–April 2005. Morb. Mortal. Wkly. Rep. 55:1–16. 15. CDC. 2006. Racial/ethnic disparities in diagnoses of HIV/AIDS—33 states, 2001–2004. Morb. Mortal. Wkly. Rep. 55:121–125. 16. CDC. 2006. Trends in HIV-related risk behaviors among high school students—United States, 1991–2005. Morb. Mortal. Wkly. Rep. 55:851–854. 17. CDC. 2006. Twenty-five years of HIV/AIDS—United States, 1981–2006. Morb. Mortal. Wkly. Rep. 55:585–589. 18. Deeks, S. G., T. Wrin, T. Liegler, R. Hoh, M. Hayden, J. D. Barbour, N. S. Hellmann, C. J. Petropoulos, J. M. McCune, M. K. Hellerstein, and R. M. Grant. 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 344:472–480. 19. Do, A. N., C. A. Ciesielski, R. P. Metler, T. A. Hammett, J. Li, and P. L. Fleming. 2003. Occupationally acquired human immunodeficiency virus (HIV) infection: national case surveillance data during 20 years of the HIV epidemic in the United States. Infect. Control Hosp. Epidemiol. 24:86–96. 20. Fenton, K. A., and J. Imrie. 2005. Increasing rates of sexually transmitted diseases in homosexual men in Western Europe and the United States: why? Infect. Dis. Clin. N. Am. 19:311–331. 21. Fleming, P. L., J. W. Ward, J. M. Karon, D. L. Hanson, and K. M. De Cock. 1998. Declines in AIDS incidence and deaths in the USA: a signal change in the epidemic. AIDS 12(Suppl. A):S55–S61. 22. Gallagher, K., P. S. Sullivan, A. Lansky, and I. Onorato. 2007. Behavioral surveillance among persons at risk for HIV infection in the U.S.: The National HIV Behavioral Surveillance System. Public Health Rep. 122(Suppl. 1):32–38. 23. Glynn, M. K., L. M. Lee, and M. T. McKenna. 2007. The status of national HIV case surveillance, United States 2006. Public Health Rep. 122(Suppl. 1):63–71. 24. Glynn, M. K., and P. Rhodes. Estimated HIV prevalence in the United States at the end of 2003, abstr. T1–B1101. 2005 Natl. HIV Prev. Conf., Atlanta, GA, 14 June 2005. 25. Hall, H. I., K. McDavid, Q. Ling, and A. Sloggett. 2006. Determinants of progression to AIDS or death after HIV diagnosis, United States, 1996 to 2001. Ann. Epidemiol. 16:824–833. 26. Health Research and Education Trust. 2006, posting date. Cross-state comparison tables. Health Research and Education Trust, Chicago, IL. http: //www.hret.org/hret/about/crossstate.html. 27. Hogg, R. S., B. Yip, C. Kully, K. J. Craib, M. V. O’Shaughnessy, M. T. Schechter, and J. S. Montaner. 1999. Improved survival among HIV-infected patients after initiation of triple-drug antiretroviral regimens. CMAJ 160:659–665. 28. Holtgrave, D. R. 2005. Causes of the decline in AIDS deaths, United States, 1995–2002: prevention, treatment or both? Int. J. Sex Transm. Dis. Acquir. Immune Defic. Syndr. 16:777–781. 29. Hoyert, D. L., M. P. Heron, S. L. Murphy, and H. Kung. April 2006, posting date. Deaths: final data for 2003. National vital statistics reports, 2006, vol. 54. National Center for Health Statistics, Hyattsville, MD. http://www.cdc .gov/nchs/data/nvsr/nvsr54/nvsr54_13.pdf. 30. Institute of Medicine. 2001, posting date. No time to lose. Institute of Medicine, Washington, DC. http://www.iom.edu/Object.File/Master/4/131 /HIV8pager.pdf. 31. Janssen, R. S., G. A. Satten, S. L. Stramer, B. D. Rawal, T. R. O’Brien, B. J. Weiblen, F. M. Hecht, N. Jack, F. R. Cleghorn, J. O. Kahn, M. A. Chesney, and M. P. Busch. 1998. New testing strategy to detect early HIV-1 infection for use in incidence estimates and for clinical and prevention purposes. JAMA 280:42–48. 32. Johnson, J., J. F. Li, X. Wei, J. Lipscomb, A. Smith, C. Stone, D. Irlbeck, M. Monsour, E. Lanier, and W. Heneine. 2007. Low frequency mutations substantially increase the prevalence of transmitted drug resistance and greatly CLIN. MICROBIOL. REV. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. strengthen the relationship between resistance mutations and virologic failure, abstr. 639. 14th Conf. Retrovir. Opportun. Infect., Los Angeles, CA, February 2007. Karon, J. M., P. L. Fleming, R. W. Steketee, and K. M. De Cock. 2001. HIV in the United States at the turn of the century: an epidemic in transition. Am. J. Public Health 91:1060–1068. Lansky, A., A. S. Abdul-Quader, M. Cribbin, T. Hall, T. J. Finlayson, R. S. Garfein, L. S. Lin, and P. S. Sullivan. 2007. Developing an HIV behavioral sruveillance system for injecting drug users: a national application of venuebased, time-space sampling. Public Health Rep. 122(Suppl. 1):48–55. Lee, L. M., J. M. Karon, R. Selik, J. J. Neal, and P. L. Fleming. 2001. Survival after AIDS diagnosis in adolescents and adults during the treatment era, United States, 1984–1997. JAMA 285:1308–1315. Lee, L. M., and M. T. McKenna. 2007. Monitoring the incidence of HIV infection in the United States. Public Health Rep. 122(Suppl. 1):72–79. Marks, G., N. Crepaz, and R. S. Janssen. 2006. Estimating sexual transmission of HIV from persons aware and unaware that they are infected with the virus in the USA. AIDS 20:1447–1450. Maslow, C. B., S. R. Friedman, T. E. Perlis, R. Rockwell, and J. D. C. Des. 2002. Changes in HIV seroprevalence and related behaviors among male injection drug users who do and do not have sex with men: New York City, 1990–1999. Am. J. Public Health 92:382–384. McQuillan, G. M., D. Kruszon-Moran, B. J. Kottiri, L. A. Kamimoto, L. Lam, M. F. Cowart, M. Hubbard, and T. J. Spira. 2006. Prevalence of HIV in the US household population: the National Health and Nutrition Examination Surveys, 1988 to 2002. J. Acquir. Immune Defic. Syndr. 41:651–656. Montessori, V., N. Press, M. Harris, L. Akagi, and J. S. Montaner. 2004. Adverse effects of antiretroviral therapy for HIV infection. CMAJ 170:229– 238. Nakashima, A. K., and P. L. Fleming. 2003. HIV/AIDS surveillance in the United States, 1981–2001. J. Acquir. Immune Defic. Syndr. 32(Suppl. 1): S68–S85. National Center for Health Statistics. October 2006, posting date. National Health and Nutrition Examination Survey (NHANES). National Center for Health Statistics, Hyattsville, MD. http://www.cdc.gov/nchs/about/major /nhanes/nhanes01-02.htm. Palella, F. J., Jr., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, and S. D. Holmberg. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338:853–860. Panlilio, A. L., D. M. Cardo, L. A. Grohskopf, W. Heneine, and C. S. Ross. 2005. Updated U.S. Public Health Service guidelines for the management of occupational exposures to HIV and recommendations for postexposure prophylaxis. Morb. Mortal. Wkly. Rep. Recomm. Rep. 54:1–17. Parekh, B. S., M. S. Kennedy, T. Dobbs, C. P. Pau, R. Byers, T. Green, D. J. Hu, S. Vanichseni, N. L. Young, K. Choopanya, T. D. Mastro, and J. S. McDougal. 2002. Quantitative detection of increasing HIV type 1 antibodies after seroconversion: a simple assay for detecting recent HIV infection and estimating incidence. AIDS Res. Hum. Retrovir. 18:295–307. Pomerantz, R. J., and D. L. Horn. 2003. Twenty years of therapy for HIV-1 infection. Nat. Med. 9:867–873. Tang, J. W., and D. Pillay. 2004. Transmission of HIV-1 drug resistance. J. Clin. Virol. 30:1–10. UNAIDS. December 2006, posting date. AIDS epidemic update. UNAIDS, Geneva, Switzerland. http://data.unaids.org/pub/EpiReport/2006/2006_EpiUpdate_en .pdf. Wheeler, W., K. Mahle, U. Bodnar, R. Kline, I. Hall, and M. McKenna. Antiretroviral drug-resistance mutations and subtypes in drug-naive persons newly diagnosed with HIV-1 infection, US, March 2003-October 2006, abstr. 648. 14th Conf. Retrovir. Opportun. Infect. Los Angeles, CA, February 2007. World Health Organization. 2006, posting date. Second generation surveillance for HIV/AIDS. World Health Organization, Geneva, Switzerland. http://www.who.int/hiv/topics/surveillance/2ndgen/en/. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 489–510 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00005-07 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Current Status of Veterinary Vaccines 1 Els N. T. Meeusen, * John Walker,2 Andrew Peters,3 Paul-Pierre Pastoret,4 and Gregers Jungersen5 Animal Biotechnology Research Laboratories, Department of Physiology, Monash University, Victoria 3800, Australia1; VMRD Pfizer Australia, 52 Poplar Rd., Parkville, Victoria 3052, Australia2; University of Edinburgh, Royal (Dick) School of Veterinary Studies, Easter Bush, Roslin, Midlothian EH25 9RG, United Kingdom3; Publications Department, World Organisation for Animal Health, 12 rue de Prony, 75017 Paris, France4; and National Veterinary Institute, Technical University of Denmark, Bülowsvej 27, 1790 Copenhagen, Denmark5 cowpox virus to confer protection against the related human smallpox virus and illustrates the close relationship between human and animal infectious disease sciences. The criteria for successful animal or veterinary vaccines can be very different from those for human vaccines depending on the animal groups under consideration. For example, criteria for companion animal vaccines are similar to those for human vaccines in that the health and welfare of the individual animal are primary concerns. The main objective of livestock vaccines, on the other hand, is to improve overall production for the primary producers, and the cost-benefit resulting from vaccination is the bottom line for this industry. Vaccination against zoonotic or food-borne infections is aimed at reducing or eliminating INTRODUCTION In its original concept, vaccination aims to mimic the development of naturally acquired immunity by inoculation of nonpathogenic but still immunogenic components of the pathogen in question, or closely related organisms. The term “vaccine” (from the Latin term “vacca,” meaning cow) was first coined by Edward Jenner to describe the inoculation of humans with the * Corresponding author. Mailing address: Department of Physiology, Building 13f, Monash University, Clayton, Victoria 3800, Australia. Phone: 61 3 99052513. Fax: 61 3 99052547. E-mail: els.meeusen @med.monash.edu.au. 489 m INTRODUCTION .......................................................................................................................................................489 VETERINARY VIRAL VACCINES ..........................................................................................................................491 Conventional Live and Inactivated Viral Vaccines ............................................................................................491 DIVA Vaccines.........................................................................................................................................................492 Molecularly Defined Subunit Vaccines ................................................................................................................493 Genetically Engineered Viral Vaccines ................................................................................................................493 Live Viral Vector Vaccines.....................................................................................................................................494 DNA Vaccines ..........................................................................................................................................................495 VETERINARY BACTERIAL VACCINES................................................................................................................495 Conventional Live Vaccines...................................................................................................................................495 Conventional Inactivated Vaccines.......................................................................................................................496 Gene-Deleted Vaccines ...........................................................................................................................................496 Subunit Vaccines.....................................................................................................................................................497 Vaccines against Zoonotic Bacteria......................................................................................................................497 Rickettsia Vaccines .................................................................................................................................................498 VETERINARY PARASITE VACCINES ...................................................................................................................499 Protozoal Vaccines ..................................................................................................................................................499 Live protozoal parasite vaccines.......................................................................................................................499 (i) Vaccines based on complete life cycle infections ..................................................................................499 (ii) Vaccines based on drug-abbreviated infections ...................................................................................500 (iii) Vaccines based on infections with parasites with a truncated life cycle.........................................500 (iv) Vaccines based on infection with virulence-attenuated strains ........................................................500 Killed or subunit protozoal parasite vaccines ................................................................................................500 Helminth and Ectoparasite Vaccines ...................................................................................................................501 VETERINARY VACCINES FOR NONINFECTIOUS DISEASES.............................................................................502 Allergy Vaccines ......................................................................................................................................................502 Cancer Vaccines ......................................................................................................................................................502 VETERINARY VACCINES FOR FERTILITY AND PRODUCTION CONTROL ............................................502 Design of Reproduction Control Vaccines ..........................................................................................................503 Vaccines against Reproductive Hormones ..........................................................................................................503 Vaccines against Gamete Antigens: Wildlife Control ........................................................................................504 Sperm antigens....................................................................................................................................................504 Oocyte antigens ...................................................................................................................................................504 Vaccines To Increase Fertility...............................................................................................................................505 CONCLUSION AND FUTURE DIRECTIONS......................................................................................................505 ACKNOWLEDGMENTS ...........................................................................................................................................506 REFERENCES ............................................................................................................................................................506 490 MEEUSEN ET AL. CLIN. MICROBIOL. REV. FIG. 1. Simplified schematic representation of immune mechanisms that can act to protect animals against invading viral, bacterial, and protozoal pathogens or against multicellular helminth parasites. Viral, bacterial, or protozoal pathogens (red ovals) that infect non-antigenpresenting cells can be killed by cytotoxic T cells (CTL) that recognize pathogen-derived epitopes presented in conjunction with major histocompatibility complex (MHC) class I on infected cells or by antibody-dependent lysis or opsonization of infected cells expressing pathogen molecules. Extracellular pathogens, or intracellular pathogens on their way to infect other cells, can be attacked by specific circulating antibodies and either killed by lysis or agglutination or phagocytosed by macrophages and neutrophils. Both antibody and CTL induction requires help from pathogenspecific CD4 helper T cells that are activated after interaction with pathogen-derived epitopes presented in conjunction with MHC class II molecules on the surface of MHC class II⫹ antigen-presenting cells. If pathogens infect antigen-presenting cells, they can be killed directly by CD4 T cells as well as CD8 CTL through the induction of mediators such as gamma interferon (IFN-␥), reactive oxygen and nitrogen species, and indoleamine 2,3-dioxygenase (IDO). Toxins released by pathogens (red circles) can be neutralized by circulating antibodies, thereby decreasing clinical signs of infection. Multicellular helminth parasites generally do not reside within host cells and are too large to be phagocytosed; therefore, they usually require alternative immune killer mechanisms mediated by antibody-directed actions of mast cells and eosinophils. Essential secreted proteins and toxins derived from the worms (brown circles) may also be neutralized by antibodies and thereby interfere with parasite growth. the risk for the consumer and in some cases to improve the productivity of the individual animal. Vaccination of wildlife is generally considered only with respect to infections that are transmittable to humans (zoonotic diseases), although welfare concerns are of increasing importance. While veterinary vaccines comprise only approximately 23% of the global market for animal health products, the sector has grown consistently due mainly to new technological advances in vaccine development, the continuous development of drug resistance by pathogens, and the emergence of new diseases. Apart from improving animal health and productivity, veterinary vaccines have a significant impact on public health through reductions in the use of veterinary pharmaceuticals and hormones and their residues in the human food chain. This will be an increasing impetus for activity with the more stringent requirements of regulatory agencies and consumer groups, particularly in the major markets of Europe and the United States (166). For example, the use of antibiotics in animal production has already been severely restricted, and the European Union has recently banned the use of coccidiostats for poultry. In addition, vaccines contribute to the well-being of livestock and companion animals, and their use is favored by the growing animal welfare lobby. The process of developing veterinary vaccines has both advantages and disadvantages over human vaccine development. On the one hand, the potential returns for animal vaccine producers are much less than those for human vaccines, with lower sales prices and smaller market sizes, resulting in a much lower investment in research and development in the animal vaccine area than in the human vaccine area, although the complexity and range of hosts and pathogens are greater. For example, the market size for the recently launched human vaccine (Gardasil) against papillomavirus and cervical cancer is estimated to be greater than 1 billion U.S. dollars, while the most successful animal health vaccines (e.g., against foot-andmouth disease [FMD] virus in cattle and Mycoplasma hyopneumoniae in pigs) enjoy a combined market size that is 10 to 20% of this figure. On the other hand, veterinary vaccine development generally has less stringent regulatory and preclinical trial requirements, which can make up the largest cost in human vaccine development, and a shorter time to market launch and return on investment in research and development. In contrast to human vaccine development, veterinary scientists are also able to immediately perform research in the relevant target species. This is an obvious advantage over human vaccine development, as experimental infections, dose-response studies, and challenge inoculations need not be carried out in less relevant rodent models. Immunity acquired through natural infection can take on several forms depending on the type and life cycle of the pathogen, as schematically represented in Fig. 1. Vaccines may be used to prevent clinical signs of disease after infection or to help control, eliminate, or even eradicate an infection at the population level (e.g., the expected global eradication of rinderpest virus through vaccination). Both vaccine effectiveness and mechanism of action may vary depending on the VOL. 20, 2007 VETERINARY VACCINES 491 TABLE 1. Second-generation licensed/commercialized veterinary viral vaccines Target pathogen Target animal Brand name(s)a Distributor PCV2 Pigs Porcilis-PCV2 Intervet PCV2 Pigs Suvaxyn PCV2 Fort Dodge Pseudorabies virus Pigs Suvaxyn Aujeszky Fort Dodge Classical swine fever virus Classical swine fever virus BHV-1 Pigs Porcilis Pesti Intervet Pigs Bayovac CSF E2 Bayer Leverkusen Cattle Bovilis IBR Marker Intervet Equine influenza virus Horses Merial WNV WNV WNV MDV (HTV) and IBDV Horses Horses Horses Poultry PROTEQ-FLU (European Union), Recombitek (United States) PreveNile West Nile-Innovator DNA RECOMBITEKEquine WNV Vaxxitek HVT⫹IBD Newcastle disease virus Poultry NA Dow AgroSciences Newcastle disease virus Avian influenza virus (H5N1) and NDV Avian influenza virus Poultry Poultry Vectormune FP-ND Biomune Intervet Poultry Poulvac FluFend I AI H5N3 RG Fort Dodge Trovac AI H5 Raboral Merial Merial Rabies virus Rabies virus Feline leukemia virus Canine parvovirusl Canine coronavirus Canine distemper virus Poultry Wildlife, canines Cats Cats Cats Dogs Dogs Dogs Purevax Feline Rabies PUREVAX Feline Rabies EURIFEL FeLV RECOMBITEK Canine Parvo RECOMBITEK Corona MLV RECOMBITEK rDistemper Merial Merial Merial Merial Merial Merial Canine distemper virus IHN virus Fur animals Salmon PUREVAXFerret Distemper Apex-IHN Merial Novartis (Aqua Health) Avian influenza virus Rabies virus a Intervet Fort Dodge Merial Merial Characteristic(s) Inactivated baculovirus expressed PCV2 ORF2 protein; adjuvanted Inactivated PCV1-2 chimera; adjuvanted gE- and thymidine kinase-deleted marker vaccine Baculovirus recombinant E2 protein without emulsion Baculovirus recombinant E2 protein without emulsion Live or inactivated gE-deleted marker vaccine Canarypox virus-vectored vaccine Live flavivirus chimera vaccine DNA vaccine Canarypox virus-vectored vaccine Live recombinant chimera virus expressing VP2 gene of IBD on HTV virus HN recombinant produced in plant cell lines (registered but not on market) Fowlpox virus vectored Chimera virus on NDV backbone; field trials in 2007 Chimera H5N3 virus, inactivated in oil-based adjuvant Fowlpox virus-vectored H5 Vaccinia virus recombinant Canarypox virus-vectored Canarypox virus-vectored Canarypox virus-vectored Modified live virus Modified live virus Canarypox virus-vectored (HA and F antigens) Canarypox virus-vectored DNA vaccine Reference(s) 20 55 56 181 116 185 113 114 45 113 43 133, 187 29 26, 100, 152, 170 vaccine vaccine vaccine 132 vaccine vaccine Brand names may differ between countries. NA, not applicable. required outcome. New technologies to achieve the selective induction of effective immune responses in the development of new vaccines are becoming available to vaccine researchers and have been extensively reviewed in recent papers (33, 136, 163, 166). Notwithstanding the scientific advances, the single factor that determines the success of an experimental vaccine is its successful commercialization and/or use in the field. This outcome requires a combination of basic research, commercial imperatives, local requirements, and global perspectives depending on the particular disease under investigation. These four aspects are represented by the authors of this review, which will concentrate on recent advances in veterinary vaccines that have reached the marketplace or are actively produced by veterinary institutes for use by local farming communities. VETERINARY VIRAL VACCINES As there are no broad-spectrum antiviral pharmaceuticals available, hygienic measures to limit exposure and vaccination are the only means to prevent or control viral infections. Viruses (especially RNA viruses) are highly variable, and many viral infections are due to viruses with multiple serotypes (e.g., FMD virus, bluetongue virus, and influenza viruses). As a consequence, many of the existing viral vaccines are often unable to cope with the prevailing strains in the field, and new ones have to be generated from field strains with new outbreaks. Numerous conventional live and inactivated viral vaccines have been produced by animal health companies and have been used for many decades in routine vaccination protocols for both companion and production animals. Increasingly, a number of rationally designed and subunit vaccines are reaching the market, and this section will concentrate mainly on these “second-generation” viral vaccines (summarized in Table 1). Conventional Live and Inactivated Viral Vaccines As with the first vaccine for human smallpox, most live veterinary viral vaccines induce mild infections with live organisms derived from nontarget hosts or attenuated through passage in different cell line cultures or chicken embryos (eggs). Attenuated viral strains are also obtained by inducing random mutations and selecting for reduced virulence. As the live organism can still infect target cells, these vaccines can replicate and induce both cellular and humoral immunity and generally do not require an adjuvant to be effective. Live products also offer the advantage of ease of administration, potentially in drinking water, intranasally, intraocularly, etc. However, they can pose a risk of residual virulence and reversion to 492 MEEUSEN ET AL. pathogenic wild types as well as provide a potential source of environmental contamination. Although modern regulatory processes require data to provide assurance on these issues, problems in the field can arise. This was highlighted during a program to control porcine respiratory and reproductive syndrome (PRRS) in Denmark. This disease first emerged in North America in the late 1980s and spread quickly in Europe in the early 1990s. The two main types of PRRS virus, European and North American, are only 55 to 80% identical at the nucleotide level (122) and cause distinguishable serological responses. Following vaccination with the live, attenuated North American PRRS vaccine against the European PRRS virus type present in Denmark in 1996, the vaccine virus reverted and spread within vaccinated herds as well as from vaccinated to nonvaccinated herds, leaving both virus types in the Danish pig population (120). Despite such drawbacks of live viral vaccines, they have played a major role in successful disease control and eradication. For example, the virtual eradication of rinderpest virus from the globe is widely believed to have been critically dependent on the use of the “Plowright” vaccine (12, 150). This is an attenuated vaccine produced from the Kabete O strain passaged 90 times in tissue culture (141). The vaccine virus was recently found to have attenuating mutations in most of its genes, none of which are sufficiently debilitating to induce strong pressure for reversion (11). Although there are examples of stable attenuations from a single point mutation in the polymerase gene, the high rate of spontaneous mutations of RNA viruses increases the risk for reversion to virulence. Safe live viral vaccines are therefore likely to require a number of attenuating mutations distributed throughout the genome. Whole inactivated or killed viral vaccines are generally more stable and do not pose the risk of reversion to virulence compared to live vaccines, but their inability to infect cells and activate cytotoxic T cells makes them much less protective. Consequently, they generally require strong adjuvants and several injections to induce the required level of immunity and are usually effective in controlling only clinical signs rather than infection (113). Inactivated adjuvanted vaccines also pose a greater risk of causing autoimmune diseases, allergic disorders, and vaccine injection site sarcomas (46). Viral inactivation is commonly achieved through heat or chemicals (e.g., formaldehyde, thiomersal, ethylene oxide, and -propriolactone). The higher production cost and need for adjuvants make these vaccines more expensive to manufacture. Inactivated viral vaccines for a wide range of viral diseases have been available for several decades (reviewed in references 113 and 121) and are still being developed for some recently emergent diseases. For example, a one-dose inactivated porcine circovirus type 2 (PCV2) vaccine has recently been licensed in the United States for the prevention of postweaning multisystemic wasting syndrome in pigs (Table 1). Much of the recent research in this area has concentrated on the development of improved adjuvanted formulations to overcome the effects of maternal antibodies on young animals (see, for example, reference 17). Inactivated vaccines for several viral diseases need to be continuously adapted to contain the appropriate serotypes, as exemplified by equine influenza virus vaccines. Vaccines for equine influenza virus, mostly inactivated, have been available since the 1960s (28). The most important equine subtypes are CLIN. MICROBIOL. REV. H7N7 and H3N8, although H7N7 has not been detected for several decades and is no longer included in vaccines, at least in Europe and the United States (130). Conversely, vaccination against H3N8 has been less effective, possibly due to antigenic drift, and there are now considered to be two distinct lineages, European and United States, and vaccines therefore tend to contain both. Over the years, improvements have been attempted, and more potent adjuvants have been used. Several European vaccines now produce high antibody responses that last for up to 1 year (113). Until recently, equine influenza virus vaccines produced in the United States have been considered to be of limited efficacy and sometimes lacking the relevant H3N8 strains (M. Mellencamp and A. Schultze, presented at the Proceedings Quality Control of Equine Influenza Vaccines, Budapest, Hungary, 2001). DIVA Vaccines For several viral infections of livestock, effective conventional vaccines are available but cannot be used, as they would interfere with disease surveillance based on serological testing and may result in the loss of a country’s disease-free status. A classic example is FMD in cattle. Although inactivated FMD vaccines have been available for many years and are quite effective in controlling clinical disease (49), they are not used in FMD-free countries, as this would compromise this status and hence international trade. Nevertheless, conventional vaccines have reduced the prevalence of disease in enzootic areas, and in a recent outbreak in The Netherlands, vaccination was used to reduce the spread of the disease (142), although the vaccinates were subsequently slaughtered to enable the rapid reestablishment of the FMD-free status of the country. The ability to identify and selectively delete genes from a pathogen has allowed the development of “marker vaccines” that, combined with suitable diagnostic assays, allow differentiating infected from vaccinated animals (DIVA) by differentiation of antibody responses induced by the vaccine (no antibodies generated to deleted genes) from those induced during infection with the wild-type virus (e.g., see Fig. 2). Such DIVA vaccines and their companion diagnostic tests are now available or in development for several diseases including infectious bovine rhinotracheitis (IBR), pseudorabies, classical swine fever (CSF), and FMD, as detailed below. IBR, caused by bovine herpesvirus type 1 (BHV-1) infection of cattle, and pseudorabies (Aujeszky’s disease) in pigs have been identified internationally as being candidates for eradication from national herds, and so there has been an impetus for the development of DIVA vaccines and diagnostics. The demand for a marker (DIVA) vaccine for IBR in Europe was met by the development of a glycoprotein E (gE)-deleted vaccine using conventional methodology (reviewed in reference 185). The gE protein is not essential for viral replication, but it plays a major role in intercellular spread, particularly along nerves. Specific diagnostic tests based on gE deletion have been developed using both gE-blocking enzyme-linked immunosorbent assay (ELISA) techniques and PCR amplification (138, 159). Deletion of the gE gene has also been used to enable a DIVA approach for an Aujeszky’s disease vaccine (137). The gene for thymidine kinase is also deleted in some formulations VOL. 20, 2007 VETERINARY VACCINES 493 proaches for FMD (reviewed in references 8 and 66). Subunit antigen approaches to vaccination have been largely ineffective, as they present only a limited number of epitopes to the animal’s immune system, and multiple antigens are generally required for protection. Current research is focused largely on combinations of capsid proteins, including empty capsid delivered by various expression systems, and the development of sensitive tests (ELISA) for antibodies against nonstructural proteins (66). Other diseases for which a DIVA approach is highly desirable but currently unavailable include bluetongue virus in cattle, Newcastle disease virus and avian influenza virus in poultry, bovine viral diarrhea, and equine viral arteritis. Molecularly Defined Subunit Vaccines FIG. 2. Simplified representation of the reverse genetic approach used to construct the chimera vaccine Poulvac FluFend i AI H5N3 RG to protect poultry against the pathogenic H5N1 virus. The HA gene was removed from an H5N1 virus (from a recent Asian outbreak), inactivated by removing the polybasic amino acid sequences, and combined with the NA gene from an H2N3 virus onto an H1N1 “backbone” virus. An immunoassay able to specifically detect antibodies against N3 and N1 proteins could be used for DIVA (i.e., N3⫹ N1⫺ indicates vaccinated, and N3⫺ N1⫹ indicates infected). (Modified from Fort Dodge Poulvac FluFend i AI H5N3 RG promotional flyer with permission.) (e.g., Suvaxyn Aujesky), adding to the degree of attenuation (e.g., see reference 56). These deletion vaccines have been available since the 1980s, and their use has contributed to disease control and eradication in the United States and several European countries (e.g., see reference 24). CSF is on the World Organization for Animal Health list of notifiable diseases and is one of the most important contagious diseases of pigs worldwide. In its classical clinical form, it is an acute hemorrhagic disease accompanied by high fever, depression, anorexia, and conjunctivitis. Morbidity and mortality are both very high and may reach 100%. However, it can also present as a subacute, chronic, or even subclinical condition. Countries in which the disease is enzootic tend to vaccinate with a very effective live, attenuated vaccine, while those that are free of disease do not (reviewed in reference 13). Two subunit vaccines based on the viral envelope glycoprotein E2 produced in a baculovirus/insect cell system, formulated in a water-in-oil adjuvant, and accompanied by discriminatory ELISA tests are available (116, 181). These vaccines will allow a DIVA approach to emergency vaccination and disease control in the case of new outbreaks, although these have not yet been used widely in the field and appear to be less protective than conventional live, attenuated CSF vaccines (13). As there are now major doubts about the sustainability of “stamping-out” policies in areas of high animal population density, there is considerable investment in DIVA vaccine ap- Identification of the protective viral antigens potentially allows their isolation and/or recombinant production so