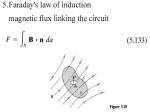
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cytokinesis wikipedia , lookup
Cell growth wikipedia , lookup
Extracellular matrix wikipedia , lookup
Signal transduction wikipedia , lookup
Tissue engineering wikipedia , lookup
Cell culture wikipedia , lookup
Organ-on-a-chip wikipedia , lookup
Cell encapsulation wikipedia , lookup
Cellular differentiation wikipedia , lookup
Doctoral thesis from the Department of Molecular Biosciences The Wenner-‐Gren Institute, Stockholm University Stockholm, Sweden Activation, adhesion and motility of B lymphocytes in health and disease Natalija Gerasimčik Stockholm, 2013 Cover: Scanning electron microscopy of Cdc42 knockout B cells by Natalija Gerasimčik © Natalija Gerasimčik, Stockholm 2013 ISBN 978-‐91-‐7447-‐704-‐7 Printed in Sweden by Universitetsservice AB, Stockholm 2013 Distributor: Stockholm University Library 2 “B cells means the Best” (PhD, Associate Professor Mikael Karlsson) To my Parents 3 Summary B cells can be activated by T cell-‐dependent stimuli, such as CD40 ligation and cytokines, which induce extensive proliferation, class switch recombination and somatic hypermutation. Epstein-‐Barr virus (EBV) can also induce B cell activation by mimicking T cell help through its main oncoprotein, latent membrane protein 1 (LMP-‐1). It is regulated by another EBV-‐encoded protein, EBV nuclear antigen 2 (EBNA-‐2), which is absent in Hodgkin and Burkitt lymphomas. We have studied LMP-‐1 induction by cytokines in vitro and shown that LMP-‐1 is induced through the transcription factor signal transducer and activator of transcription (STAT6) and a newly defined high-‐affinity STAT6-‐binding site. When IL-‐4 is added together with lipopolysaccharide (LPS) or α-‐CD40 to B cells, it induces homotypic round and tight aggregates in vitro, whereas LPS alone does not induce such morphological changes. I describe here attempts to identify the molecules that regulate these responses. I have shown that the Rho GTPase Cdc42 controls the spreading of B cells, whereas two other molecules in the same family, Rac1 and Rac2, control homotypic adhesion. Further, I have shown by conditional deletion of Cdc42 in B cells that it is important in the humoral immune response. Dock10 is a guanosine nucleotide exchange factor (GEF) for Cdc42. It is expressed through all differentiation stages of B cell development. However, targeted deletion of Dock10 in B cells does not result in an aberrant phenotype. Furthermore, by studying conditional knockout mice for Dock10, Cdc42, Rac1 and Rac2, I have elucidated the mechanism of cytoskeletal changes during B cell activation, leading to adhesion and motility. My results may lead to a better understanding of normal B cell activation and of EBV infection, which is associated with many human tumours and may help to understand cancer development and progression in B cells. 4 List of Publications This thesis is based on the results presented in the following paper and manuscripts: I. Kis, L.L., Gerasimcik, N., Salamon, D., Persson, E.K., Nagy, N. Klein, G., Severinson, E. and Klein, E. The STAT6 signaling pathway activated by the cytokines IL-‐4 and IL-‐13 induces expression of the Epstein-‐Barr virus-‐encoded protein LMP-‐1 in absence of EBNA-‐2: implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 2011; 117:165-‐174. II. Gerasimcik, N., Dahlberg, C., Baptista, M., Westerberg, L. and Severinson, E. B cells devoid of the Rho GTPase Cdc42 coordinate the actin and microtubule cytoskeleton less effectively and form an extrafollicular antibody response. Manuscript. III. Gerasimcik, N., Baptista, M., Westerberg, L. and Severinson, E. The guanine nucleotide exchange factor Dock10: expression and function in B lymphocytes. Manuscript. IV. Gerasimcik, N. and Severinson, E. Investigation of the role of the small Rho GTPases Rac1 and Rac2 in B cell activation. Preliminary results. Publication not included in this thesis: Cernysiov V., Gerasimcik N., Mauricas M., Girkontaite I. Regulation of T-‐cell-‐independent and T-‐cell-‐dependent antibody production by circadian rhythm and melatonin. International Immunology 2010; 22(1):25-‐34. 5 List of abbreviations APRIL Arp2/3 B BAFF(R) Bcl-‐6 BCR BL Btk Cdc42 CD40L CDM CFSE cHL CD CIP4 CLL CSR CZH Dbl DC DH DHR Dock DZ EBER Ebi2 EBNA EBV EMSA EDL1 F-‐BAR FCH FDC FOB GAP GBD GC γc GDI GEF GL GTPase HL HRS swIg IL ICAM-‐1 ITAM iNKT JAK JNK LCL 6 A proliferation-‐inducing ligand Actin-‐related proteins 2 and 3 Basic region B cell activating factor (receptor) B cell lymphoma 6 B cell receptor Burkitt lymphoma Bruton tyrosine kinase Cell division control protein 42 CD40 ligand Ced-‐5/Dock180/Myoblast city 6-‐Carboxyfluorescein succinimidyl ester Classical Hodgkin lymphoma Cluster of differentiation Cdc42-‐interacting protein 4 Chronic lymphocytic leukaemia Class switch recombination CDM-‐zizimin homology Diffuse B cell lymphoma Dendritic cell Dbl-‐homology Dock homology region Dedicator of cytokinesis Dark zone EBV-‐encoded RNA EBV-‐induced receptor 2 EBV-‐nuclear antigen Epstein Barr virus Electrophoretic mobility shift assay EcoRI D leftward 1 Fes/CIP4 homology-‐Bin/Amphyphysin/Rvsp Fes/CIP4 homology Follicular dendritic cell Follicular B cell GTPase activating protein GTPase-‐binding domain Germinal centre Common gamma chain Rho GTP dissociation inhibitor Guanosine nucleotide exchange factor Germline transcripts Guanine triphosphatase Hodgkin lymphoma Hodgkin Reed-‐Sternberg cells Isotype switched immunoglobulin Interleukin Intercellular adhesion molecule-‐1 Immunoreceptor tyrosine-‐based activation motif Invariant natural killer cell Janus kinase c-‐Jun N-‐terminal kinase Lymphoblastoid cell line LFA-‐1 LMP-‐1 LPS LRS LZ MAPK MZB NF-‐κB NK NPC PC PD-‐1 PHA PI3K PRD Rac RICH SEB SHM SH3 STAT Syk S1P T1, T2 MZP TFR TFH TLR TNF TR TRAF VCA VCAM-‐1 V(D)J VLA-‐4 WASP WH1 WIP Leukocyte function-‐associated antigen-‐1 Latent membrane protein 1 Lipopolysaccharide LMP-‐1 regulatory sequences Light zone Mitogen-‐activated protein kinase Marginal zone B cell Nuclear factor kappa beta Natural killer cell Nasopharyngeal carcinoma Plasma cell Programmed death-‐1 Phytohemagglutinin Phosphatidylinositol 3-‐kinase Proline-‐rich domain Ras-‐related C3 botulinum toxin substrate Rho GAP-‐interacting with CIP4 homologues Staphylococcus enterotoxin B Somatic hypermutation Src homology 3 Signal transducer and activator of transcription Spleen tyrosine kinase Sphingosine 1 phosphate Transitional 1 or 2 B cells Marginal zone precursor B cells Follicular T regulatory cell Follicular T helper cell Toll-‐like receptor Tumour necrosis factor Terminal repeat TNF-‐receptor associated factor Veprolin-‐, central-‐, acidic region domain Vascular cell adhesion mediator-‐1 Variable, Diversity, Joining Very late antigen-‐4 Wiskott-‐Aldrich syndrome protein WASP homology domain WH1-‐interacting protein 7 Contents Summary..................................................................................................................................................4 List of publications ...............................................................................................................................5 List of abbreviations ............................................................................................................................6 Contents ...................................................................................................................................................8 Introduction............................................................................................................................................9 Early B cell differentiation .......................................................................................................................... 9 Late B cell differentiation ..........................................................................................................................10 B1 and B2 cells and their localization .................................................................................................................. 10 Transitional B cells ....................................................................................................................................................... 11 Chemokine guidance of follicular and marginal zone B cells..................................................................... 12 Marginal zone B cells and their activation ......................................................................................................... 13 Follicular B cells and their further differentiation.......................................................................................... 14 Plasma cell decisions ................................................................................................................................................... 15 Memory B cells ............................................................................................................................................................... 16 The germinal centre reaction...................................................................................................................17 T cell help: CD40-CD40L interaction......................................................................................................21 When activation leads to disease: the Epstein-Barr virus..............................................................22 Signalling similarity between LMP-1 and CD40: Mimicking T cell help.....................................25 Interleukin-4 and its signalling pathway .............................................................................................26 Other interleukins........................................................................................................................................28 Regulation of B cell adhesion and motility ..........................................................................................29 Guanine nucleotide exchange factors................................................................................................................... 31 Small Rho GTPases, their effectors and effects................................................................................................. 33 The F-‐BAR protein CIP4 ............................................................................................................................................. 36 Regulation in vitro........................................................................................................................................36 Matherials and Methods .................................................................................................................. 39 Tamoxifen preparation and administration for Mb1-Cre-ERT2 induction ..............................39 Results and Discussions................................................................................................................... 40 Paper I ..............................................................................................................................................................40 Paper II.............................................................................................................................................................42 Paper III ...........................................................................................................................................................44 Part IV (preliminary results)....................................................................................................................46 Conclusions and perspectives ....................................................................................................... 48 Acknowledgements ........................................................................................................................... 51 References............................................................................................................................................ 53 8 Introduction Early B cell differentiation B lymphocytes develop in the foetal liver during embryogenesis and in the bone marrow in adults (Hardy and Hayakawa, 2001; Mackay et al., 2010). During development in the bone marrow, murine B cells express the chemokine receptor CXCR4 in order to ensure that the cells are maintained, and thus attracted to reticular stromal cells that express the ligand CXCL12 (Allende et al., 2010; Mackay et al., 2010). In early development, the murine common lymphoid progenitor (CLP) cells differentiate to pro-‐B cells (Mackay et al., 2010). Upon IL-‐7 stimulation, pro-‐B cells are induced to rearrange the immunoglobulin heavy (IgH) chain V(D)J gene segments (Herzog et al., 2009; Mackay et al., 2010). The recombination-‐activating gene products 1, 2 (Rag1 and Rag2) generate double-‐stranded DNA breaks between recombinational signal sequences (RSS) that flank the V, D and J gene segments, and join cleaved ends by non-‐homologous end joining (NHEJ). After rearrangement is complete, cells become pre-‐B cells, expressing the Igμ heavy (H) chain. Igμ associated with surrogate light (L) chains (VpreB and λ5), Igα (CD79a or Mb-‐1) and Igβ subunits, form the pre-‐B cell receptor (pre-‐BCR) complex (Hardy and Hayakawa, 2001; Herzog et al., 2009; Kurosaki et al., 2010; Gonzalez et al., 2011). This is one of the checkpoints for B cells – only B cells with a properly functioning pre-‐BCR can mature further (Herzog et al., 2009). Later, when V(D)J recombination of another IgH allele is suppressed, B cells start to produce their surface receptors with a single specificity. Several kinases become activated upon pre-‐BCR signalling – the Src-‐family protein kinase Lyn and the spleen tyrosine kinase (Syk), inducing phosphorylation of the immunoreceptor tyrosine-‐based activation motifs (ITAMs) on the cytoplasmic parts of Igα/Igβ. Syk activates phosphatidylinositol 3-‐kinase (PI3K), which regulates survival, proliferation and differentiation (Herzog et al., 2009). In addition, Bruton tyrosine kinase (Btk) activates the mitogen-‐activated protein kinase (MAPK) cascade, leading to activation of nuclear factor κB (NF-‐κB) (Pieper et al., 2013). Signalling via pre-‐BCR leads to λ5 downregulation. Rag1 and Rag2 subsequently induce Ig light chain (L) gene rearrangement (Hardy and Hayakawa, 2001; Herzog et al., 2009). After successful light chain rearrangement, cells start to express a functional B cell receptor. 