

The Trouble with Sliding Windows and the Selective Pressure in
... Simply from visual inspection, we were unable to distinguish the plots in Figure 1A for the real data from those in Figure 2A&B for the simulated data. The peaks and valleys in d̂S and d̂N in Figure 2 are random and differ between simulated replicates. However, like the real data, the simulated data ...
... Simply from visual inspection, we were unable to distinguish the plots in Figure 1A for the real data from those in Figure 2A&B for the simulated data. The peaks and valleys in d̂S and d̂N in Figure 2 are random and differ between simulated replicates. However, like the real data, the simulated data ...

Tutorial 7: Constructing new databases using ARB
... ARB is most frequently utilized for management and analysis of SSU rRNA gene data, but it can be a very useful tool to align, manage, and compare sequence data from other genes. The features used for analysis of SSU rRNA genes are very similar as to working with other genes, but one difference is th ...
... ARB is most frequently utilized for management and analysis of SSU rRNA gene data, but it can be a very useful tool to align, manage, and compare sequence data from other genes. The features used for analysis of SSU rRNA genes are very similar as to working with other genes, but one difference is th ...


Proceedings Template - WORD
... Since the training data set in the KDDCup case was small, patient level bootstrapping was used to validate solutions. Bootstrap implies taking out one sample from the training set for testing and repeating until all samples were used for testing. In this specific task which involves multiple candida ...
... Since the training data set in the KDDCup case was small, patient level bootstrapping was used to validate solutions. Bootstrap implies taking out one sample from the training set for testing and repeating until all samples were used for testing. In this specific task which involves multiple candida ...

DATA ANALYSIS - DCU School of Computing
... • For a large sample, variance of MLE can be approximated by ...
... • For a large sample, variance of MLE can be approximated by ...




Lower Bounds for the Relative Greedy Algorithm for Approximating
... The second lower bound is obtained by constructing an instance G k,l which places the instance Gk into a grid. The instance Gk,l consists of an 4k × l grid of terminals where the last terminals of each column have been identified as one terminal. For each column of terminals the graph Gk − Tb − Tc i ...
... The second lower bound is obtained by constructing an instance G k,l which places the instance Gk into a grid. The instance Gk,l consists of an 4k × l grid of terminals where the last terminals of each column have been identified as one terminal. For each column of terminals the graph Gk − Tb − Tc i ...

Python XML Element Trees
... The function calls at lines 14 and 15 is apply indentation to branch nodes (head recursion); and line 20 does it to leaf nodes (recursion end). So all subelements get the the same text and tail indention. Unfortunately, this applies the wrong value to the last sub-element’s tail field (!); but a cor ...
... The function calls at lines 14 and 15 is apply indentation to branch nodes (head recursion); and line 20 does it to leaf nodes (recursion end). So all subelements get the the same text and tail indention. Unfortunately, this applies the wrong value to the last sub-element’s tail field (!); but a cor ...

13058_2014_424_MOESM2_ESM
... In general, if there are a total of P features, then in the first step of stepwise feature selection the performance of each of P features is evaluated using Wilks’ lambda, and the feature with the best performance is selected. In the subsequent steps, assuming that m is the number of features that ...
... In general, if there are a total of P features, then in the first step of stepwise feature selection the performance of each of P features is evaluated using Wilks’ lambda, and the feature with the best performance is selected. In the subsequent steps, assuming that m is the number of features that ...





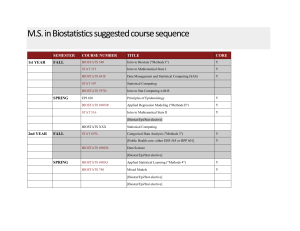


No Slide Title - Brigham Young University
... Clustering algorithms offer useful visual descriptions of microarray data. Genes may be clustered, or samples, or both. ...
... Clustering algorithms offer useful visual descriptions of microarray data. Genes may be clustered, or samples, or both. ...

Dagstuhl-Seminar
... of supervised learning? These questions have been discussed during this seminar which brought together neural modellers, statisticians, computational learning theorists (“COLT people”) and theoretical computer scientists and physicists. The field of machine learning with its broad range of pattern r ...
... of supervised learning? These questions have been discussed during this seminar which brought together neural modellers, statisticians, computational learning theorists (“COLT people”) and theoretical computer scientists and physicists. The field of machine learning with its broad range of pattern r ...

1 Divide and Conquer with Reduce
... reduce combine emptyVal (map base S) Merge Sort. As you have seen from previous classes, merge sort is a popular divide-and-conquer sorting algorithm with optimal work. It is based on a function merge that takes two already sorted sequences and returns a sorted sequence that combines all inputs from ...
... reduce combine emptyVal (map base S) Merge Sort. As you have seen from previous classes, merge sort is a popular divide-and-conquer sorting algorithm with optimal work. It is based on a function merge that takes two already sorted sequences and returns a sorted sequence that combines all inputs from ...

Longest Common Substring
... different Hashing techniques such as roller hash in conjunction with above techniques to aim to see if there could be any improvement in time complexity and reduce basic operations from current levels. 5. Look at problems that can be solved using Fast Exact Algorithms (Heuristic) for the Closest Str ...
... different Hashing techniques such as roller hash in conjunction with above techniques to aim to see if there could be any improvement in time complexity and reduce basic operations from current levels. 5. Look at problems that can be solved using Fast Exact Algorithms (Heuristic) for the Closest Str ...

Mining Frequent Patterns Without Candidate generation
... Most of the previous studies adopt an Apriori-like candidate set generation-and-test approach. However, candidate set generation is still costly, especially when there exist a large number of patterns and/or long patterns. In this study, we propose a novel frequent-pattern tree (FP-tree) structure, ...
... Most of the previous studies adopt an Apriori-like candidate set generation-and-test approach. However, candidate set generation is still costly, especially when there exist a large number of patterns and/or long patterns. In this study, we propose a novel frequent-pattern tree (FP-tree) structure, ...

Module 8: Horizontal Gene Transfer
... The interpretation of the tree will vary depending on how many organisms closely taxonomically related your organism have been sequenced. Hits in the same or Family or Order will might be considered very close relatives for organisms who have not had genomes of close relatives sequenced, and those i ...
... The interpretation of the tree will vary depending on how many organisms closely taxonomically related your organism have been sequenced. Hits in the same or Family or Order will might be considered very close relatives for organisms who have not had genomes of close relatives sequenced, and those i ...