that they can be administered as safe, nonreplicating vaccines. However, as isolated antigens generally induce poor protective immunity, subunit vaccines usually require repeated administration with strong adjuvants, making them less competitive. Notwithstanding these limitations, there are some examples of effective subunit vaccines. PCV2 is considered to be the major pathogen in the etiology of postweaning multisystemic wasting syndrome (2). A recombinant baculovirus producing the protective ORF2 protein of PCV2 has recently become available as a vaccine for pigs (20). Dow AgroSciences successfully registered the first plantbased vaccine for Newcastle disease virus in poultry in the United States in 2005. Recombinant viral HN protein was generated in plant cell lines via Agrobacterium transformation and could successfully protect chickens from viral challenge (G. A. Cardineau, H. S. Mason, J. Van Eck, D. D. Kirk, and A. M. Walmsley, 2004, PCT patent application 60/467,998, WO 2004/098533; C. A. Mihaliak, S. Webb, T. Miller, M. Fanton, D. Kirk, G. Cardineau, H. Mason, A. Walmsley, C. Arntzen, and J. Van Eck, presented at the 108th Annual Meeting of the United States Animal Health Association, Greensboro, NC, 2005). This process was a proof-of-concept exercise designed to test regulatory feasibility, and the product is not on the market. Genetically Engineered Viral Vaccines The availability of complete DNA sequences and a better understanding of gene function have allowed specific modifications or deletions to be introduced into the viral genome, with the aim of producing well-defined and stably attenuated live or inactivated viral vaccines. Gene-deleted vaccines (glycoprotein I and/or glycoprotein X) against pseudorabies allowed a DIVA approach and control of Aujeszky’s disease in swine (96); however, the potential for recombination between pseudorabies virus strains has raised concern (101). Similarly, thymidine kinase deletion in BHV-1 vaccines has been associated with latency and reactivation after treatment with dexamethasone (194), and deletion of multiple genes has been proposed in order to improve safety (14). An interesting development in genetically engineered viral 494 MEEUSEN ET AL. vaccines is the production of chimera viruses that combine aspects of two infective viral genomes. A chimera PCV1-2 vaccine has the immunogenic capsid gene of PCV2 cloned into the backbone of the nonpathogenic PCV1 and induces protective immunity to wild-type PCV2 challenge in pigs (55). A further sophistication of this approach is a recently developed vaccine against avian influenza virus (Poulvac FluFend), where the hemagglutinin (HA) gene has been removed from an H5N1 virus, inactivated by removing the polybasic amino acid sequences, and combined with the NA gene from an H2N3 virus onto an H1N1 “backbone” virus (Fig. 2). A vaccine containing the resultant inactivated H5N3-expressing virus administered in a water-in-oil emulsion protects chickens and ducks against the highly pathogenic H5N1 strain. Similarly, a live Flavivirus chimera vaccine against West Nile virus (WNV) in horses (PreveNile) was registered in the United States in 2006. In this chimera vaccine, the structural genes of the attenuated yellow fever YF-17D backbone virus have been replaced with structural genes of the related WNV. The resulting chimera vaccine express the PreM and E proteins of WNV, while the nucleocapsid (C) protein, nonstructural proteins, and nontranslated termini responsible for virus replication remain those of the original yellow fever 17D virus (114). After a single shot, the vaccine stimulates both cellmediated and humoral responses without causing any clinical illness or spreading to sentinel horses and provides protection against WNV challenge for up to 12 months (PreveNile package insert). A similar vaccine could be a candidate for a human WNV vaccine (115). Live Viral Vector Vaccines Poxviruses including vaccinia virus, fowlpox virus, and canarypox virus have been used as vectors for exogenous genes, as first proposed in 1982 (131), both for the delivery of vaccine antigens and for human gene therapy. Poxviruses can accommodate large amounts of foreign genes and can infect mammalian cells, resulting in the expression of large quantities of encoded protein. For example, modified vaccinia virus Ankara is a highly attenuated strain produced by several hundred passages of the virus in chicken cells. Modified vaccinia virus Ankara lacks about 10% of the vaccinia virus genome, including the ability to replicate in mammalian cells (reviewed in reference 139). A particular success story has been the development of an oral recombinant vaccinia-rabies vaccine in bait for wild carnivores such as foxes in Europe (26) and foxes, raccoons, and coyotes in the United States (152, 170). Rabies is caused by a negative-stranded Rhabdoviridae RNA virus transmitted mainly via saliva following a bite from an infected animal. The main source of infection for humans is domestic reservoir species including dogs and cats. There are seven rabies virus genotypes, all of which, excluding type 2, produce similar effects in humans. Rabies can infect most if not all mammals. The virus enters the central nervous system, causing an encephalomyelitis that is always fatal once symptoms develop. Worldwide, the disease causes many thousands of human deaths each year. One type of oral vaccine is in the form of a bait containing a recombinant vaccinia virus vector expressing the protective glycoprotein G of rabies virus (100, 135). After CLIN. MICROBIOL. REV. several years of vaccination campaigns against fox rabies virus in several Western European countries, rabies could be eliminated from its wildlife terrestrial reservoir, as exemplified by the successful elimination of terrestrial rabies virus from Belgium and France (26, 135, 136). The canarypox virus vector system ALVAC has been used as a platform for a range of veterinary vaccines including WNV, canine distemper virus, feline leukemia virus, rabies virus, and equine influenza virus (176) (Table 1). Canarypox virus was originally isolated from a single pox lesion in a canary and serially passaged 200 times in chicken embryo fibroblasts and serially plaque purified under agarose (Merial bulletin TSB-40019-FTB). Canarypox viruses and fowlpox viruses have the advantage of being more host restricted than vaccinia virus. While they produce an abortive infection in mammalian cells, canarypox virus recombinants still effectively express inserted foreign genes. Several veterinary viral vaccines have been produced using the ALVAC vector system (Table 1). Most notably, a novel equine influenza virus vaccine using the canarypox vector to express the hemagglutinin genes of the H3N8 Newmarket and Kentucky strains has recently been registered in the European Union (Proteq-Flu) (113) and the United States (Recombitek). It contains a polymer adjuvant (Carbopol; Merial Ltd.), and through the induction of both cell-mediated and humoral immunity, it is claimed to produce sterile immunity 2 weeks after the second of two doses. The new vaccine is also designed to protect horses against the highly virulent N/5/03 American strain of equine influenza virus and to prevent the virus from spreading through the elimination of viral shedding. Trovac AI H5 is a recombinant fowlpox virus expressing the H5 antigen of avian influenza virus. This product has had a conditional license for emergency use in the United States since 1998 and has been widely used in Central America, with over 2 billion doses administered (29). As the vaccinated birds will not develop antibodies against matrix protein/nucleoprotein, this vaccine can also be used with a DIVA approach. Several vaccines are available based on inactivated adjuvanted formulations for equine herpesvirus type 1 and equine herpesvirus type 4, equine herpesviruses that are major causes of abortion and respiratory disease. None of these vaccines are considered to provide complete clinical or virological protection (113). A canarypox virus-vectored vaccine containing the genes for gB, gC, and gD has been developed, but the latest reports suggested that it did not completely protect against challenge (113). A further application of vectored vaccines is the use of an attenuated viral pathogen as the vector, with the aim of inducing protection against two diseases, as with the live recombinant vaccine against both Marek’s disease virus (MDV) and infectious bursal disease virus (IBDV) in chickens (Vaxxitek HVT ⫹ IBD). MDV is a highly contagious neoplastic disease of poultry caused by gallid herpesvirus type 2, while IBDV replicates in the bursa of Fabricius, the primary lymphoid organ in birds, and causes a serious immunosuppressive condition in poultry flocks worldwide. Turkey herpesvirus (HVT) is nonpathogenic in chickens but confers cross-protection against MDV and has traditionally been used in live vaccines against MDV. The new vaccine is based on a recombinant parent HVT virus expressing the VP2 gene of IBDV (44) and can be given to embryonated eggs or 1-day-old chicks without interference VOL. 20, 2007 VETERINARY VACCINES from maternally derived antibodies. Data from large-scale field trials for this vaccine have not yet been reported, but those studies may encounter difficulties in maintaining high efficacy. This is because these tightly regulated recombinant vaccines cannot easily adapt to meet the emergence of very virulent strains of both IBDV and MDV, apparently induced by the comprehensive numbers of vaccinations performed against these diseases (81, 146). Chimera avian influenza virus vaccines have also recently been produced on a backbone of an existing, attenuated Newcastle disease virus vaccine strain. Both Asian H5N1 and the pathogenic H7N7 strain, responsible for the chicken influenza virus outbreak in The Netherlands in 2003, were produced as chimeras with the Newcastle disease virus strain. This chimera vaccine induced strong protection against the respective wildtype influenza virus as well as against Newcastle disease virus (133, 187). DNA Vaccines Immunization of animals with naked DNA encoding protective viral antigens would in many ways be an ideal procedure for viral vaccines, as it not only overcomes the safety concerns of live vaccines and vector immunity but also promotes the induction of cytotoxic T cells after intracellular expression of the antigens. Furthermore, DNA vaccines are very stable and do not require a cold chain. While DNA vaccination of large animals has not been as effective as initially demonstrated in mice, several groups have obtained significant improvements in immune responses using innovative technologies such as specific targeting of the vaccine antigen to antigen-presenting cells (85), priming-boosting with stimulating CpG oligodeoxynucleotides (85, 97), and in vivo electroporation of DNA (155). Considerable research into DNA vaccines for fish viruses, where this approach seems to be particularly effective (73, 99), has been ongoing. Notably, the first DNA vaccine for an edible species (Apex-IHN) was registered in 2005 in Canada to protect Atlantic salmon from infectious hematopoietic necrosis (IHN) (167). IHN disease is enzootic in wild salmon populations and can cause devastating outbreaks in farm-raised salmon that have had no prior exposure. The DNA vaccine encodes a surface glycoprotein of IHN virus and is administered intramuscularly (99). A DNA vaccine to protect horses against viremia caused by WNV (West Nile-Innovator DNA) received a license from the USDA at approximately the same time as the fish DNA vaccine. WNV infection is caused by a flavivirus belonging to the Japanese encephalitis virus complex. It is enzootic in parts of Africa and Asia but was first detected in the United States in 1999 in an outbreak involving birds, horses, and humans in New York, and it subsequently spread rapidly to many states (62). The DNA plasmid codes for the WNV outer coat proteins and is administered with a proprietary adjuvant (143). The vaccine was, however, produced as part of a buildingplatform technology rather than as a commercial product, as the manufacturer already has a WNV vaccine on the market. The success of these two DNA vaccines may be due more to good fortune than to any specific technological advances, as DNA uptake into fish muscle seems to be unusually efficient (99), and the WNV viral protein may be particularly effective 495 because it naturally produces highly immunogenic virus-like particles (161). It is likely that a wider application of DNA vaccines will require further improvements and optimization for each host-pathogen combination. VETERINARY BACTERIAL VACCINES Many attenuated live or inactivated (killed) bacterial vaccines have been available for decades as prophylaxis against bacterial diseases in veterinary medicine. For most of the attenuated bacterial strains, the nature of the attenuation is not known, and since they have a proven track record, little is done to characterize underlying genetics. In some cases, however, the old and well-recognized live strains are not highly protective, and continued research is being performed to improve and develop new vaccines or vaccination strategies against, e.g., bovine tuberculosis, paratuberculosis, and brucellosis, as described below. Inactivated vaccines generally consist of bacterins of one or more bacterial species or serotypes (i.e., crude formalin-killed whole bacterial cultures and supernatants) or more well-defined subunit antigens formulated most often in an oil or aluminum hydroxide adjuvant. Many of the established bacterial vaccines are highly efficacious, but since the technology has been available for many years, these conventional vaccines will not be dealt with in this review, and the reader is referred to company websites for information on specific diseases and vaccines. Both live and inactivated autogenous bacterial vaccines are also produced by local veterinary institutions or specialized companies for on-farm specific demands where no commercial vaccines are available. This section will review some of the more recent additions to bacterial veterinary vaccines (summarized in Table 2), with particular focus on the more molecularly defined vaccines. Conventional Live Vaccines In spite of modern technological advances, new live vaccines based on strains without identification of the attenuating characteristics continue to reach the market. One such example is a new live vaccine (Enterisol Ileitis) against porcine proliferative enteropathy caused by the obligate intracellular bacterium Lawsonia intracellularis. Identification of L. intracellularis as the cause of this disease was established in 1993 (107), and many features of the causal bacteria as well as the immunopathogenesis remain to be elucidated. The vaccine strain was cultivated from a clinical isolate, and there are no phenotypic or genotypic characteristics to separate this strain from wild-type strains. Following oral administration of the vaccine, there appear to be no or delayed fecal shedding of bacteria and a low or absent induction of systemic humoral or cell-mediated immunity, but fecal shedding upon challenge is reduced and weight gain is increased compared to unvaccinated pigs (67, 90). The vaccine has been licensed to improve weight gain and to reduce growth variability associated with ileitis in pigs and is administered through drinking water. 496 MEEUSEN ET AL. CLIN. MICROBIOL. REV. TABLE 2. Recently commercialized veterinary bacterial vaccines Target pathogen(s) Target animal Brand name Lawsonia intracellularis Pigs Enterisol Ileitis Porphyromonas gulae, P. denticanis, and P. salivosa Yersinia ruckeri Dogs Periovac Fish AquaVac ERM Aeromonas salmonicida Fish Vibrio anguillarum Fish Streptococcus equi Chlamydophila abortus Distributor Characteristic(s) Boehringer-Ingelheim Vetmedica Pfizer Animal Health Live oral vaccine Killed oral vaccine Horses Sheep AquaVac Furuvac AquaVac Vibrio Equilis StrepE Ovilis Enzovax Schering-Plough Animal Health Schering-Plough Animal Health Schering-Plough Animal Health Intervet Intervet Mycoplasma synoviae Chickens Vaxsafe MS Bioproperties Mycoplasma gallisepticum Bordetella avium Chickens Vaxsafe MG Bioproperties Turkeys Art Vax Actinobacillus pleuropneumoniae Actinobacillus pleuropneumoniae Salmonella Pigs PleuroStar APP Schering-Plough Animal Health Novartis Animal Health Pigs Porcilis APP Intervet Chickens and hens Cattle Megan Vac1 MeganEgg RB-51 Lohman Animal Health International Colorado Serum Company CZ Veterinaria Brucella abortus Conventional Inactivated Vaccines An interesting new addition to the repertoire is a vaccine consisting of inactivated bacterins of Porphyromonas gulae, P. denticanis, and P. salivosa for vaccination against periodontal disease in dogs (Periovac). The vaccine is based on research identifying these three bacteria as being the most common black-pigmenting anaerobic bacteria in periodontal pockets of dogs and were all pathogenic in a mouse model (70). A vaccine prepared from P. gulae and administered subcutaneously to mice was able to significantly reduce alveolar bone loss in this model (71). There are no published details on the performance of the trivalent vaccine in dogs, but efficacy and potency trials are ongoing with, e.g., a clinical trial running at the University of Minnesota Veterinary Medical Center. Despite the lack of published efficacy data, the vaccine is currently fully licensed in New Zealand and conditionally licensed in the United States. A killed oral vaccine (AquaVac ERM) against enteric redmouth disease caused by Yersinia ruckeri in rainbow trout has been available in United Kingdom since 2001 and is now further approved for a number of European countries. Enteric redmouth disease is a serious infectious disease of farmed rainbow trout in many countries characterized by congestive or hemorrhagic zones in various tissues and organs, particularly around the mouth and in the intestines. The disease has a very high mortality rate, and Y. ruckeri is able to form biofilms on fish tank surfaces and thus persist and remain infective in the aquatic environment, with the possibility of recurrent infections (39). Immersion of fry for 30 s into a vaccine soup at the hatchery provides initial protection for fingerlings but rarely lasts throughout the production cycle. Follow-up booster vaccination by injection is effective, but this is time-consuming and labor-intensive and may be stressful for the handled fish. The oral vaccine protocol recommends a primary immersion vac- Reference(s) 67, 90 Killed vaccine against periodontitis 64 Killed oral vaccine Killed oral vaccine Live submucosal vaccine; deletions in aroA gene Live temperature-sensitive mutant strain for subcutaneous or intramuscular injection Live temperature-sensitive mutant strain; eye drop administration Live temperature-sensitive mutant strain; eye drop administration Live temperature-sensitive mutant strain; spray inhalation or drinking water Recombinant ApxII, TbpB, CysL, OmlA(1), and OmlA(2) proteins Extracted ApxI, ApxII, ApxIII, and outer membrane proteins Double gene-deleted S. enterica serovar Typhimurium strain Spontaneous rifampin-resistant rough mutant 80 34 119 10 79 186 35 5 118 cination followed 4 to 6 months later by an oral booster of the vaccine, which is mixed and absorbed into the feed pellets. Both the primary and booster vaccine formulations are inactivated bacterial cultures, but for oral vaccination, the bacteria are incorporated into an “antigen protection vehicle,” bypassing the acidic environment of the gut and delivering the antigens to the area of the hindgut (64). There are no data available on the nature of the antigen protection vehicle, but the product résumé claims the presence of lecithin and fish oil, indicating that killed bacteria are likely incorporated into liposome structures (61). Similar vaccines against furunculosis caused by Aeromonas salmonicida and vibriosis caused by Vibrio anguillarum have also been developed, and an oral vaccine against infectious pancreatic necrosis virus is registered in Chile for use in salmon. To our knowledge, these are the only licensed inactivated mucosal vaccines against bacterial diseases in veterinary medicine. Gene-Deleted Vaccines Traditionally, attenuation of bacteria for the preparation of live vaccines has been performed by multiple passages in various media in the hope that some random mutation would deliver a nonvirulent, but replicable, type of the agent. With currently used molecular methods, the obtained deletions/mutations can be identified, but this technology also allows a more targeted design of live vaccines with specific deletions of predetermined known genes. Good targets for these deletions are genes responsible for key metabolic processes that inhibit the spread of the infection but allow the development of immune responses against virulence factors. Alternatively, deletions of virulence-associated genes are targets, but this may be more problematic when a protective immune response is desired. VOL. 20, 2007 Gene-deleted vaccines have been produced against strangles, a highly contagious disease in horses caused by infection with Streptococcus equi subsp. equi. The disease is characterized by fever, profuse nasal discharge, and abscess formation in the lymph nodes of the head and the neck. The pus discharged from bursting abscesses is highly infectious, and the swelling of involved lymph nodes may, in severe cases, cause airway restriction, hence the name. Commercial bacterin or protein extract vaccines for parenteral administration can induce high levels of serum bactericidal antibodies, but the protective effects of these antibodies are questionable (177), and the protective efficacy of inactivated vaccines in the field has been disappointing (174). A live intranasal vaccine based on a nonencapsulated attenuated strain (Pinnacle IN) has been widely used in North America since it was launched in 1998. However, the attenuating mutations of this strain have not been defined, and the vaccine strain sometimes reverts to an aggressive mucoid phenotype indistinguishable from that of wild-type strains. The Pinnacle strain has since been refined into a more stable hyaluronate synthase-defective mutant (191). It is, however, not clear if this new strain has replaced the original vaccine strain in the commercial product. Recently, the Equilis StrepE vaccine, a live recombinant bacterial vaccine prepared from the S. equi TW928 deletion mutant lacking bp 46 to 978 of the aroA gene (80, 84), was licensed in Europe. This mutant was constructed by the electroporation of gene knockout and gene deletion constructs. No foreign DNA such as antibiotic resistance markers was introduced, but the vaccine strain can allegedly be identified by an aroA PCR identifying the partial gene deletion (84). The live gene-deleted attenuated vaccine strain was originally developed for intranasal application, but protection was accomplished only by intramuscular injections, which in turn resulted in the local swelling of muscle tissue and the eventual formation of abscesses at the vaccination site (80). However, submucosal administration of the vaccine in the upper lip was shown to confer protection comparable to that of intramuscular administration but with only minimal local reactions (80), and it is with this unusual route of administration that the vaccine is now licensed. Chlamydiae are obligate intracellular bacteria with a wide host range and with a wide spectrum of diseases, several of which are zoonotic. The most important veterinary species are Chlamydophila psittaci, causing respiratory infections in poultry (psittacosis/ornithosis), and Chlamydophila abortus (formerly Chlamydia psittaci serotype 1), causing ovine enzootic abortion, one of the most important causes of ovine and caprine abortion worldwide. Both infections are zoonotic. While no vaccines are available for birds and poultry, inactivated vaccines against ovine enzootic abortion have been available for many years (reviewed in reference 145). More recently, a temperature-sensitive mutant strain, TS1B, of the C. abortus reference strain AB7 obtained by nitroguanidine mutagenesis (148) is used to prevent abortion in sheep (Ovilis Enzovax). The temperature-sensitive mutant strain has an optimal growth temperature at 38°C, but at the restrictive temperature, 39.5°C, growth is impaired. The normal body temperature of adult sheep is 38.5 to 40.0°C. The vaccine induces good and long-lasting protection in sheep (34), goats (149), and mice (even though the body temperature of mice is within the permissive growth range). However, the vaccine is licensed only VETERINARY VACCINES 497 for sheep, not goats, and there is continued research into the development of an effective subunit-based vaccine (59, 190). Temperature-sensitive-mutant vaccines have also been developed and marketed as eye drop, spray, or inhalation vaccines. These include vaccines against Mycoplasma synoviae and M. gallisepticum in chickens (Vaxsafe MS and Vaxsafe MG, respectively) (10, 119) and Bordetella avium rhinotracheitis (coryza) in turkeys (Art Vax) (79). Gene-deleted live bacteria with well-defined targeted attenuations also offer an attractive option as mucosal vectors for passenger antigens, with many potential advantages over traditionally injectable vaccines. Unlike viral vaccines, there are at present no commercialized vector vaccines based on a bacterial backbone carrier delivering antigens from other pathogens, although several bacterial vectors have shown very promising results (reviewed in references 61 and 108). Subunit Vaccines Porcine contagious pleuropneumonia is a widespread and severe disease of pigs with hemorrhagic necrotizing pneumonia and high mortality in the acute form. The disease is caused by Actinobacillus pleuropneumoniae, and prevention by vaccination with whole-cell bacterin vaccines has been severely restricted by the prevalence of 15 different serotypes. A second generation of acellular A. pleuropneumoniae subunit vaccines has been developed with four extracted (Porcilis APP) or five recombinant (PleuroStar APP) proteins, which confer some degree of cross-protection against all serotypes. Most of the pathological consequences of A. pleuropneumoniae infection are caused by pore-forming RTX (repeats in the structural toxin) exotoxins ApxI, ApxII, ApxIII, and APxIV, of which at least a combination of two are expressed in all serotypes. The serotypes expressing both ApxI and ApxII are particularly virulent (58). Vaccination with RTX toxins alone protects against mortality but does not reduce the typical lung lesions. The pentavalent recombinant vaccine containing only the ApxII toxin but supplemented with other common antigens such as transferring-binding proteins appears to provide protection that is at least as good as or better than that of the vaccine with three extracted Apx toxins supplemented with a single outer membrane protein (35, 69, 186). However, the precautions needed when such subunit vaccines are to be designed is evidenced by a study of vaccination against PalA of the peptidoglycan-associated protein family and the most “immunopredominant” outer membrane protein of A. pleuropneumoniae (and related to, e.g., the P6 protein of Haemophilus influenza). Antibodies induced against PalA alone aggravated the consequences of a challenge infection, and PalA vaccination in combination with RTX toxins even counteracted the protective effect of anti-ApxI and anti-ApxII antibodies (182). Vaccines against Zoonotic Bacteria Clinical salmonellosis in animals is often due to host-restricted serotypes such as Salmonella enterica serovar Choleraesuis in pigs, Salmonella enterica serovar Gallinarum in poultry, and Salmonella enterica serovar Dublin in young cattle with severe systemic infections, which may result in the death of the animal. In contrast, non-host-specific Salmonella serotypes 498 MEEUSEN ET AL. usually induce a self-limiting gastrointestinal infection but with the capability of causing systemic infections in a wide range of host animals, including humans. The desired immunity of vaccines against zoonotic infections not only requires the induction of a local mucosal immunity, preventing colonization of the gut of the individual animal, but should ideally also prevent or eliminate the presence of the bacteria in the flock as a whole to prevent cross-contamination of meat products at the slaughterhouse. This is a very difficult task, and available vaccines have so far yielded variable success rates (reviewed for poultry in reference 184). It is generally recognized that cell-mediated immunity is more important than humoral responses in protection against Salmonella, and together with the need for local mucosal immunity, this calls for live, attenuated vaccines as the most effective type. This is supported by a comparison of a live double-gene-deleted Salmonella enterica serovar Typhimurium vaccine (MeganVac 1) with an inactivated Salmonella enterica serovar Enteritidis vaccine (Poulvac SE), which showed reduced fecal shedding following live vaccination, while chickens receiving a killed vaccine experienced inhibited cell-mediated immune responses, enhanced antibody responses, and an increased bacterial load (5). The MeganVac 1 organism has recently been reformulated for immunization of laying hens (MeganEgg). The Megan vaccines for broilers and hens were licensed by the USDA in 1998 and 2003, respectively, but worldwide, there are at least 10 other live Salmonella vaccines available for Salmonella enterica serovar Enteritidis, Salmonella enterica serovar Typhimurium, or Salmonella enterica serovar Gallinarum infection in poultry. Campylobacter jejuni is one of the most important causes of food-borne human bacterial gastroenteritis. Although several vaccines aimed at preventing human disease are in the pipeline, an effective vaccination intervention strategy for infected poultry flocks would be the most effective means of preventing human disease. Similar to the requirements of a vaccine against non-host-specific Salmonella serotypes, such a vaccine must, however, be able to provide a very high degree of protection in the flock to eliminate the subsequent contamination of meat products. Experimental vaccines, mainly killed wholecell cultures or flagellum preparations, have been tested in poultry but provide only partial protection against a challenge with Campylobacter, and the development of an attenuated live strain may be more promising although not yet commercially available. Brucellosis continues to be a major zoonotic threat to humans and a common cause of animal disease, especially in developing countries. In many industrialized countries, a testand-slaughter policy has been effective for the eradication of the disease, while vaccines, although providing a fairly high level of protection, also induce antibodies that interfere in subsequent surveillance programs. Numerous attempts to produce a protective killed vaccine have so far been disappointing, and the most successful vaccines against brucellosis have been those employing live, attenuated Brucella spp. (158). Of these, Brucella abortus strain 19 (first described in 1930) and Brucella melitensis Rev.1 (first described in 1957) vaccines have been widely used in cattle and in small ruminants, respectively. The S19 and Rev.1 vaccines are, however, far from perfect, as absolute protection is not achieved, allowing for subclinical CLIN. MICROBIOL. REV. carrier animals, and both strains have retained some virulence and may induce abortions with variable frequency. Furthermore, both of these vaccines are infectious for humans (Rev.1 is also resistant to streptomycin), and they will induce antibodies against smooth lipopolysaccharide, making them incompatible with test-and-slaughter procedures in countries with an ongoing eradication program. More recently, a vaccine based on a stable spontaneous rifampin-resistant rough mutant of B. abortus, named RB-51 (157), has replaced S19 in many countries including the United States. RB-51 carries IS711 inserted into the wboA glycosyl transferase gene (188), but experimental data with other wboA mutants indicate that additional unknown defects are carried in this strain (118). Rough strains do not carry smooth lipopolysaccharide, and therefore, vaccination with RB-51 does not induce antibodies that are detectable in routine serological tests. This is an obvious advantage in many cases but may also result in the late diagnosis of accidental human infections, although RB-51 appears to be much less virulent for humans than S19 and Rev.1 vaccines (3). At present, several million animals have been vaccinated with the RB-51 mutant strain, but the protective efficacy in cattle compared to S19 remains controversial (118, 124, 158), and the protection against Brucella suis in pigs (172) and against B. abortus in elk (89) is very limited, if present at all. Rickettsia Vaccines The rickettsiae Ehrlichia, Anaplasma, and Coxiella are all small obligate intracellular pathogens that cause significant animal diseases. With the exception of Coxiella, all are transmitted by arthropod vectors (e.g., ticks, mites, lice, or fleas). Heartwater is the most important tick-borne disease of domestic and wild ruminants in sub-Saharan Africa and the West Indies, which is caused by Ehrlichia ruminantium. The only commercially available vaccination procedure is based on the controlled infection of animals with cryopreserved infected sheep blood, followed by antibiotic treatments with tetracyclines when fever develops. A nonvirulent strain has recently been generated through in vitro cultivation and shown to confer good protection (198). Progress in developing cost-effective in vitro cultivation processes may lead to the development of inactivated vaccines (103). Bovine anaplasmosis is another tick-borne disease caused by Anaplasma marginale infection of red blood cells. Transmission can occur by mechanical means via blood contamination or through blood-sucking arthropods and transplacentally from cow to calf. Cattle that survive acute infection are resistant to the disease but develop persistent, cyclic, low-level infections and therefore remain as “carriers.” Calves are less susceptible to infection and clinical disease than adult cattle. Infected blood containing a less pathogenic isolate or subspecies of A. marginale, generally referred to as Anaplasma centrale, remains the most widely used live vaccine in Africa, Australia, Israel, and Latin America. Infection with A. marginale followed by treatment of the patent infections with low doses of tetracycline drugs has also been used but requires close supervision for timely treatment. Following large-scale production of A. marginale antigen from infected bovine blood, a killed vaccine was effectively marketed and used in the United States until withdrawal in 1999 (88). Apart from its higher cost and need VOL. 20, 2007 VETERINARY VACCINES 499 TABLE 3. Available veterinary live protozoal vaccines Host Brand name(s) Distributor(s) Characteristic(s) Referencea Eimeria spp. Poultry Coccivac, Immucox, Paracox, Advent, Nobilis Cox ATM Sporulated oocysts of several or all of the avian species 164 Eimeria spp. Poultry Inovocox Shering-Plough, Vetech Labs, Novus International, Intervet Embrex 164 E. tenella Poultry Livacox BIOPHARM Theileria parva Cattle Theileria annulata and T. hirci Toxoplasma gondii Cattle In ovo delivery using proprietary platform injection system Precocious and egg-passaged lines Infection followed by drug treatment Culture-derived schizonts 30 Babesia bovis and B. bigemina Cattle S48 strain with a lost ability to form cysts after passages in mice Infected blood from splenectomized calves Pathogen(s) a Sheep Centre for Ticks and Tickborne Disease, Malawi Local veterinary institutes Ovilis Toxovax Intervet Local veterinary institutes 