9 After all maturation steps, cells that are highly reactive to the self-‐antigens die by apoptosis. However, B cells that have low reactivity to self-‐antigens are allowed to leave the bone marrow and enter the periphery (Pieper et al., 2013). Positively selected immature B cells downregulate expression of their receptor CXCR4, resulting in disruption of CXCR4-‐CXCL12 interaction, and are released from the bone marrow to the blood (Mackay et al., 2010). This process is also regulated by a family of G protein coupled receptors – the sphingosine 1 phosphate (S1P1) receptors (Allende et al., 2010). Later, these immature cells will differentiate into B2 conventional B cells, which are involved in the adaptive immune response. Late B cell differentiation B1 and B2 B cells and their localization B1 B cells arise from progenitors that differ from those from which conventional B2 B cells arise, and migrate to the peritoneal and pleural cavities (Montecino-‐Rodriguez and Dorshkind, 2012). They can be divided into two subsets – B1a (CD5+) and B1b (CD5-‐). B1 cells are part of the innate immune system, able to recognize self-‐antigens and carbohydrates. They are responsible for the early reaction to an antigen with IgM responses (Hardy and Hayakawa, 2001; Montecino-‐Rodriguez and Dorshkind, 2012). However, the B1b population can switch to IgA production and has a high rate of somatic hypermutations in its VH regions (Roy et al., 2009). B2 B cells, in the form of immature transitional B cells that have left the bone marrow migrate to the secondary lymphoid organs such as the spleen and lymph nodes. The spleen has a specialized structure that promotes the appropriate immune responses against many different blood-‐borne antigens, and consists of two compartments: red and white pulp. In the red pulp destruction of erythrocytes takes place. The white pulp contains white blood cells: B and T lymphocytes, macrophages, dendritic cells, and other cells (Oracki et al., 2010). 10 Transitional B cells Transitional B cells enter the red pulp of the spleen from the blood through the marginal sinus. They then enter the follicles, which are surrounded by the marginal zone. Here B cells acquire IgD expression and complete their maturation (Oracki et al., 2010). There are different ways to further sub-‐divide transitional B cells, into two or more distinct populations. According to one classification, there are two types of transitional B cells, which are defined by their expressions of the complement receptor CD21 and of the low-‐affinity Fcε receptor (FcεRII or CD23). Early immature (T1) B cells are defined as IgMhighIgDnegCD23-‐ CD21low, whereas late immature (T2) B cells are defined as IgMhighIgDnegCD23+CD21high (Loder et al., 1999; Carsetti et al., 2004; Allman and Pillai, 2008). According to another classification there are three transitional stages of immature B cells: T0, T1 and T2. All three stages express CD93 and IgM, but differ in their expressions of IgD and CD23 (Henderson et al., 2010). T0 transitional B cells (IgD-‐CD23-‐) migrate from the bone marrow via the bloodstream to the red pulp of the spleen, but are unable to enter the white pulp before they mature into the T1 (IgD+CD23-‐) and T2 (IgD+CD23+) stages (Henderson et al., 2010). For transitional T1 and T2 B cells to be able to enter the white pulp of the spleen, the GTPases Rac1 and Rac2, as well the integrins leukocyte function-‐associated antigen-‐1 (LFA-‐1), very late antigen-‐4 (VLA-‐4) and chemokine receptors, are required (Henderson et al., 2010). In mice, whose B cells lack these Rac GTPases, the transitional B cells accumulate in the blood. In addition, in the absence of the tyrosine kinase, Syk, B cell development is arrested at the same stage as in the absence of Rac1 and Rac2 (Henderson et al., 2010), and this leads to the disappearance of the most mature cells (Schweighoffer et al., 2013). One more population of transitional B cells, T3 (CD23+IgMlo), has been suggested, but appears to be an anergic population in the spleen, and these cells do not mature further (Vossenkämper and Spenser, 2011). Eventually, some of the naïve transitional cells home to the marginal zone (MZ) and become marginal zone B cells (MZB), while the majority differentiate into follicular B cells (FOB) (Radbruch et al., 2006; Vossenkämper and Spenser, 2011). All naïve B cells must encounter antigen and become activated via the BCR, which is a second checkpoint essential for their survival (Reth, 1994; Reth and Wienands, 1997). 11 Other signals are also necessary for cell survival. One such signal is the binding of B cell-‐ activating factor (BAFF) to its receptor (BAFF-‐R). Both receptors induce signalling through the transcription factor NF-‐κB, although the B cell receptor signalling leads to the classical pathway, while BAFF signalling leads to alternate pathways. In addition, PI3K, Btk and many other molecules play important roles in this signalling (Mackay et al., 2010; Pieper et al., 2013). The BCR signal-‐strength model, which describes the fates of follicular and marginal zone B cells, has been proposed: Intermediate BCR signalling to self-‐antigens, together with Btk signalling, results in T2 B cell development into follicular B cells. Weak signal via BCR and poor Btk signalling give rise to T2-‐marginal zone precursor (T2-‐MZP) cells, which, after Notch2 engagement by its ligand delta-‐like 1 (DLL1) on the epithelial cells, leads to differentiation into marginal zone B cells (Allman and Pillai, 2008; Pillai and Cariappa, 2009; Cerutti et al., 2013). Chemokine guidance of follicular and marginal zone B cells Follicular B cells. Mature B cells express the chemokine receptor CXCR5 and high levels of integrins LFA-‐1 and VLA-‐4. After B cells encounter an antigen, they become attracted to the follicles by their response to the chemokine CXCL13, which is secreted by follicular dendritic cells (FDCs) (Goodnow et al., 2010; Pereira et al., 2010; Cyster, 2010). Intercellular adhesion molecule-‐1 (ICAM-‐1) and vascular cell adhesion mediator-‐1 (VCAM-‐ 1), the ligands for LFA-‐1 and VLA-‐4, respectively, also play important roles in this interaction between B cells and FDCs, promoting their cell-‐to-‐cell contacts (Harwood and Batista, 2010). In addition to CXCL13, the cellular Epstein-‐Barr virus-‐induced receptor 2 (Ebi2, also known as GPR183) and its ligand 7α,25-‐dihydroxycholesterol (7α,25-‐OHC) guide naïve and activated B cells to the outer follicular niche(s) of secondary lymphoid organs (Pereira et al., 2009; Gatto et al., 2009; Gatto and Brink, 2013). However, germinal centres (GC) are still formed in their normal locations when Ebi2 is absent (Green et al., 2011). Moreover, S1P2 is important for inhibiting the response of GC B cells to chemoattractants, and helps to confine these cells to the middle of the follicle (Green et al., 2011). The chemokine receptor CCR7 is expressed by naïve T cells and (at a low level) in mature B cells, but its expression is greatly upregulated after encountering an antigen (Goodnow et al., 2010; Pereira et al., 2010). CCR7 helps B cells to move towards the T cell zone, where Ebi2 helps to distribute them in the border between the T cell area and the midline of the follicular B cell zone. Here, after B cell interaction with T cells through CD40 12 engagement, expression of Ebi2 is upregulated, whereas CCR7 is downregulated (Pereira et al., 2010; Kelly et al., 2011; Gatto and Brink, 2013). The shuttling of the follicular B cells between the interfollicular and outer follicular regions is very important for FOB proliferation and GC formation (Gatto and Brink, 2013). Marginal zone B cells. The cannabinoid receptor 2 (CR2) guides and positions MZB cells to the marginal zone and prevents their elution to the blood (Basu et al., 2011; Muppidi et al., 2011). Sphingosine 1 phosphate (S1P), which comes to the marginal zone via the blood stream, binds to the sphingosine 1 phosphate receptor 1 (S1P1) or sphingosine 1 phosphate receptor 3 (S1P3) on marginal zone B cells. It interferes with signals from CXCL13, and retains MZB cells in the marginal zone (Cerutti et al., 2013). The adhesive interactions between MZ B cells and stroma cells also play an important role in the retention of the former cells. In this case, LFA-‐1 and VLA-‐4 expressed on B cells interact with ICAM-‐1 and VCAM-‐1 on stromal cells. Later, marginal zone macrophages retain MZB cells by the macrophage receptor with collagenous structure (MARCO) (Pillai and Cariappa, 2009; Cerutti et al., 2013). However, marginal zone B cells are highly motile and migrate constantly between the MZ and follicles (Cinamon et al., 2008; Arnon et al., 2012). The chemokine CXCR5 is required for migration into the follicle. To return to the marginal zone, MZB cells again use S1P1 and S1P3, which are responsible for the attraction and retention of these cells. Shuttling of marginal zone B cells between the MZ and follicles ensures that they can capture an antigen more efficiently, and deliver more of it to the FDCs (Cinamon et al., 2008). In addition, Ebi2 is essential for activated MZB cell movements into the extrafollicullar areas during the primary immune response (Gatto et al., 2009). Marginal zone B cells and their activation Marginal zone B lymphocytes (MZB) are a minor population of the conventional B cells localized in the outer zone of the splenic white pulp. These cells can be identified as IgMhiIgDloCD23-‐CD21hiCD1dhi and are the first to respond to blood-‐borne pathogens. High levels of CD1d in marginal zone B cells make possible antigen (lipid) presentation by these cells to invariant natural killer cells (iNKT) (Pillai and Cariappa, 2009; Cerutti et al., 2013). MZ B cells possess polyreactive BCR and can therefore bind many microbial patterns. These cells are a link between innate and adaptive immune systems. MZ B cells express high levels of Toll-‐like receptors (TLRs) in the same way as other types of cells, such as 13 dendritic cells or macrophages (Cerutti et al., 2013). After antigen encounter, MZB cells migrate to the extra-‐follicular areas between the T cell zone and the red pulp of the spleen, rapidly proliferate and differentiate into plasmablasts with low-‐affinity antibodies (Oracki et al., 2010; Mackay et al., 2010; Vinuesa et al., 2010; Cerutti et al., 2013). However, marginal zone B cells are very diverse and can also generate long-‐lived plasma cells with high-‐affinity antibodies, which can be achieved both along pathways that are T cell-‐dependent and along those that are T cell-‐independent. In addition, MZB cells can undergo class switch recombination, to produce IgG and some IgA (Chappell et al., 2012; Puga et al., 2012; Cerutti et al., 2013). Follicular B cells and their further differentiation Follicular B cells (FOB) are the major population of the B cell pool in the spleen, which home to the follicles. They can be identified as IgMloIgDhiCD23+CD21intCD1dlo (Cerutti et al., 2013). Upon binding an antigen that is presented on FDCs, follicular B cells migrate and localize in the interfollicular zone, at the boundary between B cell follicles and the T cell zone, where ligands of CCR7, CCL21 and CCL19, are expressed (Oracki et al., 2010; Goodnow et al., 2010; Pereira et al., 2010; Cyster 2010; Kerfood et al., 2011). Here, B cells receive the necessary stimulation signals from T helper cells. Major histocompatibility complex (MHC) class II-‐antigen peptides on B cells interact with the T cell receptor (TCR) on T cells, while CD40 on B cells interacts with CD40L on T cells and provide additional stimulation from cytokines secreted by T cells. After they receive these signals, B cells can undergo one of two fates. Either they continue migration to the extra-‐follicular areas, where they differentiate into short-‐lived plasma cells located in the extra-‐follicular foci where they produce early IgM and IgG, or they re-‐enter the follicles, proliferate and form germinal centres. This occurs as early as Day 4 after infection, and is discussed below in more detail. In this case, LFA-‐1 and ICAM-‐1 interaction helps them to survive by preventing apoptosis (Allen et al., 2007; Vinuesa et al., 2010; Mackay et al., 2010; Oracki et al., 2010; Kurosaki, 2010; Cyster, 2010; Gatto and Brink, 2010; Harwood and Batista, 2010; Kerfood et al., 2011; Chu and Berek, 2012). However, B cells cannot enter the germinal centre and differentiate, until Ebi2 expression has been downregulated by the transcriptional repressor B cell lymphoma 6 (Bcl-‐6) (Chan et al., 2010; Goodnow et al., 2010; Pereira et al., 2010; Victora and Nussenzweig, 2012). While GC B cells downregulate Ebi2, they maintain 14 expression of CXCR5, which keeps them in the follicle, where CXCL13 is expressed (Chan et al., 2010; Gatto and Brink, 2010). The antibody-‐producing GC B cells have high specificity for an antigen and differentiate either into long-‐lived and non-‐dividing plasma cells or into memory B cells (Good-‐Jacobson and Shlomchik, 2010; Mackay et al., 2010; Vinuesa et al., 2010; Yoshida et al., 2010; Chu and Berek, 2012). Plasma cell decisions The location at which cells receive activation signals will later determine the ability of plasmablasts to migrate to specific locations (Radbruch et al., 2006; Mackay et al., 2010). There are two ways for B cells to become plasma cells. After activation, B cells either differentiate into short-‐lived plasma cells, which are found in the extrafollicular areas of secondary lymphoid tissue, or go through the germinal centre reaction and become long-‐ lived plasma cells, which migrate to the bone marrow (Shapiro-‐Shelef and Calame, 2005; Oracki et al., 2010; McHeyzer-‐Williams et al., 2012). It has been suggested that the affinity of the BCR for an antigen regulates the capacity of the B cells to present the antigen to follicular T helper cells (McHeyzer-‐Williams et al., 2012). With increased help from follicular T helper cells, the fate of B cells is directed towards the germinal centre reaction. Moreover, the follicular T helper cells direct B cell commitment towards either non-‐GC or GC plasma cells and determine the class of antibody produced (Schwickert et al., 2011; McHeyzer-‐Williams et al., 2012). It has, however, been suggested that the plasma cell pool in the bone marrow contains not only long-‐lived cells, but also short-‐lived cells (Bortnick and Allman, 2013). In addition, recent observations suggest that B cells that have responded to a T cell-‐independent antigen, such as lipopolysaccharide (LPS), are also able to generate long-‐lived plasma cells, even though they are not able to maintain a germinal centre response (Bortnick and Allman, 2013). Due to affinity maturation in the germinal centres, the long-‐lived plasma cells produce IgG antibodies with high affinity and with hypermutated variable regions (Radbruch et al., 2006; Chu and Berek, 2012). Plasma cells that are terminally differentiated, non-‐dividing 15 and are secreting antibodies can be identified by surface expression of CD138 (Syndecan-‐ 1) (Smith et al., 1996). For differentiation into long-‐lived plasma cells, the transcriptional repressor B lymphocyte-‐ induced maturation protein-‐1 (Blimp-‐1), which represses both Bcl-‐6 and Pax5, is essential (Shapiro-‐Shelef and Calame et al., 2005; Oracki et al., 2010; Chu and Berek, 2012). During differentiation into plasma cells, B cells downregulate CXCR5 and upregulate Ebi2 and CXCR4. This allows plasmablasts or plasma cell precursors to home to the bone marrow in response to CXCL12, which is produced by stromal cells (Chan et al., 2010; Gatto and Brink, 2010; Mackay et al., 2010; Pereira et al., 2010; Yoshida et al., 2010). Additionally, S1P1 receptors are also important for the egress of lymphocytes from the secondary lymphoid organs (Allende et al., 2010; Pereira et al., 2010; Cyster, 2010). S1P1 blockage causes plasmablast accumulation in the spleen, thereby inhibiting the migration of plasmablasts to the bone marrow (Yoshida et al., 2010). Proper homing is critical for the survival of long-‐lived plasma cells, since failure to enter the bone marrow may compromise long-‐lived humoral immunity (Bortnick and Allman, 2013). In the bone marrow, the interaction of APRIL and/or BAFF, which are produced by stromal cells, with BAFF-‐R, which is present on plasma cells, is essential for plasma cells to be sustained as long-‐lived plasma cells and thus keep antibody titres high for a long time without the need of the memory B cell pool to be activated (Ahuja et al., 2008; Allman and Pillai, 2008). Memory B cells Memory B cells play an essential role in long-‐term immunity maintenance. They can be defined as antigen-‐primed cells that express high-‐affinity antibodies, which can quickly differentiate into plasma cells during antigen recall. Memory B cells may remain in a resting state long after stimulation, and do not need antigen or T cell help for survival (Klein and Dalla-‐Favera, 2008; Shlomchik and Weisel, 2012). Memory B cells do not secrete and can be generated via germinal centres either as IgM+ or as isotype-‐switched (swIg+) types. The latter make up more than 95% of the cells. They can also be generated by a GC-‐ independent pathway with non-‐mutated receptors (Pape et al., 2011; Taylor et al., 2012). The B cell receptors on swIg+ memory B cells have higher affinity than that of the IgM+ 16 memory B cells (Pape et al., 2011). Highly mutated GC-‐derived memory B cells with either IgM+ and swIg+ receptors express the surface receptor CD73. Early memory B cells can be detected even before the formation of GCs, and they most probably come from the same precursors as the cells of the germinal centre, since they express the memory B cell markers CD38, Bcl-‐2 and CCR6, together with the GC markers GL7 and CD95 (FAS) (Taylor et al., 2012a; Taylor et al., 2012b). A key player that determines whether the precursors will differentiate into memory cells or enter the germinal centre reaction is CD40. In mice treated with anti-‐CD40 antibodies, the germinal centre differentiation was completely blocked, while generation of GC-‐ independent memory B cells was not affected (Erickson et al., 2002; Taylor et al., 2012). After antigen challenge, IgM+ memory B cells proliferate and differentiate via the GC reaction, but swIg+ can quickly generate large amounts of the plasma cells without entering GCs (Dogan et al., 2009; Pape et al., 2011). This rapid expansion of plasma cells requires help from T cells, most probably T follicular helper memory cells (Taylor et al., 2012). The function of IgM+ memory B cells has not been fully elucidated since they respond poorly. They might be important for re-‐infection when the pathogen has mutated. In this case, memory B cells can enter germinal centres, mutate their receptors, and produce a high-‐ affinity response. This contrasts with swIg+ cells, which cannot re-‐enter the germinal centres (Taylor et al., 2012). Comparing the long-‐lived plasma cell response with the memory B cell response, Purtha et al. (2011) have shown by studying virus infections in mice that the polyclonal pool of swIg memory B cells can recognize and neutralize mutated pathogens equally well as wild-‐type pathogens. Long-‐lived plasma cells, however, were specific only for the original pathogen. The Germinal Centre reaction The germinal centre (GC) (Fig. 1) is the structure within the follicle in which B cells rapidly proliferate in response to T cell-‐dependent antigen stimulation. Shortly after the germinal centre is formed, it starts to resolve into two functionally distinct compartments – the dark zone (DZ) and the light zone (LZ). In the latter, B cells undergo class switch recombination 17 and somatic hypermutation. Only B cells with high-‐affinity receptors for the antigen will be selected (in the LZ) and differentiate either to long-‐lived memory B cells or antibody-‐ producing plasma cells (MacLennan, 1994; Hauser et al., 2007; Schwickert et al., 2007; Kurosaki, 2010; Vinuesa et al., 2010; Gatto and Brink, 2010; Gonzalez et al., 2011). The germinal centre compartmentalisation into DZ and LZ is mediated by opposing gradients of CXCL12 and CXCL13 (Gatto and Brink, 2010; Victora and Nussenzweig, 2012). The DZ is densely packed with proliferating B cells, whereas the LZ is populated with B cells, follicular T helper cells (TFH) and follicular dendritic cells (FDC). The DZ is located close to the T cell zone, while the LZ is located close to the marginal sinus, where antigens enter the tissue (Hauser et al, 2007; Allen et al., 2007; Gatto and Brink, 2010; Cyster, 2010; Victora and Nussenzweig, 2012). Figure 1. The dynamic Germinal Centre model (from Victora and Nussenzweig, 2012). FDCs are stromal cells that accumulate an antigen and form a network in the LZ of the germinal centre. Ablation of FDCs from the germinal centre leads to its disappearance (Vinuesa et al., 2010; Wang et al., 2011). These cells express high levels of integrin ligands (VCAM-‐1 and ICAM-‐1), and they catch and retain antigens in the form of immune complexes through Fc and complement receptors (Allen et al., 2007; Allen and Cyster, 2008; Hauser et al., 2007; Gatto and Brink, 2010). FDCs are the source of the CXCL13 18 chemokine, the ligand for CXCR5 and which is expressed on T and B cells (Vinuesa et al., 2010). Immune complexes, presented on FDCs, are strong stimuli of B cells. B cells activation results in the expression of activation-‐induced cytidine deaminase (AID), and the induction of class switch recombination (CSR) and somatic hypermutation (SHM) (Allen and Cyster, 2008; Victora and Nussenzweig, 2012). TFH cells are a minor population (5-‐20%) in the LZ of the GC, but are crucial for the induction of GC responses by providing survival signals to GC B cells (Gatto and Brink, 2010). They can be distinguished by their high expression of programmed death-‐1 (PD-‐1). Also, TFH cells express CD40L and produce high amounts of IL-‐4 and IL-‐21, which are essential for GC B cell survival and differentiation. They are thus essential for the full development and maintenance of mature germinal centres (Goodnow et al., 2010; Vinuesa et al., 2010; Hauser et al., 2010). Additionally, follicular T regulatory (TFR) cells control the GC reaction and the humoral immune response, since the absence of these cells leads to a greater GC reaction, but with only few antigen-‐specific GC B cells (Chung et al., 2011; Linterman et al., 2011; Wollenberg et al., 2011). The dark and light zone compartments of the germinal centre have different gene expression patterns. DZ cells have upregulated expressions of genes involved in mitosis, whereas genes that control lymphocyte activation, cell surface receptors and regulators of apoptosis are elevated in LZ cells (Victora et al., 2010). B cells in both zones have similar DNA synthesis levels, but cell division occurs mainly in the DZ of the germinal centre (Victora et al., 2010). Several markers have been identified that can be used in flow cytometry to distinguish DZ and LZ B cells: CXCR4hiCD83loCD86lo (DZ cells) and CXCR4loCD83hiCD86hi (LZ cells) (Victora et al., 2010). In addition, GL7 and CD95 (FAS) can be used as markers of germinal centres (Taylor et al., 2012). During the affinity maturation process, germinal centre B cells move continuously within the LZ zone, searching for FDCs that carry an antigen, or within the DZ during division and mutation. They also move between these compartments (Hauser et al., 2007; Allen et al., 2007). It has been shown that 50% of cells from the DZ can migrate to the LZ in 6 hours in vivo, whereas cells from the LZ are less motile: only 15% migrate into the DZ during the same period (Victora et al., 2010; Victora and Nussenzweig, 2012). 19 There are several hypotheses about how cells move within the GC. The dynamic germinal centre model (Fig. 1) has recently been proposed, in which B cells in the LZ capture an antigen and compete for T cell help. Those B cells that present peptide-‐MHC class II to TFH cells obtain an activation signal and migrate to the DZ, where they rapidly divide. On the other hand, LZ cells that fail to be selected undergo apoptosis. In addition, this selection process is highly synchronized with the cycling between the DZ and LZ (Victora et al., 2010; Victora and Nussenzweig, 2012). This agrees with the finding that germinal centre B cells move preferentially from the DZ to the LZ (Beltman et al., 2011). Meyer-‐Hermann et al. have presented another, mathematical model (called LEDA), in which they combine the previous theory with an experimental mathematical approach to integrate large amounts of data. This model predicts a cyclic re-‐entry path for the B cells that have been positively selected on FDCs and can successfully compete for help from TFH cells. The more peptide-‐MHC B cells express, the more they will divide, but fewer mutations will be introduced. Cells that do not compete will die by apoptosis. The model was confirmed, since B cells enter the S phase when still in the LZ, just after they are selected, and immediately move to the DZ, just before the G2/M cell cycle phases. The model predicts that intracellular antigen is distributed to the daughter cells unequally in the dark zone, which is compatible with the asymmetric cell division described by Thaunat et al. (2012). LEDA predicts that those daughter cells that retain the antigen will enter final differentiation. In addition, the model predicts that the plasmablasts will exit via the DZ, where they must first divide and then leave the GC towards the T cell zone. Daughter cells that did not have a sufficient amount of an intracellular antigen will return to the LZ for more cycles (Meyer-‐Hermann et al., 2012). The model predicts that this is the mechanism by which the long-‐lived plasma cells are generated, but not memory B cells. Generation of memory B cells remains to be investigated separately, because it has different dynamics. In summary, Meyer-‐Herman et al. (2012) have analysed previous results and present new details of the B cell selection process. They also propose new models for division and the pathway of GC exit. In the majority of cases, GCs are formed in T cell-‐dependent B cell responses, but certain T cell-‐independent antigens, such as bacterial polysaccharides, can also induce GC formation (Sverremark and Fernandez, 1998; Good-‐Jacobson and Shlomchik, 2010; Oracki et al., 2010; Vinuesa et al., 2010). A T cell-‐independent GC response is very poor, the structures 20 are short-‐lived and somatic hypermutation of the IgV region cannot take place (Vinuesa et al., 2010; McHeyzer-‐Williams et al., 2012). Interestingly, T cells seem to be required, at least for the maintenance of the GCs, even when induced by a T cell-‐independent antigen (Sverremark and Fernandez, 1998; Vinuesa et al., 2000). There is growing evidence that cells of the innate immune system present antigens to B cells and induce a fast and highly diverse antibody response, also providing survival signals to plasma cells (Cerutti et al., 2012). T cell help: CD40-CD40L interaction The CD40-‐CD40L interaction plays a critical role in the development of humoral and cellular immune responses. It is involved in the activation and proliferation of B cells, the formation of germinal centres, antibody production, isotype switching, somatic hypermutation, and the generation of memory B cells and plasma cells (Elgueta et al., 2009; Graham et al., 2010). CD40L (also known as CD154) belongs to the TNF family, and is mainly expressed on activated T cells (Elgueta et al., 2009; Kurosaki et al., 2010; Vinuesa et al., 2010; Graham et al., 2010). It is expressed also on monocytes, macrophages, platelets, mast cells, basophils, eosinophils, epithelial cells and NK cells (Elgueta et al., 2009; Graham et al., 2010). CD40L is a very important co-‐stimulus for the initiation of the GC reaction, and plays an important role in T cell-‐independent GC formation. In the latter case, CD40L is expressed on non-‐T cells (Vinuesa et al., 2010; Cerutti et al., 2012). CD40 belongs to the tumour necrosis factor receptor (TNF-‐R) family. It was first found as a cell surface antigen, restricted to human urinary bladder carcinomas and B cells (Paulie et al., 1985). CD40 is constitutively expressed on almost all B cells (not in plasma cells), DCs, platelets, monocytes and macrophages (Kurosaki et al., 2010; Vinuesa et al., 2010; Graham et al., 2010). It is expressed also on non-‐hematopoietic cells such as fibroblasts, epithelial and endothelial cells (Elgueta et al., 2009). CD40 is associated with TNF receptor-‐ associated factors (TRAFs), and it initiates NF-‐κB, c-‐Jun N-‐terminal kinase (JNK) and p38 signalling pathways (Kurosaki et al., 2010; Graham et al., 2010). B cell activation via BCR and CD40 leads to inhibition of the transcription factor Bcl-‐6, a critical regulator of GCs (Klein and Dalla-‐Favera, 2008; Kurosaki et al., 2010). In T helper cells, Bcl-‐6 expression 21 induced by IL-‐21 leads to differentiation to TFH cells, which are, in turn, are very important for GC B cell formation (Vinuesa et al., 2010). CD40 signalling upregulates activation markers, such as MHC class II and adhesion molecules on B cells. It can also induce the production of chemokines and cytokines, which enhance B cell-‐mediated T cell activation (Elgueta et al., 2009; Graham et al., 2010). In addition, B cells cultured in vitro with anti-‐CD40 antibodies escape apoptosis (Graham et al., 2010). Interestingly, signalling through both BCR and CD40 is necessary not only for GC development, but also for termination of the GC reaction to become either plasma cells or memory B cells (Elgueta et al., 2009; Bolduc et al., 2010; Kishi et al., 2010; Kurosaki et al., 2010; Vinuesa et al., 2010). Mice deficient in either CD40 or CD40L do not have a sufficient response to T cell-‐ dependent antigens, do not form GCs, and cannot undergo class switch recombination (Kurosaki et al., 2010; Good-‐Jacobson and Shlomchik, 2010). Humans deficient in either CD40 or CD40L develop hyper-‐IgM syndrome. Their B cells cannot form GCs and have impaired productions of IgG, IgA and IgE, whereas the level of IgM antibodies is normally elevated. Upon in