196 23 165 48 See also company websites. for yearly boosters, the killed vaccine was generally less effective in inducing protective immunity than live vaccines. VETERINARY PARASITE VACCINES Protozoal Vaccines Protozoal infections in animals cause significant production losses and are a major impediment to the introduction of high-productivity breeds in poorer, mainly tropical areas around the world. Many also cause zoonotic diseases in humans or have close relationships to human parasites, increasing their significance as infection reservoirs or animal models for human diseases. While no vaccines for human protozoa are available as yet, several veterinary vaccines have been on the market or have been produced by agriculture/veterinary departments for local use for many decades. Most of these vaccines are based on live organisms; however, an increasing number of killed subunit vaccines have been developed and commercialized in recent years. The following overview will exemplify currently used protozoal vaccines according to in- creasing sophistication of vaccine production. A list of currently used protozoal vaccines is provided in Tables 3 and 4. Live protozoal parasite vaccines. Protozoal parasites have a high degree of genetic complexity. The difficulty of vaccine development for these organisms is further exacerbated by the antigenic diversity displayed by their different life cycle stages within the host as well as between different species and strains and, in the case of hemoprotozoal parasites, even within the same life cycle stage. While most protozoal infections induce various degrees of immunity after previous infections, the immunological mechanisms involved in protection and the stages involved have mostly not been defined. It is therefore not surprising that most vaccines make use of the live organism itself to elicit the required protective immune response. Depending on the characteristics of infection, these vaccines can take several formats, as discussed below. (i) Vaccines based on complete life cycle infections. Vaccination with low doses of infective organisms has been used extensively in the poultry industry to combat coccidiosis, the major economic parasitic disease of poultry worldwide. Coc- TABLE 4. Available veterinary killed/subunit protozoal vaccines Pathogen(s) Host Brand name(s) Distributor(s) Neospora caninum Giardia duodenalis Cattle Dogs Bovilis, Neoguard Giardiavax Intervet Fort Dodge Sarcocystis neurona Horses Epm vaccine Fort Dodge Babesia canis Dogs Leishmania donovani Dogs Pirodog and Nobivac Piro Leishmune Merial and Intervet (respectively) Fort Dodge Eimeria maxima Poultry CoxAbic Novartis AH a Reference or sourcea Characteristic(s) Killed tachyzoites, reduces abortion Cultured trophozoites, reduces disease and cyst shedding In vitro-cultured merozoites, chemically inactivated In vitro-cultured supernatant antigens, reduce clinical disease Native fucose-mannose-ligand antigen complex Gametocyte antigen(s); transmission blocking through maternal antibody transfer Heuer et al.,b 151 128 104 117, 156 21 192 See also company websites. C. Heuer, C. Nicholson, D. Russell, and J. Weston, presented at the 19th International Conference of the World Association for the Advancement of Veterinary Parasitology, New Orleans, LA, 2003. b 500 MEEUSEN ET AL. cidiosis in poultry is caused by the obligate intracellular protozoal parasite Eimeria species, which undergoes a defined number of asexual cycles of merozoite production in gut epithelial cells (three to four merogenic cycles) before the final sexual stages develop and produce the infective oocysts. The infection is therefore self-limiting, and vaccination with small doses of oocysts, while producing minimal pathology, induces solid protection against homologous challenge. More recently developed live vaccines contain oocysts selected from naturally occurring “precocious” Eimeria strains that produce less merogenic cycles and are therefore safer to use. Although there are many problems with this kind of vaccine, including the need for simultaneous administration to prevent infection of susceptible birds by vaccine-produced oocysts and species- and strain-specific immunity, live coccidian vaccines have been used successfully for over 50 years and are produced as a commercial product by many animal health companies (Table 3) (reviewed in reference 164). The commercial success lies primarily in breeder and layer flocks where anticoccidial drugs have to be withdrawn to prevent the carryover of drugs into eggs. (ii) Vaccines based on drug-abbreviated infections. Hemoprotozoal parasite infections are not self-limiting, and parasites can proliferate continuously in the blood stages if not checked by the immune response or drug treatment. In contrast to most hemoprotozoal pathogens, including the closely related Theileria annulata, Theileria parva causes a highly fatal disease in cattle by transforming infected lymphocytes, while the erythrocyte stage of this parasite is much less pathogenic. Solid, sterile immunity develops after primary infection, and vaccination of cattle by infection with pathogenic wild-type T. parva followed by drug treatment (long-acting tetracyclines) has been used for many years to control East Coast fever. This vaccination regimen confers solid protection against homologous challenge and limited protection against heterologous challenge but is expensive to administer. Resistance is thought to be conferred mainly by cell-mediated immunity, more specifically, CD8⫹ cytotoxic T cells, against the intracellular schizont (106), and the targets of the protective cytotoxic-Tlymphocyte (CTL) response are currently being defined (65). (iii) Vaccines based on infections with parasites with a truncated life cycle. Several protozoal parasites produce cysts within the host that are a persistent source of infection when eaten by carnivores. These cysts can also cause reinfection when the immune system is compromised or can be reactivated during pregnancy, causing congenital disease and abortion. Toxoplasma gondii infects a wide variety of hosts, including humans, and is the major cause of abortion in sheep and goats. As immunity to primary infection develops, the intracellularly replicating tachyzoites become encysted in a dormant stage (zoitocysts), which can persist for several years, containing hundreds of infective bradyzoites. T. gondii parasites that were continuously passaged in mice to produce diagnostic antigens were later found to have lost their ability to form cysts. The “incomplete” S48 strain of T. gondii now forms the basis of a commercial vaccine conferring long-lasting immunity (⬃18 months) of susceptible ewes against Toxoplasma-induced abortion when administered prior to mating (30). (iv) Vaccines based on infection with virulence-attenuated strains. Continuous passage of the tick-borne piroplasms Babesia bovis and Babesia bigemina in splenectomized calves was shown CLIN. MICROBIOL. REV. to result in attenuated infections while still inducing immunity in young calves. Live vaccines using infected blood collected from acute infections of splenectomized calves were developed in Australia several decades ago (31, 41) and are still used in most countries to protect against babesiosis, usually produced by local departments of agriculture or veterinary institutions (reviewed in reference 48). In some cases, this is supplemented with A. centrale-infected blood where A. marginale is enzootic. To increase shelf life and allow more rigorous safety testing, several veterinary institutes now produce frozen-blood vaccines stored in liquid nitrogen using either dimethyl sulfoxide or glycerol as the cryoprotectant. For reasons that are still unknown, young cattle up to 9 months of age are more resistant to Babesia infections, but vaccination of susceptible adult cattle often requires additional drug treatment even using the attenuated strains. Continuous exposure to natural tick infections is generally required to ensure continuous and longlasting immunity. A live, attenuated Theileria annulata vaccine has been produced by continuous in vitro passaging of the intracellular macroschizont stage and is used in many tropical and subtropical countries for the control of tropical theileriosis in cattle. In contrast to T. parva, immunity to the erythrocytic pathogen T. annulata is short-lived and wanes after 6 months in the absence of natural challenge infections (reviewed in references 140 and 165). Killed or subunit protozoal parasite vaccines. Several inactivated vaccines consisting of crude whole organisms or, more recently, defined antigenic structures have been registered and target mostly the companion animal market (Table 4). In general, these vaccines are not as effective as live organisms but can ameliorate disease or transmission to various degrees. They may also form the basis for the development of recombinant vaccines. The final host of the coccidial parasite Neospora caninum is the dog, but its economic impact is felt mostly in the intermediate cattle host, where it is a major cause of abortion (51, 78). A crude N. caninum vaccine has been licensed in the United States to aid in the reduction of N. caninum-induced abortion in healthy pregnant cattle and prevent the transmission of the parasite to calves in utero. The vaccine consists of inactivated N. caninum tachyzoites with an adjuvant administered subcutaneously. A large field study in Costa Rica (151), where infection is highly prevalent in diary herds, demonstrated an overall twofold (46%) reduction in abortion rates through vaccination (49/438 versus 91/438 in saline-injected controls). As also reported in a multiherd vaccination trial in New Zealand (C. Heuer, C. Nicholson, D. Russell, and J. Weston, presented at the 19th International Conference of the World Association for the Advancement of Veterinary Parasitology, New Orleans, LA, 2003), there was a high variability in efficacy between farms, which is likely due to abortions being caused by other infections or by noninfectious causes (87). Timing of vaccination is also likely to play a role in preventing abortion and transmission (78). A vaccine to alleviate a neurological disease in horses caused by infection with Sarcocystis neurona, equine protozoal myeloencephalitis, has recently been released and is being tested under conditional USDA license by Fort Dodge Animal Health. It consists of in vitro-cultured merozoites, originally VOL. 20, 2007 obtained from the spinal cord of a horse, which are chemically inactivated and mixed with a proprietary adjuvant for intramuscular injection (104). Giardia lamblia (synonyms, Giardia duodenalis and Giardia intestinalis) is an enteric parasite of many animal species. Infection is generally self-limiting, but severe gastrointestinal disease can develop in young and immunocompromised individuals. Its importance is mainly in animal-to-human transmission, and Giardia is a major cause of outbreaks of waterborne infections. Only one commercial vaccine has been licensed for use in dogs and cats in the United States (GiardiaVax). It is licensed to prevent clinical disease in dogs and significantly reduce the incidence, severity, and duration of cyst shedding. The vaccine consists of a crude preparation of disrupted, axenically cultured G. duodenalis trophozoites (sheep isolate) and has been shown to eliminate most clinical signs of infection and significantly reduce the total number of cysts shed in the feces in puppies and, to a lesser extent, in kittens. Some efficacy in the clearing of chronic infections resistant to chemotherapeutic agents may also be achieved through vaccination, but this requires more extensive testing (128, 129). It is thought that the vaccine acts mainly through the neutralization of parasite toxins by antibodies. Two subunit vaccines have been developed to protect dogs against canine babesiosis caused by Babesia canis (Table 4). Both vaccines consist of soluble parasite antigens (SPA) released into the culture supernatant by in vitro-cultured parasites, combined with adjuvant. The first vaccine released, Pirodog, contains SPA from B. canis cultures only (117), while the recently released NobivacPiro contains SPA from B. canis and Babesia rossi in an attempt to broaden the strain-specific immunity. The protective effect of this vaccine seems to be based on the antibody-dependent neutralization of a soluble parasite substance that causes hypotension and clinical disease, rather than acting through reducing parasitemia per se (156). This vaccine approach was also evaluated in cattle but did not confer sufficient protection (165). A killed subunit vaccine has been developed against coccidiosis in poultry by ABIC Veterinary Products, Israel, particularly for use in the broiler industry (192, 193). Interestingly, this vaccine does not target the merozoite stages, as most live vaccines are thought to do, but rather targets the final sexual, macrogametocyte stages that develop to form the diseasetransmitting oocysts. The principle behind this vaccine strategy is that it will still allow immunity against the asexual stages to be generated by natural infections while reducing oocyst shedding and parasite transmission. Added advantages of this approach are that the laying hens, rather than the chicks, are immunized, transferring protective immunoglobulins into the egg yolk and subsequently the hatchlings. Considering that each hen lays more than 100 eggs in her lifetime, this considerably reduces the number of vaccinations and animal handling. In contrast to the species and strain specificity of live vaccines, this gametocyte vaccine was shown to confer partial protection across the three major Eimeria species. A major disadvantage of the vaccine is that it is expensive to produce, consisting of affinity-purified native gametocyte antigens derived from infected chickens, and is still a fairly complex preparation (15). Three major components of affinity-purified native gametocyte antigens have recently been cloned and VETERINARY VACCINES 501 characterized with a view to identifying the protective components and develop a recombinant vaccine (16). It remains to be seen if the translation of native vaccine to recombinant vaccine will be successful, as this has been a major stumbling block for much of the parasite development area (123). Human visceral leishmaniasis, or kala-azar, is a devastating human disease caused by the intracellular parasite Leishmania chagasi or Leishmania infantum and transmitted through sand flies. Dogs are the principal carriers of the disease and are also clinically affected. Recently, a subunit vaccine against canine visceral leishmaniasis, based on a strongly antigenic surface glycoprotein complex, fucose mannose ligand, or FML antigen, from Leishmania donovani and saponin adjuvant has been developed in Brazil. Vaccine efficacy was reported to be 76 to 80% against both homologous and heterologous challenge with L. chagasi and to last for at least 3.5 years (21, 44). A concomitant reduction in human incidence of the disease was also reported, which is likely due to the transmission-blocking properties of the vaccine (42, 125). The vaccine may also have a therapeutic effect on infected dogs (22). Helminth and Ectoparasite Vaccines Multicellular parasites are the most complex pathogens, with genome sizes that approach those of their hosts. Apart from their genetic complexity, they are also the only pathogens that, due to their physical size, cannot be internalized by phagocytic cells of the immune system or killed by classical cytotoxic T cells (Fig. 1). In fact, the immune system had to develop a whole new mechanism to deal with these parasites, which is generally referred to as the type 2 or allergic-type immune response, typified by the recruitment and activation of potent effector leukocytes, mast cells, and eosinophils (9, 102). There are three different families of helminths or worms, nematodes (roundworms), trematodes (flatworms), and cestodes (tapeworms), that infect both animals and humans. At present, only one worm vaccine is on the market in Europe for the cattle lung nematode Dictyocaulus viviparous (Bovilis Lungworm), consisting of irradiated infective L3 larvae that cannot develop into the adult stage (7). Vaccination with irradiated L3 larvae of the economically important gastrointestinal nematodes has been attempted but was not successful due mainly to their lack of efficacy in inducing immunity in young animals (reviewed in reference 6). The increasing drug resistance of gastrointestinal nematodes has renewed intense interest in developing vaccines for these important veterinary pathogens (reviewed in references 19, 123, and 189). Tapeworms have a larval stage in intermediate hosts that is uniquely susceptible to immune killing after a single infection. Antigens from this early larval stage (oncospheres) were the first to confer protection against a multicellular pathogen as recombinant proteins (82). Commercial or field application of anticestode vaccines is, however, still in progress (reviewed in reference 98). The most important veterinary trematode species are liver flukes (Fasciola hepatica and Fasciola gigantica). Vaccine development against these parasites is hindered by the fact that they do not seem to induce immunity in their natural ruminant hosts, even after repeated infections. Recently, a unique breed of sheep (Indonesian thin tail) was shown to develop immunity 502 MEEUSEN ET AL. against F. gigantica, and further dissection of this protective mechanism may offer new approaches to vaccine development (109). Ectoparasitic arthropods would seem to be the ultimate challenge in vaccine development, as they not only are large and complex but also spend most of their life outside or on the surface of the host. Interestingly, the only recombinant parasite antigen vaccine commercially available is against a tick parasite, Boophilus microplus, and was first introduced commercially in Australia in 1994 (TickGUARD; Fort Dodge Australia) and later in Cuba and a few South American countries (Gavac; Heber Biotec SA, Cuba). This vaccine is unique in that it is not based on natural antigens recognized by the immune system during infection but takes advantage of the ferocious blood-feeding habits of the tick. High antibody levels are generated by vaccinating cattle against a tick gut membrane-bound protein, Bm86, using a recombinant protein in a potent adjuvant. These antibodies bind to the tick’s gut surface when taking a blood meal, causing the rupture of gut wall and tick death. The vaccine induces significant levels of protection against tick infestation and, in some cases, against tick-borne diseases (195). However, as the molecule is not seen during natural infection (“hidden” or “concealed” antigen), antibody levels are not boosted by infection and need to be maintained at high levels by repeated immunization. The vaccine is best used in conjunction with drug administration, which limits its practical and commercial appeal. The presence of a tick immunoglobulin excretion system seems to hamper the effectiveness of this vaccine approach in other ticks (126). VETERINARY VACCINES FOR NONINFECTIOUS DISEASES Allergy Vaccines As is the case in humans, there is a genetic predisposition in some animals, especially cats, dogs, and horses, to develop allergic skin disease or atopic dermatitis in response to environmental allergens such as grass pollen, weeds, mold spores, and house dust mites. This can be exacerbated by secondary bacterial or yeast infections resulting in urticaria. The most common treatment against atopic dermatitis is vaccination with an allergen extract to which the animal has been shown to react, as determined by intradermal injection or allergen-specific serum immunoglobulin E assays. This “allergen-specific immunotherapy” (ASIT) consists of administering gradually increasing amounts of the allergen extract, either aqueous or precipitated with alum, over a period of several months, followed by yearly boosters. The reported effectiveness of this treatment varies widely, from 20% to close to 100% in dogs, depending on such factors as study design, parameters used, source of vaccine, and concurrent treatment for secondary infections (37). It is clear that a more rigorous evaluation of ASIT vaccine effectiveness is required, which would be aided greatly by a better understanding of the mechanisms by which ASIT works to reduce allergies. In parallel to human studies, it is likely that this involves the induction of specific regulatory T cells and/or immunoglobulin G antibodies that compete with and mask the allergens (94, 180). The identification of the exact mechanisms and mediators associated with successful CLIN. MICROBIOL. REV. ASIT may provide more reliable correlates of vaccine effectiveness and more rational and standardized vaccination protocols. Cancer Vaccines With longer life spans of domestic pets and higher value placed on the animals by their owners, treatment of spontaneous cancers has become of increasing interest. Canine malignant melanoma (CMM) is the most common oral tumor in dogs. CMM is similar to some malignant melanomas in humans, and despite treatment, most dogs die within a year of diagnosis. Several groups have anticancer vaccines against CMM in phase III clinical trials, and Merial launched a CMM DNA vaccine under conditional license from the USDA in 2006. These experimental vaccines are based primarily on studies for human cancer vaccines and include immunizations with canine tumor cell lines transfected with human granulocytemacrophage colony-stimulating factor (75) or human gp100 (1) or DNA vaccination with human tyrosinase (18). The latter two immunizations with human melanocyte-specific proteins are based on the demonstration that immune tolerance against self-antigens can be broken through cross-reaction between a xenogeneic antigen and a self-antigen. The overall response rate in these studies was estimated to be around 17%, with occasional complete remission in individual dogs and prolongation of survival times. The experimental designs of the studies are, however, limited by small sample sizes, differences in breeds and clinical status, and comparisons to historical, stagematched controls. No clear correlations between immune parameters and the likelihood of tumor control could be established. Local vaccination with bacillus Calmette-Guérin (BCG) has long been used as a therapeutic treatment for superficial cancer of the urinary tract in humans and has been shown to be effective in the treatment of equine sarcoids and, to a lesser extent, bovine ocular squamous cell carcinoma (105, 153). The mode of action of this vaccination regimen is unknown but may involve the upregulation of tumor-specific antigens through local inflammation and activation of the innate immune system (162). VETERINARY VACCINES FOR FERTILITY AND PRODUCTION CONTROL Immunocontraceptive vaccination is a fast-moving area of vaccine research and development in the human and animal health areas, with a number of products for livestock and companion animals recently brought to the market. Since before written history, humans have practiced neutering of animals used for food and transport and to be kept as pets. This has been termed “man’s first attempt at bioengineering” (175). Following the discovery of the reproductive hormone system, attempts were made to control reproduction by immunization against key hormones in both humans and animals. Many early attempts gave encouraging results but were variable due to a lack of knowledge of how to consistently elicit an effective neutralizing immune response against self-proteins. There are two goals of reproductive control vaccines that VOL. 20, 2007 VETERINARY VACCINES 503 responses can be induced with high efficacy and potency, provided that the elements of T-cell help against a foreign antigen, in conjunction with self-epitopes, are presented in combination with an appropriate and strong adjuvant. Vaccines against Reproductive Hormones FIG. 3. The key hormones of the hypothalmic-pituitary-gonadal axis. Tissues are in orange, and hormones are in green. *, hormones and gametes that have been targeted in constructing experimental and commercial vaccines. can be simply categorized as (i) immunocontraception and (ii) immunoneutering. Immunocontraceptive vaccines aim to prevent either fertilization of the oocyte by sperm or implantation of the fertilized egg yet retain sexual behavior patterns and competition in mating; this approach is most suited to the control of feral animal pests and native wildlife. Immunoneutering vaccines aim to prevent all sexual behaviors in both male and female animals as well as controlling fertility; these outcomes are suitable for companion animals, livestock, and, in some instances, feral animal pest control. Design of Reproduction Control Vaccines The hormone cascade involved in reproduction is shown in Fig. 3. T-cell help has been incorporated into vaccine formulations by using whole proteins or defined T-cell helper epitopes as peptides (36, 63, 92); however, all commercialized vaccines to date have been based on carrier protein-peptide conjugates. Commercialized peptide-carrier protein vaccines have used bacterial toxoids, including tetanus and diphtheria toxoids or ovalbumin as carriers, to which peptides of the self-antigens being targeted are conjugated. The main criterion for the selection of a carrier has been to provide strong antibody responses and potency, and the relative effectivenesses of different carriers have been identified (57). Other important factors for the selection of carrier proteins are abundance for large-scale manufacture, cost, and compliance with regulatory requirements. In some instances, the target sequence and carrier have been produced as a recombinant fusion protein and have shown good efficacy in species as diverse as cats and cattle (38, 147). Variable results from studies using anti-luteinizing hormone-releasing hormone (LHRH) vaccines under late-stage commercial development in pigs (52) and horses (173) have shown efficacies as low as 66 to 75%, leading to the view that it remains difficult to consistently stimulate a strong immune response to self-antigens. While this may still be the case for the efficacious induction of anti-self-T-cell responses due to tolerance and apoptosis of autoreactive T cells, recently commercialized vaccines have shown that strong anti-self-antibody The targeting of specific hormones involved in sexual development and function has resulted in the most scientifically and commercially successful approach for the control of reproduction by vaccination. The key hormone targets have been those of the hypothalamic-pituitary-gonad axis (Fig. 3). The best-studied and best-characterized hormone, used as a vaccine target, has been LHRH, also known as gonadotropin-releasing hormone and gonadotropin-releasing factor. LHRH is the key hormone controlling reproductive function and development and is released from the hypothalamus. It is a simple 10-amino-acid peptide that is conserved across all species of mammals, with variants identified in other organisms from lampreys to birds and fish. Immunoneutralization of this pivotal hormone of the pituitary-gonad axis prevents reproductive function, provides contraception in all mammals, and controls estrus behavior in females and sexual and aggressive behaviors in males. Because of its simple structure and central controlling role, LHRH was the target of vaccine research soon after its discovery (36). Since then, there have been many research programs for the development of anti-LHRH vaccines both in academia and commercially; however, the commercial successes have been relatively few, as summarized in Table 5, and will be discussed further here. The first commercial vaccine developed against LHRH was Vaxstrate, comprising a conjugate of ovalbumin and LHRH peptide, presented in an oil emulsion adjuvant (76). It was sold for use as an immunospaying vaccine for extensively grazed female cattle in northern Australia. It was launched in the late 1980s and withdrawn from the market in 1996 due to poor sales, resulting mainly from being highly reactogenic (about 40% of animals with abscesses) and poor efficacy in the field. The two doses required for the administration of Vaxstrate prevented its wider use, as this did not fit well with the single annual mustering of cattle in northern Australia. A more advanced anti-LHRH vaccine, Improvac, was developed for use in entire male pigs to control boar taint (Table 5). The formulation comprises a carrier protein conjugated to a modified form of LHRH peptide with a water-soluble adjuvant. This vaccine was launched in 1998 and has been sold since then in Australia and New Zealand and was recently launched in the Philippines, Mexico, Brazil, and South Africa. To date, it is the most successful of all the reproduction control vaccines. Improvac is given as two doses, with the first dose at least 8 weeks prior to slaughter and the second dose 4 weeks before slaughter, which is sufficient to induce an anamnestic anti-LHRH antibody response that in turn suppresses LHRH production, levels of gonadotrophins, and testicular function and allows the washout of the taint steroid androstenone and other taint compounds such as skatole, which are fat soluble. The main feature that distinguishes Improvac from the many noncommercialized vaccines is that it achieves a very high level of efficacy in the field (53). Another key feature that has ensured commercial success is that there is no impact on growth 504 MEEUSEN ET AL. CLIN. MICROBIOL. REV. TABLE 5. Commercialized reproduction control vaccines Target antigen Target species Brand name Distributor Characteristic(s) LHRH Female cattle Vaxstrate LHRH Male pigs Improvac Websters Animal Health (withdrawn in 1996) CSL Ltd. (now Pfizer Animal Health) CSL Ltd. (now Pfizer Animal Health) LHRH Female horses Equity LHRH Male dogs Pfizer Animal Health LHRH Deer, bison, horses Canine gonadotropinreleasing factor immunotherapeutic GonaCon ZP (ZPC/ZP3) antigen Several species SpayVac Androstenedione Ewes Fecundin Androstenedione Androstenedione Ewes Ewes Androvax Ovastim ImmunoVaccine Technologies, Canada (no longer available) Coopers Animal Health (withdrawn) Agvax Pty. Ltd. (now Intervet) Virbac USDA (registration reported to be in progress) rates and more efficient use of feed over the last 4 weeks after the second dose. This is despite highly suppressed levels of the anabolic hormone testosterone for this period (53). The production advantages are probably a result of behavior modification resulting from the suppression of testosterone and have been shown to be significant compared to raising boars and are more pronounced than those of castrated boars, such as those that are raised in most pig-producing countries. An anti-LHRH vaccine, Equity, has also been developed for use in female horses (Table 5). This product is used for the control of estrus and estrus-related behavior during the breeding season and was commercialized in 2001 in Australia. It comprises a peptide-protein conjugate and the Iscom-related immunostimulating complex adjuvant. This formulation is an advance over other commercial formulations trialed in mares and stallions that were reactogenic (50, 178). An anti-LHRH vaccine was also conditionally licensed in the United States in 2004 for the treatment of benign prostatic hyperplasia in entire male dogs and together with Improvac are the only anti-LHRH vaccines commercialized outside of Australia and New Zealand; the dog vaccine is labeled canine gonadotropin-releasing factor immunotherapeutic under a USDA conditional license. Benign prostatic hyperplasia is very common in entire male dogs over 4 to 5 years of age, and the condition is dependent on the conversion of testosterone to dihydrotestosterone by cells in the prostate gland. Suppression of testosterone via an anti-LHRH response leads to a reduction in dihydrotestosterone and indirectly controls prostatic hyperplasia. A related application for anti-LHRH vaccines in men is for treatment of prostatic cancer, and a number of phase I/II studies have shown some degree of efficacy in men with this tumor (168). Control of wildlife through an anti-LHRH formulation has been pursued by researchers at the National Wildlife Research Center of the USDA (86, 112). This vaccine, termed GonaCon, is based on a peptide-keyhole limpet hemocyanin carrier protein conjugate antigen formulated in a commercially available vaccine for Johne’s disease in an oil-based adjuvant (AdjuVac). This formulation has the effect of making the skin of LHRH peptide conjugated to ovalbumin; oil emulsion adjuvant Peptide-protein conjugate and water-miscible adjuvant; control of boar taint Peptide-protein conjugate and proprietary adjuvant (immunostimulating complex); control of estrus and estrus-related behavior Peptide-protein conjugate and water-miscible adjuvant; treatment of benign prostatic hyperplasia Peptide conjugated to keyhole limpet hemocyanin in mycobacterium-oil adjuvant (AdjuVac); contraceptive vaccine to control wildlife Crude ZPA preparation; supplied to research groups only Reference or source 76 53 54 USDA 112 27 Linked to human serum albumin; increased ovulation/twinning Increased ovulation/twinning Increased ovulation/twinning vaccinated animals test positive for Mycobacterium avium. Its advantage is that it is effective with a single vaccination, and efficacy has been shown in valued wildlife such as deer, bison, and horses (112) and in controlling feral pigs (86). Registration of this formulation is reportedly being undertaken by the Wildlife Research Group of the USDA through the EPA, as this agency is able to register products that would be restricted to use in wildlife. Vaccines against Gamete Antigens: Wildlife Control For the control of wildlife, the widely held view is that the maintenance of libido and sexual behavior would be optimal to achieve this goal, and hence, the hormone system that drives those behaviors has not been generally targeted. The exception to this is the control of native animal species by an anti-LHRH vaccine (see above). Alternate strategies to control wildlife have been to develop vaccines that prevent the fertilization of the oocyte by sperm or to prevent the implantation of the embryo and allow immunized animals to continue to compete in mating rituals. With this approach, antigens of the gametes (sperm and oocytes) have been widely targeted to prevent fertilization. Sperm antigens. Over 20 sperm antigens have been identified and characterized, and many have been tested as vaccine candidates in animals. Most of these are surface proteins and include sperm antigen 10 (SP10), SP17, FA-1, LDH-C4, and PH-20 (47). While some effect could be expected in vaccinated male animals, the large number of sperm in the male reproductive tract and observed autoimmune-mediated orchitis have focused efforts instead on vaccinating the female. Fertility levels in vaccinated females are generally reduced from levels around 75 to 80% to 25 to 30% in a range of species including mice (95), baboons (127, 171), and guinea pigs (179). The potency of the range of antigens is similar, and no one sperm antigen gives an exceptional contraceptive effect. Oocyte antigens. A family of surface antigens from the zona pellucida (ZP) has been identified as providing effective immunocontraception. These surface antigens have been re- VOL. 20, 2007 VETERINARY VACCINES ferred to by a variety of terms that may vary between species. The major antigens are ZPA (also termed ZP2), ZPB (ZP1), and ZPC (ZP3). Most work has focused on ZPC, with a range of approaches. Vaccine studies have used formulations containing porcine ZPC, as it cross-reacts with the ZPC of many other species. For a period between approximately 2002 and 2005, a vaccine called SpayVac was commercially available (Table 5), which was based on a crude porcine ZP antigen preparation, probably purified from pig ovaries. This was shown to have efficacy in a number of species (27). SpayVac was supplied to researchers for experimental wildlife population control and should not be considered to be a major commercialized vaccine product. Many of the experimental ZP-based vaccines have induced reasonably high levels of efficacy sufficient to engender strong interest in further development and commercialization, with ZP antigens alone or in combination with sperm antigens to increase efficacy. Such combination vaccines as recombinant fusion antigens have been tested in a range of species to good effect (95). Despite considerable scientific advances, there has been only very limited commercial success of gamete antigen-based vaccines. This approach has some major difficulties for wild or feral animal populations, including developing a delivery system to mass vaccinate wild animal populations without capture or restraint and being capable of delivering a booster dose, i.e., allow revaccination; ensuring the specificity of the delivery system to ensure vaccination of the target species only and to prevent unintentional vaccination and downregulation of fertility in bystander and native animal species; ensuring reasonable duration of efficacy (the duration required would be related to the frequency and effectiveness of boost vaccinations); and being able to induce and maintain high levels of antibody in the female reproductive tract. Currently, these issues remain unresolved, and it is unlikely that fertility control vaccines will be used in wildlife management programs or commercialized until such technical hurdles are overcome. For application of gamete antigen vaccines in humans, the safety of the formulation would need to be demonstrated. Many constructs of ZP-based vaccines resulted in inflammation and immunopathology of the ovaries. The use of sperm antigens in the female would be less likely to induce safety problems. Vaccines To Increase Fertility There have been three fecundity vaccines for sheep that have been commercialized, all based on stimulating an immune response to the steroid androstenedione. Vaccination of ewes against this intermediate steroid leads to a reduction in estrogen levels, and estrogen (-estradiol) has a negative-feedback effect on the production of follicle-stimulating hormone. Thus, immunoneutralization of androstenedione leads to the increased production of follicle-stimulating hormone, and this has the effect of increasing the frequency of multiple ovulations. The immunogen in the first available vaccine, Fecundin, was polyandroalbumin that contained androstenedione linked to human serum albumin. Similar vaccines, Androvax and Ovastim, are now marketed in New Zealand and Australia, 505 respectively. Vaccination is carried out 5 and 2 weeks before mating for 6 to 8 weeks; in subsequent years, a booster dose is given to the flock 2 weeks before mating. The claimed increase in twinning is about 20 to 25% across a flock of ewes. The actual increased yield of lambs achieved with Fecundin was variable, and the vaccine was withdrawn from market. For these vaccines that increase fecundity, the correct nutritional maintenance of ewes with twins is not always easily managed, and vaccination will not correct underlying fertility problems. CONCLUSION AND FUTURE DIRECTIONS Vaccinology has become a recognized science that combines disciplines of immunology, microbiology, protein chemistry, and molecular biology with practical considerations of production costs, regulatory affairs, and commercial returns. The ultimate aim of any new vaccine is to provide a product that will be used to protect animals and humans against disease. More recently, vaccines have also found applications in animal production and reproduction processes. Veterinary vaccines have already made enormous impacts not only on animal health, welfare, and production but also on human health. A continuous interchange between animal and human disease control agencies and scientists will be essential to be prepared for the ever-present threat of new, emerging diseases (83). This is exemplified most recently with the advent of avian influenza virus, where poultry and wildfowl are identified as the major carriers of the disease, but recent data have shown that both wild and domestic cats can also become infected and may present a source of disease for humans (91). Pigs are susceptible to both avian and human influenza viruses, and it is speculated that coinfection of pigs with highly pathogenic avian influenza virus and human influenza virus may create viral reassortant strains with the ability for human-to-human transmission (40). Increasing animal travel and wildlife-human interactions promoted by global climate changes will also require sustained surveillance for the spread of diseases in different parts of the world, with both domestic, production, and wild animals forming important reservoirs of many vectorborne human diseases; e.g., the emergence of WNV in the United States and Europe requires continuous surveillance and control programs for the presence of the virus in birds and horses as well as humans (72). A novel addition to veterinary vaccines for human disease is a cattle vaccine against Escherichia coli O157:H7 that recently received a conditional license for distribution from the Canadian Food Inspection Agency. E. coli O157:H7 is a leading cause of food-borne disease in humans worldwide, and ruminant livestock are considered to be its major reservoir. As highlighted in this review, much progress has been made in expanding the range of veterinary vaccines available as well as increasing efficacy and reducing side effects of existing vaccines. Many problems remain to be resolved, and there is ample scope to incorporate new knowledge and technologies into vaccine design. In particular, most vaccines are still based on live, attenuated pathogen strains. Apart from the obvious dangers involved with this type of immunization, this approach is not generally desirable for commercial companies, as it exposes them to risks of mitigation, and the short shelf life and strain/region specificity of many vaccines make them uneco- 506 MEEUSEN ET AL. nomical to produce. While several variably defined subunit vaccines are available on the veterinary market, they are generally much less protective than live organisms. A better understanding of the molecular and immunological disease processes is likely to be required to improve the effectiveness of killed or subunit vaccines. In particular, while it is well established that the immune system has several effector mechanisms to deal with different pathogens depending on their individual life cycles and microenvironments (Fig. 1), most killed and subunit vaccines still rely predominantly on the induction of neutralizing antibodies (93). An increased ability to target pathogens at different stages of their life cycle is likely to open up new avenues for antigen discovery and increase the effectiveness of killed or subunit vaccines. One way that this may be achieved is through novel delivery systems such as plasmid DNA, liposomes, nano- or microparticles, and live vectors that introduce the vaccine antigens into the intracellular compartment (reviewed in references 4, 25, 61, and 154). Another major advance in immunology that will have an impact on an often neglected part of vaccine development is the increased awareness of the central role that innate immunity plays in the action of vaccine adjuvants (144). The recently discovered innate immune receptors are currently being screened for active novel adjuvant compounds (134), and their corresponding ligands (pathogen-associated molecular patterns) are being used to increase or modulate vaccine responses (77, 85, 169). The use of adjuvants in veterinary vaccinology is much less restricted than that in human vaccines, and a large number of different types and formulations of adjuvants are currently used in licensed veterinary vaccines, compared to only three adjuvants licensed for human vaccine use (134). In many cases, the details of the veterinary adjuvants are unfortunately withheld as proprietary information, but hopefully, this area will be reviewed in the near future. Apart from the scientific challenges that are being addressed, the development of a commercially successful veterinary vaccine also needs to meet the regulatory hurdles that pave the route to the marketplace. For example, under current U.S. law, vaccines that target noninfectious disease (e.g., production gains and reproduction) come under the more stringent jurisdiction of the FDA and are treated as pharmaceuticals, whereas most animal vaccines come under the USDA, with more rapid and lower-cost routes to registration. In the European Union, regulatory matters are based on European Union legislation, and company dossiers are assessed and legalized by the European Medicines Evaluation Agency. In principle, three different procedures can be used to register a vaccine. During the centralized procedure, new and innovative vaccines are assessed and legalized in all member states in one procedure. During the mutual recognition procedure, the company selects a single reference country to evaluate the vaccine dossier, which is followed by an application in the relevant countries to have the vaccine registered afterwards. The third possibility is to apply for the recently introduced decentralized procedure, which can be chosen when a more expedient registration is desired. In this case, the vaccine dossier is reviewed by all selected countries at the same time to save the first step in the mutual recognition procedure. The Veterinary International Committee for Harmonization brings together the regulatory authorities of the European Union, Japan, and the CLIN. MICROBIOL. REV. United States and representatives from the animal health industry in the three regions to harmonize technical requirements for the registration of veterinary products. The Veterinary International Committee for Harmonization harmonizes guidelines that represent scientific consensus regarding regulatory requirements for the three regions. Expert working groups, under the supervision of the Steering Committee, are created to draft and recommend the harmonized guidelines. Research and development form the basis for the generation of new and improved veterinary vaccines. Animal scientists can borrow heavily from medical research, particularly in the areas of welfare and geriatric medicine for companion animals, which are becoming increasingly lucrative markets for animal health companies (166). On the other hand, animal research scientists can also significantly contribute to human vaccine development, as they are able to bridge the gap between results obtained in small-rodent models, which are often not directly translatable to humans. Due to their similar sizes and anatomies, large-animal models are particularly useful for the testing of different delivery systems (160, 197) and have been extensively used to optimize the uptake of plasmid DNA for effective DNA vaccination (85, 155, 183). New animal health vaccines are also likely to be therapeutic rather than prophylactic, with cancer and osteoarthritis in longer-lived companion animals being obvious targets. Expected reductions in the cost of recombinant antibodies should make the passive immunotherapy of dogs and cats feasible. Due to their less stringent regulatory requirements and quicker route to the market, veterinary vaccines are also at the forefront of the testing and commercialization of innovative technologies, as exemplified by the recent successful licensing of two DNA vaccines for horses and fish and the conditional license of a DNA vaccine against CMM. ACKNOWLEDGMENTS We thank all the companies that provided up-to-date information for their products and the scientific reviewers of the manuscript for their useful comments and corrections. REFERENCES 1. Alexander, A. N., M. K. Huelsmeyer, A. Mitzey, R. R. Dubielzig, I. D. Kurzman, E. G. Macewen, and D. M. Vail. 2006. Development of an allogeneic whole-cell tumor vaccine expressing xenogeneic gp100 and its implementation in a phase II clinical trial in canine patients with malignant melanoma. Cancer Immunol. Immunother. 55:433–442. 2. Allan, G. M., F. McNeilly, J. Ellis, S. Krakowka, A. Botner, K. McCullough, H. Nauwynck, S. Kennedy, B. Meehan, and C. Charreyre. 2004. PMWS: experimental model and co-infections. Vet. Microbiol. 98:165–168. 3. Ashford, D. A., J. di Pietra, J. Lingappa, C. Woods, H. Noll, B. Neville, R. Weyant, S. L. Bragg, R. A. Spiegel, J. Tappero, and B. A. Perkins. 2004. Adverse events in humans associated with accidental exposure to the livestock brucellosis vaccine RB51. Vaccine 22:3435–3439. 4. Azad, N., and Y. Rojanasakul. 2006. Vaccine delivery—current trends and future. Curr. Drug Deliv. 3:137–146. 5. Babu, U., R. A. Dalloul, M. Okamura, H. S. Lillehoj, H. Xie, R. B. Raybourne, D. Gaines, and R. A. Heckert. 2004. Salmonella enteritidis clearance and immune responses in chickens following Salmonella vaccination and challenge. Vet. Immunol. Immunopathol. 101:251–257. 6. Bain, R. K. 1999. Irradiated vaccines for helminth control in livestock. Int. J. Parasitol. 29:185–191. 7. Bain, R. K., and G. M. Urquhart. 1988. Parenteral vaccination of calves against the cattle lungworm Dictyocaulus viviparus. Res. Vet. Sci. 45:270– 271. 8. Balamurugan, V., R. M. Kumar, and V. V. Suryanarayana. 2004. Past and present vaccine development strategies for the control of foot-and-mouth disease. Acta Virol. 48:201–214. 9. Balic, A., V. M. Bowles, and E. N. Meeusen. 2000. The immunobiology of VOL. 20, 2007 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. gastrointestinal nematode infections in ruminants. Adv. Parasitol. 45:181– 241. Barbour, E. K., S. K. Hamadeh, and A. Eidt. 2000. Infection and immunity in broiler chicken breeders vaccinated with a temperature-sensitive mutant of Mycoplasma gallisepticum and impact on performance of offspring. Poult. Sci. 79:1730–1735. Baron, M. D., A. C. Banyard, S. Parida, and T. Barrett. 2005. The Plowright vaccine strain of rinderpest virus has attenuating mutations in most genes. J. Gen. Virol. 86:1093–1101. Barrett, T., P.-P. Pastoret, and W. Taylor. 2006. Rinderpest and peste des petits ruminants, virus plagues of large and small ruminants. Elsevier, London, United Kingdom. Beer, M., I. Reimann, B. Hoffmann, and K. Depner. 4 January 2007, posting date. Novel marker vaccines against classical swine fever. Vaccine doi: 10.1016/j.vaccine.2006.12.036. Belknap, E. B., L. M. Walters, C. Kelling, V. K. Ayers, J. Norris, J. McMillen, C. Hayhow, M. Cochran, D. N. Reddy, J. Wright, and J. K. Collins. 1999. Immunogenicity and protective efficacy of a gE, gG and US2 gene-deleted bovine herpesvirus-1 (BHV-1) vaccine. Vaccine 17:2297–2305. Belli, S. I., M. Lee, P. Thebo, M. G. Wallach, B. Schwartsburd, and N. C. Smith. 2002. Biochemical characterisation of the 56 and 82 kDa immunodominant gametocyte antigens from Eimeria maxima. Int. J. Parasitol. 32:805–816. Belli, S. I., K. Mai, C. D. Skene, M. T. Gleeson, D. M. Witcombe, M. Katrib, A. Finger, M. G. Wallach, and N. C. Smith. 2004. Characterisation of the antigenic and immunogenic properties of bacterially expressed, sexual stage antigens of the coccidian parasite, Eimeria maxima. Vaccine 22:4316–4325. Bergman, J. G., M. Muniz, D. Sutton, R. Fensome, F. Ling, and G. Paul. 2006. Comparative trial of the canine parvovirus, canine distemper virus and canine adenovirus type 2 fractions of two commercially available modified live vaccines. Vet. Rec. 159:733–736. Bergman, P. J., J. McKnight, A. Novosad, S. Charney, J. Farrelly, D. Craft, M. Wulderk, Y. Jeffers, M. Sadelain, A. E. Hohenhaus, N. Segal, P. Gregor, M. Engelhorn, I. Riviere, A. N. Houghton, and J. D. Wolchok. 2003. Longterm survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin. Cancer Res. 9:1284–1290. Bethony, J. M., A. Loukas, P. J. Hotez, and D. P. Knox. 2006. Vaccines against blood-feeding nematodes of humans and livestock. Parasitology 133:S63–S79. Blanchard, P., D. Mahe, R. Cariolet, A. Keranflec’h, M. A. Baudouard, P. Cordioli, E. Albina, and A. Jestin. 2003. Protection of swine against postweaning multisystemic wasting syndrome (PMWS) by porcine circovirus type 2 (PCV2) proteins. Vaccine 21:4565–4575. Borja-Cabrera, G. P., N. N. Correia Pontes, V. O. da Silva, E. Paraguai de Souza, W. R. Santos, E. M. Gomes, K. G. Luz, M. Palatnik, and C. B. Palatnik de Sousa. 2002. Long lasting protection against canine kala-azar using the FML-QuilA saponin vaccine in an endemic area of Brazil (Sao Goncalo do Amarante, RN). Vaccine 20:3277–3284. Borja-Cabrera, G. P., A. Cruz Mendes, E. Paraguai de Souza, L. Y. H. Okada, F. A. de A Trivellato, J. K. Kawasaki, A. C. Costa, A. B. Reis, O. Genaro, L. M. Batista, M. Palatnik, and C. B. Palatnik-de-Sousa. 2004. Effective immunotherapy against canine visceral leishmaniasis with the FML-vaccine. Vaccine 22:2234–2243. Boulter, N., and R. Hall. 1999. Immunity and vaccine development in the bovine theilerioses. Adv. Parasitol. 44:41–97. Bouma, A. 2005. Determination of the effectiveness of Pseudorabies marker vaccines in experiments and field trials. Biologicals 33:241–245. Bramwell, V. W., and Y. Perrie. 2006. Particulate delivery systems for vaccines: what can we expect? J. Pharm. Pharmacol. 58:717–728. Brochier, B., M. P. Kieny, F. Costy, P. Coppens, B. Bauduin, J. P. Lecocq, B. Languet, G. Chappuis, P. Desmettre, K. Afiademanyo, et al. 1991. Largescale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature 354:520–522. Brown, R. G., W. D. Bowen, J. D. Eddington, W. C. Kimmins, M. Mezei, J. L. Parsons, and B. Pohajdak. 1997. Temporal trends in antibody production in captive grey, harp and hooded seals to a single administration immunocontraceptive vaccine. J. Reprod. Immunol. 35:53–64. Bryans, J. T., E. R. Doll, J. C. Wilson, and W. H. McCollum. 1966. Immunization for equine influenza. J. Am. Vet. Med. Assoc. 148:413–417. Bublot, M., N. Pritchard, D. E. Swayne, P. Selleck, K. Karaca, D. L. Suarez, J. C. Audonnet, and T. R. Mickle. 2006. Development and use of fowlpox vectored vaccines for avian influenza. Ann. N. Y. Acad. Sci. 1081:193–201. Buxton, D., and E. A. Innes. 1995. A commercial vaccine for ovine toxoplasmosis. Parasitology 110:S11–S16. Callow, L. L., R. J. Dalgliesh, and A. J. de Vos. 1997. Development of effective living vaccines against bovine babesiosis—the longest field trial? Int. J. Parasitol. 27:747–767. Reference deleted. Carter, P. B. 2003. The current state of veterinary vaccines: is there hope for the future? J. Vet. Med. Ed. 30:152–154. Chalmers, W. S., J. Simpson, S. J. Lee, and W. Baxendale. 1997. Use of a VETERINARY VACCINES 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 507 live chlamydial vaccine to prevent ovine enzootic abortion. Vet. Rec. 141: 63–67. Chiers, K., I. van Overbeke, P. De Laender, R. Ducatelle, S. Carel, and F. Haesebrouck. 1998. Effects of endobronchial challenge with Actinobacillus pleuropneumoniae serotype 9 of pigs vaccinated with inactivated vaccines containing the Apx toxins. Vet. Q. 20:65–69. Clarke, I. J., H. M. Fraser, and A. S. McNeilly. 1978. Active immunization of ewes against luteinizing hormone releasing hormone, and its effects on ovulation and gonadotrophin, prolactin and ovarian steroid secretion. J. Endocrinol. 78:39–47. Colombo, S., P. B. Hill, D. J. Shaw, and K. L. Thoday. 2005. Effectiveness of low dose immunotherapy in the treatment of canine atopic dermatitis: a prospective, double-blinded, clinical study. Vet. Dermatol. 16:162–170. Cook, R. B., J. D. Popp, J. P. Kastelic, S. Robbins, and R. Harland. 2000. The effects of active immunization against gnRH on testicular development, feedlot performance, and carcass characteristics of beef bulls. J. Anim. Sci. 78:2778–2783. Coquet, L., P. Cosette, L. Quillet, F. Petit, G. A. Junter, and T. Jouenne. 2002. Occurrence and phenotypic characterization of Yersinia ruckeri strains with biofilm-forming capacity in a rainbow trout farm. Appl. Environ. Microbiol. 68:470–475. Cyranoski, D. 2005. Bird flu spreads among Java’s pigs. Nature 435:390– 391. Dalgliesh, R. J., L. L. Callow, L. T. Mellors, and W. McGregor. 1981. Development of a highly infective Babesia bigemina vaccine of reduced virulence. Aust. Vet. J. 57:8–11. Dantas-Torres, F. 2006. Leishmune vaccine: the newest tool for prevention and control of canine visceral leishmaniosis and its potential as a transmission-blocking vaccine. Vet. Parasitol. 141:1–8. Darteil, R., M. Bublot, E. Laplace, J. F. Bouquet, J. C. Audonnet, and M. Riviere. 1995. Herpesvirus of turkey recombinant viruses expressing infectious bursal disease virus (IBDV) VP2 immunogen induce protection against an IBDV virulent challenge in chickens. Virology 211:481–490. da Silva, V. O., G. P. Borja-Cabrera, N. N. Correia Pontes, E. P. de Souza, K. G. Luz, M. Palatnik, and C. B. Palatnik de Sousa. 2000. A phase III trial of efficacy of the FML-vaccine against canine kala-azar in an endemic area of Brazil (Sao Goncalo do Amaranto, RN). Vaccine 19:1082–1092. Davis, B. S., G. J. Chang, B. Cropp, J. T. Roehrig, D. A. Martin, C. J. Mitchell, R. Bowen, and M. L. Bunning. 2001. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzymelinked immunosorbent assays. J. Virol. 75:4040–4047. Day, M. J. 2006. Vaccine side effects: fact and fiction. Vet. Microbiol. 117:51–58. Delves, P. J., T. Lund, and I. M. Roitt. 2002. Antifertility vaccines. Trends Immunol. 23:213–219. de Waal, D. T., and M. P. Combrink. 2006. Live vaccines against bovine babesiosis. Vet. Parasitol. 138:88–96. Doel, T. R. 2003. FMD vaccines. Virus Res. 91:81–99. Dowsett, K. F., W. A. Pattie, L. M. Knott, A. E. Jackson, R. M. Hoskinson, R. P. Rigby, and B. A. Moss. 1991. A preliminary study of immunological castration in colts. J. Reprod. Fertil. Suppl. 44:183–190. Dubey, J. P., and D. S. Lindsay. 1996. A review of Neospora caninum and neosporosis. Vet. Parasitol. 67:1–59. Dufour, R., C. Roulet, C. Chouvet, and M. B. Bonneau. December 1996. Method for improving the organoleptic qualities of the meat from uncastrated male domestic animals, vaccines which are usable in this method, new peptide, in particular, for producing these vaccines and vaccination kit relating thereto. U.S. patent 5573767. Dunshea, F. R., C. Colantoni, K. Howard, I. McCauley, P. Jackson, K. A. Long, S. Lopaticki, E. A. Nugent, J. A. Simons, J. Walker, and D. P. Hennessy. 2001. Vaccination of boars with a GnRH vaccine (Improvac) eliminates boar taint and increases growth performance. J. Anim. Sci. 79:2524–2535. Elhay, M., A. Newbold, A. Britton, P. Turley, K. Dowsett, and J. Walker. 2007. Suppression of behavioural and physiological oestrus in the mare by vaccination against GnRH. Aust. Vet. J. 85:35–45. Fenaux, M., T. Opriessnig, P. G. Halbur, F. Elvinger, and X. J. Meng. 2004. A chimeric porcine circovirus (PCV) with the immunogenic capsid gene of the pathogenic PCV type 2 (PCV2) cloned into the genomic backbone of the nonpathogenic PCV1 induces protective immunity against PCV2 infection in pigs. J. Virol. 78:6297–6303. Ferrari, M., A. Brack, M. G. Romanelli, T. C. Mettenleiter, A. Corradi, N. Dal Mas, M. N. Losio, R. Silini, C. Pinoni, and A. Pratelli. 2000. A study of the ability of a TK-negative and gI/gE-negative pseudorabies virus (PRV) mutant inoculated by different routes to protect pigs against PRV infection. J. Vet. Med. B Infect. Dis. Vet. Public Health 47:753–762. Ferro, V. A., and W. H. Stimson. 1998. Investigation into suitable carrier molecules for use in an anti-gonadotrophin releasing hormone vaccine. Vaccine 16:1095–1102. Frey, J. 1995. Virulence in Actinobacillus pleuropneumoniae and RTX toxins. Trends Microbiol. 3:257–261. 508 MEEUSEN ET AL. 59. Garcia de la Fuente, J. N., C. B. Gutierrez-Martin, N. Ortega, E. F. Rodriguez-Ferri, M. L. del Rio, O. R. Gonzalez, and J. Salinas. 2004. Efficacy of different commercial and new inactivated vaccines against ovine enzootic abortion. Vet. Microbiol. 100:65–76. 60. Reference deleted. 61. Gerdts, V., G. K. Mutwiri, S. K. Tikoo, and L. A. Babiuk. 2006. Mucosal delivery of vaccines in domestic animals. Vet. Res. 37:487–510. 62. Gerhardt, R. 2006. West Nile virus in the United States (1999–2005). J Am. Anim. Hosp. Assoc. 42:170–177. 63. Ghosh, S., J. Walker, and D. C. Jackson. 2001. Identification of canine helper T-cell epitopes from the fusion protein of canine distemper virus. Immunology 104:58–66. 64. Gould, C. 2003. Successful oral vaccination. Skretting Outlook 20:10–11. 65. Graham, S. P., R. Pelle, Y. Honda, D. M. Mwangi, N. J. Tonukari, M. Yamage, E. J. Glew, E. P. de Villiers, T. Shah, R. Bishop, E. Abuya, E. Awino, J. Gachanja, A. E. Luyai, F. Mbwika, A. M. Muthiani, D. M. Ndegwa, M. Njahira, J. K. Nyanjui, F. O. Onono, J. Osaso, R. M. Saya, C. Wildmann, C. M. Fraser, I. Maudlin, M. J. Gardner, S. P. Morzaria, S. Loosmore, S. C. Gilbert, J. C. Audonnet, P. van der Bruggen, V. Nene, and E. L. Taracha. 2006. Theileria parva candidate vaccine antigens recognized by immune bovine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 103:3286–3291. 66. Grubman, M. J. 2005. Development of novel strategies to control foot-andmouth disease: marker vaccines and antivirals. Biologicals 33:227–234. 67. Guedes, R. M., and C. J. Gebhart. 2003. Onset and duration of fecal shedding, cell-mediated and humoral immune responses in pigs after challenge with a pathogenic isolate or attenuated vaccine strain of Lawsonia intracellularis. Vet. Microbiol. 91:135–145. 68. Reference deleted. 69. Haesebrouck, F., F. Pasmans, K. Chiers, D. Maes, R. Ducatelle, and A. Decostere. 2004. Efficacy of vaccines against bacterial diseases in swine: what can we expect? Vet. Microbiol. 100:255–268. 70. Hardham, J., K. Dreier, J. Wong, C. Sfintescu, and R. T. Evans. 2005. Pigmented-anaerobic bacteria associated with canine periodontitis. Vet. Microbiol. 106:119–128. 71. Hardham, J., M. Reed, J. Wong, K. King, B. Laurinat, C. Sfintescu, and R. T. Evans. 2005. Evaluation of a monovalent companion animal periodontal disease vaccine in an experimental mouse periodontitis model. Vaccine 23:3148–3156. 72. Hayes, E. B., and D. J. Gubler. 2006. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 57:181–194. 73. Heppell, J., and H. L. Davis. 2000. Application of DNA vaccine technology to aquaculture. Adv. Drug Deliv. Rev. 43:29–43. 74. Reference deleted. 75. Hogge, G. S., J. K. Burkholder, J. Culp, M. R. Albertini, R. R. Dubielzig, N. S. Yang, and E. G. MacEwen. 1999. Preclinical development of human granulocyte-macrophage colony-stimulating factor-transfected melanoma cell vaccine using established canine cell lines and normal dogs. Cancer Gene Ther. 6:26–36. 76. Hoskinson, R. M., R. D. Rigby, P. E. Mattner, V. L. Huynh, M. D’Occhio, A. Neish, T. E. Trigg, B. A. Moss, M. J. Lindsey, G. D. Coleman, et al. 1990. Vaxstrate: an anti-reproductive vaccine for cattle. Aust. J. Biotechnol. 4:166–170, 176. 77. Huleatt, J. W., A. R. Jacobs, J. Tang, P. Desai, E. B. Kopp, Y. Huang, L. Song, V. Nakaar, and T. J. Powell. 2007. Vaccination with recombinant fusion proteins incorporating Toll-like receptor ligands induces rapid cellular and humoral immunity. Vaccine 25:763–775. 78. Innes, E. A., S. Wright, P. Bartley, S. Maley, C. Macaldowie, I. EstebanRedondo, and D. Buxton. 2005. The host-parasite relationship in bovine neosporosis. Vet. Immunol. Immunopathol. 108:29–36. 79. Jackwood, M. W., and Y. M. Saif. 1985. Efficacy of a commercial turkey coryza vaccine (Art-Vax) in turkey poults. Avian Dis. 29:1130–1139. 80. Jacobs, A. A., D. Goovaerts, P. J. Nuijten, R. P. Theelen, O. M. Hartford, and T. J. Foster. 2000. Investigations towards an efficacious and safe strangles vaccine: submucosal vaccination with a live attenuated Streptococcus equi. Vet. Rec. 147:563–567. 81. Jarosinski, K. W., B. K. Tischer, S. Trapp, and N. Osterrieder. 2006. Marek’s disease virus: lytic replication, oncogenesis and control. Expert Rev. Vaccines 5:761–772. 82. Johnson, K. S., G. B. Harrison, M. W. Lightowlers, K. L. O’Hoy, W. G. Cougle, R. P. Dempster, S. B. Lawrence, J. G. Vinton, D. D. Heath, and M. D. Rickard. 1989. Vaccination against ovine cysticercosis using a defined recombinant antigen. Nature 338:585–587. 83. Kahn, L. H. 2006. Confronting zoonoses, linking human and veterinary medicine. Emerg. Infect. Dis. 12:556–561. 84. Kelly, C., M. Bugg, C. Robinson, Z. Mitchell, N. Davis-Poynter, J. R. Newton, K. A. Jolley, M. C. Maiden, and A. S. Waller. 2006. Sequence variation of the SeM gene of Streptococcus equi allows discrimination of the source of strangles outbreaks. J. Clin. Microbiol. 44:480–486. 85. Kennedy, N. J., T. W. Spithill, J. Tennent, P. R. Wood, and D. Piedrafita. 2006. DNA vaccines in sheep: CTLA-4 mediated targeting and CpG motifs CLIN. MICROBIOL. REV. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. enhance immunogenicity in a DNA prime/protein boost strategy. Vaccine 24:970–979. Killian, G., L. Miller, J. Rhyan, and H. Doten. 2006. Immunocontraception of Florida feral swine with a single-dose GnRH vaccine. Am. J. Reprod. Immunol. 55:378–384. Kirkbride, C. A. 1993. Diagnoses in 1,784 ovine abortions and stillbirths. J Vet. Diagn. Investig. 5:398–402. Kocan, K. M., J. de la Fuente, A. A. Guglielmone, and R. D. Melendez. 2003. Antigens and alternatives for control of Anaplasma marginale infection in cattle. Clin. Microbiol. Rev. 16:698–712. Kreeger, T. J., W. E. Cook, W. H. Edwards, P. H. Elzer, and S. C. Olsen. 2002. Brucella abortus strain RB51 vaccination in elk. II. Failure of high dosage to prevent abortion. J. Wildl. Dis. 38:27–31. Kroll, J. J., M. B. Roof, and S. McOrist. 2004. Evaluation of protective immunity in pigs following oral administration of an avirulent live vaccine of Lawsonia intracellularis. Am. J. Vet. Res. 65:559–565. Kuiken, T., G. Rimmelzwaan, D. van Riel, G. van Amerongen, M. Baars, R. Fouchier, and A. Osterhaus. 2004. Avian H5N1 influenza in cats. Science 306:241. Ladd, A. E., C. Y. Wang, and T. J. Zamb. January 1998. Immunogenic LHRH peptide constructs and synthetic universal immune stimulators for vaccines. U.S. patent 5843446. Lambert, P. H., M. Liu, and C. A. Siegrist. 2005. Can successful vaccines teach us how to induce efficient protective immune responses? Nat. Med. 11:S54–S62. Larche, M., C. A. Akdis, and R. Valenta. 2006. Immunological mechanisms of allergen-specific immunotherapy. Nat. Rev. Immunol. 6:761–771. Lea, I. A., E. E. Widgren, and M. G. O’Rand. 2002. Analysis of recombinant mouse zona pellucida protein 2 (ZP2) constructs for immunocontraception. Vaccine 20:1515–1523. Lehman, J. R., R. M. Weigel, A. M. Siegel, L. G. Herr, A. C. Taft, and W. F. Hall. 1993. Progress after one year of a pseudorabies eradication program for large swine herds. J. Am. Vet. Med. Assoc. 203:118–121. Liang, R., J. V. van den Hurk, L. A. Babiuk, and S. van Drunen Littel-van den Hurk. 2006. Priming with DNA encoding E2 and boosting with E2 protein formulated with CpG oligodeoxynucleotides induces strong immune responses and protection from bovine viral diarrhea virus in cattle. J. Gen. Virol. 87:2971–2982. Lightowlers, M. W., and D. D. Heath. 2004. Immunity and vaccine control of Echinococcus granulosus infection in animal intermediate hosts. Parasitologia 46:27–31. Lorenzen, N., and S. E. LaPatra. 2005. DNA vaccines for aquacultured fish. Rev. Sci. Tech. 24:201–213. Mackowiak, M., J. Maki, L. Motes-Kreimeyer, T. Harbin, and K. Van Kampen. 1999. Vaccination of wildlife against rabies: successful use of a vectored vaccine obtained by recombinant technology. Adv. Vet. Med. 41:571–583. Maes, R. K., M. D. Sussman, A. Vilnis, and B. J. Thacker. 1997. Recent developments in latency and recombination of Aujeszky’s disease (pseudorabies) virus. Vet. Microbiol. 55:13–27. Maizels, R. M., A. Balic, N. Gomez-Escobar, M. Nair, M. D. Taylor, and J. E. Allen. 2004. Helminth parasites—masters of regulation. Immunol. Rev. 201:89–116. Marcelino, I., M. F. Sousa, C. Verissimo, A. E. Cunha, M. J. Carrondo, and P. M. Alves. 2006. Process development for the mass production of Ehrlichia ruminantium. Vaccine 24:1716–1725. Marsh, A. E., J. Lakritz, P. J. Johnson, M. A. Miller, Y. W. Chiang, and H. J. Chu. 2004. Evaluation of immune responses in horses immunized using a killed Sarcocystis neurona vaccine. Vet. Ther. 5:34–42. Martens, A., A. De Moor, L. Vlaminck, F. Pille, and M. Steenhaut. 2001. Evaluation of excision, cryosurgery and local BCG vaccination for the treatment of equine sarcoids. Vet. Rec. 149:665–669. McKeever, D. J., E. L. Taracha, W. I. Morrison, A. J. Musoke, and S. P. Morzaria. 1999. Protective immune mechanisms against Theileria parva: evolution of vaccine development strategies. Parasitol. Today 15:263–267. McOrist, S., S. Jasni, R. A. Mackie, N. MacIntyre, N. Neef, and G. H. Lawson. 1993. Reproduction of porcine proliferative enteropathy with pure cultures of ileal symbiont intracellularis. Infect. Immun. 61:4286–4292. Medina, E., and C. A. Guzman. 2001. Use of live bacterial vaccine vectors for antigen delivery: potential and limitations. Vaccine 19:1573–1580. Meeusen, E. N., and D. Piedrafita. 2003. Exploiting natural immunity to helminth parasites for the development of veterinary vaccines. Int. J. Parasitol. 33:1285–1290. Reference deleted. Reference deleted. Miller, L. A., J. C. Rhyan, and M. Drew. 2004. Contraception of bison by GnRH vaccine: a possible means of decreasing transmission of brucellosis in bison. J. Wildl. Dis. 40:725–730. Minke, J. M., J. C. Audonnet, and L. Fischer. 2004. Equine viral vaccines: the past, present and future. Vet. Res. 35:425–443. Monath, T. P., J. Arroyo, C. Miller, and F. Guirakhoo. 2001. West Nile virus vaccine. Curr. Drug Targets Infect. Disord. 1:37–50. VOL. 20, 2007 115. Monath, T. P., J. Liu, N. Kanesa-Thasan, G. A. Myers, R. Nichols, A. Deary, K. McCarthy, C. Johnson, T. Ermak, S. Shin, J. Arroyo, F. Guirakhoo, J. S. Kennedy, F. A. Ennis, S. Green, and P. Bedford. 2006. A live, attenuated recombinant West Nile virus vaccine. Proc. Natl. Acad. Sci. USA 103:6694–6699. 116. Moormann, R. J., A. Bouma, J. A. Kramps, C. Terpstra, and H. J. De Smit. 2000. Development of a classical swine fever subunit marker vaccine and companion diagnostic test. Vet. Microbiol. 73:209–219. 117. Moreau, Y., E. Vidor, G. Bissuel, and N. Dubreuil. 1989. Vaccination against canine babesiosis: an overview of field observations. Trans. R. Soc. Trop. Med. Hyg. 83(Suppl.):95–96. 118. Moriyon, I., M. J. Grillo, D. Monreal, D. Gonzalez, C. Marin, I. LopezGoni, R. C. Mainar-Jaime, E. Moreno, and J. M. Blasco. 2004. Rough vaccines in animal brucellosis: structural and genetic basis and present status. Vet. Res. 35:1–38. 119. Morrow, C. J., J. F. Markham, and K. G. Whithear. 1998. Production of temperature-sensitive clones of Mycoplasma synoviae for evaluation as live vaccines. Avian Dis. 42:667–670. 120. Mortensen, S., R. Stryhn, A. Boklund, K. D. Stark, J. Christensen, and P. Willeberg. 2002. Risk factors for infection of sow herds with porcine reproductive and respiratory syndrome (PRRS) virus. Prev. Vet. Med. 53:83–101. 121. Murphy, F. A., E. P. J. Gibbs, M. C. Horzinek, and M. J. Studdert. 1999. Veterinary virology, 3rd ed. Academic Press, London, United Kingdom. 122. Murtaugh, M. P., M. R. Elam, and L. T. Kakach. 1995. Comparison of the structural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS virus. Arch. Virol. 140:1451–1460. 123. Newton, S. E., and E. N. Meeusen. 2003. Progress and new technologies for developing vaccines against gastrointestinal nematode parasites of sheep. Parasite Immunol. 25:283–296. 124. Nielsen, K., D. R. Ewalt, B. Garin-Bastuji, A. P. MacMillan, J. Stack, and P. Wright. 2004. Bovine brucellosis, p. 409–438. In Office International des Epizooties (ed.), Manual of diagnostic tests and vaccines for terrestrial animals. Office International des Epizooties, Paris, France. 