vitro stimulation with CD40 agonists and cytokines, B cells from patients who lack CD40L can proliferate and switch their Ig class. However, B cells from patients who lack CD40 cannot respond (Kracker et al., 2010; Davies and Thrasher, 2010), providing evidence that the CD40 deficiency is intrinsic to B cells. When activation leads to disease: the Epstein-Barr virus About 95% of all lymphomas are of B cell origin, coming from different stages of B cell development (Küppers et al., 1999; Küppers, 2005; Klein and Favera, 2008). Even though the majority of B cell lymphomas originate either from the germinal centre itself or from post-‐germinal centre B cells, they need very different clinical treatments (Teitell and Pandolfi, 2004; Küppers, 2005; Natkunam, 2007). Hodgkin, Burkitt and follicular lymphomas, for example, originate from the germinal centre, where IL-‐4 signalling and CD40 ligation are very important for B cells. CD40 signalling is essential for the development of a normal immune response, but is also needed for the proliferation and 22 survival of the majority of B cell lymphomas (Teitell and Pandolfi, 2004; Küppers, 2005; Graham et al., 2010). B lymphocytes are the main target of the Epstein-‐Barr virus (EBV), which is strongly associated with certain B cell malignancies. In Burkitt lymphoma, 98% of the B cells carry EBV (Thorley-‐Lawson, 2005). In approximately 40% of classical Hodgkin lymphoma (HL) cases, the Hodgkin Reed-‐Sternberg (HRS) cells are infected with this virus. HRS cells attract T helper cells by secreting T cell-‐attracting cytokines that are usually restricted to DCs. Even though the HRS cells have downregulated many genes, they still express molecules important for interaction with T cells, including CD40. This shows that T cell help is important of for HRS survival (Küppers, 2005). The Epstein-‐Barr virus is a γ-‐herpes virus (also known as human herpes virus 4 or HHV4), which preferentially infects resting B lymphocytes. It can infect also epithelial, T and NK cells. During infection, EBV binds to the CD21 (also known as the CR2) molecule on the surface of B lymphocytes, using its major envelope glycoprotein gp350. It binds also to human leukocyte antigen (HLA) class II as a co-‐receptor, using the glycoprotein gp42 (Cohen, 1999; Young and Rickinson, 2004). More than 90% of the human population is infected with EBV. Infection usually occurs during childhood and is asymptomatic. If the primary infection is delayed until adulthood, about 50% of patients develop infectious mononucleosis. The virus is carried for lifetime by an infected person (Thorley-‐Lawson, 2001; Young and Rickinson, 2004; Kis et al., 2006; Hislop et al., 2007; Luzuriaga and Sullivan, 2010). To escape an immune response in vivo, EBV remains latent in the resting memory B cells (Cohen, 1999; Faulkner et al., 2000; Babcock et al., 2000; Young and Rickinson, 2004), using a highly restricted transcription program without the expression of any immunogenic viral proteins (Faulkner et al., 2000; Babcock et al., 2000). EBV can be reactivated during immunosupression or autoimmunity (Young and Rickinson, 2004). In vitro EBV infection of resting B lymphocytes results in their transformation and immortalization into continuously proliferating lymphoblastoid cell lines (LCLs) (Young and Rickinson, 2004, Kis et al., 2006). Moreover, EBV-‐infected B cells show high levels of expression of several activation markers, such as CD23, CD30, CD39, and CD70, and high levels of expression of various adhesion molecules, such as LFA-‐1, LFA-‐3 and ICAM-‐1 (Young and Rickinson, 2004). Interestingly, LCLs have a typical growing pattern of large 23 and tight aggregates (Rowe et al., 1987; Gregory et al., 1990), which resembles the morphology of anti-‐CD40+IL-‐4 stimulated primary B cells. The Epstein-‐Barr virus is associated with both non-‐malignant diseases, such as infectious mononucleosis (IM), and malignant diseases, such as Burkitt lymphoma (BL), Hodgkin lymphoma (HL), nasopharyngeal carcinoma (NPC), post-‐transplant lymphoproliferative disorders (PTLD), and many others (Faulkner et al., 2000; Thorley-‐Lawson, 2001; Hislop et al., 2007). In various tumours, EBV expresses a different set of latent genes (Table 1 and Fig. 2) (Young et al., 2000; Thorley-‐Lawson, 2001; Young and Rickinson, 2004; Klein et al., 2007). Table 1. Examples of EBV latent gene expression patterns EBV-associated tumours EBV gene expression* Type of latency BL, NPC, PTLD EBNA-‐1 (Qp) Type I cHL, NPC, PTLD EBNA-‐1 (Qp), LMP-‐1, LMP-‐2A, LMP-‐2B Type II PTLD EBNA-‐1 (Cp), -‐2, -‐3, -‐4, -‐5, -‐6, LMP-‐1, LMP-‐2A, LMP-‐2B Type III BL -‐ Burkitt lymphoma, NPC – nasopharyngeal carcinoma, PTLD – post-‐transplant lymphoproliferative disorders, cHL – classical Hodgkin lymphoma *EBERs are expressed in all types of latency Figure 2. The EBV genome is a double-stranded DNA episome (note that the EBNAs also have an alternative nomenclature: here EBNA-‐3A = EBNA-‐3, EBNA-‐3B = EBNA-‐4, EBNA-‐LP = EBNA-‐5 and EBNA-‐3C = EBNA-‐6) (from Young et al., 2000) All cells that are latently infected with EBV constitutively express small, non-‐coding RNAs known as EBERs (EBER-‐1 and EBER-‐2) (Fig. 2) and miRNAs. Every EBV-‐transformed LCL cell carries multiple extra-‐chromosomal copies of the viral episome and constitutively 24 expresses viral latent proteins (Young et al., 2000; Young and Rickinson, 2004). One of the EBV-‐encoded nuclear antigens (EBNAs), EBNA-‐1 is required for the maintenance of the viral episome in proliferating cells. LCLs maintain the viral genome as an episome and express a full set of EBV latent genes (nine virally encoded proteins): six EBV nuclear antigens (EBNA1-‐6) and three latent membrane proteins (LMP-‐1, LMP-‐2A and LMP-‐2B) (Fig. 2) (Young et al., 2000; Babcock et al., 2000; Klein et al., 2007). This gene expression pattern in LCLs corresponds to what is known as “Type III” latency (Table 1). It correlates with the primary infection in humans, when a very strong immune response is induced (Kurth et al., 2000; Hislop et al., 2007). Burkitt lymphomas express only EBNA-‐1 (Type I latency), whereas classical Hodgkin lymphomas express EBNA-‐1 and LMPs: LMP-‐1, LMP-‐ 2A and LMP-‐2B (Type II latency) (Table 1) (Klein et al., 2007). In addition, under the influence of LMP-‐1 and LMP-‐2, EBV-‐infected B cells undergo a germinal centre reaction. Whereas LMP-‐2 forces B cells to start such a reaction, LMP-‐2 and LMP-‐1 can induce class switching and mutations of the Ig genes, respectively. Moreover, LMP-‐1 can downregulate Bcl-‐6 expression, and memory B cells that carry EBV leave the germinal centre. In the periphery, latently infected memory B cells immediately switch to the Type I latency, in which only EBNA-‐1 is expressed (Thorley-‐Lawson, 2005). Signalling similarity between LMP-1 and CD40: Mimicking T cell help The main EBV-‐encoded transforming oncoprotein LMP-‐1 contains six transmembrane sequences (of length 162 aa) with a short (24 aa) cytoplasmic amino terminal domain and a large (200 aa) carboxy terminal domain (Busch and Bishop, 1999; Young et al., 2000; Thorley-‐Lawson, 2001). LMP-‐1 acts as a constitutively active pseudo-‐receptor and is critical for the in vitro transformation and proliferation of EBV-‐infected human B cells (Dirmeier et al., 2003). It has a pleiotropic effect in B cells, by inducing the expression of cell-‐surface adhesion and activation molecules, and by upregulating the expression of several anti-‐apoptotic proteins (Thorley-‐Lawson, 2001; Young and Rickinson, 2004). LMP-‐1 mimics T helper cell signals in a ligand-‐independent manner and has functional homology with CD40, although their structures are different (Young et al., 2000; Thorley-‐ Lawson, 2001). In addition, when LMP-‐1 needs to amplify and maintain its signal, CD40 signalling is tightly regulated (Graham et al., 2010). Interestingly, LMP-‐1 can partially restore the defects when expressed in B cells of CD40-‐/-‐ mice (Uchida et al., 1999). 25 Deregulation of this signalling pathway is sufficient to induce B cell transformation, and can lead to tumour formation (Hömig-‐Hölzel et al., 2008; Klein and Dalla-‐Favera, 2008; Kishi et al., 2010). LMP-‐1 acts as a constitutively active TNFR and can induce several signalling pathways. CD40 and LPM-‐1 both signal via TRAFs, and induce similar early pathways with the activation of different kinase cascades (Graham et al., 2010). As a CD40, LMP-‐1 signals through a TRAF-‐binding domain and induces the NF-‐κB pathway. This is followed by B cell proliferation and survival (Young et al., 2000; Thorley-‐Lawson, 2001; Graham et al., 2010). Even though both CD40 and LMP-‐1 act through the same adaptor proteins, the pathways are regulated in different ways. TRAF3, for example, regulates CD40 negatively, while it induces LMP-‐1 signals (Graham et al., 2010). Interestingly, LMP-‐1 itself can also induce the JAK/STAT pathway, when ectopically expressed in an embryonic kidney cell line. It interacts with Janus-‐activated kinase 3 (JAK3), induces its autophosphorylation, and thereafter activates the signal transducer and activator of transcription 1 (STAT1) (Gires et al., 1999; Young et al., 2000). In addition, LMP-‐1 can activate the small Rho GTPase Cdc42 when expressed in fibroblasts, and it can induce filopodia formation (Puls et al., 1999; Young et al., 2000). Activation of the LMP-‐1 promoter and expression of the LMP-‐1 protein are needed for in vitro proliferation of EBV-‐infected B cells, and are regulated by the EBV-‐encoded protein EBNA-‐2 (Klein et al., 2007). However, LMP-‐1 is still expressed in the absence of EBNA-‐2 in Hodgkin lymphomas. Goormachtigh et al. (2006) proposed that EBV has at least one more way to express LMP-‐1 in the absence of EBNA-‐2, and discovered an alternative mechanism of JNK-‐dependent LMP-‐1 auto-‐activation. Furthermore, Kis et al. (2005) showed that IL-‐4 and CD40 ligation in EBV-‐infected Hodgkin lymphoma cell lines can induce LMP-‐1 and in this way replace EBNA-‐2. However, Chen et al. had proposed as early as 2001, that STATs are responsible for EBNA-‐2-‐independent LMP-‐1 expression (Chen et al., 2001). Interleukin-4 and its signalling pathways There are many different signal molecules, such as cytokines, that transmit signals from the outside of the cell to the inside and induce specific gene transcription. One such that is relevant to the work described here is the pleiotropic Type I cytokine IL-‐4, which is 26 produced mainly by CD4+ T helper cells. It is produced also by basophils, mast cells, NK-‐T cells and γ/δ T cells (Nelms et al., 1999). IL-‐4 plays a critical role in the regulation of the immune responses, and it controls lymphocyte differentiation, proliferation and apoptosis (Boothby et al., 2001; Lu et al., 2005). It regulates the differentiation of antigen-‐stimulated naïve T cells. In proliferating B cells, it acts as a differentiation factor by regulating class switching to IgE and IgG1 (in mice) or to IgE and IgG4 (in humans) (Snapper et al., 1988; Lundgren et al., 1994; Nelms et al., 1999; Stavnezer et al., 2008). It has been shown that IL-‐4 causes large changes in B cell morphology and induces B cell polarization. It causes also B cell spreading with long protrusions, and microvilli adhesion and motility (Davey et al., 1998 and 2000). IL-‐4 increases MHC class II expression in B cells and enhances CD23 expression. It also upregulates the expression of its own receptor (IL-‐4R) and acts as a co-‐mitogen for B cell proliferation (Howard et al., 1982; Vitetta et al., 1985; Nelms et al., 1999; Boothby et al., 2001). It is also important for tissue adhesion and inflammation. Together with TNF, IL-‐4 induces the expression of VCAM-‐1, and downregulates the expression of E-‐selectin on endothelial cells (Nelms et al., 1999; Boothby et al., 2001). Figure 3. JAK-STAT6 pathway activation (adapted from Kis, 2009) IL-‐4 binds to its surface receptor, which consists of the high-‐affinity binding chain IL-‐4Rα and the common γ chain (γc) (Type I receptor complex). In the cytoplasm, IL-‐4Rα associates with the tyrosine kinase JAK1, while γc associates with JAK3 (Nelms et al., 1999; Wills-‐Karp and Finkelman, 2008). IL-‐4 binding to its receptor causes heterodimerization of the receptor components and activation of the JAKs, which are constitutively associated 27 with the IL-‐4 receptor (Boothby et al., 2001; Murray, 2007). Activated JAKs phosphorylate specific tyrosine residues of the intracellular part of the IL-‐4 receptor, which creates binding sites for the Src-‐homology 2 (SH2) domains of STAT6 and allows recruitment of the latter (Lu et al., 2005; Wills-‐Karp and Finkelman, 2008). JAKs phosphorylate STAT6 (Y641), which leads to its homodimerization and translocation to the nucleus, where it binds to the specific DNA sequence TTC(N)4GAA in various promoters and activates the transcription of target genes (Fig. 3) (Nelms et al., 1999; Ehret et al., 2001; Wills-‐Karp and Finkelman, 2008; Chen and Reich, 2010). The JAK-‐STAT signalling pathway is essential for the signal transduction of many cytokines. There are approximately 38 cytokines and 36 cytokine receptor combinations that use this pathway (Bromberg and Darnell, 2000; Murray, 2007). STATs are negatively regulated by the dephosphorylation of signalling components by protein tyrosine phosphatases (suppressor of cytokine signalling -‐ SOCS) or by the induction of a protein inhibitor of activated STAT (PIAS) (Chen et al., 2001; Murray, 2007). The transcription factor STAT6 itself is crucial for the development of protective immunity, but an imbalance in its activity or in any downstream component of the JAK-‐STAT pathway is associated with pathogenesis of different human diseases (Bromberg and Darnell, 2000; Chen et al., 2001; Lu et al., 2005; Chen and Reich, 2010). Other Interleukins The common gamma-‐chain (γc) is a subunit of the functional receptor complexes not only of IL-‐4, but also of IL-‐2, IL-‐7, IL-‐9, IL-‐15 and IL-‐21 (Spolski and Leonard, 2008; Kovanen and Leonard, 2009). Mutations in γc lead to X-‐linked severe combined immunodeficiency (XSCID) (Spolski and Leonard, 2008). Interleukin-2 (IL-‐2) was one of the first cytokines to be described, and plays an important role in the immune system as a growth, differentiation and survival factor (Nelson, 2002). It was discovered as a T cell factor, promoting T cell-‐dependent immune responses. More recent studies have shown a crucial role of this cytokine in the maintenance of T regulatory cells, which suggests an important role of IL-‐2 in the control of the immune responses (Malek, 2008; Dooms and Abbas, 2010). It has also been suggested that IL-‐2 plays an essential role in immune memory (Malek, 2008). 