125. Nogueira, F. S., M. A. Moreira, G. P. Borja-Cabrera, F. N. Santos, I. Menz, L. E. Parra, Z. Xu, H. J. Chu, C. B. Palatnik-de-Sousa, and M. C. Luvizotto. 2005. Leishmune vaccine blocks the transmission of canine visceral leishmaniasis: absence of Leishmania parasites in blood, skin and lymph nodes of vaccinated exposed dogs. Vaccine 23:4805–4810. 126. Nuttall, P. A., A. R. Trimnell, M. Kazimirova, and M. Labuda. 2006. Exposed and concealed antigens as vaccine targets for controlling ticks and tick-borne diseases. Parasite Immunol. 28:155–163. 127. O’Hern, P. A., C. S. Bambra, M. Isahakia, and E. Goldberg. 1995. Reversible contraception in female baboons immunized with a synthetic epitope of sperm-specific lactate dehydrogenase. Biol. Reprod. 52:331–339. 128. Olson, M. E., H. Ceri, and D. W. Morck. 2000. Giardia vaccination. Parasitol. Today 16:213–217. 129. Olson, M. E., D. W. Morck, and H. Ceri. 1997. Preliminary data on the efficacy of a Giardia vaccine in puppies. Can. Vet. J. 38:777–779. 130. Paillot, R., D. Hannant, J. H. Kydd, and J. M. Daly. 2006. Vaccination against equine influenza: quid novi? Vaccine 24:4047–4061. 131. Paoletti, E. 1996. Applications of pox virus vectors to vaccination: an update. Proc. Natl. Acad. Sci. USA 93:11349–11353. 132. Pardo, M. C., and M. Mackowiak. 1999. Efficacy of a new canine-origin, modified-live virus vaccine against canine coronavirus. Canine Practice 24:6–8. 133. Park, M. S., J. Steel, A. Garcia-Sastre, D. Swayne, and P. Palese. 2006. Engineered viral vaccine constructs with dual specificity: avian influenza and Newcastle disease. Proc. Natl. Acad. Sci. USA 103:8203–8208. 134. Pashine, A., N. M. Valiante, and J. B. Ulmer. 2005. Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 11:S63–S68. 135. Pastoret, P. P., and B. Brochier. 1999. Epidemiology and control of fox rabies in Europe. Vaccine 17:1750–1754. 136. Pastoret, P. P., and P. Jones. 2004. Veterinary vaccines for animal and public health. Dev. Biol. (Basel) 119:15–29. 137. Pensaert, M., G. Labarque, H. Favoreel, and H. Nauwynck. 2004. Aujeszky’s disease vaccination and differentiation of vaccinated from infected pigs. Dev. Biol. (Basel) 119:243–254. 138. Perrin, B., M. Perrin, A. Moussa, and M. Coudert. 1996. Evaluation of a commercial gE blocking ELISA test for detection of antibodies to infectious bovine rhinotracheitis virus. Vet. Rec. 138:520. 139. Philippon, V. 2006. Modified vaccinia Ankara (MVA) vector vaccines. University of California San Francisco School of Medicine, San Francisco, CA. http://chi.ucsf.edu/vaccines/vaccines?page⫽vc-01-04. 140. Pipano, E., and V. Shkap. 2000. Vaccination against tropical theileriosis. Ann. N. Y. Acad. Sci. 916:484–500. 141. Plowright, W. 1972. The production and use of rinderpest cell culture vaccine in developing countries. World Anim. Rev. 1:14–18. 142. Pluimers, F. H. 2004. Foot-and-mouth disease control using vaccination: the Dutch experience in 2001. Dev. Biol. (Basel) 119:41–49. 143. Powell, K. 2004. DNA vaccines—back in the saddle again? Nat. Biotechnol. 22:799–801. 144. Pulendran, B., and R. Ahmed. 2006. Translating innate immunity into VETERINARY VACCINES 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. 509 immunological memory: implications for vaccine development. Cell 124: 849–863. Rank, R. G. 1999. Models of immunity, p. 239–295. In R. S. Stephens (ed.), Chlamydia: intracellular biology, pathogenesis, and immunity. American Society for Microbiology, Washington, DC. Rautenschlein, S., C. Kraemer, J. Vanmarcke, and E. Montiel. 2005. Protective efficacy of intermediate and intermediate plus infectious bursal disease virus (IBDV) vaccines against very virulent IBDV in commercial broilers. Avian Dis. 49:231–237. Robbins, S. C., M. D. Jelinski, and R. L. Stotish. 2004. Assessment of the immunological and biological efficacy of two different doses of a recombinant GnRH vaccine in domestic male and female cate (Felis catus). J. Reprod. Immunol. 64:107–119. Rodolakis, A. 1983. In vitro and in vivo properties of chemically induced temperature-sensitive mutants of Chlamydia psittaci var. ovis: screening in a murine model. Infect. Immun. 42:525–530. Rodolakis, A., and A. Souriau. 1986. Response of goats to vaccination with temperature-sensitive mutants of Chlamydia psittaci obtained by nitrosoguanidine mutagenesis. Am. J. Vet. Res. 47:2627–2631. Roeder, P. 2006. Rinderpest eradication—is it feasible? p. 61–62. In I. Olsen and T. Gjøen (ed.), Proceedings of the International Veterinary Vaccine and Diagnostics Conference, Oslo, Norway. Reprosentralen, University of Oslo, Oslo, Norway. Romero, J. J., E. Perez, and K. Frankena. 2004. Effect of a killed whole Neospora caninum tachyzoite vaccine on the crude abortion rate of Costa Rican dairy cows under field conditions. Vet. Parasitol. 123:149–159. Rupprecht, C. E., C. A. Hanlon, and D. Slate. 2004. Oral vaccination of wildlife against rabies: opportunities and challenges in prevention and control. Dev. Biol. (Basel) 119:173–184. Rutten, V. P., W. R. Klein, W. A. De Jong, W. Misdorp, P. A. Steerenberg, W. H. De Jong, W. Den Otter, and E. J. Ruitenberg. 1991. Immunotherapy of bovine ocular squamous cell carcinoma by repeated intralesional injections of live bacillus Calmette-Guerin (BCG) or BCG cell walls. Cancer Immunol. Immunother. 34:186–190. Scheerlinck, J. P., and D. L. Greenwood. 2006. Particulate delivery systems for animal vaccines. Methods 40:118–124. Scheerlinck, J. P., J. Karlis, T. E. Tjelle, P. J. Presidente, I. Mathiesen, and S. E. Newton. 2004. In vivo electroporation improves immune responses to DNA vaccination in sheep. Vaccine 22:1820–1825. Schetters, T. 2005. Vaccination against canine babesiosis. Trends Parasitol. 21:179–184. Schurig, G. G., R. M. Roop II, T. Bagchi, S. Boyle, D. Buhrman, and N. Sriranganathan. 1991. Biological properties of RB51; a stable rough strain of Brucella abortus. Vet. Microbiol. 28:171–188. Schurig, G. G., N. Sriranganathan, and M. J. Corbel. 2002. Brucellosis vaccines: past, present and future. Vet. Microbiol. 90:479–496. Schynts, F., E. Baranowski, M. Lemaire, and E. Thiry. 1999. A specific PCR to differentiate between gE negative vaccine and wildtype bovine herpesvirus type 1 strains. Vet. Microbiol. 66:187–195. Sedgmen, B. J., S. A. Lofthouse, and E. N. Meeusen. 2006. The ovine nasal mucosa: an alternative tissue site for mucosal immunization. Methods 38: 112–116. Seregin, A., R. Nistler, V. Borisevich, G. Yamshchikov, E. Chaporgina, C. W. Kwok, and V. Yamshchikov. 2006. Immunogenicity of West Nile virus infectious DNA and its noninfectious derivatives. Virology 356:115–125. Seya, T., T. Akazawa, J. Uehori, M. Matsumoto, I. Azuma, and K. Toyoshima. 2003. Role of Toll-like receptors and their adaptors in adjuvant immunotherapy for cancer. Anticancer Res. 23:4369–4376. Shams, H. 2005. Recent developments in veterinary vaccinology. Vet. J. 170:289–299. Shirley, M. W., A. L. Smith, and F. M. Tomley. 2005. The biology of avian Eimeria with an emphasis on their control by vaccination. Adv. Parasitol. 60:285–330. Shkap, V., and E. Pipano. 2000. Culture-derived parasites in vaccination of cattle against tick-borne diseases. Ann. N. Y. Acad. Sci. 916:154–171. Shryock, T. R. 2004. The future of anti-infective products in animal health. Nat. Rev. Microbiol. 2:425–430. Simard, N., C. Lyngoy, V. Funk, G. Traxler, S. LaPatra, and K. Salonius. 2006. Research to market: meeting safety and efficacy requirements for a DNA vaccine used in Atlantic salmon, p. 46. In I. Olsen and T. Gjøen (ed.), Proceedings of the International Veterinary Vaccine and Diagnostics Conference, Oslo, Norway. Reprosentralen, University of Oslo, Oslo, Norway. Simms, M. S., D. P. Scholfield, E. Jacobs, D. Michaeli, P. Broome, J. E. Humphreys, and M. C. Bishop. 2000. Anti-GnRH antibodies can induce castrate levels of testosterone in patients with advanced prostate cancer. Br. J. Cancer 83:443–446. Singh, M., and D. T. O’Hagan. 2003. Recent advances in veterinary vaccine adjuvants. Int. J. Parasitol. 33:469–478. Slate, D., C. E. Rupprecht, J. A. Rooney, D. Donovan, D. H. Lein, and R. B. Chipman. 2005. Status of oral rabies vaccination in wild carnivores in the United States. Virus Res. 111:68–76. 510 MEEUSEN ET AL. 171. Stevens, V. C. 1997. Some reproductive studies in the baboon. Hum. Reprod. Update 3:533–540. 172. Stoffregen, W. C., S. C. Olsen, and B. J. Bricker. 2006. Parenteral vaccination of domestic pigs with Brucella abortus strain RB51. Am. J. Vet. Res. 67:1802–1808. 173. Stout, T. A., and B. Colenbrander. 2004. Suppressing reproductive activity in horses using GnRH vaccines, antagonists or agonists. Anim. Reprod. Sci. 82–83:633–643. 174. Sweeney, C. R., J. F. Timoney, J. R. Newton, and M. T. Hines. 2005. Streptococcus equi infections in horses: guidelines for treatment, control, and prevention of strangles. J. Vet. Intern. Med. 19:123–134. 175. Taylor, G. 2000. Castration—an abbreviated history of western manhood. Routledge, New York, NY. 176. Taylor, J., B. Meignier, J. Tartaglia, B. Languet, J. VanderHoeven, G. Franchini, C. Trimarchi, and E. Paoletti. 1995. Biological and immunogenic properties of a canarypox-rabies recombinant, ALVAC-RG (vCP65) in non-avian species. Vaccine 13:539–549. 177. Timoney, J. F., and D. Eggers. 1985. Serum bactericidal responses to Streptococcus equi of horses following infection or vaccination. Equine Vet. J. 17:306–310. 178. Tshewang, U., K. F. Dowsett, L. M. Knott, and T. E. Trigg. 1997. Preliminary study of ovarian activity in fillies treated with a GnRH vaccine. Aust. Vet. J. 75:663–667. 179. Tung, K. S., P. Primakoff, L. Woolman-Gamer, and D. G. Myles. 1997. Mechanism of infertility in male guinea pigs immunized with sperm PH-20. Biol. Reprod. 56:1133–1141. 180. Umetsu, D. T., and R. H. DeKruyff. 2006. The regulation of allergy and asthma. Immunol. Rev. 212:238–255. 181. van Aarle, P. 2003. Suitability of an E2 subunit vaccine of classical swine fever in combination with the E(rns)-marker-test for eradication through vaccination. Dev. Biol. (Basel) 114:193–200. 182. van den Bosch, H., and J. Frey. 2003. Interference of outer membrane protein PalA with protective immunity against Actinobacillus pleuropneumoniae infections in vaccinated pigs. Vaccine 21:3601–3607. 183. van Drunen Littel-van den Hurk, S., S. Babiuk, and L. A. Babiuk. 2006. Needle-free delivery of veterinary DNA vaccines. Methods Mol. Med. 127: 91–105. 184. Van Immerseel, F., U. Methner, I. Rychlik, B. Nagy, P. Velge, G. Martin, N. Foster, R. Ducatelle, and P. A. Barrow. 2005. Vaccination and early protection against non-host-specific Salmonella serotypes in poultry: exploitation of innate immunity and microbial activity. Epidemiol. Infect. 133:959– 978. 185. van Oirschot, J. T., M. J. Kaashoek, and F. A. Rijsewijk. 1996. Advances in the development and evaluation of bovine herpesvirus 1 vaccines. Vet. Microbiol. 53:43–54. CLIN. MICROBIOL. REV. 186. Van Overbeke, I., K. Chiers, R. Ducatelle, and F. Haesebrouck. 2001. Effect of endobronchial challenge with Actinobacillus pleuropneumoniae serotype 9 of pigs vaccinated with a vaccine containing Apx toxins and transferrinbinding proteins. J. Vet. Med. B Infect. Dis. Vet. Public Health 48:15–20. 187. Veits, J., D. Wiesner, W. Fuchs, B. Hoffmann, H. Granzow, E. Starick, E. Mundt, H. Schirrmeier, T. Mebatsion, T. C. Mettenleiter, and A. RomerOberdorfer. 2006. Newcastle disease virus expressing H5 hemagglutinin gene protects chickens against Newcastle disease and avian influenza. Proc. Natl. Acad. Sci. USA 103:8197–8202. 188. Vemulapalli, R., J. R. McQuiston, G. G. Schurig, N. Sriranganathan, S. M. Halling, and S. M. Boyle. 1999. Identification of an IS711 element interrupting the wboA gene of Brucella abortus vaccine strain RB51 and a PCR assay to distinguish strain RB51 from other Brucella species and strains. Clin. Diagn. Lab. Immunol. 6:760–764. 189. Vercruysse, J., D. P. Knox, T. P. Schetters, and P. Willadsen. 2004. Veterinary parasitic vaccines: pitfalls and future directions. Trends Parasitol. 20:488–492. 190. Vretou, E., E. Psarrou, M. Kaisar, I. Vlisidou, V. Salti-Montesanto, and D. Longbottom. 2001. Identification of protective epitopes by sequencing of the major outer membrane protein gene of a variant strain of Chlamydia psittaci serotype 1 (Chlamydophila abortus). Infect. Immun. 69:607–612. 191. Walker, J. A., and J. F. Timoney. 2002. Construction of a stable non-mucoid deletion mutant of the Streptococcus equi Pinnacle vaccine strain. Vet. Microbiol. 89:311–321. 192. Wallach, M. 1997. The importance of transmission-blocking immunity in the control of infections by apicomplexan parasites. Int. J. Parasitol. 27: 1159–1167. 193. Wallach, M., N. C. Smith, M. Petracca, C. M. Miller, J. Eckert, and R. Braun. 1995. Eimeria maxima gametocyte antigens: potential use in a subunit maternal vaccine against coccidiosis in chickens. Vaccine 13:347–354. 194. Whetstone, C. A., J. M. Miller, B. S. Seal, L. J. Bello, and W. C. Lawrence. 1992. Latency and reactivation of a thymidine kinase-negative bovine herpesvirus 1 deletion mutant. Arch. Virol. 122:207–214. 195. Willadsen, P. 2004. Anti-tick vaccines. Parasitology 129:S367–S387. 196. Williams, R. B. 2002. Fifty years of anticoccidial vaccines for poultry (1952– 2002). Avian Dis. 46:775–802. 197. Yen, H. H., J. P. Scheerlinck, S. Gekas, and P. Sutton. 2006. A sheep cannulation model for evaluation of nasal vaccine delivery. Methods 38: 117–123. 198. Zweygarth, E., A. I. Josemans, M. F. Van Strijp, L. Lopez-Rebollar, M. Van Kleef, and B. A. Allsopp. 2005. An attenuated Ehrlichia ruminantium (Welgevonden stock) vaccine protects small ruminants against virulent heartwater challenge. Vaccine 23:1695–1702. CLINICAL MICROBIOLOGY REVIEWS, July 2007, p. 511–532 0893-8512/07/$08.00⫹0 doi:10.1128/CMR.00004-07 Copyright © 2007, American Society for Microbiology. All Rights Reserved. Vol. 20, No. 3 Laboratory Diagnostic Techniques for Entamoeba Species R. Fotedar,1 D. Stark,1* N. Beebe,2 D. Marriott,1 J. Ellis,3 and J. Harkness1 St. Vincent’s Hospital, Department of Microbiology, Sydney, Australia1; University of Technology Sydney, Institute for the Biotechnology of Infectious Diseases, Broadway, Australia2; and University of Technology Sydney, Department of Medical and Molecular Biosciences, Broadway, Australia3 INTRODUCTION .......................................................................................................................................................511 Entamoeba histolytica...............................................................................................................................................512 Entamoeba dispar .....................................................................................................................................................512 Entamoeba moshkovskii ...........................................................................................................................................512 IMPORTANCE OF DIAGNOSIS .............................................................................................................................512 CLINICAL MANIFESTATIONS ..............................................................................................................................513 Asymptomatic Colonization...................................................................................................................................513 Dysentery/Amebic Colitis .......................................................................................................................................513 Extraintestinal Amebiasis......................................................................................................................................513 LABORATORY DIAGNOSIS....................................................................................................................................514 Microscopy ...............................................................................................................................................................514 Culture Methods .....................................................................................................................................................514 Isoenzyme Analysis .................................................................................................................................................516 Antibody Detection Tests .......................................................................................................................................516 Antigen Detection Tests .........................................................................................................................................517 Immunochromatographic Assays..........................................................................................................................519 DNA-BASED DIAGNOSTIC TESTS........................................................................................................................519 Complexity of Fecal Samples ................................................................................................................................519 Methods of DNA Extraction..................................................................................................................................519 Manual methods .................................................................................................................................................519 Automated methods ............................................................................................................................................520 Conventional PCR ..................................................................................................................................................520 Real-Time PCR .......................................................................................................................................................523 Microarray Development .......................................................................................................................................523 Typing Methods ......................................................................................................................................................524 CONCLUSION............................................................................................................................................................527 REFERENCES ............................................................................................................................................................527 imately 50 million people have invasive disease, resulting in 100,000 deaths per year (81, 210). Although the parasite has a worldwide distribution, high prevalence rates of more than 10% of the population have been reported from various developing countries (173). Entamoeba histolytica-related diarrheal illnesses have recently been reported to have a negative impact on the growth of children (114). Despite the availability of effective therapy, morbidity and mortality associated with amebic infection have persisted, suggesting that interventions designed to limit or to eliminate disease are ineffective. As humans appear to be the only host, an appropriate control program could potentially eradicate amebiasis. New approaches to the identification of E. histolytica are based on detection of E. histolytica-specific antigen and DNA in stool and other clinical samples. Several molecular diagnostic tests, including conventional and real-time PCR, have been developed for the detection and differentiation of E. histolytica, E. dispar, and E. moshkovskii in clinical samples. These molecular methods have led to a reevaluation of the epidemiology of amebiasis in terms of prevalence and morbidity, particularly in those geographical areas with high endemic rates. The purpose of this review is to discuss the methods that exist for the identification of E. histolytica, E. dispar, and E. moshkovskii which are available to the clinical diagnostic lab- INTRODUCTION The genus Entamoeba contains many species, six of which (Entamoeba histolytica, Entamoeba dispar, Entamoeba moshkovskii, Entamoeba polecki, Entamoeba coli and Entamoeba hartmanni) reside in the human intestinal lumen. Entamoeba histolytica is the only species definitely associated with pathological sequelae in humans; the others are considered nonpathogenic (31, 57). Although recent studies highlight the recovery of E. dispar and E. moshkovskii from patients with gastrointestinal symptoms (52, 73, 130, 189, 201), there is still no definitive evidence of a causal link between the presence of these two species and the symptoms of the host. Entamoeba histolytica is the causative agent of amebiasis and is globally considered a leading parasitic cause of human mortality (77, 81, 210). Clinical features of amebiasis due to E. histolytica range from asymptomatic colonization to amebic dysentery and invasive extraintestinal amebiasis, which is manifested most commonly in the form of liver abscesses. Approx- * Corresponding author. Mailing address: Department of Microbiology, St. Vincent’s Hospital, Sydney, Darlinghurst, NSW 2010, Australia. Phone: 61 2 8382 9196. Fax: 61 2 8382 2989. E-mail: dstark @stvincents.com.au. 511 512 FOTEDAR ET AL. oratory. To address the need for a specific diagnostic test for amebiasis, a substantial amount of work has been carried out over the last decade in different parts of the world, and molecular diagnostic tests are increasingly being used for both clinical and research purposes. Entamoeba histolytica Entamoeba histolytica was first described by Fedor Lösch in 1875 in St. Petersburg, Russia. He described intestinal amebiasis in detail, and the species name E. histolytica was first coined by Fritz Schaudinn in 1903 (155). Entamoeba histolytica is the pathogenic species of Entamoeba that causes amebic dysentery and a wide range of other invasive diseases, including amebic liver abscess, respiratory tract infections, and cerebral and genitourinary amebiasis. Entamoeba dispar In 1925, Brumpt formulated the theory that the difference between many asymptomatic amebic infections and those of individuals with amebic disease could be correlated with the existence of two distinct but morphologically identical species, namely, E. histolytica (which is capable of causing invasive disease) and E. dispar (which never causes disease). This hypothesis was dismissed at that time, but subsequently evidence which gave support to Brumpt’s findings began accumulating. In 1993, 68 years after the original discovery of E. dispar, E. histolytica and E. dispar were formally accepted as different yet closely related species on the basis of extensive genetic, immunological, and biochemical analyses (43, 177, 188). Although E. dispar was previously considered to be nonpathogenic E. histolytica and was regarded as a commensal species, intestinal symptoms in patients infected with this species have been reported (95). In a recent study from India by Parija and Khairnar (130), 68 fecal specimens in which Entamoeba species were demonstrated on microscopy were tested using PCR. Eleven patients positive for E. dispar and E. moshkovskii (in association) had mild gastrointestinal discomfort; however, the study failed to clarify whether other parasites or bacterial or viral pathogens were detected in these 11 samples. Entamoeba dispar can produce variable focal intestinal lesions in animals (28, 48, 202) and can destroy epithelial cell monolayers in vitro (49). There is also some evidence that following infection with E. dispar, pathological changes may occur in some humans (111). However Koch’s postulates have not been fulfilled, and no large case-controlled studies have been undertaken to assess the true pathogenic potential of this organism. Entamoeba moshkovskii Entamoeba moshkovskii is another species of Entamoeba and is morphologically indistinguishable from E. histolytica and E. dispar. This species was first described from Moscow sewage by Tshalaia in 1941 (193) and was thereafter reported to occur in many different countries (30, 160). Entamoeba moshkovskii was initially thought to be a free-living environmental strain. However in 1961 an E. histolytica-like strain was isolated from a resident of Laredo, TX, who presented with diarrhea, weight CLIN. MICROBIOL. REV. loss, and epigastric pain (46). This strain was named the E. histolytica Laredo strain and shared many biological features with E. moshkovskii. Both the Laredo strain and E. moshkovskii grow at room temperature, are osmotolerant, and are resistant to emetine. These characteristics distinguished them from E. histolytica and E. dispar (30, 34). Subsequent molecular studies have confirmed that the E. histolytica Laredo strain is a strain of E. moshkovskii (30). The exact taxonomic classification of the species has yet to emerge, as E. moshkovskii seems to be a complex of at least two species (30). Although the early isolations of this species have been from sewage, recent studies have reported the recovery of E. moshkovskii from human feces (8, 30, 52, 73, 130, 189). Reports on detection of E. moshkovskii from human specimens to date have come from North America, Italy, South Africa, Bangladesh, India, Iran, Australia, and Turkey (8, 30, 52, 73, 130, 171, 176, 189). Although previous reports on the identification of E. moshkovskii in fecal samples have not shown any association with clinical illness (30), recent studies from Bangladesh and India have reported E. moshkovskii as a sole potential enteropathogen in patients presenting with gastrointestinal symptoms and/or dysentery, highlighting the need for further study to investigate the pathogenic potential of this organism (73, 130). IMPORTANCE OF DIAGNOSIS The epidemiology of E. histolytica, E. dispar, and E. moshkovskii parasitoses remains uncertain, because most of the existing data were obtained using methods incapable of distinguishing among the three morphologically identical species. Entamoeba dispar appears to be about 10 times more common than E. histolytica, with most of the 500 million people infected with E. histolytica/E. dispar carrying E. dispar (91). Little is known about the epidemiology and incidence of E. moshkovskii infections, as only a few studies have used molecular methods to identify this parasite. Most morbidity and mortality due to amebiasis occur in developing regions such as Central and South America, Africa, and the Indian subcontinent (203). In Bangladesh, where diarrheal diseases are the leading cause of childhood death, approximately 50% of children have serological evidence of exposure to E. histolytica by 5 years of age (74). In developed countries, high-risk groups include travelers, immigrants from areas of endemicity, and men who have sex with men (MSM) (122, 125–127, 185, 186). It is estimated that 20% to 30% of MSM are colonized with E. dispar in Western countries, which is attributed to oral-anal sex practices (10). In addition, a few reports describe cases of invasive amebiasis in homosexual men from Taiwan and Korea (88, 124) and Australia (52, 175). Early detection of infection in these high-risk individuals by using molecular diagnostic methods will improve understanding of the public health issues and expedite the initiation of control measures (125–127, 175, 176). The existence of these morphologically indistinguishable species of Entamoeba led the World Health Organization (WHO) to recommend the development and application of improved methods for the specific diagnosis of E. histolytica infection (210). Epidemiological surveys of amebiasis should include tools to diagnose E. histolytica and E. dispar individu- LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 ally, simultaneously, and accurately. Identification of E. histolytica remains an important goal of the clinical parasitology laboratory, and molecular diagnostics represent an important confirmatory diagnostic step in the management of patients who may be infected with E. histolytica and require specific therapy (210). Techniques developed for the identification of E. histolytica include the detection of E. histolytica-specific antibodies and specific antigen in stool and other clinical samples. In addition, several molecular diagnostic tests, including conventional, nested, and real-time PCR, have been developed for diagnosis of E. histolytica, E. dispar, and E. moshkovskii by clinical laboratories. CLINICAL MANIFESTATIONS Asymptomatic Colonization Asymptomatic cyst passage, with 90% of human infections either asymptomatic or mildly symptomatic, is considered to be the most common manifestation of E. histolytica. However, these studies have been based on the microscopic examination of fecal samples (203, 210). Patients can clear their infection without any signs of disease. In stool samples, cysts are usually detected, and trophozoites, which are rarely seen, lack ingested red blood cells (RBCs). Individuals harboring E. histolytica (asymptomatic carriers) can develop antibody titers in the absence of invasive disease (60, 93, 145). Asymptomatic colonization with E. histolytica, if left untreated, can lead to amebic dysentery and a wide range of other invasive diseases, but more often the infection resolves spontaneously without the development of diseases (19, 20, 60, 75). Dysentery/Amebic Colitis When followed for 1 year, 4 to 10% of asymptomatic individuals colonized with E. histolytica developed colitis or extraintestinal disease (60, 75); therefore, it is recommended that asymptomatic cyst carriers should be treated. Symptoms commonly attributed to E. histolytica colitis include abdominal pain or tenderness with watery, bloody, or mucous diarrhea. Eighty percent of patients complain of localized abdominal pain; some patients may have only intermittent diarrhea alternating with constipation. Microscopically, trophozoites are readily detected in submucosal tissue or fecal samples by permanent stains. Since E. histolytica invades the colonic mucosa, feces are almost universally positive for occult blood. The presence of Charcot-Leyden crystals and blood is the most common finding in the acute stage. In addition to the RBCs, macrophages and polymorphonuclear cells (PMNs) can also be seen on microscopy in cases of amebic dysentery. Fever is unusual, occurring in ⬍40% of patients (4). Occasionally individuals develop fulminant amebic colitis, with profuse bloody diarrhea, fever, pronounced leukocytosis, and widespread abdominal pain, often with peritoneal signs and extensive involvement of the colon (184). Toxic megacolon, ameboma (5), cutaneous amebiasis (112), and rectovaginal fistulae (108) can occur as complications of intestinal amebiasis. 513 Extraintestinal Amebiasis The most common extraintestinal manifestation is amebic liver abscess (ALA), which is associated with significant morbidity and mortality. This was a progressive and almost invariably fatal disease little more than a century ago, but since the introduction of effective medical treatment and rapid diagnosis, mortality rates have fallen to 1 to 3% (22, 166). ALA is caused by hematogenous spread of the invasive trophozoites from the colon, which reach the liver via the portal vein. This explains the frequent occurrence of abscesses in the right hepatic lobe, which receives most of the blood draining the cecum and ascending colon (154). Some individuals presenting with ALA have concurrent amebic colitis, but more often they have no bowel symptoms, and stool microscopy is usually negative for E. histolytica trophozoites and cysts (5, 152, 190). Individuals can present with ALA months to years after travel or residency in an area of endemicity, so a careful travel history is mandatory (14, 102, 166). The disease should be suspected in anyone with an appropriate exposure history (residency or travel in an area of endemicity) presenting with fever, right upper quadrant pain, and substantial hepatic tenderness. Cough may be present, and dullness and rales in the right lung base are not infrequent (5, 14, 166, 190). Jaundice is unusual. Symptoms are usually acute (⬍10 days in duration) but can be chronic, with anorexia and weight loss as prominent features. Leukocytosis without eosinophilia, mild anemia, a raised concentration of alkaline phosphatase, and a high rate of erythrocyte sedimentation are the most common laboratory findings (5, 14, 166, 190). The most serious complication of ALA is rupture, particularly into the pericardium, and superinfection with bacteria. Rupture into the pleura is relatively common and usually has a good prognosis. With early diagnosis and therapy, the mortality from uncomplicated ALA is less than 1% (5). Complications of extraintestinal amebiasis include pleuroplumonary amebiasis secondary to ALA rupture through the diaphragm, brain abscess, and genitourinary amebiasis. Diagnosis of brain abscess is usually made by the microscopic detection of parasites on brain biopsy or at autopsy; however, a recent study has highlighted the first diagnosis of E. histolytica encephalitis using PCR (172). Diagnosis of liver abscess is confirmed by a positive serological test, as amebic serology is highly sensitive (⬎94%) and highly specific (⬎95%) for diagnosis. A false-negative serological test can be obtained early during infection (within the first 7 to 10 days), but a repeat test is usually positive. Abdominal ultrasound or computed tomography scan does not provide specificity for ALA. However, a positive serological test in combination with abdominal imaging is helpful for diagnosis where PCR is not routinely available. A recent study confirms that in the majority of successfully treated ALA patients the abscess completely resolves; however, in 7.1% of patients residual lesions are detected, with the unique sonographic appearance of round or oval hypo- or isoechoic areas surrounded by the hyperchoic wall (21). The successful use of PCR methods in detection of E. histolytica DNA in patients with ALA has shown high sensitivity (100%) (52, 182, 214, 215). 514 FOTEDAR ET AL. CLIN. MICROBIOL. REV. FIG. 1. Cysts and trophozoites of Entamoeba species. LABORATORY DIAGNOSIS Microscopy Microscopic techniques employed in a diagnostic clinical laboratory include wet preparation, concentration, and permanently stained smears for the identification of E. histolytica/E. dispar/E. moshkovskii in feces. Microscopic examination of a direct saline (wet) mount is a very insensitive method (⬍10%) which is performed on a fresh specimen (90). The sample should be examined within 1 h of collection to search for motile trophozoites which may contain RBCs. However, in patients who do not present with acute dysentery, trophozoites will not contain RBCs. Patients with asymptomatic carriage generally have only cysts in the fecal sample. Although the concentration technique is helpful in demonstrating cysts, the use of permanently stained smears (trichrome or iron hematoxylin) is an important method for recovery and identification of Entamoeba species. Microscopy is a less reliable method of identifying Entamoeba species than either culture or antigen detection tests (80, 104). The sensitivity of microscopy can be poor (60%) and confounded with false-positive results due to misidentification of macrophages as trophozoites, PMNs as cysts (especially when lobed nuclei of PMNs break apart), and other Entamoeba species (67, 72, 76, 80, 188) (Fig. 1; Table 1). As Entamoeba trophozoites generally degenerate rapidly in unfixed fecal specimens (137) and refrigeration is not recommended, specimens should be preserved with a fixative which prevents the degradation of the morphology of the parasite and allows concentration and permanent smears to be performed. Fixatives used for the concentration procedure include Schaudinn’s fluid, merthiolate iodine-formalin, sodium acetate-acetic acid-formalin (SAF), or 5% or 10% formalin. The fixatives for the permanently stained smears include trichrome, iron hematoxylin, Ziehl-Neelsen stains, modified polyvinyl alcohol (PVA) (containing mercury compounds), and SAF. Examination for ova and parasites in a minimum of three stool samples over no more than 10 days is recommended, as these organisms may be excreted intermittently or may be unevenly distributed in the stool. This improves the detection rate to 85 to 95% (107). The presence of RBCs in the cytoplasm is still considered diagnostic for E. histolytica in patients with dysentery and may be used to distinguish between E. histolytica and E. dispar. However, trophozoites containing ingested RBCs are not present in the majority of patients (67, 178). The specificity of this finding was further reduced when it was demonstrated that in some patients E. dispar also contains RBCs (80). In vitro studies have also confirmed the ability of E. dispar to ingest RBCs (191). In one study, the specificity of E. histolytica/E. dispar as determined by microscopy (formalinether concentrates and permanent stains) was only 9.5% in community laboratories compared with the Entamoeba test and ProSpecT enzyme immunoassay (EIA) antigen detection tests (134). Culture Methods Culture techniques for the isolation of Entamoeba species have been available for over 80 years. Culture media include xenic (diphasic and monophasic) and axenic systems. Xenic cultivation is defined as the growth of the parasite in the presence of an undefined flora (35). The xenic culture of E. LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 515 TABLE 1. Characteristics of trophozoites and cysts of common intestinal Entamoeba speciesa Characteristics Size, nuclei, and motility Trophozoites Cysts Other features Trophozoites Chromatin (stained) E. hartmanni E. coli E. polecki 15–20 m, 1 nucleus (difficult to see in unstained preparations), actively motile with finger shaped pseudopodia 10–15 m, mature cyst with 4 nuclei, immature cyst has 1 or 2 nuclei (nuclear characters difficult to see on wet prepn) 8–10 m, 1 nucleus (usually not seen in unstained prepn), usually unprogressive 20–25 m, 1 nucleus (often visible in unstained prepn); sluggish, short, and blunt pseudopodia 15–20 m, 1 nucleus (occasionally seen on wet prepn), sluggish 6–8 m, mature cyst with 4 nuclei, immature cyst has 1 or 2 nuclei, two nucleated cysts very common 15–25 m, mature cyst has 8 nuclei, occasionally 16 or more nuclei 10–15 m, mature cyst with 1 nucleus, rarely 2 or 4 nuclei Chromatin peripheral, may have beaded appearance Nucleus may stain more darkly than E. histolytica, chromatin may appear as solid ring rather than beaded (trichrome) Karyosome usually small and compact; centrally located or eccentric Chromatin clumped and unevenly arranged, appears as solid ring with no beads Chromatin finely granular, chromatin may also be clumped at one or both edges of membrane Karyosome large, not compact, may or may not be eccentric, may be diffuse or darkly stained Cytoplasm granular with differentiation into cytoplasm and endoplasm, vacuolated; bacteria, yeast, and other debris may be present Karyosome small and usually centrally located Karyosome (stained) Karyosome small, compact, centrally located but may be eccentric Cytoplasm (stained) Cytoplasm is fine, granular, may contain bacteria; presence of RBCs diagnostic for E. histolytica, although some E. dispar strains may very occasionally contain RBCs Cytoplasm finely granular, may contain bacteria, no RBCs Chromatin peripheral with fine uniform granules, evenly distributed Chromatin granules evenly distributed (nuclear characteristics may be difficult to see) Karyosome is small, compact, usually centrally located Chromatin coarsely granular, may be clumped and unevenly arranged Chromatin finely granular Karyosome large, eccentric, occasionally centrally located Karyosome small and usually centrally located Usually present; chromatoidal bodies usually elongate with blunt, rounded, smooth edges; may be round or oval; chromatin may or may not be present May be present (less frequent than in E. histolytica), splinter shaped with rough pointed ends, may be diffuse or absent in mature cysts, clumped mass occasionally seen in mature cysts Abundant chromatoidal bodies with angular pointed ends, thread-like chromatoidal bodies may also be present, half of the cysts contain spherical or ovoidal inclusion mass Cysts Chromatin (stained) Karyosome (stained) Cytoplasm (stained) a E. histolytica/E. dispar/ E. moshkovskii Karyosome is small, compact, usually centrally located but occasionally eccentric May be present; chromatoidal bodies usually elongate with blunt, rounded, smooth edges; may be round or oval; chromatin may be diffuse or absent in mature cyst; clumped chromatin mass may be present in early cysts Cytoplasm is finely granular, may contain ingested bacteria Data are from references 57, 73, and 188. histolytica was first introduced by Boeck and Drbohlav in 1925 in a diphasic egg slant medium, and a modification of this medium (Locke-egg) is still used today (35). Different monophasic media that were developed for E. histolytica are the egg yolk infusion medium of Balamuth (12), Jones’s medium (96), and TYSGM-9 (41). Of the different media developed for the xenic cultivation of E. histolytica, only three media, diphasic Locke-egg, Robinson’s medium (150), and the monophasic TYSGM-9 (41), are in common use (for details, see reference 35). Axenic cultivation involves the cultivation of parasites in the absence of any other metabolizing cells (35). The axenic cultivation of E. histolytica was first achieved by Diamond in 1961 (39). The monophasic medium TP-S-1 was developed and used widely for culture of E. histolytica in different research laboratories (35, 40). Currently TYI-S-33 (45) and YI-S (44) are the most widely used media for axenic cultivation of E. histolytica (35). Culture of E. histolytica can be performed from fecal specimens, rectal biopsy specimens, or liver abscess aspirates. As the liver abscess aspirates of ALA patients are usually sterile (98% cases) (19), addition of a bacterium or a trypanosomatid is necessary before inoculation of amebae into xenic culture (35, 53, 204). The success rate for culture of E. histolytica is between 50 and 70% in reference laboratories (35). As culture of E. histolytica from clinical samples such as feces or liver abscesses has a significant false-negative rate and is technically difficult, it is not undertaken in a routine clinical laboratory. Entamoeba dispar can be grown in xenic culture; however, most isolates grow poorly in monoxenic culture, and the growth of only a few strains has been reported to be viable in 516 FOTEDAR ET AL. axenic culture, suggesting that E. dispar may be less able than E. histolytica to obtain nutrients in a particle-free medium (29, 103). The use of different media for the culture of E. dispar has been investigated, and these studies indicate that YI-S may not be a suitable medium for the culture of E. dispar (35, 103). For E. moshkovskii strains, culture media employed include TTY-SB-monophasic with the trypanosomatid, TP-S-1-GM monophasic for the axenic culture of amebae (40), and the TP-S-1-GM monophasic medium (42). Other media containing bovine serum used for culture of E. moshkovskii include axenic medium TYI-S-33 with 10% bovine serum at 24°C (45) or xenic medium TYSGM-9 with 5% bovine serum at either 24°C or 37°C (41). Culture of E. histolytica in a clinical diagnostic laboratory is not feasible as a routine procedure and is less sensitive than microscopy as a detection method (35). Parasite cultures are difficult, expensive, and labor-intensive to maintain in the diagnostic laboratory (35). Overgrowth of bacteria, fungi, or other protozoans during culture is the main problem encountered, and therefore culture is not recommended as a routine diagnostic procedure for the detection of Entamoeba species (35). Isoenzyme Analysis The pioneering work of Sargeaunt et al. (158) demonstrated that isoenzyme analysis of cultured amebae would enable the differentiation of Entamoeba species. A zymodeme is defined as a group of ameba strains that share the same electrophoretic pattern and mobilities for several enzymes. Zymodemes consist of electrophoretic patterns of malic enzyme, hexokinase, glucose phosphate isomerase, and phosphoglucomutase isoenzyme (159). A total of 24 different zymodemes have been described, of which 21 are from human isolates (9 of E. histolytica and 12 of E. dispar). The presence of starch in the medium influences the most variable zymodeme patterns (16), and many zymodemes “disappear” upon removal of bacterial floras, suggesting that at least some of the bands are of bacterial rather than amebal origin (94). If the zymodemes defined by stable bands alone are counted, only three remain for E. histolytica (II, XIV, and XIX) and one for E. dispar (I). Isoenzyme (zymodeme) analysis of cultured amebae enables differentiation of E. histolytica from E. dispar and was considered the gold standard for diagnosing amebic infection prior to development of newer DNA-based techniques. Zymodeme analysis has a number of disadvantages, including the difficulty of performing the test. It is a time-consuming procedure and relies on establishing the amebae in culture, with a large number of cells needed for the enzyme analysis. This process is not always successful. The cultivation of amebae may lead to selection bias, and one species or strain may outgrow the other, which is not desirable when studying zymodemes. Furthermore, the amebic cultures and therefore isoenzyme analyses are negative for many microscopy-positive stool samples (67, 76, 80, 178). Zymodeme analysis is not easily incorporated into routine clinical laboratory work because of the expertise required to culture the parasites, the complexity of the diagnostic process, and the cost. Isoenzyme analysis has been superseded by DNA-based methods as the method of choice for studying Entamoeba species. CLIN. MICROBIOL. REV. Antibody Detection Tests Serological tests for the identification of E. histolytica infection may be helpful from a diagnostic perspective in industrialized nations, where infections due to E. histolytica are not common (127, 207). However, in areas where infection is endemic and people have been exposed to E. histolytica, the inability of serological tests to distinguish past from current infection makes a definitive diagnosis difficult (26, 60). Detection of antibodies can be helpful in the case of ALA where patients do not have detectable parasites in feces. The sensitivity for detection of antibodies to E. histolytica in serum in patients with ALA is reported to be about 100% (215). In contrast, a study from a area of high endemicity, Hue in Vietnam, revealed that 82.6% (38/46) of individuals who were infected with E. histolytica even when asymptomatic had significant antiameba antibody titers. These results were confirmed by real-time PCR studies (18, 19). Many different assays have been developed for the detection of antibodies, including indirect hemagglutination (IHA), latex agglutination, immunoelectrophoresis, counterimmunoelectrophoresis (CIE), the amebic gel diffusion test, immunodiffusion, complement fixation, indirect immunofluorescence assay (IFA), and enzyme-linked immunosorbent assay (ELISA). A variety of antibody assays for detection of E. histolytica antibodies in human serum are also commercially available (Table 2). Complement fixation tests appear to be less sensitive than others, cost more to perform, and are not used by most laboratories. IHA is simple to perform and has been shown to be a highly specific (99.1%) diagnostic tool in human immunodeficiency virus-infected patients presenting with gastrointestinal symptoms (88). However, the lower sensitivity may lead to false-negative results compared to ELISA (156). The latex agglutination test appears to detect the same antibody as IHA. Commercial kits are available, and the test can be performed in 10 min. However, due to nonspecific reactions, the specificity of this test appears to be disappointing (156). Immunoelectrophoresis, CIE, and immunodiffusion use the property of antibody and antigen precipitation in agar gel membrane. Sheehan et al. (168) reported that detection of antibody to extraintestinal E. histolytica by CIE is time-consuming but has a high sensitivity (100%) in patients with invasive amebiasis. Detection of antibodies using the IFA test was shown to be rapid, reliable, and reproducible and helps to differentiate ALA from other nonamebic etiologies (56). In addition to this, IFA tests have been shown to differentiate between past (treated) and present disease (56). A study conducted by Jackson et al. (92) indicated that monitoring of immunoglobulin M (IgM) levels using the IFA can be of clinical value in cases of invasive amebiasis. The IgM levels become negative in a short period of time after infection, with more than half of the subjects having negative results at 6 months or 100% becoming negative by 46 weeks after treatment. In ALA the sensitivity of the IFA is reported to be 93.6%, with a specificity of 96.7%, making it more sensitive than the ELISA (165). A negative test therefore indicates that a patient never had invasive amebiasis. However, this test requires skills in culture and subsequent antigen preparation, making it difficult to undertake in a routine clinical laboratory (131). ELISA is the most popular assay in diagnostic laboratories LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 517 TABLE 2. Commercially available antibody assays for diagnosis of amebiasis Sensitivity, % (reference) Antibody assay Specificity, % (reference) Manufacturer Cellognost-Amoebiasis (IHA) 100a (134), 99 (84) 90.9–100a (134), 99.8 (84) Novagnost Entamoeba IgG ⬎95b ⬎95b Bichro-Latex Amibe 93.3 (194), 98.3 (149) 95.5 (194), 96.1 (149) I.H.A. Amoebiasis 93.4 (149) 97.5 (149) Amoeba-Spot IF Amebiasis Serology microplate ELISA Amebiasis Serology microwell EIA (HK-9 antigen, axenic) RIDASCREEN Entamoeba (IgG detection) NAc (61) 95b NA (61) 97b Dade Behring Marburg GmbH, Marburg, Germany NovaTec Immundiagnostica GmbH, Dietzenbach, Germany Fumouze Diagnostics, Levallois-Perret Cedex, France Fumouze Diagnostics, Levallois-Perret Cedex, France bioMérieux, Marcy-l’Etoile, France Light Diagnostics 97.9 (84), 92.5 (169) 94.8 (84), 91.3 (169) LMD Laboratories, Inc., Carlsbad, CA b 100 , 97.7–100 (100) b 95.6 , 97.4 (100) R-Biopharma AG, Darmstadt, Germany For the titer of ⱖ1:64, 100% sensitive and 90. 9% specific; for the titer of ⱖ1:512, 100% sensitive and 100% specific. As recommended by the manufacturer. c NA, not available. a b throughout the world and has been used to study the epidemiology of asymptomatic disease (66) and the diagnosis of symptomatic amebiasis after fecal examination. This method is widely thought to be sufficient for clinical purposes, particularly for diagnosis of patients with ALA, and can be easily performed in a clinical laboratory. It may also be useful in the evaluation of intestinal and extraintestinal infections where amebiasis is suspected but organisms cannot be detected in feces (152). A microtiter ELISA to detect antibodies to E. histolytica (LMD Laboratories, Inc., Carlsbad, CA) has been shown to be 97.9% sensitive and 94.8% specific for detection of E. histolytica antibodies in ALA patients (84). Serum IgG antibodies persist for years after E. histolytica infection, whereas the presence of IgM antibodies is shortlived and can be detected during the present or current infection. An ELISA for detection of serum IgM antibodies to amebic adherence lectin was successfully used with patients suffering from acute colitis for less than 1 week, as 45% had detectable antilectin IgM antibodies (1). In another study, it was shown that an assay based on the detection of anti-LC3 (recombinant cysteine-rich portion of the E. histolytica galactose-inhibitable lectin’s 170-kDa subunit) antibodies in saliva is a more sensitive and specific test for diagnosis of ALA and acute amebic colitis than detection of serum anti-LC3 IgG antibodies (2). Of the recommended serological tests such as ELISA, those that demonstrate the presence of serum antilectin antibodies are the most frequently used for diagnosis of patients with ALA (145). A high ELISA antibody titer is helpful in the diagnosis of amebiasis in patients with detectable parasites in stool, as it has a sensitivity of 95%. Since there is no cross-reaction with other, non-E. histolytica parasites, it is a useful test for the diagnostic clinical laboratory (23, 64, 68, 144, 165, 188). Antigen Detection Tests Several investigators have developed ELISAs for the detection of antigens in fecal samples. These antigen detection tests have a sensitivity approaching that of stool culture and are rapid to perform. Antigen-based ELISA kits that are specific for E. histolytica use monoclonal antibodies against the Gal/ GalNAc-specific lectin of E. histolytica (E. histolytica II; TechLab, Blacksburg, VA) or monoclonal antibodies against serine-rich antigen of E. histolytica (Optimum S kit; Merlin Diagnostika, Bornheim-Hersel, Germany). Other ELISA kits for antigen detection include the Entamoeba CELISA PATH kit (Cellabs, Brookvale, Australia), which uses a monoclonal antibody specific for lectin of E. histolytica, and the ProSpecT EIA (Remel Inc.; previously manufactured by Alexon-Trend, Inc., Sunnyvale, CA), which detects E. histolytica-specific antigen in fecal specimens (Table 3). In addition to the abovementioned clinical assays, research-based detection tests have included the use of monoclonal antibodies against a lectin-rich surface antigen (132), a lipophospholglycan (113), a 170-kDaadherence lectin amebic antigen detected in saliva (2), and an uncharacterized antigen (209). The E. histolytica TechLab kit was designed in 1993 to detect specifically E. histolytica in feces (72, 76). This antigen detection test captures and detects the parasite’s Gal/GalNAc lectin in stool samples. The lectin is conserved and highly immunogenic, and because of the antigenic differences in the lectins of E. histolytica and E. dispar, the test enables specific identification of the disease-causing E. histolytica. The level of detection of amebic antigens is quite high, requiring approximately 1,000 trophozoites per well (78, 113). However, this test suffers from the disadvantage that the antigens detected are denatured by fixation of the stool sample, therefore limiting testing to fresh or frozen samples. Nevertheless, this test has demonstrated good sensitivity and specificity for detection of E. histolytica antigen in stool specimens of people suffering from amebic colitis and asymptomatic intestinal infection (72, 76, 80). The TechLab ELISA for detection of E. histolytica antigen in stool specimens from people suffering from diarrhea was shown to have an excellent correlation with nested PCR (72), and in other studies this test was found to be more sensitive (80 to 94%) and specific (94 to 100%) than microscopy and culture (76, 80). In contrast, Gonin and Trudel (65) found that TechLab ELISA was less sensitive than microscopy (concen- 518 FOTEDAR ET AL. CLIN. MICROBIOL. REV. TABLE 3. Commercially available antigen assays for the diagnosis of amebiasis Sensitivity, % (reference) Specificity, % (reference) 96.9–100,b 14.2 (61),c 87.5 (76),d 86 (76),e 71 (201), 95 (80),f 79 (153)g 95–100b 94.7–100,b 98.3 (61),c 100 (76),d 98 (76),e 100 (201), 93.0 (80),f 96 (153)g 93–100b Optimum S Entamoeba histolytica antigen ELISAa 100 (134) NPh Triage parasite paneli 96.0 (58),j 68.3 (133),k 100 (167)l 87,m 54.5 (61),c 78 (128)n 99.1 (58),j 100 (133),k 100 (167)l 99,m 94 (61),c 99 (128)n Test TechLab E. histolytica IIa Entamoeba CELISA-PATHa ProSpecT Entamoeba histolytica microplate assayi Manufacturer Detection limit TechLab, Blacksburg, VA 0.2–0.4 ng of adhesion per well Cellabs Pty Ltd., Brookvale, Australia Merlin Diagnostika, Berheim-Hersel, Germany BIOSITE Diagnostics, San Diego, CA REMEL Inc., Lenexa, KSo 0.2–0.4 ng of adhesion per well Not given Not given 40 ng/ml of E. histolyticaspecific antigen a Specific for E. histolytica. Sensitivity and specificity compared to culture/zymodeme, as cited by the manufacturer. Sensitivity and specificity compared to culture and microscopy. d Compared to isoenzyme analysis. e Compared to culture. f Compared to culture and microscopy. g Compared to real-time PCR. h NP, not published. i Cannot distinguish between E. histolytica and E. dispar. j Compared to permanent staining with trichrome and modified acid-fast stains. k Compared to ProSpecT Entamoeba histolytica microplate assay. l Compared to ovum and parasite examination. m As mentioned by the manufacturer, related to ovum and parasite identifications. n Compared to microscopy (wet mounts and concentration). o Previously manufactured by Alexon-Trend, Inc., Sunnyvale, CA. b c tration and permanent staining) and PCR in differentiating E. histolytica from E. dispar from fecal samples. In another comparative study on the use of ELISA and PCR for the detection of E. histolytica and E. dispar, PCR was found to be 100 times more sensitive than ELISA for the differentiation of the two species (113). This kit has been discontinued by the manufacturer and has been replaced by a second-generation TechLab E. histolytica II kit, which has been found to be sensitive (86% to 95%) and specific (93% to 100%) compared with microscopy (wet mount with 0.9% saline and Lugol’s iodine) and culture for identification of E. histolytica as a screening method in areas of Bangladesh with high endemicity (76, 80). The TechLab II ELISA compared to real-time PCR as a reference test also demonstrated good levels of sensitivity and specificity for the diagnosis of E. histolytica (71 to 79% and 96 to 100%, respectively) (153, 201). However, another study demonstrated much lower sensitivity (14.3%) and specificity (98.4%) for E. histolytica compared to culture and zymodeme identification (61). In addition to this, cross-reactivity of samples is an issue with this assay, since samples positive by PCR for E. dispar may give false-positive results (55, 201). No specific antigen tests are available for the detection of E. dispar and E. moshkovskii from clinical samples. The TechLab E. histolytica II kit can also be used for the detection of E. histolytica lectin antigen in the serum and liver abscess pus of patients with liver disease (79). In Bangladesh, 96% (22/23) and 100% (3/3) of patients with ALA had detectable levels of lectin antigen in their serum and liver abscess pus samples, respectively, before treatment with metronidazole. However, the sensitivities of this method were only 33% (32/ 98) and 41% (11/27) for serum and liver abscess pus, respectively, after a few days of treatment with metronidazole (79), which is probably associated with a decrease in the amount of antigen in the serum or pus following therapy. Results of antigen detection using both the TechLab kits suggest that more specific and sensitive diagnostic tests, such as PCR, are needed to establish the actual worldwide distribution of E. histolytica and E. dispar (61, 65). Detection of specific antigens of E. histolytica and E. dispar in feces by ELISA could be useful for clinical and epidemiological studies where molecular assays cannot be used (76, 78). Importantly, of the four diagnostic methods, i.e., antigen detection, antibody detection, microscopy, and isoenzyme analysis, only antigen detection using ELISA is both rapid and technically simple to perform and can be used in laboratories that do not have molecular facilities, thus making it appropriate for use in the developing world, where amebiasis is most prevalent. In all cases, the combination of serological tests with detection of the parasite (by antigen detection or PCR) offers the best approach to diagnosis. However, as reported by Mirelman et al. (113), improvements in automation and simplification of PCR procedures for clinical sampling directly from feces suggest that a comparison with ELISA needs to be performed. The ProSpecT EIA (Remel Inc.) is a microplate EIA which detects both E. histolytica and E. dispar. However, this assay cannot differentiate between E. histolytica and E. dispar. The advantage of this test is that it can be performed on fresh, frozen, or Cary-Blair specimens but not on formalin-fixed fecal samples. The sensitivity of the ProSpecT EIA was compared with that of conventional microscopy (using wet mounts and concentration methods) for the diagnosis of E. histolytica/E. dispar, and a sensitivity of 78% and specificity of 99% were reported (128). In another study, by Gatti et al. (61), the reported sensitivity and specificity of ProSpecT ELISA were LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 54.5% and 94%, respectively, compared to culture and zymodeme identification for E. histolytica/E. dispar. Immunochromatographic Assays The Triage parasite panel (TPP) (Biosite Diagnostic Inc., San Diego, CA) is the first immunochromatographic assay for the simultaneous detection of antigens specific for Giardia lamblia, E. histolytica/E. dispar, and Cryptosporidium parvum. The immunochromatographic strip used in this assay is coated with monoclonal antibodies specific for the 29-kDa surface antigen (E. histolytica/E. dispar), alpha-1-giardin (G. lamblia), and protein disulfide isomerase (C. parvum). By using specific antibodies, antigens specific for these organisms from the stool samples are captured and immobilized on a membrane. A high sensitivity (96% to 100%) and specificity (99.1% to 100%) of the TPP kit compared to microscopy (stool ovum and parasite examination) for E. histolytica/E. dispar were reported (58, 167). In another study, although the specificity of the Triage kit was high (100%), the sensitivity was low (68.3%) compared to that of the ProSpecT test (133). A recent study from Sweden has compared the TPP test with PCR and demonstrated a low sensitivity for TPP assay (106). The advantage of the TPP method is that it can be performed in approximately 15 min with fresh or frozen, unfixed human fecal specimens. The TPP provides diagnostic laboratories with a simple, convenient alternative method for performing simultaneous, discrete detection of Giardia-, Cryptosporidium-, and E. histolytica/E. dispar-specific antigens in patient fecal specimens. However, the inability of this test to differentiate between E. histolytica, E. dispar, and E. moshkovskii makes it not a method of choice for the diagnostic laboratory. Only fresh or fresh-frozen unpreserved stool samples can be tested by the Triage assay, and to maintain the integrity of the specimens, they need to be frozen or transported to the laboratory for testing as soon as possible after collection. 519 methods for the isolation of parasitic DNA from feces have been developed, which enhance detection and increase the sensitivity of the PCR assay when used directly from clinical samples. Recent approaches that attempt to eliminate fecal inhibitors which copurify with the DNA consist of purification methods prior to DNA extraction and/or direct removal of inhibitors during DNA extraction (83, 206). However, many of these methods include multiple steps that are time-consuming and expensive, and so only a limited number of samples can be processed at a time. The QIAamp DNA stool kit (QIAGEN, Hilden, Germany) has proved to be a successful and reliable method for the recovery of DNA from fecal material (196). Improvement in reproducibility and sensitivity has been obtained by modifying the extraction kit method by optimizing the duration and temperature of proteinase K digestion and by adding an additional wash step prior to DNA elution (153). Transportation of fecal samples containing parasites at ambient temperatures may result in the rapid degeneration of parasite DNA, especially for highly labile stages such as trophozoites. Consequently, the sensitivity of DNA assays using unpreserved fecal specimens is time dependent (105). Specimens may be preserved by refrigeration or in PVA fixative, SAF, or formalin. PVA and SAF preserve trophozoites and cysts, and formalin preserves cysts for examination in wet mounts. However, methods of fixation of feces with fixatives or preservatives may result in a decreased sensitivity of PCR with time (143, 192). A few groups have, however, shown good results using formalin-fixed samples for PCR (147, 157). Ethanol is a simple transport medium that preserves amebic DNA. The most widely used reagent for the preservation of fecal samples is 10% buffered formalin solution (120); however, this solution is reported to hamper product amplification by PCR because of the interfering nature of the fixative, which perfuses the organisms and reacts with DNA (143). Consequently, freezing a fresh fecal specimen at ⫺20°C before extraction of DNA is a better strategy, as it does not affect the sensitivity of the molecular assays (52, 105, 123). DNA-BASED DIAGNOSTIC TESTS DNA-based assays are limited to research laboratories and clinical laboratories in developed countries. In most tropical and subtropical countries where amebiasis is responsible for significant morbidity and mortality, the diagnosis is still made by microscopic examination due to the lack of facilities to conduct DNA-based tests. Complexity of Fecal Samples Over the last decade, many DNA-based methods for the detection of viral, bacterial, and parasite DNA have been published. Fecal samples are considered to be among the most complex specimens for direct PCR testing because of the presence of PCR inhibitors, such as heme, bilirubins, bile salts, and complex carbohydrates, which are often coextracted along with pathogen DNA (85). Therefore, optimization of the fecal DNA extraction procedure is critical to the success of PCR studies. In the past, the isolation of DNA directly from fecal samples was problematic and laborious. Recently, simple and effective Methods of DNA Extraction Manual methods. Earlier methods of DNA extraction relied on the culture of microscopy-positive fecal samples in Robinson’s medium followed by subsequent extraction of DNA from cultured trophozoites by the phenol-chloroform method (135, 181, 183). Later methods included direct extraction of DNA from microscopy-positive fecal samples (151), and with modification this proved to be a successful strategy for formalinether-concentrated samples (3, 116, 147, 148, 179) and for unpreserved stool samples and stool samples stored at 4°C or ⫺20°C (123, 141). QIAamp tissue kit spin columns (QIAGEN, Hilden, Germany) have been used for the purification of DNA from microscopy-positive samples stored at ⫺20°C in phosphate-buffered saline (196, 199) and for DNA isolation using other modifications (such as treatment with 2% polyvinylpolypyrrolidone [Sigma]) which improve the sensitivity of the PCR (197). The use of the QIAamp stool kit for the extraction of DNA from fecal samples was a major advance, and this has proven to be the most widely accepted method for DNA ex- 520 FOTEDAR ET AL. traction. Formalin-fixed fecal samples have also been used for DNA extraction without a reduction in the ability to perform amplification of E. histolytica and E. dispar (143). Other kits used for the direct extraction of DNA from fecal samples include the XTRAX DNA extraction kit (Gull Laboratories, Salt Lake City, UT) (50), the Extract MasterFaecal DNA extraction kit (Epicenter Biotechnologies, WI), and the Genomic DNA Prep Plus kit (A&A Biotechnology, Gdansk, Poland) (118). Of the methods for DNA extraction from feces, those based on the QIAamp stool kit (QIAGEN) have predominated (50, 51, 54, 65, 82, 129, 196, 199) and are now used widely in clinical research laboratories in developed nations, as they minimize the extraction time and the DNA can be extracted directly from the feces without the need to culture the parasites. Automated methods. A number of automated methods are available for the extraction of DNA from fecal samples. The MagNA Pure LC workstation is an automated “walkaway” system for nucleic acid extraction. With a MagNA Pure LC DNA isolation kit, genomic DNA from organisms lysed in buffer containing guanidine isothiocyanate is bound to magnetic glass particles under chaotropic conditions. The magnetic particles are washed to remove unbound substances and impurities. The washed DNA is eluted from the magnetic particles under conditions of low salt concentration and elevated temperature. MagNA Pure LC total nucleic acid isolation kit (Roche Applied Sciences) extraction technology has successfully been used for DNA extraction from microsporidia in fecal specimens (208) and for extraction of bacterial and viral DNA from clinical samples (87, 101). However, a reduction in PCR sensitivity was reported using DNA extracted from wholeblood samples for detection of viral pathogens (161). This reduction of the PCR activity was related to problems with retrieval of DNA from the magnetic glass particles, where up to 60% of the DNA could not be retrieved by use of the MagNA Pure extraction system (161). Other available automated methods include the QIAGEN automated BioRobot M48 (QIAGEN) and Nuclisens easyMAG (bioMerieux, Marcy, l⬘Etoile, France), but so far there have been no published protocols using these automated systems for the successful recovery of Entamoeba DNA from feces. Conventional PCR PCR-based approaches are the method of choice for clinical and epidemiological studies in the developed countries (27, 71, 81, 212) and have been strongly endorsed by the WHO. Entamoeba histolytica can be identified in a variety of clinical specimens, including feces, tissues, and liver abscess aspirate (188). PCR of the small-subunit rRNA gene (18S rDNA) is reported to be approximately 100 times more sensitive than the best ELISA kit currently available (113, 192). Edman et al. (47) used PCR to amplify the gene which encodes the 125-kDa surface antigen, and this was subsequently adapted to distinguish among Entamoeba species by restriction digestion (187). The initial studies by Edman et al. (47) and Tannich and Burchard (187) were performed with DNA extracted from laboratory-maintained control isolates of Entamoeba species. PCR was subsequently used in an epidemiological study of E. histolytica/E. dispar infection, using CLIN. MICROBIOL. REV. DNA extracted from cultured trophozoites from feces, and the PCR was performed using primers specific for highly repetitive sequences present in pathogenic and nonpathogenic E. histolytica (now identified as E. dispar) strains defined previously through their respective isoenzyme patterns (59, 151). There is now a wide variety of PCR methods, targeting different genes, which have been described for detection and differentiation of the three Entamoeba species (Table 4). The consistent genetic diversity detected between the 18S rDNAs of E. histolytica and E. dispar initiated the use of 18S rDNA as a target for differentiation of the two species (31, 32, 36, 138). DNA extracted from laboratory-cultured trophozoites and DNA recovered directly from microscopy-positive fecal samples using the manual and automated methods were tested, and the PCR methods proved to be highly sensitive and specific for detecting Entamoeba DNA (33, 34, 82, 116, 117, 141, 142, 192, 196, 199). PCR assays targeting 18S rDNA are widely used for the detection and differentiation of Entamoeba species, as these targets are present on multicopy, extrachromosomal plasmids in the amebae (15), making the 18S rDNA more easily detected than a DNA fragment of a single-copy gene. The successful use of PCR in studying the epidemiology of Entamoeba infection was first reported by Acuña-Soto et al. (3). Those authors used DNA extracted directly from feces, avoiding the need to culture trophozoites, and the primers were targeted to amplify the extrachromosomal circular DNA. This gene