28 Interleukin-5 (IL-‐5) is a T cell-‐derived cytokine. It was identified as a cytokine that plays an important role in the proliferation and differentiation of mouse and human B cells and eosinophils in vitro. IL-‐5 plays an essential role in terminal B cell differentiation to IgM-‐ secreting and IgG1-‐secreting plasma cells, but the major targets of IL-‐5 in humans are eosinophils. It is an important player in the pathogenesis of asthma and other eosinophil-‐ dependent inflammatory diseases (Kouro and Takatsu, 2009; Takatsu et al., 2009). Interleukin-13 (IL-‐13) does not use γc as the other receptors mentioned here, but its specific receptor, IL-‐13Rα1, forms heterodimers with IL-‐4Rα. Another IL-‐13Rα2 functions as a trap receptor. Whereas IL-‐4 can signal through IL-‐4Rα with γc and also through this IL-‐ 13Rα1/IL-‐4Rα heterodimer, IL-‐13 can do so only through IL-‐13Rα1/IL-‐4Rα. In addition, both can signal through JAK1/JAK3 and STAT6. IL-‐13 is a Th2 cell-‐derived immunoregulatory cytokine that has many diverse functions, one of which is to act as a key mediator of allergic inflammation. Similarly to IL-‐4, IL-‐13 induces B cell proliferation when combined with CD40-‐CD40L, with the subsequent Ig class switching to IgE and IgG4 in humans, but not in mice. It also has important functions in non-‐hematopoietic cells (Jiang et al., 2000; Khurana Hershey, 2003). Interleukin-21 (IL-‐21) is produced by NKT and TFH cells and has pleiotropic effects on both innate and adaptive immune responses. In addition, IL-‐21 has potent anti-‐tumour activity and is associated with autoimmune diseases (Spolski and Leonard, 2008). IL-‐21 acts directly on B cells to induce proliferation, class switching to IgG1, and it keeps B cells in the GCs and promotes their longevity. It also drives TFH cells that are important for an efficient B cell response in the GCs. IL-‐21R signalling in vivo increases the expression of transcription factor Bcl-‐6 (Linterman et al., 2010, Zotos et al., 2010; Goodnow et al., 2010). Without IL-‐21R, Bcl-‐6 expression decreases and B cells exit from the GC (Good-‐Jacobson and Shlomchik, 2010). Linterman et al. (2010) proposed that IL-‐21 contributes to GC formation and the affinity maturation of GC B cells because of the induction/maintenance of Bcl-‐6 (Linterman et al., 2010). In contrast, IL-‐4 plays an important role in the T cell-‐ dependent selection of GC B cells and downregulates Bcl-‐6 (Vinuesa et al., 2010). In addition, Bcl-‐6 suppresses Ebi2, which is in turn responsible for guiding and retaining B cells in the outer follicles (Zotos et al., 2010). 29 Regulation of B cell adhesion and motility Many animal cells have extensions known as microvilli on their surfaces. These are formed by bundles of filamentous actin, held together by actin-‐bundling proteins. Actin is widely expressed in all eukaryotic cells and supports many cell surface structures such as microvilli, microspikes, filopodia and lamellipodia (Campellone and Welch, 2010). Microvilli express several adhesion molecules on their surfaces, and these molecules are important for cell contacts (Revenu et al., 2004). Surface molecules, such as integrins play an essential role in B cell adhesion. They mediate dual activation modes, such as “outside in” or “inside out” signalling. The former reflects the events happening in the cytoplasm and the nucleus after the interaction of integrins with their ligands. The latter acts in the reverse direction, and leads to increased affinity of integrins for their extracellular matrix ligands (Qin et al., 2004; Anthis and Campbell, 2011). Contacts between lymphocytes and stromal cells are initiated mainly by β1 integrins, such as VLA-‐4 on B cells, which binds to VCAM-‐1. VCAM-‐1 is expressed on the stromal cells in bone marrow and on follicular dendritic cells in GCs. β2 integrins, in contrast, induce interactions mainly in the germinal centres, where LFA-‐1, expressed on B cells interacts with its ligand ICAM-‐1, expressed on neighbouring B cells, DCs and FDCs (Harwood and Batista, 2010; Vinuesa et al., 2010). LFA-‐1 interaction with its ligand ICAM-‐1 prevents GC B cells undergoing apoptosis (Harwood and Batista, 2010; Pereira et al., 2010). Furthermore, LFA-‐1-‐induced adhesion depends on B cell activation through BCR cross-‐linking, which in turn induces Rac2-‐ dependent B cell spreading. In addition, CD19 enhances BCR signalling and enables cytoskeletal re-‐organization. One of the downstream signalling molecules involved in these processes is Vav, a guanine nucleotide exchange factor (GEF) for Rho GTPases (Harwood and Batista, 2010; Batista et al., 2010). In the processes, B cells undergo re-‐ organization/re-‐distribution of the molecules on the surface of their membrane, resulting in immunological synapse formation. Fully matured immunological synapses have central and peripheral supramolecular activation clusters (cSMAC and pSMAC), where the cSMAC region is responsible for antigen internalization, and where pSMAC contains LFA-‐1 and VLA-‐4 (Harwood and Batista, 2010). In addition, LFA-‐1 clustering during immunological synapse formation depends on the Wiskott-‐Aldrich syndrome protein and on GEF Dock8 30 (Batista et al., 2010). In general, conformational changes in the integrins lead to changes in affinity for their ligands. Moreover, the binding avidity of integrins is induced by rearrangements of the cytoskeleton (van Kooyk and Figdor, 2000; Kucik, 2002). The cytoskeleton is a highly dynamic structure that allows cell shape to change. It is crucial for the survival of cells, being responsible for cell contact, signalling, movement and division. Reorganization of the cytoskeleton depends on actin polymerization and de-‐ polymerization. Globular actin (G-‐actin) is a monomeric ATP-‐binding protein that can undergo self-‐assembly to form filamentous actin (F-‐actin). Actin filaments are polar and dynamic. They polymerize (grow) at the barbed end and depolymerize at the pointed end. Turnover of the filaments is regulated by actin-‐binding proteins (Revenu et al., 2004; Campellone and Welch, 2010). Microtubules are also responsible for cell shape, polarity and the organization of intracellular organelles (Jaffe and Hall, 2005). They also have a minus end and a plus end, and are localized at the centrosome (MTOC) and at the periphery, respectively. Microtubules are very dynamic and can shrink (catastrophe) and grow (rescue) (Jaffe and Hall, 2005). Actin, tubulin and other proteins (such as Rho guanine nucleotide exchange factors (GEFs) and small Rho GTPases and their effectors) are all involved in the regulation of B cell cytoskeleton reorganization. Guanine nucleotide exchange factors Guanine nucleotide exchange factors (GEFs) are several proteins that respond to extracellular stimuli and activate Rho GTPases. These then regulate many cellular responses that require cytoskeletal changes (Rossman et al., 2005). GEFs can be divided into two groups: classical and non-‐conventional (atypical). The classical GEFs share a common Dbl-‐homology (DH) domain, whereas non-‐conventional GEFs contain either Ced-‐5/Dock180/Myoblast city (CDM)-‐zizimin homology 2 (CZH2) or Dock homology region 2 (DHR2) catalytic domains (Meller et al., 2005; Rossman et al., 2005; Tybulewicz and Henderson, 2009). These non-‐conventional (CZH/DHR) GEF proteins can activate cell division control protein 42 (Cdc42) and Ras-‐related C3 botulinum toxin substrate (Rac), and they can regulate the polymerization of actin (Meller et al., 31 2005). The CZH/DHR GEF family consists of 11 dedicator of cytokinesis (Dock) members that belong to four sub-‐families (Table 2) (Miyamoto and Yamauchi, 2010). Table 2. CZH/DHR GEF family members and their binding partners Sub-family Dock-A Dock-B Dock-C Dock-D (Zir) (Zizimin) Members Dock1(180), 2, 5 Dock3, 4 Dock6, 7, 8 Dock9, 10, 11 Binds to Rac Rac Rac, Cdc42 Cdc42 Dock8 (Zir3) is expressed mostly in hematopoietic cells, but also in other cells. It is highly expressed in germinal centre cells and in tumour cells of different origin (McGhee and Chatila, 2010). Dock8 functions in B cells as an adaptor protein in the TLR9-‐MyD88 signalling pathway (Jabara et al., 2012). Mutations in Dock8 lead to immunodeficiency with susceptibility to cutaneous infections, eosinphilia and high IgE levels. Dock8 is a GEF for both Rac and Cdc42, and it may be related to other immunodeficiencies, such as Wiskott-‐ Aldrich syndrome. Dock9 (Zizimin1) is expressed in several tissues of non-‐hematopoietic cells. It regulates the dendritic growth of hypocampal neurons (Miyamoto and Yamauchi, 2010). Dock9 is a dimer with two Cdc42-‐binding sites, and dimerizes through the CZH2 domain. With higher Cdc42 concentrations, the binding affinity of Dock9 increases. This might represent a mechanism for the regulation of Dock9 activity (Meller et al., 2004; Meller et al., 2005). Dock11 (Zizimin2) has 60% homology with Dock9, but is expressed mainly in B and T lymphocytes. In addition, Dock11 expression is higher in GC B cells after immunization with T cell-‐dependent antigen than its expression in non-‐GC B cells. Its expression in COS-‐1 cells induces the activation of Cdc42 (Nishikimi et al., 2005). Also, Dock11 induces the formation of filopodia in bone marrow-‐derived dendritic cells in response to FcγR or TLR4 signalling. This response is Cdc42-‐dependent. In addition, over-‐expression of the CZH2 domain of Dock11 has a dominant negative effect on 293T cell migration (Sakabe et al., 2012). Dock10 (Zizimin3) is expressed both in hematopoietic and non-‐hematopoietic tissues and has 50% homology with Dock9 and Dock11 (Nishikimi et al., 2005). It is an IL-‐4-‐inducable gene in chronic lymphocytic leukaemia cells (CLLs) and in human peripheral blood B and T 32 cells (Yelo et al., 2008; Alcaraz-‐Garcia et al., 2011). In addition, Dock10 plays an important role in amoeboid migration in melanoma cells (Gadea et al., 2008). Furthermore, it may be involved in invasion and metastasis during the epithelial-‐mesenchymal transition of squamous cell carcinoma (HNSCC) (Humtsoe et al. 2012). Small Rho GTPases, their effectors and effects Rho GTPases are members of a large sub-‐family, belonging to the Ras superfamily. They play roles in very diverse processes, such as transcription activation, adhesion, polarization, cytoskeletal rearrangement, migration, cell cycle progression, cell proliferation, survival/apoptosis and the maintenance of genomic stability (Williams et al., 2008; Mulloy et al., 2010). Small Rho GTPases transduce signals from many different receptors, and act as crossroads in various signalling pathways. Figure 4. The molecular events necessary for B cell adhesion and motility. GEF – guanine nucleotide exchange factor; GAP – GTPase activating protein; GDI – guanine nucleotide dissociation inhibitors; WH1 domain – WASP homology domain; B – basic region; GBD – GTPase binding domain (also called CRIB – Cdc42 and Rac interactive binding); PRD – proline-‐rich domain; VCA – veprolin-‐, central-‐, acidic region domain; Arp2/Arp3 – actin-‐related proteins 2 and 3. WASP – Wiskott-‐Aldrich syndrome protein Rho GTPases are guanine nucleotide-‐binding proteins and thus they are inactive when bound to GDP and active when bound to GTP. The cycling between these two states is regulated by guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs) and guanine nucleotide dissociation inhibitors (GDIs). Since many of the effectors are membrane-‐associated proteins, Rho GTPases can be modified at the carboxy terminus to restrict their locations to the plasma membrane. GAPs stimulate GTP hydrolysis, which leads to inactivation of Rho GTPases. In addition, GDIs can bind to the carboxy terminus of 33 Rho GTPases and thereby inhibit their binding to the membrane. In this case, Rho GTPases cannot be activated to interact with their effectors. GEFs induce a switch from GDP-‐binding to GTP-‐binding and Rho GTPases can bind their target proteins when they are in the active GTP-‐bound state. This is the way in which they induce many processes such as cytoskeleton regulation, microtubule dynamics, cell division, migration and adhesion (Fig. 4) (Meller et al., 2005; Tybulewicz and Henderson, 2009; Feng and Cerione, 2010; Mulloy et al., 2010). Several GEFs, GAPs and GDIs can act on the same Rho GTPase, and each Rho GTPase can activate many effectors, thus inducing many signals simultaneously and affecting various cellular functions. Cdc42 and Racs, members of small Rho GTPases that are key regulators of cytoskeleton reorganization, control actin polymerization and microtubule dynamics in B cells (Jaffe and Hall, 2005). Cdc42 is involved in many different cellular systems. It is clear that Cdc42 regulates signalling pathways in a cell-‐specific and tissue-‐specific manner, especially in activities that involve the actin cytoskeleton (Melendez et al., 2011). Cdc42 deletion, using the CD19-‐Cre-‐ expressing mouse strain, results in ablation of Cdc42 from the pro-‐/pre-‐B cell stage and later differentiation stages. B cell development is impaired and blocked at the early transitional stage. The numbers of mature B cells are lower, as is the strength of the antigen-‐specific humoral immune response. This indicates that Cdc42 is importance for B cell differentiation and activation (Guo et al., 2009). There are three Rac GTPases (Rac1, Rac2 and Rac3), which have reasonably degrees of homology. However, their expression patterns differ. Rac1 is expressed ubiquitously, and mice that lack it are embryonic lethal. Rac2 is expressed only in hematopoietic cells, while Rac3 is expressed mainly in the brain (Gu et al., 2003). Rac1 and Rac2 are crucial regulators of hematopoiesis (Gu et al., 2003; Cancelas et al., 2005; Mulloy et al., 2010). Both GTPases are critical for B cell development. When mouse B cells lack both Rac1 and Rac2, mature B cells are absent from the spleen, due to a developmental block in T1 B cells. The absence of Rac2 alone leads to a milder phenotype than the phenotype that develops in the absence of both Rac1 and Rac2, with more a 34 pronounced deficiency in marginal zone B cells and B1a cells. There is no obvious effect when only Rac1 is deleted (Walmsley et al., 2003; Henderson et al., 2010). Deletion of either Rac1 or Rac2 does not affect T cell development, but development stopps at the pre-‐ TCR checkpoint when both Racs are lacking (Tybulewicz and Henderson, 2009). In summary, Rac1 and Rac2 are redundant and sometimes can compensate for each other. There are many Cdc42 and Rac1 effectors, including several kinases, lipases, oxidases and scaffold proteins (Thrasher and Burns, 2010). Two of the target proteins, Wiskott-‐Aldrich syndrome protein (WASP) and N-‐WASP, have 50% homology, and a much higher homology in the functional domains (Blundell et al., 2010). WASP is expressed in all hematopoietic cells. Mutations in WASP in humans cause defects in cell migration, leading to the combined X-‐linked recessive primary immunodeficiency Wiskott-‐Aldrich syndrome (WAS) (Blundell et al., 2010; Campellone and Welch, 2010; Thrasher and Burns, 2010). Mutations