target was subsequently used by other researchers (6, 24). This PCR target, with colorimetric detection of the product, was also used with DNA extracted from fecal samples, using a modification of the QIAGEN kit (6, 196, 199). Primers for the 29-kDa/30-kDa antigen gene have been used for distinguishing among pathogenic and nonpathogenic species of Entamoeba using conventional PCR (183). In research laboratories, this target has been used for analyses of microscopy-positive feces which have been cultured in the laboratory (135, 181) as well as formalin-fixed fecal samples (146, 147, 148, 179). Other gene targets for PCR include two protein-encoding genes which have been shown to exhibit polymorphism in the coding region. These are the serine-rich E. histolytica protein (SREPH) gene (174) and the chitinase gene (38). SREPH as a target was reported for the amplification of DNA recovered from laboratory cultures and microscopypositive feces concentrated by the zinc-sulfate gradient floatation technique (141). A nested SREPH PCR approach was recently used to investigate E. histolytica diversity in a single human population, using DNA extracted from microscopypositive feces (11). PCR using the cysteine proteinase gene and actin genes as targets was also used to study the epidemiology of amebiasis (54). In addition, a novel PCR assay based on the E. histolytica hemolysin gene HLY6 (hemoPCR) was developed for the detection of E. histolytica DNA with fecal and ALA samples and was shown to have 100% sensitivity and specificity (216). In an attempt to increase the sensitivity of the PCR assay, a nested multiplex PCR was developed for the simultaneous detection and differentiation of E. histolytica and E. dispar from DNA extracted from microscopy-positive fecal samples (50, 72, 89, 97). Utilizing this multiplex technique, the sensitivity and specificity were increased to 94% and 100%, respectively (123). This method has been successfully used for detection of E. LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 521 TABLE 4. Primers used for conventional PCR for E. histolytica, E. dispar, and E. moshkovskii PCR assay Single tube Gene target Amplification product (bp) Primers Sequence (5⬘33⬘) M17 482 P1-S17a P1-AS20a GCAACTAGTGTTAGTTA CCTCCAAGATATGTTTTAAC 63, 187, 214 30-kDa protein 100 P11a P12a GGAGGAGTAGGAAAGTTGAC TTCTTGCAATTCCTGCTTCGA 69, 119, 135, 146, 147, 148, 157, 179, 181, 182, 183, 214, 216 101 P13b P14b AGGAGGAGTAGGAAAATTAGG TTCTTGAAACTCCTGTTTCTAC 145 EHP1a EHP2a TCAAAATGGTCGTCGTCTAGGC CAGTTAGAAATTATTGTACTTTGTA 133 EHNP1b EHNP2b GGATCCTCCAAAAAATAAAGT CCACAGAACGATATTGGATACC 876 Psp Fa Psp Ra GGCCAATTCATTCAATGAATTGAG CTCAGATCTAGAAACAATGCTTCTC NPspFb NPspRb GGCCAATTTATGTAAGTAAATTGAG CTTGGATTTAGAAACAATGTTTCTTC 145 P1a P2a TCAAAATGGTCGTCGTCTAGGC CAGTTAGAAATTATTGTACTTTGTA 133 NP1b NP2b GGATCCTCCAAAAAATAAAGTTT ATGATCCATAGGTTATAGCAAGACA 1,950 RD5c GGAAGCTTATCTGGTTGATCCTGCC AGTA GGGATCCTGATCCTTCCGCAGGTTCAC CTAC 141, 214 82, 192 DNA highly repetitive sequences Small-subunit rRNA Extrachromosomal circular DNA Small-subunit rRNA c RD3 Nested 151 32, 105, 106, 116, 117 118, 119, 141, 142, 196 3, 6, 24, 214 880 Eh5a Eh3a GTACAAAATGGCCAATTCATTCAATG CTCAGATCTAGAAACAATGCTTCTCT 880 Ed5b Ed5b GTACAAAGTGGCCAATTTATGTAAGT ACTTGGATTTAGAAACAATGTTTCTTC Hemolysin gene (HLY6) LSU rRNA 256 EH6Fa Eh6Ra GACCTCTCCTAATATCCTCGT GCAGAGAAGTACTGTGAAGG 216 30-kDa protein 374 HFc HRc AAGAAATTGATATTAATGAATATA ATCTTCCAATTCCATCATCAT 86 Cysteine proteinase 242 Ehcp6Fa Ehcp6Ra GTTGCTGCTGAAGAAACTTG GTACCATAACCAACTACTGC 54 Actin gene 300 Act3Fc Act5Rc GGGACGATATGGAAAAGATC CAAGTCTAAGAATAGCA TGTG Small-subunit rRNA 135 EH1a ED1b EHD 2c GTACAAAATGGCCAATTCATTCAATG TACAAAGTGGCCAATTTATGTAAGTA ACTACCAACTGATTGATAGATCAG 27, 65 Small-subunit rRNA 900 EH-1c EH-2c TTTGTATTAGTACAAA GTA(A/G)TATTGATATACT 11, 72, 97, 214, 216 Small-subunit rRNA Duplex single step Reference(s) Continued on following page 522 FOTEDAR ET AL. CLIN. MICROBIOL. REV. TABLE 4—Continued PCR assay Gene target Amplification product (bp) Sequence (5⬘33⬘) Reference(s) EHP-1a EHP-2a EHN-1b EHN-2b AATGGCCAATTCATTCAATG TCTAGAAACAATGCTTCTCT AGTGGCCAATTTATGTAAGT TTTAGAAACAATGTTTCTTC E1c E2c TGCTGTGATTAAAACGCT TTAACTATTTCAATCTCGG 427 Eh-La Eh-Ra ACATTTTGAAGACTTTATGTAAGTA CAGATCTAGAAACAATGCTTCTCT 195 Ed-Lb Ed-Rb GTTAGTTATCTAATTTCGATTAGAA ACACCACTTACTATCCCTACC 823 Outer 1Fc Outer 1Rc GAAATTCAGATGTACAAAGA CAGAATCCTAGAATTTCAC 447 Eh1a Eh2a AAGCATTGTTTCTAGATCTG CACGTTAAAAGAGGTCTAAC 603 Ed1b Ed2b AAACATTGTTTCTAAATCCA ACCACTTACTATCCCTACC 553 SRPEh F SRPEh Rc CCTGAAAAGCTTGAAGAAGCTG AACAATGAATGGACTTGATGCA 452 nSRPEh Fa nSRPEh Ra TGAAGATAATGAAGATGATGAAGATG TATTATTATCGTTATCTGAACTACTT CCTG 567 SRPEd Fb SRPEd Rb GTAGTTCATCAAACACAGGTGA CAATAGCCATAATGAAAGCAA Small-subunit rRNA 260 Em-1d Em-2d nEm-1d nEm-2d CTCTTCACGGGGAGTGCG TCGTTAGTTTCATTACCT GAATAAGGATGGTATGAC AAGTGGAGTTAACCACCT 8, 52, 130, 171 Small-subunit rRNA 166 752 EntaFd Ehrd ATGCACGAGAGCGAAAGCAT GATCTAGAAACAATGCTTCTCT 71 580 Edrd Emrd CACCACTTACTATCCCTACC TGACCGGAGCCAGAGACAT 132 EhP1a EhP2a CGATTTTCCCAGTTAGAAATTA CAAAATGGTCGTCGTCTAGGC 96 EdP1b EdP2b ATGGTGAGGTTGTAGCAGAGA CGATATTGGATACCTAGTACT 900 900 Small-subunit rRNA Small-subunit rRNA Multiplex Primers Tandem repeats in extrachromosomal circular rDNA 1,076 50, 51, 129 89 141 123 a Specific for E. histolytica. Specific for E. dispar. c Specific for E. histolytica and E. dispar. d Specific for E. moshkovskii. b histolytica and E. dispar in formalin-fixed stool samples (129). A PCR solution hybridization enzyme-linked immunoassay targeting extrachromosomal circular DNA from E. histolytica and E. dispar with specific primers and a biotin-conjugated probe was shown to be sensitive for detection and differentiation of the two Entamoeba species in clinical samples (7, 11, 199). PCR for the detection of E. histolytica DNA from liver abscess samples was first employed using the gene encoding the 30-kDa antigen, and 100% sensitivity was reported (182). In another study, PCR performed on liver samples demonstrated only 33% sensitivity for the presence of E. histolytica using primers specific for 18S rDNA of E. histolytica, whereas the second pair, specific for the 30-kDa antigen gene (182), showed a sensitivity of 100% (215). Direct amplification for detection of E. histolytica DNA (without the extraction of DNA) from ALA pus was reported using 10 different previously published primer pairs (used for amplification of E. his- LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 tolytica from liver and stool samples) (214). Of the 10 different primer pairs tested, two pairs, i.e., P1-P2, targeting extrachromosomal circular DNA of E. histolytica (3), and P11-P12, targeting the 30-kDa antigen gene (182), gave 100% sensitivity. Another PCR assay (hemo-PCR), based on the novel hemolysin gene HLY6 of E. histolytica, was analyzed for the liver abscess samples. The hemo-PCR gave a positive result for 89% of ALA samples, compared to 77% and 28% for the 30-kDa antigen gene and 18S rDNA, respectively (216). The hemoPCR was found to be a valuable diagnostic tool for identification of E. histolytica in liver and fecal samples. For the identification of E. moshkovskii in fecal specimens, a riboprinting method was first reported by Haque et al. (72). Subsequently, a PCR for the identification of E. moshkovskii in fecal samples was developed as a nested 18S rDNA PCR followed by restriction endonuclease digestion (8). This method has a high sensitivity and specificity (100%) with DNA extracted directly from stool samples using the QIAGEN stool extraction kit (52). Although PCR-based methods have been successfully used for detection of all three Entamoeba species, their application in routine diagnosis is still very limited. The introduction of PCR-based methods has been hindered by difficulties in DNA extraction from fecal samples (115). Moreover, the amplification and detection of DNA are time-consuming and expensive. The shortcomings of PCR-based assays become apparent during practical applications. The generation of nonspecific DNA fragments from environmental and clinical samples poses a significant problem that often results in false-positive results. Real-Time PCR Real-time PCR is a new and a very attractive methodology for laboratory diagnosis of infectious diseases because of its characteristics that eliminate post-PCR analysis, leading to shorter turnaround times, a reduction in the risk of amplicon contamination of laboratory environments, and reduced reagent costs (99). This approach allows specific detection of the amplicon by binding to one or two fluorescencelabeled probes during PCR, thereby enabling continuous monitoring of amplicon (PCR product) formation throughout the reaction. An important aspect of real-time PCR is enhanced sensitivity compared to conventional PCR, with an ability to detect 0.1 cell per gram of feces (18). In addition, real-time PCR is a quantitative method and allows the determination of the number of parasites in various samples. Distinct real-time PCR protocols have recently been published for identification and differentiation of E. histolytica from E. dispar (Table 5). These include a Light Cycler assay utilizing hybridization probes to detect amplification of the 18S rDNA from fecal samples (18, 27) and two TaqMan assays, one targeting the 18S rDNA (98, 195, 198) and another targeting the episomal repeats, using DNA extracted from fecal samples collected from primates and humans (198, 200). A molecular beacon-based real-time PCR targeting 18S rDNA of E. histolytica for use on fecal and ALA specimens was described (153). A SYBR green real-time assay targeting the 18S rDNA was described by Qvarnstrom et al. (139). The sequences selected in the majority of these real-time studies have included rDNA as the target for PCR. A recent 523 evaluation of three real-time PCR assays, focusing on the weaknesses and strengths of each assay and their usefulness for clinical laboratory diagnosis, was published by Qvarnstrom et al. (139). This study highlighted major differences in detection limits and assay performance that were observed among the evaluated tests. Two of the assays in this study could not reliably distinguish E. histolytica from E. dispar, including the Light Cycler assay (17) and the TaqMan assay targeting episomal repeats (198, 200). A multiplex real-time assay was subsequently developed for detection of different intestinal parasites with 100% sensitivity and specificity (195). This assay allows detection of E. histolytica, G. lamblia, and C. parvum and offers the possibility of introducing DNA detection in the routine diagnosis of intestinal parasitic infections. The implementation of such multiplex assays and the development of automated DNA isolation procedures could have a tremendous impact on routine parasitology practice. Accurate diagnosis necessitates that the same reaction conditions are used for a standard and for the sample. Duplex or multiplex approaches with internal standardization provide a solution for this problem. A real-time PCR for detection of E. moshkovskii in clinical samples has not yet been reported. Further research is therefore required to develop these methods for the detection of E. moshkovskii. Although real-time PCR assays are sufficiently sensitive to detect a single cell, the limited number of probes that can be applied in one reaction hinders its utility for confident multitarget detection and genotyping analysis (139). The overabundance of one species to be detected in a real-time PCR can mask the ability to detect a second species when the same amplification primers are shared in a duplex assay. Such duplex (or multiplex) assays that distinguish between targets only by use of different probes are not suitable for simultaneous detection of more than one microorganism in a single reaction. In addition to this, real-time PCR is a costly procedure compared with fecal microscopy and antigen-based detection tests. Thus, poor regions of the world, where E. histolytica is most prevalent, will unfortunately be less likely to benefit from real-time PCR. Instead, this technique will be feasible primarily in clinical laboratories in developed countries that need to diagnose amebiasis in high-risk groups such as MSM, travelers, and immigrants from regions of the world where E. histolytica is endemic. Microarray Development One application that has revolutionized the postgenomic era is the development and use of microarray technology. DNA microarrays are a newly developed technology used for the detection of pathogens and are rapid and sensitive. The method involves four steps: extraction of genomic DNA, amplification of targeted DNA, hybridization of labeled DNA with oligonucleotide probes immobilized on a microarray, and data analysis. Oligonucleotide microarrays have been successfully applied to the diagnosis of many pathogens in recent years. Microarray-based approaches represent an attractive diagnostic tool for detection and identification of parasitic species in clinical and epidemiological investigations. The first oligonucleotide microarray developed for parallel detection of E. histolytica, E. dispar, G. lamblia assemblages A 524 FOTEDAR ET AL. CLIN. MICROBIOL. REV. TABLE 5. Published real-time PCR assays for E. histolytica and E. dispar Assay Light cycler TaqMan 1 TaqMan 2 Multiplex real time PCR Gene target 18S rRNA 18S rRNA Episomal repeats (SREPH gene) 18S rRNA Artus (Hamburg, Germany) real-time LC-PCR kitd SYBER green Molecular beacon 18S rRNA 18S rRNA Amplicon (bp) Primer or probe Sequence (5⬘33⬘) Nucleotide position Reference(s) Eh-S26Ca Ed-27 Cb Eh-Ed-AS25c GTACAAAATGGCCAATTCATTCAACG GTACAAAGTGGCCAATTTATGTAAGCA GAATTGATTTTACTCAACTCTAGAG 190–216 191–217 497–473 Eh/Ed-24LC-Red 640 Eh-Ed-25-Fc LC-Red-640-TCGAACCCCAATTCCTCGTTA TCCp FL-GCCATCTGTAAAGCTCCCTCTCCGAX 373–350 Eh-d-239Fa Ehd-88Rb ATTGTCGTGGCATCCTAACTCA GCGGACGGCTCATTATAACA 260–239 88–107 Histolytica-96Ta VIC-TCATTGAATGAATTGGCCATTTnonfluorescent quencher 217–197 Dispar-96Tb FAM-TTACTTACATAAATTGGCCACTTTGnonfluorescent quencher 218–194 Histolytica-50Fa Histolytica-132Ra CATTAAAAATGGTGAGGTTCTTAGGAA TGGTCGTCGTCTAGGCAAAATATT 50–76 132–109 Histolytica-78Ta FAM-TTGACCAATTTACACCGTTGATTTTCG GA-Eclipse Dark quencher 106–78 137 Dispar-1Fb Dispar-137Rb Dispar-33b GGATCCTCCAAAAAATAAAGTTTTATCA ATCCACAGAACGATATTGGATACCTAGTA HEX-UGGUGAGGUUGUAGCAGAGAUAUUA AUU-TAMRA 1–28 137–109 33–60 172 Ehd-239Fc Ehd-88Rc Histolytica-96Ta ATTGTCGTGGCATCCTAACTCA GCGGACGGCTCATTATAACA VIC-TCATTGAATGAATTGGCCATTTnonfluorescent quencher 260–239 88–107 217–197 195 230 E. histolytica NDe 55 877 PSP5a PSP3a GGCCAATTCATTCAATGAATTGAG CTCAGATCTAGAAACAATGCTTCTC 200–223 1076–1052 139 878 NPSP5b NPSP3b GGCCAATTTATGTAAGTAAATTGAG CTTGGATTTAGAAACAATGTTTCTTC 200–224 1077–1052 134 Ehfa Ehra Molecular beacon probe AACAGTAATAGTTTCTTTGGTTAGTAAAA CTTAGAATGTCATTTCTCAATTCAT Texas Red-GCGAGC-ATTAGTACAAAATGGCC AATTCATTCA-GCTCGC-dR Elle 307 231 83 18, 27, 139 400–376 104–238 98, 139, 195, 198 139, 198, 200 153 a Specific for E. histolytica. Specific for E. dispar. c Specific for E. histolytica and E. dispar. d Discontinued. e ND, not described. b and B, and C. parvum types 1 and 2 in a single assay with high specificity and sensitivity was reported by Wang et al. (205). In addition to distinguishing between the principal genotypes, this assay proved to be useful in detecting and differentiating E. moshkovskii from E. histolytica. However, this study was conducted with purified genomic DNA extracted from standard culture strains of different parasites (205). A microarray-based genotyping assay (comparative genomic hybridization) technique was later developed using sequenced genomic DNA clones from E. histolytica (HM-1:IMSS). This was the first genome-wide analysis of Entamoeba strains, and it revealed that this technology can be used to distinguish E. histolytica from E. dispar, to identify genes restricted to virulent strains, and to find potential genotypic-phenotypic associations (164). Microarray assays are at this time mostly a research tool and have seldom been used in the clinical diagnostic laboratory for detection and differentiation of parasites. However, with anticipated improvements in the microarray technology along with decreasing cost, it is possible that this technology may become placed at the forefront of parasitic research. Typing Methods The observed heterogeneity in virulence among strains, which may determine a strain’s ability to cause invasive disease, LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 525 TABLE 6. Primers for fingerprinting of E. histolytica, E. dispar, and E. moshkovskii Gene target Primer Sequence (5⬘33⬘) Reference(s) Strain-specific gene for E. histolytica SSG5 SSG3 GGTCTCAAAAAACCCACGAG CAAACGATAAAATCTAGCAAACTAC 33, 213 Serine gene for E. histolytica SREHP5 (EHF) SREPH3 (EHR) GCTAGTCCTGAAAAGCTTGAAGAAGCTG GGACTTGATGCAGCATCAAGGT 11, 33, 69, 146, 170, 213 nSREPH5 nSREPH3 TATTATTATCGTTATCTGAACTACTTCCTG TGAAGATAATGAAGATGATGAAGATG Serine gene for E. dispar SREHP (F) EDF SREHP (R) EDR AGATACTAAGATTTCAGTC CATAATGAAAGCAAAGAG 62 Chitinase gene for E. histolytica EHF EHR GGAACACCAGGTAAATGTATA TCTGTATTGTGCCCAATT 62, 69 Chitinase gene for E. dispar EDF EDR GGAACACCAGGTAAATGCCTT TCTGTATTGTGCCCAATT 62 Intergenic regions between actin gene and superoxide dismutase gene of E. histolytica and E. dispar EH/EDF EH/EDR TTGGTGGAATGTAGTCAACTG AAATCCGGCTTTACACATTCC 62 Locus 1-2 (E. histolytica and E. dispar) Dsp1 Dsp2 TTGAAGAGTTCACTTTTTATACTATA TAACAATAAAGGGGAGGG 136, 211, 212 Locus 5-6 (E. histolytica and E. dispar) Dsp5 Dsp6 CTATACTATATTCTT TTTATGTACTTCCC CTGAGAGCATTGTTTTTAAAGAA E. moshkovskii Arg gene EmR-1 EmR-2 GGCGCCTTTTTTACTTTATGG GCTAACAAGGCCAATCGATAAA has stimulated efforts through molecular epidemiological studies to determine whether some subgroups of E. histolytica are more likely than others to cause invasive disease. The parasite and host variables that contribute to the epidemiology of disease are not clear, and there is probably a complex interplay between host genetics, immunity, enteric flora, nutrition, and parasite genetics that occurs and contributes to disease. Whether there are subtypes of E. histolytica that have higher or lower virulence potential or a predilection for infection of certain organs is not known. The WHO has prioritized efforts to determine whether functional subgroups of E. histolytica exist, which may help address some of the unanswered questions surrounding the virulence of this parasite (210). The strain identification tools available to date are limited. Isoenzyme analysis provided the first markers (159), but it is now known that isoenzyme patterns are not fixed (see “Isoenzyme Analysis” above), and therefore many assigned zymodemes are unreliable (94). Intraspecific variation in E. histolytica was described by Clark and Diamond (33), and their studies on E. histolytica cultures (xenic and axenic) from different geographical areas of the world demonstrated the presence of extensive polymorphism in the SREHP gene (174) and the strain-specific gene (SSG) (25) (Table 6). The SREHP gene, which encodes an 8 immunodominant surface antigen, encodes contains 8- and 12-amino-acid tandem repeats. The existence of genetic differences among strains of E. histolytica which cause intestinal or liver disease has been demonstrated by the polymorphism exhibited in the SREHP gene using nested PCR performed on DNA extracted from stool and liver samples (11). However, these findings were later contradicted by Haghighi et al. (70). The SSG, which is a noncoding gene and contains tandemly repeated sequences ranging in size from 8 to 16 bp, has been used to differentiate strains by the number of repeats among strains of E. histolytica (25, 33). However, the complete absence of this locus in certain strains makes it a poor marker for intraspecies typing (162). The use of short tandem repeats that are linked to tRNA genes has been developed for genotyping of E. histolytica (9). This PCR-based genotyping of E. histolytica should allow the investigation of a possible association between the genotype and the outcome of infection (9). Other DNA markers to distinguish among isolates of E. histolytica include the chitinase gene, which encodes tandem repeats of a degenerate 7-amino-acid sequence (38, 69). Studies with the chitinase gene as a marker for studying populations of E. dispar have revealed the presence of different strains in Serum (acute infection) Serum (convalescent infection) Stool Stool Liver aspirate Antibody detection Culture and isoenzyme PCR-based assaysc Data are from references 77, 90, 91, 188, and 215. NA, not available. c PCR assay for cerebrospinal fluid (172). b NA 100 ⬎90 NA ⬎90 90–100 Gold standard ⬍25 75–85 ⬎90 Lower than that of antigen or PCR tests (problems in case of mixed infections) 90–100 ⬎85 Yes NA ALA fluid ⬎95 ⬎90 10–50 100 Specificity (%) 70–100 ⬎90 Usually negative 75 (late), 100 (early detection, before treatment) 100 (before treatment) ⬎95 65 (early) Antigen detection (ELISA) Stool Serum ALA ⬍10 ⬍25 Stool Liver abscess fluid a Sensitivity (%) Amebic colitis 25–60 NAb Specimen Microscopy (wet mount/permanent stain) Test 1–2 days 1–2 wk 2–3 h 3h 1–2 h Time required TABLE 7. Sensitivity and specificity of different laboratory tests for diagnosis of amebiasisa Yes Yes For certain antibody assays None Yes Technical expertise required High High, laborintensive Low Low Low Cost 526 FOTEDAR ET AL. CLIN. MICROBIOL. REV. LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES VOL. 20, 2007 different geographical areas by using DNA extracted from fecal samples (62, 140). Other typing methods targeting repeats include the use of microsatellite typing for detecting intra- and interspecies differences. Microsatellites are segments of DNA that consist of tandem repeats of very simple motifs such as (CT)n. The microsatellite typing is performed by amplifying the microsatellite by PCR using specific primers. Two minisatellite loci containing internal repeats, loci 1-2 and 5-6, have demonstrated variable polymorphism for E. histolytica and E. dispar (136, 211, 212), indicating that these loci have the potential to be used as molecular markers for investigating the epidemiology of the two Entamoeba species. Riboprinting has revealed considerable genetic divergence among isolates of E. moshkovskii (34). Detection of polymorphisms among the E. moshkovskii samples was studied using the EmR primers, and this attempt was only partially successful due to the differences in sequence of the primer-binding regions (8). With the increasing reports highlighting the recovery of E. moshkovskii from human stool samples, further studies involving typing of E. moshkovskii would be helpful for studying the epidemiology of this Entamoeba species. CONCLUSION Amebiasis caused by E. histolytica is one of the most common parasitic infections of mankind. Research on different aspects of the parasite, carried out in various parts of the world, particularly in the last two decades, has provided the basis for breakthroughs such as the discovery of the other closely related Entamoeba species, E. dispar and E. moshkovskii. The redefinition of E. histolytica, the redescription of E. dispar, and the recent studies of recovery of E. moshkovskii from patients have dramatically changed our understanding of the prevalence of different Entamoeba species in the community. This has motivated researchers to develop diagnostic tests capable of distinguishing and differentiating the three species of Entamoeba encountered in the clinical laboratory (Table 7). The diagnosis of invasive amebiasis is most commonly attempted by a combination of microscopy of a fecal specimen, serological testing, and, where indicated, by colonoscopy and biopsy of intestinal amebic lesions or by drainage of a liver abscess. The inability of microscopic examination to detect and differentiate the three species of Entamoeba has been highlighted. While serological testing remains a useful tool for the diagnosis of amebiasis, studies have demonstrated the unreliability of serological tests to correctly diagnose this infection in areas of endemicity, as the level of antiamebic antibodies remains elevated in serum for many years, resulting in the inability to distinguish past from current infection. Antigen detection using fecal ELISA is another diagnostic tool, which could be used in areas of endemicity where diarrheal diseases are common, and it has proven to be useful in the developing world, where most cases of amebiasis occur and where molecular techniques cannot be used because of cost constraints. However, the sensitivity of the fecal antigen test is about 100 times less than that of PCR, and in addition, several studies have highlighted low specificity because of cross-reaction with other Entamoeba species. The development of molecular tools, including PCR and real-time PCR, to detect E. histolytica, E. 527 dispar, and E. moshkovskii DNA in stool or liver abscess samples has led to major advances in making an accurate diagnosis during recent years. This in turn has assisted clinical diagnosis and the appropriate selection of patients for antiamebic therapy. In order to minimize undue treatment of individuals infected with other species of Entamoeba such as E. dispar and E. moshkovskii, efforts have been made for specific diagnosis of E. histolytica rather than treatment based on the microscopic examination of Entamoeba species in feces. In tropical and subtropical countries where amebiasis is endemic, the standard clinical approach is to treat all asymptomatic individuals with cysts in feces with an antiprotozoal agent. This approach to treatment causes indiscriminate use of antiamebic agents and has led to increased MICs of these therapeutic agents against E. histolytica, with a potential for resistant strains to appear (13, 110). These considerations suggest that positive fecal samples should be confirmed with reliable tests prior to initiation of therapy. Our understanding of different aspects of Entamoeba species and the disease they can cause makes this a very exciting and rewarding time for the study of amebiasis. The incorporation of many new technologies into the diagnostic laboratory will represent a challenge to us all. Such studies will then lead to a better understanding of the public health problem and measures to control the disease. REFERENCES 1. Abd-Alla, M. D., T. G. Jackson, and J. L. Ravdin. 1998. Serum IgM antibody response to the galactose-inhibitable adherence lectin of Entamoeba histolytica. Am. J. Trop. Med. Hyg. 59:431–434. 2. Abd-Alla, M. D., T. F. Jackson, S. Reddy, and J. I. Ravdin. 2000. Diagnosis of invasive amoebiasis by enzyme linked immunosorbent assay of saliva to detect amoebic lectin antigen and antilectin immunoglobulin G antibodies. J. Clin. Microbiol. 38:2344–2347. 3. Acuna-Soto, R., J. Samuelson, P. De Girolami, L. Zarate, F. MillanVelasco, G. Schoolnick, and D. Wirth. 1993. Application of the polymerase chain reaction to the epidemiology of pathogenic and nonpathogenic Entamoeba histolytica. Am. J. Trop. Med. Hyg. 48:58–70. 4. Adams, E. B., and I. N. MacLeod. 1977. Invasive amebiasis. I. Amebic dysentery and its complications. Medicine 56:315–323. 5. Adams, E. B., and I. N. MacLeod. 1977. Invasive amebiasis. II. Amebic liver abscess and its complications. Medicine 56:325–334. 6. Aguirre, A., D. C. Warhurst, F. Guhl, and I. A. Frame. 1995. Polymerase chain reaction-solution hybridization enzyme-linked immunoassay (PCRSHELA) for the differential diagnosis of pathogenic and non-pathogenic Entamoeba histolytica. Trans. R. Soc. Trop. Med. Hyg. 89:187–188. 7. Aguirre, A., S. Molina, C. Blotkamp, J. Verveij, T. Vinuesa, M. E. Valls, F. Guhl, A. Polderman, M. T. Jimenez de Anta, M. Corachan, A. GonzalezRuiz, I. A. Frame, and D. Warhurst. 1997. Diagnosis of Entamoeba histolytica and Entamoeba dispar in clinical specimens by PCR-SHELA. Arch. Med. Res. 28:285–287. 8. Ali, I. K., M. B. Hossain, S. Roy, P. F. Ayeh-Kumi, W. A. Petri, Jr., R. Haque, and C. G. Clark. 2003. Entamoeba moshkovskii infections in children, Bangladesh. Emerg. Infect. Dis. 9:580–584. 9. Ali, I. K., M. Zaki, and C. G. Clark. 2005. Use of PCR amplification of tRNA gene-linked short tandem repeats for genotyping Entamoeba histolytica. J. Clin. Microbiol. 43:5842–5847. 10. Allason-Jones, E., A. Mindel, P. Sargeaunt, and P. Williams. 1986. Entamoeba histolytica as a commensal intestinal parasite in homosexual men. N. Engl. J. Med. 31:353–356. 11. Ayeh-Kumi, P. F., I. M. Ali, L. A. Lockhart, C. A. Gilchrist, W. A. Petri, Jr., and R. Haque. 2001. Entamoeba histolytica: genetic diversity of clinical isolates from Bangladesh as demonstrated by polymorphisms in the serinerich gene. Exp. Parasitol. 99:80–88. 12. Balamuth, W. 1946. Improved egg yolk medium for cultivation of Entamoeba histolytica and other intestinal protozoa. Am. J. Clin. Pathol. 16: 380–384. 13. Bansal, D., R. Sehgal, Y. Chawla, R. C. Mahajan, and N. Malla. 2004. In vitro activity of antiamoebic drugs against clinical isolates of Entamoeba histolytica and Entamoeba dispar. Ann. Clin. Microbiol. Antimicrob. 21:3:27. 528 FOTEDAR ET AL. 14. Barnes, P. F., K. M. DeCock, T. N. Reynolds, and P. W. Tralla. 1987. A comparison of amebic and pyogenic abscess of the liver. Medicine (Baltimore) 66:472–483. 15. Bhattacharya, S., A. Bhattacharya, L. S. Diamond, and A. T. Soldo. 1989. Circular DNA of Entamoeba histolytica encodes ribosomal RNA. J. Protozool. 36:455–458. 16. Blanc, D., and P. G. Sargeaunt. 1991. Entamoeba histolytica zymodemes: exhibition of gamma and delta bands only of glucose phosphate isomerase and phosphoglucomutase may be influenced by starch content in the medium. Exp. Parasitol. 72:87–90. 17. Blessmann, J., I. K. Ali, P. A. Nu, B. T. Dinh, T. Q. Viet, A. L. Van, C. G. Clark, and E. Tannich. 2003. Longitudinal study of intestinal Entamoeba histolytica infections in asymptomatic adult carriers. J. Clin. Microbiol. 41:4745–4750. 18. Blessmann, J., H. Buss, P. A. Nu, B. T. Dinh, Q. T. Ngo, A. L. Van, M. D. Alla, T. F Jackson, J. I. Ravdin, and E. Tannich. 2002. Real-time PCR for detection and differentiation of Entamoeba histolytica and Entamoeba dispar in fecal samples. J. Clin. Microbiol. 40:4413–4417. 19. Blessmann, J., P. Van Linh, P. A. Nu, H. D. Thi, B. Muller-Myhsok, H. Buss, and E. Tannich. 2002. Epidemiology of amebiasis in a region of high incidence of amebic liver abscess in central Vietnam. Am. J. Trop. Med. Hyg. 66:578–583. 20. Blessmann, J., A. L. Van, and E. Tannich. 2006. Epidemiology and treatment of amebiasis in Hue, Vietnam. Arch. Med. Res. 37:270–272. 21. Blessmann, J., N. D. Khoa, A. L. Van, and E. Tannich. 2006. Ultrasound patterns and frequency of focal liver lesions after successful treatment of amoebic liver abscess. Trop. Med. Int. Health 4:504–508. 22. Boonyapisit, S., O. Chinapak, and U. Plengvait. 1993. Amoebic liver abscess in Thailand, clinical analysis of 418 cases. J. Med. Assoc. Thailand 76:243–246. 23. Braga, L. L., Y. Mendonca, C. A. Paiva, A. Sales, A. L. Cavalcante, and B. J. Mann. 1998. Seropositivity for and intestinal colonization with Entamoeba histolytica and Entamoeba dispar in individuals in northeastern Brazil. J. Clin. Microbiol. 36:3044–3045. 24. Britten, D., S. M. Wilson, R. McNerney, A. H. Moody, P. L. Chiodini, and J. P. Ackers. 1997. An improved colorimetric PCR-based method for detection and differentiation of Entamoeba histolytica and Entamoeba dispar. J. Clin. Microbiol. 35:1108–1111. 25. Burch, D. J., E. Li, S. Reed, T. F. Jackson, and S. L. Stanley. 1991. Isolation of a strain-specific Entamoeba histolytica cDNA clone. J. Clin. Microbiol. 29:696–701. 26. Caballero-Salcedo, A., M. Viveros-Rogel, B. Salvatierra, R. Tapia-Conyer, J. Sepulveda-Amor, G. Gutierrez, and L. Ortiz-Ortiz. 1994. Seroepidemiology of amebiasis in Mexico. Am. J. Trop. Med. Hyg. 50:412–419. 27. Calderaro, A., C. Gorrini, S. Bommezzadri, G. Piccolo, G. Dettori, and C. Chezzi. 2006. Entamoeba histolytica and Entamoeba dispar: comparison of two PCR assays for diagnosis in a non-endemic setting. Trans. R. Soc. Trop. Med. Hyg. 100:450–457. 28. Chadee, K., J. M. Smith, and E. Meerovitch. 1985. Entamoeba histolytica: electrophoretic isoenzyme patterns of strains and their virulence in the cecum of gerbils (Meriones unguiculatus). Am. J. Trop. Med. Hyg. 34:870– 878. 29. Clark, C. G. 1995. Axenic cultivation of Entamoeba dispar Brumpt 1925, Entamoeba insolita Geiman and Wichterman 1937 and Entamoeba ranarum Grassi 1879. J. Eukaryot. Microbiol. 42:590–593. 30. Clark, C. G., and L. S. Diamond. 1991. The Laredo strain and other Entamoeba histolytica-like amoebae are Entamoeba moshkovskii. Mol. Biochem. Parasitol. 46:11–18. 31. Clark, C. G., and L. S. Diamond. 1991. Ribosomal RNA genes of ‘pathogenic’ and ‘nonpathogenic’ Entamoeba histolytica are distinct. Mol. Biochem. Parasitol. 49:297–302. 32. Clark, C. G., and L. S. Diamond. 1992. Differentiation of pathogenic Entamoeba histolytica from other intestinal protozoa by riboprinting. Arch. Med. Res. 23:15–16. 33. Clark, C. G., and L. S. Diamond. 1993. Entamoeba histolytica: a method for isolate identification. Exp. Parasitol. 77:450–455. 34. Clark, C. G., and L. S. Diamond. 1997. Intraspecific variation and phylogenetic relationships in the genus Entamoeba as revealed by riboprinting. J. Eukaryot. Microbiol. 44:142–154. 35. Clark, C. G., and L. S. Diamond. 2002. Methods for cultivation of luminal parasitic protists of clinical importance. Clin. Microbiol. Rev. 15:329–341. 36. Cruz-Reyes, J. A., W. M. Spice, T. Rehman, E. Gisborne, and J. P. Ackers. 1992. Ribosomal DNA sequences in the differentiation of pathogenic and non-pathogenic isolates of Entamoeba histolytica. Parasitology 104:239–246. 37. Debnath, A., P. Das, M. Sajid, and J. H. McKerrow. 2004. Identification of genomic responses to collagen binding by trophozoites of Entamoeba histolytica. J. Infect. Dis. 190:448–457. 38. De la Vega, H., C. A. Specht, C. E. Semino, P. W. Robbins, D. Eichinger, D. Caplivski, S. Ghosh, and J. Samuelson. 1997. Cloning and expression of chitinases of Entamoebae. Mol. Biochem. Parasitol. 85:139–147. 39. Diamond, L. S. 1961. Axenic cultivation of Entamoeba histolytica. Science 134:336–337. CLIN. MICROBIOL. REV. 40. Diamond, L. S. 1968. Improved method for the monoxenic cultivation of Entamoeba histolytica Schaudinn, 1903 and E. histolytica-like amebae with trypanosomatids. J. Parasitol. 54:715–719. 41. Diamond, L. S. 1982. A new liquid medium for xenic cultivation of Entamoeba histolytica and other lumen dwelling protozoa. J. Parasitol. 68:958– 959. 42. Diamond, L. S., and I. L. Bartgis. 1970. Entamoeba moshkovskii: axenic cultivation. Exp. Parasitol. 28:171–175. 43. Diamond, L. S., and C. G. Clark. 1993. A redescription of Entamoeba histolytica Schaudinn, 1903 (Emended Walker, 1911) separating it from Entamoeba dispar Brumpt, 1925. J. Eukaryot. Microbiol. 40:340–344. 44. Diamond, L. S., C. G. Clark, and C. C. Cunnick. 1995. YI-S, a casein-free medium for axenic cultivation of Entamoeba histolytica, related Entamoeba, Giardia intestinalis and Trichomonas vaginalis. J. Eukaryot. Microbiol. 42: 277–278. 45. Diamond, L. S., D. R. Harlow, and C. C. Cunnick. 1978. A new medium for the axenic cultivation of Entamoeba histolytica and other Entamoeba. Trans. R. Soc. Trop. Med. Hyg. 72:431–432. 46. Dreyer, D. A. 1961. Growth of a strain of Entamoeba histolytica at room temperature. Texas Rep. Biol. Med. 19:393–396. 47. Edman, U., M. A. Meraz, S. Rausser, N. Agabian, and I. Meza. 1990. Characterization of an immuno-dominant variable surface antigen from pathogenic and nonpathogenic Entamoeba histolytica. J. Exp. Med. 172: 879–888. 48. Espinosa-Cantellano, M., G. Castañon Gutierrez, and A. Martı́nezPalomo. 1997. In vivo pathogenesis of Entamoeba dispar. Arch. Med. Res. 28:S204–S206. 49. Espinosa-Cantellano, M., A. González-Robles, B. Chávez, G. Castañon, C. Argüello, A. Lázaro-Haller, and A. Martı́nez-Palomo. 1998. Entamoeba dispar: ultrastructure, surface properties, and cytopathic effect. J. Eukaryot. Microbiol. 