may also cause X-‐linked trombocytopenia (XLT) or X-‐linked neutropenia (XLN). Hematopoietic cells from patients with WAS have aberrant microvilli on their surfaces (Takenawa and Suetsugu, 2007; Blundell et al., 2010). Neural (N)-‐WASP is ubiquitously expressed, and its deletion in mice is embryonic lethal. If N-‐WASP is not functioning properly, cells have multiple deficiencies in processes that require actin dynamics (Campellone and Welch, 2010). In the absence of binding partners, WASP has an auto-‐inhibited conformation in which the GTPase-‐binding domain (GBD) interacts with the VCA (verprolin-‐, central-‐, acidic region-‐) domain and prevents the actin-‐related protein 2 and 3 (Arp2/3) complex and monomeric actin binding to its carboxy terminus (Fig. 4) (Takenawa and Suetsugu, 2007; Thrasher and Burns, 2010). The inactive state of WASP is stabilized by another protein known as the WH1-‐interacting protein (WIP), which also regulates the absolute level of WASP in the cell, protects it from degradation, and localizes it to the areas of active actin polymerization (Blundell et al., 2010; Campellone and Welch, 2010; Thrasher and Burns, 2010). When Cdc42 is active, it can bind to the GBD on WASP and activate it by induction of a conformational change. This allows the recruitment and activation of the Arp2/3 complex (ARPC 1-‐5, Arp2 and Arp3), leading to actin polymerization and filopodia formation (Fig. 4) 35 (Revenu et al., 2004; Jaffe and Hall, 2005; Takenawa and Suetsugu, 2007; Thrasher and Burns, 2010; Feng and Cerione, 2010). This process also induces the elongation of microtubules, leading to the formation of membrane protrusions (Feng and Cerione, 2010). In addition, Rac1 can also bind to the GBD on WASP, but to a lesser degree (Blundell et al., 2010). Rac1, however, does not seem to be involved in WASP activation, although it may play a role in the activation of N-‐WASP (Thrasher and Burns, 2010). The F-BAR protein CIP4 Cdc42-‐interacting protein 4 (CIP4) belongs to the Fes/CIP4 homology-‐Bin/Amphyphysin/ Rvsp (F-‐BAR) multi-‐domain protein family (Aspenström, 2009). CIP4 binds the lipid membrane through the F-‐BAR domain, and becomes associated with microtubules through its highly conserved N-‐terminal Fes/CIP4 homology (FCH) domain. Its coiled-‐coiled domain subsequently interacts with active Cdc42 and its C terminal Src homology 3 (SH3) domain interacts with WASP (Aspenström, 1997; Tian et al., 2000). In addition, the RhoGAP that interacts with the CIP4 homologues (RICH-‐1) protein is a GAP for Cdc42 and Rac (Richnau and Aspenström, 2001). Interestingly, RICH-‐1 binds CIP4 through the same SH3 domain as WASP (Richnau and Aspenström, 2001). B and T cell development is not affected in CIP4-‐deficient mice, although the number of germinal centres and the levels of T cell-‐dependent antibody production and affinity maturation are reduced. Moreover, the defect that is induced by deletion of CIP4 is intrinsic to the T cells (Koduru et al., 2010). Also, CIP4 is important in NK cells, where it acts as a cytoskeletal adaptor and polarizes the microtubule-‐organizing centre (MTOC) (Banerjee et al. 2007). A recent study of cells from patients with chronic lymphocytic leukaemia (CLL) showed that CIP4 associates with active Cdc42, and is necessary for lamellipodium polarization and directed cell movement (Malet-‐Engra et al., 2013). All these results show that CIP4 is important in actin polymerization and cytoskeleton rearrangements. 36 Regulation in vitro As mentioned previously, B cells must undergo cytoskeletal changes to be able to adhere and move during their differentiation processes. Most studies about regulation of the cytoskeleton and changes in cell shape have been performed in vitro, but presumably similar mechanisms operate in vivo. When activated with various stimuli in vitro, B cells spread, express microvilli, form aggregates, become polarized and become motile (Fig. 5) (Severinson and Westerberg, 2003). Contacts between lymphocytes are necessary for the immune response. Lymphocytes may contact each other by forming microvilli, which contain many receptors (including integrins and perhaps also chemokine receptors). ICAM-‐1, MHC class II and CD86 are present on the microvilli, which suggests that these molecules are important for adhesion and antigen presentation (Greicius et al., 2003). Figure 5. Morphology responses induced in B cells (from Severinson and Westerberg, 2003) Cytokine stimulation is very important for B cell activation and proliferation. One of the T cell-‐derived cytokines, IL-‐4, changes the activation status of Cdc42. In addition, over-‐ expressed and constitutively active Cdc42 and Rac1 can induce filopodia and lamellipodia, respectively, in B cells (Westerberg et al., 2001). IL-‐4 induces cell adhesion and motility. When activated B cells are cultured in the presence of immobilized antibodies against B cell surface structures (such as anti-‐CD44), they form long dendritic protrusions. This process is coordinated by actin polymerization, microtubules and vimentin. The spreading process starts with actin polymerization, followed by the growth of microtubules and intermediate filaments that are oriented towards polymerized actin (Sumoza-‐Toledo and Santos-‐Argumedo, 2004). 37 B cells stimulated with LPS+IL-‐4 or anti-‐CD40±IL-‐4, but not LPS alone or in the presence of other cytokines, exhibit dendritic protrusions and extensive microvilli formation (Davey et al., 1998; Greicius et al., 2003). IL-‐4-‐induced cell adhesion and actin rearrangements are STAT6-‐dependent. Using STAT6-‐/-‐ mice, Davey et al. have shown that STAT6 is needed for the IL-‐4-‐induced regulation of B cell morphology and adhesion. These STAT6-‐/-‐ B cells cannot spread, polarize, or form aggregates in response to IL-‐4 (Davey et al., 2000). This indicates that the morphology responses depend on transcriptional activation. Greicius et al. have shown that B cells activated with LPS+IL-‐4 express slightly more ICAM-‐ 1 than those stimulated with LPS alone. In addition, ICAM-‐1 is preferably localized to the tips of the microvilli, whereas LFA-‐1 is expressed on the flat surface (Greicius et al., 2003). The authors suggest that receptor distribution plays a role in direct IL-‐4-‐induced cellular interactions. Interestingly, anti-‐CD40+IL-‐4 activated B lymphocytes from WASP-‐/-‐ mice or from patients with WAS have fewer long microvilli (Westerberg et al., 2001). Further, WASP-‐/-‐ B lymphocytes have impaired homing in vivo, and defective migration, adhesion, aggregation, polarization and spreading in vitro (Westerberg et al., 2001 and 2005). Anti-‐CD40 alone or LPS together with IL-‐4 can induce homotypic round and tight aggregates in B cell cultures. In contrast, LPS alone induces fewer cells to aggregate, and the aggregates have irregular shapes. Experiments with LFA-‐1-‐/-‐ mice or monoclonal antibodies to LFA-‐1 showed that homotypic B cell adhesion, induced by LPS with or without IL-‐4, is LFA-‐1-‐dependent (Greicius et al., 1998). However, the contact time between cells stimulated by LPS+IL-‐4 is longer than that of cells stimulated with LPS. In the presence of anti-‐CD40, these contacts are even longer. Greicius et al. (1998) hypothesize that tight round aggregates induce a dynamic response in response to T cell-‐dependent stimuli, resulting in aggregates with the maximal number of contacts per cell. They speculate that this response is important for the formation of germinal centres. 38 Materials and Methods Tamoxifen preparation and administration for Mb1-cre-ERT2 induction When a mouse strain that expresses a non-‐inducible cre, a recombinase enzyme, is crossed with a mouse strain that has loxP insertions, effects will accumulate through all differentiation stages in which the cre-‐gene is expressed. A strain with an inducible cre will avoid this problem, especially when used to study genes that are expressed in mature cells. For this reason, we used Mb1-‐Cre-‐ERT2, but not regular Mb1-‐Cre or the very commonly used CD19-‐Cre, to delete genes from mature B cells. Mb1-‐Cre-‐ERT2 is expressed in the cytoplasm as a fusion protein of Mb1-‐Cre with the binding domain of the estrogen receptor. After treatment with tamoxifen, an antagonist of the estrogen receptor, Cre-‐ERT2 translocates into the nucleus and induces recombination of the sequence surrounded by loxP sites, resulting in the removal of the target gene. The majority of protocols for tamoxifen administration employ quite large volumes given to mice. We have, however, used a protocol that allowed us to reduce the volume from 250 to 50 μl/mouse. To make this possible, 100 mg of tamoxifen (Sigma) was pre-‐wetted in 100 μl of 100% ethanol and dissolved in 900 μl corn oil (Sigma) to a final concentration of 100 mg/ml, by vigorous shaking at 55°C for approx. 5 hours. The dissolved tamoxifen was divided into aliquotes and stored at -‐20°C. A dose of 5 mg of tamoxifen was given per mouse at a time. It was administrated orally by gavage for 5 days in a row. Mice were killed on Day 3 after the final tamoxifen administration. Tamoxifen can cause peritonitis when injected intraperitoneally, which is harmful to mice and might affect the humoral immune response. We did not notice any side effects of the tamoxifen. Additionally, we did not notice any detectable deletion in the absence of tamoxifen treatment. 39 Results and Discussion Paper I The STAT6 signalling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-Barr virus-encoded protein LMP-1 in absence of EBNA-2: implications for the type II EBV latent gene expression in Hodgkin lymphoma. The aim of this study was to investigate the influence of IL-‐4 and IL-‐13 on the Epstein-‐Barr virus latent membrane protein (LMP)-‐1 in the absence of Epstein-‐Barr virus nuclear antigen (EBNA)-‐2, and possible involvement in Hodgkin lymphoma. Epstein-‐Barr virus (EBV) is a γ-‐herpes virus found in many tumours. EBV-‐infected B cells can give rise to lymphomas, when the immune system is compromised (Type III latency) (Young and Rickinson, 2004). LMP-‐1 is the main EBV oncoprotein necessary for B cell transformation and proliferation in vitro (Dirmeier et al., 2003). Its expression is regulated in Type III latency by EBNA-‐2 (Young and Rickinson, 2004). However, in Hodgkin-‐Reed Sternberg cells that carry EBV LMP-‐1 is expressed in the absence of EBNA-‐2 (Type II EBV latency). CD40 ligation and stimulation with IL-‐4 can induce the expression of LMP-‐1 in the absence of EBNA-‐2 in the EBV-‐converted, HL-‐derived cell line KMH2-‐EBV (Kis et al., 2005, 2006). We showed that IL-‐4 alone induces LMP-‐1 expression in the KMH2-‐EBV cells, importantly in the absence of EBNA-‐2 expression. Apart from IL-‐4, IL-‐13 was also found to be able to induce LMP-‐1 expression. Both IL-‐4 and IL-‐13 signal through STAT6 (Nelms et al., 1999; Hershey, 2003), which is constitutively active in HRS cells (Skinnider et al., 2002). We showed that both cytokines induce STAT6 phosphorylation in KMH2 and KMH2-‐EBV cells. Small interfering RNAs (siRNAs) ectopically introduced into KMH2-‐EBV cells inhibited LMP-‐1 induction by about 50% after IL-‐4 treatment and by 80% after IL-‐13 treatment, thus providing evidence that STAT6 is involved in the induction of LMP-‐1 expression by these cytokines. Two STAT-‐binding sites in the LMP-‐1 promoter had previously been identified (Chen et al., 2001), but neither of them had the TTC(N)4GAA sequence that is expected for a high-‐ affinity STAT6-‐binding site (Nelms et al., 1999; Ehret et al, 2001). Re-‐analysing the sequences of the LMP-‐1 promoter, we identified a new STAT-‐binding site with the TTCAGGCGAA sequence, which we named LRS-‐STAT6 (LMP-‐1 regulatory sequence). The 40 other two STAT binding sites consist of palindromes spaced by either two (LRS-‐EDL1) or three (LRS-‐TR) nucleotides. Furthermore, all three binding sites are conserved in multiple EBV strains. Electrophoretic mobility shift assay showed that LRS-‐STAT6 contains a functional STAT6-‐ binding site. LRS-‐STAT6-‐NPC (nucleotide sequences from an Asian EBV strain) could also work as an efficient STAT6 binding site, but a mutation introduced into the TTC/GAA palindrome prevented binding. Surprisingly, LRS-‐TR with sequences spaced by 3 nucleotides could also function as a STAT6 binding site, but its affinity was lower. In addition, LRS-‐EDL1 (TTC(N)2GAA) was not functional. Using a luciferase reporter assay and immunoprecipitations, we confirmed the preferential binding of STAT6 to LRS-‐STAT6. These results suggest that IL-‐4 activates the LMP-‐1 promoter through the newly identified high-‐affinity STAT6 binding site. CD40L alone did not induce LMP-‐1 in HL, but it did so in EBV-‐carrying BL cells. LMP-‐1 induction in BL cells was enhanced in the presence of cytokines IL-‐4 or IL-‐10, but IL-‐4 alone was not able to induce LMP-‐1, which suggests that CD40L activation can sensitize the BL cells to IL-‐4, for further induction of LMP-‐1. In tonsillar B cells infected with a non-‐transforming EBV strain in which EBNA-‐2 has been deleted, LMP-‐1 was induced weakly after CD40 ligation, but strongly after IL-‐4 stimulation, whereas the effect of both stimuli together was stronger (a synergistic effect). In addition, IL-‐4 did not upregulate expression of either LMP-‐2A or LMP2-‐B, which are co-‐expressed with LMP-‐1 in Type II latency (Thorley-‐Lawson, 2001; Young and Rickinson, 2004). Since IL-‐4, IL-‐13 and CD40L are products of T cells, we co-‐cultured peripheral blood T helper cells and tonsillar T cells with the BL cell line that carries EBV. We found that LMP-‐1 was induced only when T cells were activated by phytohemagglutinin (PHA) or Staphylococcus enterotoxin B (SEB), and that in addition cell-‐cell contacts were needed for this effect. We showed that the T cell-‐derived stimuli IL-‐4, IL-‐13 and CD40L acted as LMP-‐1 inducers in EBV-‐positive HL and BL cell lines, and also in EBV-‐infected normal B cells. Our results show that IL-‐4 and IL-‐13 induce LMP-‐1 expression in the absence of EBNA-‐2. Moreover, LMP-‐1 is induced via STAT6 binding to its newly defined high-‐affinity binding site on LRS-‐ STAT6. This mechanism might be important for the pathogenesis of EBV-‐positive HLs and other EBV-‐carrying tumours, such as the monoclonal B cell expansions that occur in angioimmunoblastic T cell lymphomas and in peripheral T cell lymphomas (Chen et al., 2001; Hömig-‐Hölzel et al., 2008; Kis et al., 2011). However, LMP-‐1 is always expressed in 41 cHL, while STAT6 is activated only in approximately 80% of the cases (Skinnider et al., 2002), giving room for additional, as yet undefined mechanisms for LMP-‐1 expression in the absence of EBNA-‐2 in HRS cells. Papers II, III and IV Identification of genes responsible for B cell adhesion and motility The aim of this study was to investigate the influence of IL-‐4 and CD40 ligation on the morphological responses of mouse B cells. Specific subsidiary aims were: • To identify genes that might be responsible for the regulation of cytoskeletal changes in B cells, such as microvilli formation, spreading, tight adhesion and motility; • To investigate the importance of these genes for B cell responses in vitro and in vivo. Paper II B cells devoid of the Rho GTPase Cdc42 coordinate the actin and microtubule cytoskeleton less effectively and form an extrafollicular antibody response. Motility and adhesion require the cytoskeleton rearrangement, and are very important for B cells throughout their lifetime. Cdc42, is a small Rho GTPase, and, is crucial for actin polymerization, adhesion, migration, proliferation and survival (Hall, 2005). It is essential for B lymphocyte development and activation (Guo et al., 2009). B cells can be stimulated in vitro in the process that mimics T cell activation using anti-‐ CD40 antibodies and the cytokine IL-‐4, which will induce the formation of large, round and tight aggregates. When cultured on antibody-‐coated monolayers, B cells spread and form finely branched, long protrusions (Severinson and Westerberg, 2003). Both of these responses depend on cytoskeleton rearrangements, and suggest that Cdc42 is important. We wished to investigate the specific role of Cdc42 for B cell activation in vitro, and for the humoral immune response in vivo. We used conditional gene targeting to induce the deletion of Cdc42 in B cells. CD21-‐Cre mice are extensively used for the conditional deletion of genes from mature B cells. However, Cdc42 deletion using the mouse strain that expresses CD21-‐Cre was embryonic lethal. CD21 and CD35 are encoded by the same gene in mice. However, CD35 is expressed 42 not only in B cells, but also in other cell types. CD35 and Cdc42 are expressed in erythrocyte precursors (Wong et al., 2011, Flygare et al., 2011). It is possible therefore, that CD21-‐Cre is expressed in the erythrocyte precursors, which explains the embryonic lethality of these mice. All the experiments in this study were carried out using the tamoxifen-‐inducible Mb1-‐Cre-‐ERT2, which allowed us to delete Cdc42 in B cells in adult mice. The deletion of Cdc42 from B cells led to greatly reduced numbers of transitional and follicular B cells, in agreement with previous results (Guo et al., 2009). However, we found that the number of marginal zone B cells also was lower. Immunized Cdc42flox/flox/mb1cre-‐ ERT2/+ mice formed smaller germinal centres, but they mounted normal IgM and IgG responses to a particulate antigen. The majority of plasma cells that produced IgG1, however, were extrafollicular in the knockout mice. In addition, the recall response in response to hapten carrier, was diminished, and the affinity of antibodies was lower that those of the wild-‐type mice and mice heterozygotes for the deletion of Cdc42. Cdc42 is one of the major players in the cytoskeleton reorganization and is crucial for B cells, which have to migrate constantly. This led us investigate the homing capacity of B cells. The B cells were able to home to B cell areas independently of their Cdc42 status, but to a lesser extent than wild-‐type cells. Migration towards the chemokine CXCL12 in vitro was normal, in agreement with previous results (Guo et al., 2009). The spreading response, which depends on actin polymerization and the presence of microtubules, was severely affected in B cells from Cdc42flox/flox/mb1cre-‐ERT2/+ mice. Instead of the long, finely branched protrusions formed in wild-‐type mice, Cdc42 knockout B cells formed only short, thin and brush-‐like protrusions, which stained only for polymerized actin, whereas protrusions of the wild-‐type stained for both polymerized actin and tubulin. CIP4 can mediate the interactions between WASP and microtubules (Aspenström, 1997, Tian et al., 2000). CIP4 was present in the cytoplasm and in the nucleus of both Cdc42-‐ deficient and Cdc42-‐sufficient cells. It was also present along the complete length of the protrusions in the wild-‐type B cells. It was, however, absent in the brush-‐like protrusions of Cdc42 knockout B cells. The spreading of fixed cells resembled the trailing uropods found in cultures stimulated by anti-‐CD40+IL-‐4, as observed using time-‐lapse microscopy. Cdc42-‐sufficient B cells formed long protrusions and seemed to use them in contacts with other cells, whereas Cdc42-‐ deficient B cells formed only short extensions and were perhaps less efficient in adhesive interactions. However, B cells activated by anti-‐CD40+IL-‐4 that lacked Cdc42 formed 43 equally large, round and tight aggregates as those formed by wild-‐type B cells. Our results show that Cdc42 is important for the GC response of B cells in vivo and the spreading response in vitro, and that these two reactions are linked. Paper III The guanine nucleotide exchange factor Dock 10: expression and function in B lymphocytes The guanine nucleotide exchange factor Dock10 is coded by a gene that is induced by IL-‐4. Dock10 was identified in human peripheral blood B and T cells and in the tumour cells of patients suffering from chronic lymphocytic leukaemias (CLLs) (Yelo et al., 2008). Dock10 is a GEF for Cdc42 and plays an important role in amoeboid cell migration (Gadea et al., 2008). However, very little is known about the function of Dock10 in B cells. STAT6 is needed for the IL-‐4-‐induced morphological responses in B cells, such as spreading and aggregation (Davey et al., 2000). This suggests that these responses depend on transcription. Therefore, we carried out microarray analysis to compare B cells stimulated by anti-‐CD40+IL-‐4 with those stimulated by LPS to find a gene that induces these morphological responses. Dock10 was a possible candidate. We confirmed that IL-‐4 induced strong Dock10 expression in mouse spleen B cells, and that it was the only stimulus/cytokine capable of this. Dock10 was located in the cytoplasm and in the long protrusions of spread B cells that had been induced by anti-‐CD40+IL-‐4. It is possible that it co-‐localizes with actin filaments. Dock10 knockout B cells cultured in vitro, however, had normal aggregation and spreading, suggesting that Dock10 acts in a redundant manner together with other closely related GEFs. It is also possible that it is not important for these responses. Dock10 expression in CLLs (Yelo et al., 2008) and its importance in the amoeboid cell motility of melanoma cells (Gadea et al., 2008) led us to test different mouse B cell tumours. Dock10 was expressed only in L10 and A20 cells. Moreover, Dock10 expression was upregulated by IL-‐4 in both cell lines that expressed Dock10 before treatment, and also in BCL1, which did not express Dock10 before stimulation. Interestingly, BCL1 is a spontaneously derived tumour cell line, corresponding to the human CLL. This is in agreement with Dock10 expression and upregulation by IL-‐4 in CLLs, and suggests that it plays a role in tumour transformation. 44 We purchased Dock10Flox/Frt mice from the European Mouse Mutant Archive (EMMA) to investigate the role of Dock10 in B lymphocytes. These mice were produced by flanking exon 4 of Dock10 by loxP sites and by inserting the LacZ-‐encoded gene, surrounded by FRT sites. By breeding with either CD21-‐Cre or Mb1-‐Cre-‐ERT2 mice, a Dock10-‐LacZ reporter mouse was created and used to investigate Dock10 expression in hematopoietic cells in various lymphoid organs. We detected LacZ in these mice by X-‐Gal analysis using FACS (Guo and Wu, 2008). We found that Dock10 was expressed at all stages during the development of B cells, and that it was expressed in other hematopoietic cells. Dock10 deletion from one allele of the B cells did not affect either B or T cell differentiation or B cell phenotype, but preliminary results suggest that immunized mice have higher numbers of GC B cells than the wild-‐types have. However, these experiments need to be repeated with more mice. When Dock10Flox/Frt mice were first bred with mice that expressed Flp-‐recombinase, the LacZ-‐gene was removed. This was followed by a second breeding with Mb1-‐Cre-‐ERT2 mice, to delete exon 4 of Dock10 from the mature B cells. Genomic Dock10 deletion was efficient in spleen B cells. Dock10 mRNA levels were reduced 5-‐fold in knockout B cells, but expressed at normal levels in heterozygotes. Interestingly, neither homozygotes nor heterozygotes expressed detectable amounts of Dock10 protein. The Cre-‐induced deletion may have resulted in a truncated protein that we cannot detect, because of the lack of antibodies. If Dock10 is a dimer (as are Dock8 and Dock9), the absence of Dock10 protein in heterozygotes may be due to dimerization of a wild-‐type and a truncated Dock10, leading to an unstable dimer and thus no detectable protein. The absence of Dock10 in B cells had no detectable effect on the B cell phenotype, or on their function in the bone marrow or in the spleen. Our preliminary results suggest that the numbers of T2-‐marginal zone precursor (MZP) cells and marginal zone (MZ) B cells in the spleen are slightly elevated. These experiments must be repeated before any conclusions can be drawn. The humoral immune response to TNP-‐SRBC, measured as IgM or IgG antibody titres in the serum, did not differ between knockout mice and wild-‐type mice. In vitro switching was also normal, but B cells with one functional allele of Dock10 had significantly higher switching to IgG3. TNP-‐SRBC is a very potent antigen, and it would therefore be interesting to use a less potent antigen, such as NP-‐KLH. It would also be interesting to use a T cell-‐ independent antigen. The motility, aggregation and spreading of B cells deficient in Dock10 was normal. 45 It is possible that we would have observed a difference if we had used a different mouse strain that express Cre, such as CD19-‐Cre, since this would result in accumulative effects. Also, it would be interesting to delete Dock10 in other cell types, such as T cells. It is possible that another closely related GEF, Dock11, which is expressed in hematopoietic cells as Dock10 is, acts in a redundant manner. If that is the case, conditional targeting of both GEFs may provide an answer to whether they play a role in B cell cytoskeletal responses. In addition, deletion of Rac1/2 together with Dock10 might give a phenotype, if there is a compensation mechanism between these pathways. Part IV (preliminary results) Investigation of the role of the small Rho GTPases Rac1 and Rac2 in B cell activation Like many other members of the Rho GTPase family, Rac1 and Rac2 have been implicated in regulating the actin cytoskeleton, and in cell survival and proliferation. Rac2 is expressed only in hematopoietic cells, while Rac 1 is ubiquitously expressed and mice that lack Rac1 are embryonic lethal (Gu et al., 2003). When both Rac1 and Rac2 are deleted from early B cell development, the cells are not able to enter the white pulp of the spleen (Henderson et al., 2010). To achieve deletion of Rac1 only in mature B cells, we used the Mb1-‐Cre-‐ERT2 mouse strain. In the absence of Rac1 and Rac2, the B cell phenotype was similar to the phenotype described previously using the CD19-‐Cre mouse, but it was less severe (Walmsley et al., 2003; Henderson et al., 2010). B cells stimulated by anti-‐CD40+IL-‐4 normally form long, thin and branched protrusions when cultured on antibody monolayer (Davey et al., 1998; Severinson and Westerberg, 2003). Our results confirmed that small Rho GTPases are essential for these morphological changes in B cells, since B cells that lack Cdc42, have an impaired spreading response (Paper II). To our surprise, neither deletion of one of the Racs, nor the double deletion of Rac1 and Rac2 from B cells, influenced the spreading response. Signals from T cells are very important during B cell activation. B cells activated in vitro with anti-‐CD40+IL-‐4 formed large, tight and round aggregates. B cells deficient in Rac1 aggregated normally, but cells deficient in Rac2 formed smaller aggregates with irregular shapes. When both Rac1 and Rac2 were deleted, on the other hand, there was almost no aggregation and the small aggregates that did form had irregular shapes. In addition, in vitro experiments showed that the degree to which B cells switched to IgG2b increased 46 significantly, when the cells stimulated with LPS. Furthermore, when activated with anti-‐ CD40+IL-‐4, Rac1 and Rac2 double-‐deficient B cells switched to IgG1 to a lower degree. In conclusion, we have found that B cell spreading depends on Cdc42, but is independent of Rac1/2. Furthermore, B cell homotypic adhesion depends on Rac1/2, but not on Cdc42. Failure in the aggregation in the absence of Rac1/2, however, might explain the surprising switching results. Cells would first need to proliferate and form tight aggregates to be able to produce T cell-‐dependent IgG1. The results presented are preliminary and should be repeated with more mice. 47 Conclusions and perspectives The work presented here has examined the activation and motility of B cells. CD40-‐CD40 ligation and signalling by cytokines (such as IL-‐4) are very important for both of these processes. In classical Hodgkin lymphoma, EBV-‐infected cells express latent protein LMP-‐1, but do not express EBNA-‐2, which is essential for LMP-‐1 expression. We investigated in Paper I the molecular mechanism behind LMP-‐1 expression in EBV-‐infected HL-‐derived cell lines. The KMH2-‐EBV cell line was derived by infection of KMH2 with EBV. This gave us a model system that resembles cHL, because exposure of these cells in vitro to CD40L and IL-‐4 in the absence of EBNA-‐2 induced LMP-‐1 expression (Kis et al., 2005). Using this cell line, we found that the cytokines IL-‐4 and IL-‐13 act through the transcription factor STAT6, binding to the viral promoter element LRS-‐STAT6. In this way, the cytokines were able to induce LMP-‐1 expression in the absence of EBNA-‐2. The same mechanism of LMP-‐1 induction might be responsible for pathogenesis in EBV-‐positive classical Hodgkin lymphoma or in some other EBV-‐carrying tumours. Therefore, results from investigation of the LMP-‐1 induction and maintenance by cytokine signalling might be useful in the treatment of EBV-‐ positive lymphomas. Papers II and III describe investigations of genes involved in B cell motility and cytoskeletal changes in vitro and in vivo. Preliminary results from a further aspect of this project are presented in Part IV. We conditionally deleted either Dock10 or the small Rho GTPases Cdc42 and Rac1 together with Rac2 in B cells. We wished to study mature B cells, so first we tried to use breedings with CD21-‐Cre mice to induce Cdc42 deletion. However, surprisingly, in crossings with Cdc42flox mice, homozygous mice died before birth. Therefore, we used the tamoxifen-‐inducible Mb1-‐Cre-‐ERT2 mouse strain instead. Although Dock10 was selectively upregulated by IL-‐4 in primary cells and CLL cells, and might be co-‐localized with polymerized actin, it was not involved in B cell spreading or aggregation. Deletion of the Dock10 Rho GTPase Cdc42 from B cells resulted in impaired spreading (Paper II), as did also deletion of the downstream effector of Cdc42, WASP (Westerberg et al., 2001). However, the expression pattern of Dock10 in cell lines suggests that it plays a role in tumour development and progression. Deletion of Dock10 in B cells did not give rise to an aberrant phenotype, even though it is expressed in the early differentiation stages of B cells (Paper III). 