45:265–272. 50. Evangelopoulos, A., G. Spanakos, E. Patsoula, N. Vakalis, and N. A. Legakis. 2000. A nested, multiplex, PCR assay for the simultaneous detection and differentiation of Entamoeba histolytica and Entamoeba dispar in faeces. Ann. Trop. Med. Parasitol. 94:233–240. 51. Evangelopoulos, A., N. Legakis, and N. Vakalis. 2001. Microscopy, PCR and ELISA applied to the epidemiology of amoebiasis in Greece. Parasitol. Int. 50:185–189. 52. Fotedar, R., D. Stark, N. Beebe, D. Marriott, J. Ellis, and J. Harkness. 2007. PCR detection of Entamoeba histolytica, Entamoeba dispar, and Entamoeba moshkovskii in stool samples from Sydney, Australia. J. Clin. Microbiol. 45:1035–1037. 53. Freedman, L., S. E. Maddison, and R. Elsdon-Dew. 1958. Monoxenic culture of Entamoeba histolytica derived from human liver abscesses. S. Afr. J. Med. Sci. 23:9–12. 54. Freitas, M. A., E. N. Vianna, A. S. Martins, E. F. Silva, J. L. Pesquero, and M. A. Gomes. 2004. A single step duplex PCR to distinguish Entamoeba histolytica from Entamoeba dispar. Parasitology 128:625–628. 55. Furrows, S. J., A. H. Moody, and P. L. Chiodini. 2004. Comparison of PCR and antigen detection methods for diagnosis of Entamoeba histolytica infection. J. Clin. Pathol. 57:264–266. 56. Garcia, L. S., D. A. Bruckner, T. C. Brewer, and R. Y. Shimizu. 1982. Comparison of indirect fluorescent-antibody amoebic serology with counterimmunoelectrophoresis and indirect hemagglutination amoebic serologies. J. Clin. Microbiol. 5:603–605. 57. Garcia, L. S., and D. A. Bruckner. 1997. Diagnostic medical parasitology. ASM Press, Washington, DC. 58. Garcia, L. S., R. Y. Shimizu, and C. N. Bernard. 2000. Detection of Giardia lamblia, Entamoeba histolytica/Entamoeba dispar, and Cryptosporidium parvum antigens in human fecal specimens using the Triage parasite panel enzyme immunoassay. J. Clin. Microbiol. 38:3337–3340. 59. Garfinkel, L. I., M. Giladi, M. Huber, C. Gitler, D. Mirelman, M. Revel, and S. Rozenblatt. 1989. DNA probes specific for Entamoeba histolytica possessing pathogenic and nonpathogenic zymodemes. Infect. Immun. 57: 926–931. 60. Gathiram, V., and T. F. Jackson. 1987. A longitudinal study of asymptomatic carriers of pathogenic zymodemes of Entamoeba histolytica. S. Afr. Med. J. 72:669–672. 61. Gatti, S., G. Swierczynski, F. Robinson, M. Anselmi, J. Corrales, J. Moreira, G. Montalvo, A. Bruno, R. Maserati, Z. Bisoffi, and M. Scaglia. 2002. Amebic infections due to the Entamoeba histolytica-Entamoeba dispar complex: a study of the incidence in a remote rural area of Ecuador. Am. J. Trop. Med. Hyg. 67:123–127. 62. Ghosh, S., M. Frisardi, L. Ramirez-Avila, S. Descoteaux, K. SturmRamirez, O. A. Newton-Sanchez, J. I. Santos-Preciado, C. Ganguly, A. Lohia, S. Reed, and J. Samuelson. 2000. Molecular epidemiology of Entamoeba spp.: evidence of a bottleneck (demographic sweep) and transcontinental spread of diploid parasites J. Clin. Microbiol. 38:3815–3821. 63. Gomes, M. A., J. B. Pesquero, C. Furst, P. R. Valle, J. L. Pesquero, and E. F. Silva. 1999. An improved method to distinguish Entamoeba histolytica and Entamoeba dispar. Parasitology 119:359–362. 64. Goncalves, M. L., V. L. da Silva, C. M. de Andrade, K. Reinhard, G. C. da VOL. 20, 2007 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES Rocha, M. Le Bailly, F. Bouchet, L. F. Ferreira, and A. Araujo. 2004. Amoebiasis distribution in the past: first steps using an immunoassay technique. Trans. R. Soc. Trop. Med. Hyg. 98:88–91. Gonin, P., and L. Trudel. 2003. Detection and differentiation of Entamoeba histolytica and Entamoeba dispar isolates in clinical samples by PCR and enzyme-linked immunosorbent assay. J. Clin. Microbiol. 41:237–241. Gonzalez, C. R., A. Isibasi, V. Ortiz-Navarrete, J. Paniagua, J. A. Garcia, A. Ramirez, B. Salvatierra, R. Tapia, J. Sepulveda, G. Gutierrez, et al. 1995. Prevalence of antibodies against Entamoeba histolytica in Mexico measured by ELISA. Epidemiol. Infect. 115:535–543. Gonzalez-Ruiz, A., R. Haque, A. Aguire, G. Castanon, A. Hall, F. Guhl, G. Ruiz-Palacios, M. A. Miles, and D. C. Warhurst. 1994. Value of microscopy in the diagnosis of dysentery associated with invasive Entamoeba histolytica. J. Clin. Pathol. 47:236–239. Grundy, M. S. 1982. Preliminary observations using a multi-layer ELISA method for the detection of Entamoeba histolytica trophozoite antigens in stool samples. Trans. R. Soc. Trop. Med. Hyg. 76:396–400. Haghighi, A., S. Kobayashi, T. Takeuchi, G. Masuda, and T. Nozaki. 2002. Remarkable genetic polymorphism among Entamoeba histolytica isolates from a limited geographic area. J. Clin. Microbiol. 40:4081–4090. Haghighi, A., S. Kobayashi, T. Takeuchi, N. Thammapalerd, and T. Nozaki. 2003. Geographic diversity among genotypes of Entamoeba histolytica field isolates, J. Clin. Microbiol. 41:3748–3756. Hamzah, Z., S. Petmitr, M. Mungthin, S. Leelayoova, and P. Chavalitshewinkoon-Petmitr. 2006 Differential detection of Entamoeba histolytica, Entamoeba dispar, and Entamoeba moshkovskii by a single-round PCR assay. J. Clin. Microbiol. 44:3196–3200. Haque, R., I. K. Ali, S. Akther, and W. A. Petri, Jr. 1998. Comparison of PCR, isoenzyme analysis, and antigen detection for diagnosis of Entamoeba histolytica infection. J. Clin. Microbiol. 36:449–452. Haque, R., I. K. M Ali, C. G. Clark, and W. A. Petri, Jr. 1998. A case report of Entamoeba moshkovskii infection in a Bangladeshi child. Parasitol. Int. 47:201–202. Haque, R., I. K. M. Ali, and W. A. Petri, Jr. 1999. Prevalence and immune response to Entamoeba histolytica infection in preschool children in Bangladesh. Am. J. Trop. Med. Hyg. 60:1031–1034. Haque, R., I. M. Ali, R. B. Sack, B. M. Farr, G. Ramakrishnan, and W. A. Petri, Jr. 2001. Amebiasis and mucosal IgA antibody against the Entamoeba histolytica adherence lectin in Bangladeshi children. J. Infect. Dis. 183: 1787–1793. Haque, R., A. S. G. Faruque, P. Hahn, D. Lyerly, and W. A. Petri, Jr. 1997. Entamoeba histolytica and Entamoeba dispar infection in children in Bangladesh. J. Infect. Dis. 175:734–736. Haque, R., C. D. Huston, M. Hughes, E. Houpt, and W. A. Petri, Jr. 2003. Current concepts: amebiasis. N. Engl. J. Med. 348:1565–1573. Haque, R., K. Kress, S. Wood, T. F. G. H. Jackson, D. Lyerly, T. Wilkins, and W. A. Petri, Jr. 1993. Diagnosis of pathogenic Entamoeba histolytica infection using a stool ELISA based on monoclonal antibodies to the galactose specific adhesin. J. Infect. Dis. 167:247–249. Haque, R., N. U. Mollah, I. K. M. Ali, K. Alam, A. Eubank, D. Luerly, and W. A. Petri, Jr. 2000. Diagnosis of amebic liver abscess and intestinal infection with the TechLab Entamoeba histolytica II antigen detection and antibody tests. J. Clin. Microbiol. 38:3235–3239. Haque, R., L. M. Neville, P. Hahn, and W. A. Petri, Jr. 1995. Rapid diagnosis of Entamoeba infection by using Entamoeba and Entamoeba histolytica stool antigen detection kits. J. Clin. Microbiol. 33:2558–2561. Haque, R., and W. A. Petri, Jr. 2006. Diagnosis of amebiasis in Bangladesh. Arch. Med. Res. 37:273–276. Heckendorn, F., E. K. NⴕGoran, I. Felgar, P. Vounatsou, A. Yapi, A. Oettli, H. P. Marti, M. Dobler, M. Traore, K. L. Lohourignon, and C. Lengeler. 2002. Species-specific field-testing of Entamoeba spp. in an area of high endemicity. Trans. R. Soc. Trop. Med. Hyg. 96:521–528. Higgins, J. A., R. Fayer, J. M. Trout, L. Xiao, A. A. Lal, S. Kerby, and M. C. Jenkins. 2001. Real-time PCR for the detection of Cryptosporidium parvum. J. Microbiol. Methods 47:323–337. Hira, P. R., J. Iqbal, F. Al-Ali, R. Philip, S. Grover, E. D’Almeida, and A. A. Al-Eneizi. 2001. Invasive amebiasis: challenges in diagnosis in a non-endemic country (Kuwait). Am. J. Trop. Med. Hyg. 65:341–345. Holland, J. L., L. Louie, A. E Simor, and M. Louie. 2000. PCR detection of Escherichia coli 5O157:H7 directly from stools: evaluation of commercial extraction methods for purifying fecal DNA. J. Clin. Microbiol. 38:4108– 4113. Hooshyar, H., M. Rezaian, B. Kazemi, M. Jeddi-Tehrani, and S. SolaymaniMohammadi. 2004. The distribution of Entamoeba histolytica and Entamoeba dispar in northern, central, and southern Iran. Trans. Parasitol. Res. 94:96–100. Hougardy, N., A. Louahabi, and P. Goffinet. 2006. Direct and fast detection of methicillin resistant Staphylococcus aureus carriage by automated nucleic acid extraction and real time PCR. Pathol. Biol. 54:461–477. Hung, C. C., P. J. Chen, S. M. Hsieh, J. M. Wong, C. T. Fang, S. C. Chang, and M. Y. Chen. 1999. Invasive amoebiasis: an emerging parasitic disease in patients infected with HIV in an area endemic for amoebic infection AIDS. 13:2421–2428. 529 89. Hung, C. C., H. Y. Deng, W. H. Hsiao, S. M. Hsieh, C. F. Hsiao, M. Y. Chen, S. C. Chang, and K. E. Su. 2005. Invasive amebiasis as an emerging parasitic disease in patients with human immunodeficiency virus type 1 infection in Taiwan. Arch. Intern. Med. 165:409–415. 90. Huston, C. D., R. Haque, and W. A. Petri, Jr. 1999. Molecular-based diagnosis of Entamoeba histolytica infection. Expert Rev. Mol. Med. 22:1–11. 91. Huston, C. D., and W. A. Petri, Jr. 1999. Amebiasis: clinical implications of the recognition of Entamoeba dispar. Curr. Infect. Dis. Rep. 1:441–447. 92. Jackson, T. F., C. B. Anderson, and A. E. Simjee. 1984. Serological differentiation between past and present infection in hepatic amoebiasis. Trans. R. Soc. Trop. Med. Hyg. 78:342–345. 93. Jackson, T. F., V. Gathiram, and A. E. Simjee. 1985. Seroepidemiological study of antibody responses to the zymodemes of Entamoeba histolytica. Lancet i:716–719. 94. Jackson, T. F., and S. Suparsad. 1997. Zymodeme stability of Entamoeba histolytica and E. dispar. Arch. Med. Res. 28:304–305. 95. Jetter, A., B. Walderich, D. Britten, O. Mete, V. Goral, G. D. Burchard, and J. Ackers. 1997. An epidemiological study of Entamoeba histolytica and E. dispar infection in eastern Turkey using a colorimetric polymerase chain reaction. Arch. Med. Res. 28(Suppl.):319–321. 96. Jones, W. R. 1946. The experimental infection of rats with Entamoeba histolytica; with a method for evaluating the anti-amoebic properties of new compounds. Ann. Trop. Med. Parasitol. 40:130–140. 97. Katzwinkel-Wladarsch, S., T. Loscher, and H. Rinder. 1994. Direct amplification and differentiation of pathogenic and non-pathogenic Entamoeba histolytica DNA from stool specimens, Am. J. Trop. Med. Hyg. 51:115–118. 98. Kebede, A., J. J. Verweij, T. Endeshaw, T. Messele, G. Tasew, B. Petros, and A. M. Polderman. 2004. The use of real-time PCR to identify Entamoeba histolytica and E. dispar infections in prisoners and primary-school children in Ethiopia. Ann. Trop. Med. Parasitol. 98:43–48. 99. Klein, D. 2002. Quantification using real-time PCR technology: applications and limitations. Trends Mol. Med. 8:257–260. 100. Knappik, M., U. Borner, and T. Jelinek. 2005. Sensitivity and specificity of a new commercial enzyme-linked immunoassay kit for detecting Entamoeba histolytica IgG antibodies in serum samples. Eur. J. Clin. Microbiol. Infect. Dis. 24:701–703. 101. Knepp, J. H., M. A. Geahr, M. S. Forman, and A. Valsamakis. 2003. Comparison of automated and manual nucleic acid extraction methods for detection of enterovirus RNA. J. Clin. Microbiol. 41:3532–3536. 102. Knobloch, J., and E. Mannweiler. 1983. Development and persistence of antibodies to Entamoeba histolytica in patients with amebic liver abscess. Analysis of 216 cases Am. J. Trop. Med. Hyg. 32:727–732. 103. Kobayashi, S., E. Imai, H. Tachibana, T. Fujiwara, and T. Takeuchi. 1998. Entamoeba dispar: cultivation with sterilized Crithidia fasciculata. J. Eukaryot. Microbiol. 45:3S–8S. 104. Krogstad, D. J., H. C. Spencer, Jr., G. R. Healy, N. N. Gleason, D. J. Sexton, and C. A. Herron. 1978. Amebiasis: epidemiologic studies in the United States, 1971–1974. Ann. Intern. Med. 88:89–97. 105. Lebbad, M., and S. G. Svard. 2005. PCR differentiation of Entamoeba histolytica and Entamoeba dispar from patients with amoeba infection initially diagnosed by microscopy. Scand. J. Infect. Dis. 37:680–685. 106. Leiva, B., M. Lebbad, J. Winiecka-Krusnell, I. Altamirano, A. Tellez, and E. Linder. 2006. Overdiagnosis of Entamoeba histolytica and Entamoeba dispar in Nicaragua: a microscopic, Triage parasite panel and PCR study Arch. Med. Res. 37:529–534. 107. Li, E., and S. L. Stanley, Jr. 1996. Protozoa. Amebiasis. Gastroenterol. Clin. N. Am. 25:471–492. 108. Lysy, J., J. Zimmerman, Y. Sherman, R. Feigin, and M. Ligumsky. 1991. Crohn’s colitis complicated by superimposed invasive amebic colitis. Am. J. Gastroenterol. 86:1063–1065. 109. MacFarlane, R. C., and U. Singh. 2006. Identification of differentially expressed genes in virulent and nonvirulent Entamoeba species: potential implications for amebic pathogenesis. Infect. Immun. 74:340–351. 110. Martinez-Palomo, A., and M. Martinez-Baez. 1983. Selective primary health care: strategies for control of disease in the developing world. X. Amebiasis. Rev. Infect. Dis. 5:1093–1102. 111. McMillan, A., H. M. Gilmour, G. McNeillage, and G. R. Scott. 1984. Amoebiasis in homosexual men. Gut 25:356–360. 112. Mhlanga, B. R., L. O. Lanoie, H. J. Norris, E. E. Lack, and D. H. Connor. 1992. Amebiasis complicating carcinomas—a diagnostic dilemma. Am. J. Trop. Med. Hyg. 46:759–764. 113. Mirelman, D., Y. Nuchamowitz, and T. Stolarsky. 1997. Comparison of use of enzyme-linked immunosorbent assay-based kits and PCR amplification of rRNA genes for simultaneous detection of Entamoeba histolytica and E. dispar. J. Clin. Microbiol. 35:2405–2407. 114. Mondal, D., W. A. Petri, Jr., R. B. Sack, B. D. Kirkpatrick, and R. Haque. 2006. Entamoeba histolytica-associated diarrheal illness is negatively associated with the growth of preschool children: evidence from a prospective study. Trans. R. Soc. Trop. Med. Hyg. 100:1032–1038. 115. Monteiro, L., D. Bonnemaison, A. Vekris, K. G. Petry, J. Bonnet, R. Vidal, 530 116. 117. 118. 119. 120. 121. 122. 123. 124. 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. FOTEDAR ET AL. J. Cabrita, and F. Megraud. 1997. Complex polysaccharides as PCR inhibitors in feces: Helicobacter pylori model. J. Clin. Microbiol. 35:995–998. Moran, P., F. Ramos, M. Ramiro, O. Curiel, E. Gonzalez, A. Valadez, A. Gomez, G. Garcia, E. I. Melendro, and C. Ximenez. 2005. Entamoeba histolytica and/or Entamoeba dispar: infection frequency in HIV⫹/AIDS patients in Mexico City. Exp. Parasitol. 110:331–334. Moran, P., F. Ramos, M. Ramiro, O. Curiel, E. Gonzalez, A. Valadez, A. Gomez, G. Garcia, E. I. Melendro, and C. Ximenez. 2005. Infection by human immunodeficiency virus-1 is not a risk factor for amebiasis. Am. J. Trop. Med. Hyg. 73:296–300. Myjak, P., J. Kur, and H. Pietkiewicz. 1997. Usefulness of new DNA extraction procedure for PCR technique in species identification of Entamoeba isolates. Wiad. Parazytol. 43:163–170. Myjak, P., J. Kur, H. Pietkiewicz, A. Kotlowski, W. Nahorski, and B. Szostakowska. 2000. Molecular differentiation of Entamoeba histolytica and Entamoeba dispar from stool and culture samples obtained from polish citizens infected in tropica and in Poland. Acta Protozool. 39:217–224. National Committee for Clinical Laboratory Standards. 1997. Procedures for the recovery and identification of parasites from the intestinal tract. Approved guideline M28-A. National Committee for Clinical Laboratory Standards, Wayne, PA. Newton-Sanchez, O. A., K. Sturm-Ramirez, J. L. Romero-Zamora, J. I. Santos-Preciado, and J. Samuelson. 1997. High rate of occult infection with Entamoeba histolytica among non-dysenteric Mexican children. Arch. Med. Res. 28:311–313. Nozaki, T., S. R. Motta, T. Takeuchi, S. Kobayashi, and P. G. Sargeaunt. 1989. Pathogenic zymodemes of Entamoeba histolytica in Japanese male homosexual population. Trans. R. Soc. Trop. Med. Hyg. 83:525. Nunez, Y. O., M. A. Fernandez, D. Torres-Nunez, J. A. Silva, I. Montano, J. L. Maestre, and L. Fonte. 2001. Multiplex polymerase chain reaction amplification and differentiation of Entamoeba histolytica and Entamoeba dispar DNA from stool samples. Am. J. Trop. Med. Hyg. 64:293–297. Oh, M. D., K. Lee, E. Kim, S. Lee, N. Kim, H. Choi, M. H. Choi, J. Y. Chai, and K. Choe. 2000. Amoebic liver abscess in HIV-infected patients. AIDS 18:1872–1873. Ohnishi, K., Y. Kato, A. Imamura, M. Fukayama, T. Tsunoda, Y. Sakaue, M. Sakamoto, and H. Sagara. 2004. Present characteristics of symptomatic Entamoeba histolytica infection in the big cities of Japan. Epidemiol. Infect. 132:57–60. Ohnishi, K., M. Murata, and E. Okuzawa. 1994. Symptomatic amebic colitis in a Japanese homosexual AIDS patient. Intern. Med. 33:120–122. Ohnishi, K., and M. Murata. 1997. Present characteristics of symptomatic amebiasis due to Entamoeba histolytica in the east-southeast area of Tokyo. Epidemiol. Infect. 119:363–367. Ong, S. J., M. Y. Cheng, K. H. Liu, and C. B. Horng. 1996. Use of the ProSpecT microplate enzyme immunoassay for the detection of pathogenic and non-pathogenic Entamoeba histolytica in faecal specimens. Trans. R. Soc. Trop. Med. Hyg. 90:248–249. Paglia, M. G., and P. Visca. 2004. An improved PCR-based method for detection and differentiation of Entamoeba histolytica and Entamoeba dispar in formalin-fixed stools. Acta Trop. 92:273–277. Parija, S. C., and K. Khairnar. 2005. Entamoeba moshkovskii and Entamoeba dispar-associated infections in Pondicherry, India. J. Health Pop. Nutr. 23:292–295. Patterson, M., and L. E. Schoppe. 1982. The presentation of amoebiasis. Med. Clin. N. Am. 66:689–705. Petri, W.A., Jr., and U. Singh. 1999. Diagnosis and management of amebiasis Clin. Infect. Dis. 29:1117–1125. Pillai, D. R., and K. C. Kain. 1999. Immunochromatographic strip-based detection of Entamoeba histolytica-E. dispar and Giardia lamblia coproantigen. J. Clin. Microbiol. 37:3017–3019. Pillai, D. R., J. S. Keystone, D. C. Sheppard, J. D. MacLean, D. W. MacPherson, and K. C. Kain. 1999. Entamoeba histolytica and Entamoeba dispar: epidemiology and comparison of diagnostic methods in a setting of non endemicity. Clin. Infect. Dis. 29:1315–1318. Pinheiro, S. M., R. M. Carneiro, I. S. Aca, J. I. Irmao, M. A. Morais, Jr., M. R. Coimbra, and L. B. Carvalho, Jr. 2004. Determination of the prevalence of Entamoeba histolytica and E. dispar in the Pernambuco state of northeastern Brazil by a polymerase chain reaction. Am. J. Trop. Med. Hyg. 70:221–224. Pinheiro, S. M., R. F. Maciel, M. A. Morais, Jr., I. S. Aca, L. B. Carvalho, Jr., and M. R. Coimbra. 2005. Genetic characterization of Entamoeba dispar isolates in Northeast Brazil. Acta Trop. 94:35–40. Proctor, E. M. 1991. Laboratory diagnosis of amebiasis. Clin. Lab. Med. 11:829–859. Que, X., and S. L. Reed. 1991. Nucleotide sequence of a small subunit ribosomal RNA (16S-like rRNA) gene from Entamoeba histolytica: differentiation of pathogenic from non-pathogenic isolates. Nucleic Acids Res. 11:5438. Qvarnstrom, Y., C. James, M. Xayavong, B. P Holloway, G. S. Visvesvara, R. Sriram, and A. J. Da Silva. 2005. Comparison of real-time PCR proto- CLIN. MICROBIOL. REV. 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. 160. 161. 162. cols for differential laboratory diagnosis of amebiasis. J. Clin. Microbiol. 43:5491–5497. Ramos, F., G. Garcia, A. Valadez, P. Moran, E. Gonzalez, A. Gomez, E. I. Melendro, O. Valenzuela, and C. Ximenez. 2005. E. dispar strain: analysis of polymorphism as a tool for study of geographic distribution. Mol. Biochem. Parasitol. 141:175–177. Ramos. F., P. Moran, E. Gonzalez, G. Garcia, M. Ramiro, A. Gomez, C. de Leon Mdel, E. I. Melendro, A. Valadez, and C. Ximenez. 2005. Entamoeba histolytica and Entamoeba dispar: prevalence infection in a rural Mexican community. Exp. Parasitol. 110:327–330. Ramos, F., E. Valdez, P. Moran, E. Gonzalez, G. Padilla, A. Gomez, M. Ramiro, E. I. Melendro, O. Munoz, C. G. Clark, and C. Ximenez. 2000. Prevalence of Entamoeba histolytica and Entamoeba dispar in a highly endemic rural population. Arch. Med. Res. 31(Suppl.):34–35. Ramos, F., R. Zurabian, P. Moran, M. Ramiro, A. Gomez, C. G. Clark, E. I. Melendro, G. Garcia, and C. Ximenez. 1999. The effect of formalin fixation on the polymerase chain reaction characterization of Entamoeba histolytica. Trans. R. Soc. Trop. Med. Hyg. 93:335–336. Randall, G. R., R. S. Goldsmith, J. Shek, S. Mehalko, and D. Heyneman. 1984. Use of the enzyme-linked immunosorbent assay (ELISA) for detection of Entamoeba histolytica antigen in faecal samples. Trans. R. Soc. Trop. Med. Hyg. 78:593–595. Ravdin, J. I., T. F. Jackson, W. A. Petri, Jr., C. F. Murphy, B. L. Ungar, V. Gathiram, J. Skilogiannis, and A. E. Simjee. 1990. Association of serum antibodies to adherence lectin with invasive amebiasis and asymptomatic infection with pathogenic Entamoeba histolytica. J. Infect. Dis. 162:768–772. Rivera, W. L., S. R. Santos, and H. Kanbara. 2006. Prevalence and genetic diversity of Entamoeba histolytica in an institution for the mentally retarded in the Philippines. Parasitol. Res. 98:106–110. Rivera, W. L., H. Tachibana, and H. Kanbara. 1998. Field study on the distribution of Entamoeba histolytica and Entamoeba dispar in the northern Philippines as detected by the polymerase chain reaction. Am. J. Trop. Med. Hyg. 59:916–921. Rivera, W. L., H. Tachibana, M. R. Silva-Tahat, H. Uemura, and H. Kanbara. 1996. Differentiation of Entamoeba histolytica and E. dispar DNA from cysts present in stool specimens by polymerase chain reaction: its field application in the Philippines. Parasitol. Res. 82:585–589. Robert, R., C. Mahaza, C. Bernard, C. Buffard, and J. M. Senet. 1990. Evaluation of a new bicolored latex agglutination test for immunological diagnosis of hepatic amoebiasis. J. Clin. Microbiol. 28:1422–1424. Robinson, G. L. 1968. The laboratory diagnosis of human parasitic amoebae. Trans. R. Soc. Trop. Med. Hyg. 62:285–294. Romero, J. L., S. Descoteaux, S. Reed, E. Orozco, J. Santos, and J. Samuelson. 1992. Use of polymerase chain reaction and nonradioactive DNA probes to diagnose Entamoeba histolytica in clinical samples. Arch. Med. Res. 23:277– 279. Rosenblatt, J. E., L. M. Sloan, and J. E Bestrom. 1995. Evaluation of an enzyme-linked immunoassay for the detection in serum of antibodies to Entamoeba histolytica. Diagn. Microbiol. Infect. Dis. 22:275–278. Roy, S., M. Kabir, D. Mondal, I. K. Ali, W. A. Petri, Jr., and R. Haque. 2005. Real-time-PCR assay for diagnosis of Entamoeba histolytica infection. Clin. Microbiol. 43:2168–2172. Rustgi, A. K., and J. M. Richter. 1989. Pyogenic and amebic liver abscess. Med. Clin. N. Am. 73:847–858. Saklatvala, T. 1993. Milestones in parasitology. Parasitol. Today 9:347–348. Sanchez-Giillen, M. C., M. Velazquez-Rojas, H Salgado-Rosas, E. TorresRasgado, R. Perez Fuentes, J. Martinez-Munguia, and P. TalamasRohana. 2000. Seroprevalence of anti-Entamoeba histolytica antibodies by IHA and ELISA assay in blood donors from Puebla, Mexico. Arch. Med. Res. 31:S53–S54. Sanuki, J., T. Asai, E. Okuzawa, S. Kobayashi, and T. Takeuchi. 1997. Identification of Entamoeba histolytica and E. dispar cysts in stool by polymerase chain reaction. Parasitol. Res. 83:96–98. Sargeaunt, P. G., J. E. Williams, and J. D. Grene. 1978. The differentiation of invasive and non-invasive Entamoeba histolytica by isoenzyme electrophoresis Trans. R. Soc. Trop. Med. Hyg. 72:519–521. Sargeaunt, P. G., T. F. Jackson, S. Wiffen, R. Bhojnani, J. E. Williams, D. Felmingham, D. Goldmeir, E. Allason-Jones, A. Mindel, and E. Phillips. 1987. The reliability of Entamoeba histolytica zymodemes in clinical laboratory diagnosis. Arch. Investig. Med. 18:69–75. Scaglia, M., S. Gatti, M. Strosselli, V. Grazioli, and M. R. Villa. 1983. Entamoeba moshkovskii (Tshalaia, 1941): morpho-biological characterization of new strains isolated from the environment, and a review of the literature. Ann. Parasitol. Hum. Comp. 58:413–422. Schuurman, T., A. van Breda, R. de Boer, M. Kooistra-Smid, M. Beld, P. Savelkoul, and R. Boom. 2005. Reduced PCR sensitivity due to impaired DNA recovery with the MagNA Pure LC total nucleic acid isolation kit. J. Clin. Microbiol. 43:4616–4622. Sehgal, D., V. Mittal, S. Ramachandran, S. K. Dhar, A. Bhattacharya, and S. Bhattacharya. 1994. Nucleotide sequence organisation and analysis of the nuclear ribosomal DNA circle of the protozoan parasite Entamoeba histolytica. Mol. Biochem. Parasitol. 67:205–214. VOL. 20, 2007 LABORATORY DIAGNOSTIC TECHNIQUES FOR ENTAMOEBA SPECIES 163. Sehgal, R., M. Abd-Alla, A. H. Moody, P. L. Chiodini, and J. P. Ackers. 1995. Comparison of two media for the isolation and short-term culture of Entamoeba histolytica and E. dispar. Trans. R. Soc. Trop. Med. Hyg. 89:394. 164. Shah, P. H., R. C. MacFarlane, D. Bhattacharya, J. C. Matese, J. Demeter, S. E. Stroup, and U. Singh. 2005. Comparative genomic hybridizations of Entamoeba strains reveal unique genetic fingerprints that correlate with virulence. Eukaryot. Cell 4:504–515. 165. Shamsuzzaman, S. M., R. Haque, S. K. Hasin, and Y. Hashiguchi. 2000. Evaluation of indirect fluorescent antibody test and enzyme-linked immunosorbent assay for diagnosis of hepatic amebiasis in Bangladesh. J. Parasitol. 86:611–615. 166. Shandera, W. X., P. Bollam, R. H. Hashmey, Jr., P. A. Athey, S. B. Greenberg, and A. C. White, Jr. 1998. Hepatic amebiasis among patients in a public teaching hospital. South Med. J. 91:829–837. 167. Sharp, S. E., C. A. Suarez, Y. Duran, and R. J. Poppiti. 2001. Evaluation of the Triage Micro Parasite Panel for detection of Giardia lamblia, Entamoeba histolytica/Entamoeba dispar, and Cryptosporidium parvum in patient stool specimens. J. Clin. Microbiol. 39:332–334. 168. Sheehan, D. J., E. J. Bottone, K. Pavletich, and M. C. Heath. 1979. Entamoeba histolytica: efficacy of microscopic, cultural, and serological techniques for laboratory diagnosis. J. Clin. Microbiol. 10:128–133. 169. Shenai, B. R., B. L. Komalam, A. S. Arvind, P. R. Krishnaswamy, and P. V. Rao. 1996. Recombinant antigen-based avidin-biotin microtiter enzymelinked imunosorbent assay for serodiagnosis of invasive amebiasis. J. Clin. Microbiol. 34:828–833. 170. Simonishvili S., S. Tsanava, K. Sanadze, R. Chlikadze, A. Miskalishvili, N. Lomkatsi, P. Imnadze, W. A. Petri, Jr., and N. Trapaidze. 2005. Entamoeba histolytica: the serine-rich gene polymorphism-based genetic variability of clinical isolates from Georgia. Exp. Parasitol. 110:313–317. 171. Solaymani-Mohammadi, S., M. Rezaian, Z. Babaei, A. Rajabpour, A. R. Meamar, A. A. Pourbabai, and W. A. Petri, Jr. 2006. Comparison of a stool antigen detection kit and PCR for diagnosis of Entamoeba histolytica and Entamoeba dispar infections in asymptomatic cyst passers in Iran. J. Clin. Microbiol. 44:2258–2261. 172. Solaymani-Mohammadi, S., M. M. Lam, J. R. Zunt, and W. A. Petri, Jr. 2007. Entamoeba histolytica encephalitis diagnosed by PCR of cerebrospinal fluid. Trans. R. Soc. Trop. Med. Hyg. 101:311–313. 173. Stanley, S. L., Jr. 2003. Amoebiasis. Lancet. 361:1025–1034. 174. Stanley, S. L., Jr., A. Becker, C. Kunz-Jenkins, L. Foster, and E. Li. 1990. Cloning and expression of a membrane antigen of Entamoeba histolytica possessing multiple tandem repeats. Proc. Natl. Acad. Sci. USA 87:4976– 4980. 175. Stark, D., R. Fotedar, J. T. Ellis, and J. L. Harkness. 2006. Locally acquired infection with Entamoeba histolytica in men who have sex with men in Australia. Med. J. Aust. 185:417. 176. Stark, D., R. Fotedar, S. van Hal, B. Nigel., M. Deborah, J. Ellis, and J. L. Harkness. 2007. Prevalence of enteric protozoa in HIV-positive and HIVnegative men who have sex with men from Sydney. Am. J. Trop. Med. Hyg. 76:549–552. 177. Stauffer, W., and J. I. Ravdin. 2003. Entamoeba histolytica: an update. Curr. Opin. Infect. 16:479–485. 178. Strachan, W. D., P. L. Chiodini, W. M. Spice, A. H. Moody, and J. P. Ackers. 1988. Immunological differentiation of pathogenic and nonpathogenic isolates of E. histolytica. Lancet i:561–563. 179. Tachibana, H., X. J. Cheng, S. Kobayashi, N. Matsubayashi, S. Gotoh, and K. Matsubayashi. 2001. High prevalence of infection with Entamoeba dispar, but not E. histolytica, in captive macaques. Parasitol. Res. 87:14–17. 180. Tachibana, H., S. Ihara, S. Kobayashi, Y. Kaneda, T. Takeuchi, and Y. Watanabe. 1991. Differences in genomic DNA sequences between pathogenic and nonpathogenic isolates of Entamoeba histolytica identified by polymerase chain reaction. J. Clin. Microbiol. 29:2234–2239. 181. Tachibana, H., S. Kobayashi, K. Nagakura, Y. Kaneda, and T. Takeuchi. 2000. Asymptomatic cyst passers of Entamoeba histolytica but not Entamoeba dispar in institutions for the mentally retarded in Japan. Parasitol. Int. 49:31–35. 182. Tachibana, H., S. Kobayashi, E. Okuzawa, and G. Masuda. 1992. Detection of pathogenic Entamoeba histolytica DNA in liver abscess fluid by polymerase chain reaction. Int. J. Parasitol. 22:1193–1196. 183. Tachibana, H., S. Kobayashi, M. Takekoshi, and S. Ihara. 1991. Distinguishing pathogenic isolates of Entamoeba histolytica by polymerase chain reaction. J. Infect. Dis. 164:825–826. 184. Takahashi, T., A. Gamboa-Dominguez, T. J. Gomez-Mendex, J. M. Remes, D. Martinez-Gonzalez, J. Gutierrez-Saldivar, J. C. Morales, J. Granados, and J. Sierra-Madero. 1997. Fulminant amebic colitis: analysis of 55 cases. Dis. Colon Rectum 40:1362–1367. 185. Takeuchi, T., E. Okuzawa, T. Nozaki, S. Kobayashi, M. Mizokami, N. Minoshima, M. Yamamoto, and S. Isomura. 1989. High seropositivity of Japanese homosexual men for amebic infection. J. Infect. Dis. 159:808. 186. Takeuchi, T., S. Kobayashi, K. Asami, and N. Yamaguchi. 1987. Correlation of positive syphilis serology with invasive amebiasis in Japan. Am. J. Trop. Med. Hyg. 36:321–324. 531 187. Tannich, E., and G. D. Burchard. 1991. Differentiation of pathogenic from nonpathogenic Entamoeba histolytica by restriction fragment analysis of a single gene amplified in vitro. J. Clin. Microbiol. 29:250–255. 188. Tanyuksel, M., and W. A. Petri, Jr. 2003. Laboratory diagnosis of amebiasis. Clin. Microbiol. Rev. 16:713–729. 189. Tanyuksel, M., M. Ulukanligil, Z. Guclu, E. Araz, O. Koru, and W. A. Petri, Jr. 2007. Two cases of rarely recognized infection with Entamoeba moshkovskii. Am. J. Trop. Med. Hyg. 76:723–724. 190. Thompson, J. E., Jr., S. Forlenza, and R. Verma. 1985. Amebic liver abscess: a therapeutic approach. Rev. Infect. Dis. 7:171–179. 191. Trissl, D., A. Martinez-Palomo, M. de la Torre, R. de la Hoz, and E. Perez de Suarez. 1978. Surface properties of Entamoeba: increased rates of human erythrocyte phagocytosis in pathogenic strains. J. Exp. Med. 148:1137– 1143. 192. Troll, H., H. Marti, and N. Weiss. 1997. Simple differential detection of Entamoeba histolytica and Entamoeba dispar in fresh stool specimens by sodium acetate-acetic acid-formalin concentration and PCR. J. Clin. Microbiol. 35:1701–1705. 193. Tshalaia, L. E. 1941. On a species of Entamoeba detected in sewage effluents. Med. Parazit. (Moscow) 10:244–252. 194. Van Doorn, H. R., H. Hofwegen, R. Koelewijn, H. Gilis, R. Peek, J. C. Wetsteyn, P. J. Van Genderen, T. Vervoort, and T. Van Gool. 2005. Use of rapid dipstick and latex agglutination tests and enzyme-linked immunosorbent assay for serodiagnosis of amebic liver abscess, amebic colitis, and Entamoeba histolytica cyst passage. J. Clin. Microbiol. 43:4801–4806. 195. Verweij, J. J., R. A. Blange, K. Templeton, J. Schinkel, E. A. Brienen, M. A. Van Rooyen, L. Van Lieshout, and A. M. Polderman. 2004. Simultaneous detection of Entamoeba histolytica, Giardia lamblia, and Cryptosporidium parvum in fecal samples by using multiplex real-time PCR. J. Clin. Microbiol. 42:1220–1223. 196. Verweij, J. J., J. Blotkamp, E. A. Brienen, A. Aguirre, and A. M. Polderman. 2000. Differentiation of Entamoeba histolytica and Entamoeba dispar cysts using polymerase chain reaction on DNA isolated from faeces with spin columns. Eur. J. Clin. Microbiol. Infect. Dis. 19:358–361. 197. Verweij, J. J., D. Laeijendecker, E. A. Brienen, L. Van Lieshout, and A. M. Polderman. 2003. Detection and identification of Entamoeba species in stool samples by a reverse line hybridization assay. J. Clin. Microbiol. 41:5041–5045. 198. Verweij, J. J., F. Oostvogel, E. A. Brienen, A. Nang-Beifubah, J. Ziem, and A. M. Polderman. 2003. Prevalence of Entamoeba histolytica and Entamoeba dispar in northern Ghana. Trop. Med. Int. Health 8:1153–1156. 199. Verweij, J. J., L. Van Lieshout, C. Blotkamp, E. A. Brienen, S. Van Duivenvoorden, M. Van Esbroeck, and A. M. Polderman. 2000. Differentiation of Entamoeba histolytica and Entamoeba dispar using PCR-SHELA and comparison of antibody response. Arch. Med. Res. 31:44–46. 200. Verweij, J. J., J. Vermeer, E. A. Brienen, C. Blotkamp, D. Laeijendecker, L. Van Lieshout, and A. M. Polderman. 2003. Entamoeba histolytica infections in captive primates. Parasitol. Res. 90:100–103. 201. Visser, L. G., J. J. Verweij, M. Van Esbroeck, W. M. Edeling, J. Clerinx, and A. M. Polderman. 2006 Diagnostic methods for differentiation of Entamoeba histolytica and Entamoeba dispar in carriers: performance and clinical implications in a non-endemic setting. Int. J. Med. Microbiol. 296:397– 403. 202. Vohra, H., H. S. Bhatti, N. K. Ganguly, and R. C. Mahajan. 1989. Virulence of pathogenic and non-pathogenic zymodemes of Entamoeba histolytica (Indian strains) in guinea pigs. Trans. R. Soc. Trop. Med. Hyg. 83:648–650. 203. Walsh, J. A. 1986. Problems in recognition and diagnosis of amebiasis: estimation of the global magnitude of morbidity and mortality. Rev. Infect. Dis. 8:228–238. 204. Wang, L. T., G. Jen, and J. H. Cross. 1973. Establishment of Entamoeba histolytica from liver abscess in monoxenic cultures with hemoflagellates. Am. J. Trop. Med. Hyg. 22:30–32. 205. Wang, Z., G. J. Vora, and D. A. Stenger. 2004. Detection and genotyping of Entamoeba histolytica, Entamoeba dispar, Giardia lamblia, and Cryptosporidium parvum by oligonucleotide microarray. J. Clin. Microbiol. 42:3262– 3271. 206. Ward L. A., and Y. Wang. 2001. Rapid methods to isolate Cryptosporidium DNA from frozen feces for PCR. Diagn. Microbiol. Infect. Dis. 41:37–42. 207. Weinke, T., B. Friedrich-Janicke, P. Hopp, and K. Janitschke. 1990. Prevalence and clinical importance of Entamoeba histolytica in two high-risk groups: travelers returning from the tropics and male homosexuals. J. Infect. Dis. 161:1029–1031. 208. Wolk, D. M., S. K. Schneider, N. L. Wengenack, L. M. Sloan, and J. E. Rosenblatt. 2002. Real-time PCR method for detection of Encephalitozoon intestinalis from stool specimens. J. Clin. Microbiol. 40:3922–3928. 209. Wonsit, R., N. Thammapalerd, S. Tharavanij, P. Radomyos, and D. Bunnag. 1992. Enzyme-linked immunosorbent assay based on monoclonal and polyclonal antibodies for the detection of Entamoeba histolytica antigens in faecal specimens. Trans. R. Soc. Trop. Med. Hyg. 86:166–169. 210. World Health Organization. 1997. World Health Organization/Pan American Health Organization/UNESCO report of a consultation of experts on 532 FOTEDAR ET AL. amoebiasis. Wkly. Epidemiol. Rec. 72:97–99. 211. Zaki, M., and C. G. Clark. 2001. Isolation and characterization of polymorphic DNA from Entamoeba histolytica. J. Clin. Microbiol. 39:897–905. 212. Zaki, M., P. Meelu, W. Sun, and C. G. Clark. 2002. Simultaneous differentiation and typing of Entamoeba histolytica and Entamoeba dispar. J. Clin. Microbiol. 40:1271–1276. 213. Zaki, M., J. J. Verweij, and C. G. Clark. 2003. Entamoeba histolytica: direct PCR-based typing of strains using faecal DNA. Exp. Parasitol. 104:77–80. 214. Zaman, S., J. Khoo, S. W. Ng, R. Ahmed, M. A Khan, R. Hussain, and V. CLIN. MICROBIOL. REV. Zaman. 2000. Direct amplification of Entamoeba histolytica DNA from amoebic liver abscess pus using polymerase chain reaction. Parasitol. Res. 86:724–728. 215. Zengzhu, G., R. Bracha, Y. Nuchamowitz, W. Cheng, and D. Mirelman. 1999. Analysis by enzyme-linked immunosorbent assay and PCR of human liver abscess aspirates from patients in China for Entamoeba histolytica. J. Clin. Microbiol. 37:3034–3036. 216. Zindrou, S., E. Orozco, E. Linder, A. Tellez, and A. Bjorkman. 2001. Specific detection of Entamoeba histolytica DNA by hemolysin gene targeted PCR. Acta Trop. 78:117–125.