48 One explanation is that compensation from other closely related guanine nucleotide exchange factors (such as Dock11) take place, or compensation from Rho GTPases (Rac1). Double knockouts would allow us to determine whether there is redundancy between Dock10 and other molecules. However, the activation status of Rac1/2, Cdc42 and Dock11 in B cells could be tested in mice deficient for Dock10, in order to gain information about the compensation possibilities. Dock10 may be is important for other hematopoietic or non-‐hematopoietic cells. When the Dock10 effector, Cdc42, was deleted from B cells, there was a much pronounced phenotype (Paper II). Mice with Cdc42-‐deficient B cells were not able to produce a proper germinal centre response with high-‐affinity antibodies. Furthermore, these mice did not respond to an antigen recall. Despite normal migration to chemokines, Cdc42-‐/-‐ B cells had reduced homing capacity in vivo. In addition, B cells that lack Cdc42 failed to spread. These B cells formed thin, short protrusions that stained for polymerized actin, instead of long, branched dendrites, positive for both polymerized actin and tubulin. This indicates that Cdc42 is crucial for the coordination of both actin-‐dependent and microtubule-‐dependent responses. Interestingly, Cdc42-‐interacting protein 4 (CIP4) can connect WASP to microtubules. It is possible that CIP4 is activated by Cdc42 and WASP, and that this induces its binding to tubulin. This suggests that in B cells in which Cdc42 has been knocked out, CIP4 cannot connect WASP and bring it to the microtubules, and thereby stabilize them. Therefore, Cdc42-‐deficient B cells do not have a proper spreading response, and only short, brush-‐like protrusions are formed. We found that CIP4 was, surprisingly, present in the nucleus. More experiments are needed to confirm this, and to determine its function in the nucleus. In addition, CIP4 acts as a FasL binding partner, which suggests that it is important for the germinal centres in vivo. In addition, our results show that Cdc42-‐dependent motility is essential for mounting an efficient humoral response in mice. Rac1 and Rac2 double knockout B cells (Part IV) are clearly deficient in aggregation. This might imply that Rac1/2 are important for the germinal centre formation in vivo. In addition, the extremely high switching rates to IgG2b, while the levels of IgG1 are lower, make it very interesting to study Ig class responses to T-‐dependent and T-‐independent antigens in vivo. Furthermore, investigation of the Cdc42 activation status in Rac1/2-‐ deficient cells may be an option, since these GTPases might influence each other’s activity. The preliminary results presented here require confirmation from further experiments, especially in the Dock10 project (Paper III), and the Rac1/2 data (Part IV). 49 Different signalling pathways communicate with each other by crosstalk, and often proteins cooperate or antagonize each other. Therefore, deleting a GEF, GTPase or a target protein might affect a whole cascade of other molecules in the same or related pathways. All these pathways are connected in one way, or another in vivo: switching one pathway off may lead to another pathway being switched on, and a subsequent pathological response. All the molecules studied here are responsible for the motility of normal cells, and thus may play roles in tumour invasion and dissemination. We hope that these results shed some light on the importance of the molecules we have investigated for activation and motility in normal B cells, and on the possible roles of these molecules in B cell malignancies. 50 Acknowledgements This work was performed at the Department of Molecular Biosciences, the Wenner-‐Gren Institute, Stockholm University. Many people have contributed and supported me during my PhD. First, I would like to thank my supervisor Eva Severinson, for her scientific guidance, her endless support and patience. Thank you for accepting me as your PhD student, giving me the fantastic opportunity to come to Sweden. Thank you for inspiring me, helping to focus on important things, for your constructive criticism and creative suggestions, especially during the writing of the manuscripts and the thesis, and sharing your huge knowledge and experience with me. I am really grateful for your role in my growth as a researcher, and as a person. Thank you for believing in me! Second, I would like to thank my co-‐supervisor Lisa Westerberg for all valuable discussions, suggestions, encouragement and help during my studies and writing of my thesis and manuscripts. I would like to thank all seniors at the former Immunology Department -‐ Eva Sverremark-‐ Ekström, Carmen Fernandez, Marita Troye-‐Blomberg and Klavs Berzins, and those at the Cell Biology Department – Roger Karlsson, Ann Kristin Östlund Farrants, Per Ljungdal and Claes Andreasson. Thank you for you support, help and discussions! I would like to thank Anna-‐Stina Höglund for her help with microscopy. I would like to thank Eva Nygren, Ellinor Ljunglöf, Solveig Sundberg and all other personnel at the Animal Facility for always being friendly and taking good care of my mice. I would like to thank all the administrative personal for their help -‐ Lina, Gelana, Anna-‐ Leena, but especially Magdalena – you are the best! I would like to thank the IT support – Bengt and Gunnar for all their help. I would like to thank all former and present people at the former Immunology Department, especially Maggan, for all your help with technical issues; Shanie, Manijeh, Pablo, Jubayer and Stephanie for discussions and help during my PhD; and of course Olga, for being my good friend, for all discussions, your help, support and all the fun we had together. I would like to thank all former and present people at the former Cell Biology department, especially Kicki, for all your help and support with technical and other issues; Javier, for lots of discussions and all suggestions; my officemates -‐ Ming, Naveen and Mats for lots of fun and discussions; Sara and Steffi for your discussions and being always kind and helpful; Kerstin, Bojana and Peter – it was nice to have you as students. I would like to thank Andrea S., in particular, for being my good friend, for all scientific and non-‐scientific discussions and all the fun we had together. Thank you once more everyone at the departments, if I haven’t mentioned your name, it does not mean you are not important! You all have contributed in one way or another! The departments would be different without all of you! Thank you for creating such a great working and non-‐working environment! I would like to thank some people from the new Department of Molecular Biosciences: Andrea E., Katarina T, Widad, Steffi Bauer and Elina for all scientific and non-‐scientific discussions, your help and fun. Thank you for your friendship! 51 I would like to thank some people from Eva Klein’s lab, MTC, Karolinska Institutet: my friend Lorand (Lori), for all help, scientific and non-‐scientific discussions; also, Noemi, Daniel, Emma and of course Eva Klein and George Klein – for the all help, discussions and support at the beginning of my PhD studies. I would like to thank people from the Translational Immunology Unit at Karolinska University Hospital, Solna for always being nice and helpful to me. I would like especially to thank my friend and co-‐author Marisa, for all your help, and for the discussions and all the fun we had together; Carin, also co-‐author in one paper, for all your help with my experiments; Liliana for your help with cryo-‐sectioning; and, of course Mikael Karlsson for help and the possibility of using the reagents. I would like to thank the Lithuanian folk dance group “BALTIJA” for all the fun moments, travel and concerts. I would especially like to thank Gedas for “taking” me there, Jurga, for accepting me, Vilma J. and Tomas, Aurelija and Valentinas, Olga O., Irute and Donatas, Kristina and Mindaugas, Paulius and Skirmante and Giedrius for being my good friends in the group and outside. “BALTIJA” helped me to feel at home here in Stockholm! I would like to thank my friend Vilma V. for my first trip to Stockholm (I knew I would come back!) and Vilma U. for 10 years of scientific and non-‐scientific friendship, which started back in Lithuania and continues in Sweden… I would like to thank Irute Girkontaite, from the Centre For Innovative Medicine, State Research Institute, for introducing me to the B cell world and sharing all your knowledge with me. Also, I would like to thank my other friends and colleagues from Vilnius, Lithuania: Neringa, Siga, Tania, Lauryte, Virga, Ieva, Danute Davidoniene, Ingrida, Ritute and Aida – thank you for your friendship and support during these years! And last, but not the least, I would like to thank my family in Lithuania, especially my parents for endless love and support! Thank you for believing in me! This work was supported by the Swedish Research Council. 52 References Ahuja A, Anderson SM, Khalil A, Shlomchik MJ. Maintenance of the plasma cell pool is independent of memory B cells. PNAS 2008; 105(12):4802-‐4807. Alcaraz-‐Garcia MJ, Ruiz-‐Lafuente N, Sebastian-‐Ruiz S, Majado MJ, Gonzalez-‐Garcia C, Bernardo MV, Alvarez-‐Lopez MR. Human and mouse DOCK10 splicing isoforms with alternative first coding exon usage differentially expressed in T and B lymphocytes. Human Immunology 2011; 72:531-‐537. Allen CDC, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Seminars in Immunology 2008; 20:14-‐25. Allen CDC, Okada T, Cyster JG. Germinal-‐Centre organization and cellular dynamics. Immunity 2007; 27: 190-‐202. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of germinal centre selection events during affinity maturation. Science 2007; 315:528-‐531. Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med 2010; DOI: 10.1084/jem.20092210. Allman D, Pillai S. Peripheral B cell subsets. Current Opinion in Immunology 2008; 20:149-‐157. Anthis NJ, Campbell ID. The tail of integrin activation. Trends in Biochemical Sciences 2011; 36(4):191-‐198. Arnon TI, Horton RM, Grigorova IL, Cyster JG. Visualization of splenic marginal zone B-‐cell shuttling and follicular B-‐cell egress. Nature 2013; 493(7434):684-‐688. Aspenström P. A Cdc42 target protein with homology to the non-‐kinase domain of FER has a potential role in regulating the actin cytoskeleton. Current Biology 1997; 7(7):479-‐487. Aspenström P. Roles of F-‐BAR/PCH proteins in the regulation of membrane dynamics and actin reorganization. Int Rev Cell Mol Biol 2009; 272:1-‐31. Babcock GJ, Hochberg D, Thorley-‐Lawson DA. The Expression Pattern of Epstein-‐Barr Virus Latent Genes In Vivo Is Dependent upon the Differentiation Stage of the Infected B Cell. Immunity 2000; 13:497–506. Banerjee PP, Pandey R, Zheng R, Suhoski MM, Monaco-‐Shawver L, Orange JS. J Exp Med 2007; 204(10):2305-‐2320. Basu S, Ray A, Dittel BN. Cannabinoid receptor 2 is critical for the homing and retension of marginal zone B lineage cells and for efficient T-‐independent immune responses. The Journal of Immunology 2011; 187:5720-‐5732. Batista FD, Treanor B, Harwood NE. Vizualizing a role for the actin cytoskeleton in the regulation of B-‐cell activation. Immunological Reviews 2010; 237:191-‐204. Beltman JB, Allen CDC, Cyster JG, de Boer RJ. B cells within germinal centers migrate preferentially from dark to light zone. PNAS 2011; 108(21):8755-‐8760. 53 Blundell MP, Worth A, Bouma G, Thrasher AJ. The Wiskott-‐Aldrich syndrome: the actin cytoskeleton and immune cell function. Disease Markers 2010; 157-‐175. Bolduc A, Long E, Stapler D, Cascalho M, Tsubata T, Koni PA, Shimoda M. Constitutive CD40L expression on B cells prematurely terminates germinal centre response and leads to augmented plasma cell production in T cell areas. Journal of Immunology 2010; 185:220-‐230. Boothby M, Mora AL, Aronica MA, Youn J, Sheller JR, Goenka S, Stephenson L. IL-‐4 Signalling, Gene Transcription Regulation, and the Control of Effector T Cells. Immunologic Research 2001; 23(2-‐3):179–191. Bortnik A, Allman D. What is and what should always have been: long-‐lived plasma cells induced by T cell-‐independent antigens. The Journal of Immunology 2013; 190:5913-‐5918. Bromberg J, Darnell JE Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000; 19:2468-‐2473. Busch LK, Bishop GA. The EBV Transforming Protein, Latent Membrane Protein 1, Mimics and Cooperates with CD40 Signaling in B Lymphocytes. The Journal of Immunology 1999; 162:2555–2561. Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 2010; 11:237-‐251. Cancelas JA, Lee AW, Prabhakar R, Stringer KF, Zheng Yi, Williams DA. Rac GTPases differentially integrate signals regulating hematopoietic stem cell localization. Nature Medicine 2005; 11(8):886-‐891. Carsetti R, Rosado MM, Wardemann H. Peripheral development of B cells in mouse and man. Immunological Reviews 2004; 197:179-‐191. Cerutti A, Cols M, Puga I. Activation of B cells by non-‐canonical helper signals. EMBO reports 2012; 13(9):798-‐810. Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-‐like antibody-‐producing lymphocytes. Nat Rev Immunology 2013; 13:118-‐132. Chan TD, Gardam S, Gatto D, Turner VM, Silke J, Brink R. In vivo control of B-‐cell survival and antigen-‐specific B-‐cell responses. Immunological Reviews 2010; 237:90-‐103. Chappell CP, Draves KE, Giltiay NV, Clark EA. Extrafollicular B cell activation by marginal zone dendritic cells drives T cell-‐dependent antibody responses. J Exp Med 2012; DOI: 10.1084/jem.20120774. Chen H, Lee JM, Zong Y, Borowitz M, NG MH, Ambinder RF, Hayward SD. Linkage between STAT Regulation and Epstein-‐Barr Virus Gene Expression in Tumors. Journal of Virology 2001; 75(6):2929–2937. Chen H-‐C, Reich NC. Live Cell Imaging Reveals Continuous STAT6 Nuclear Trafficking. The Journal of Immunology 2010; 185:64–70. Chu VT, Berek C. The establishment of the plasma cell survival niche in the bone marrow. Immunological Reviews 2013; 251:177-‐188. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, Wang Yi-‐H, Lim H, Reynolds JM, Zhou X-‐ H, Fan H-‐M, Liu Z-‐M, Neelapu SS, Dong C. Follicular regulatory T cells expressing Foxp3 and 54 Bcl-‐6 suppress germinal center reactions. Nature Medicine 2011; 17(8):983-‐988. Cinamon G, Zachariah MA, Lam OM, Foss FW Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nature Immunology 2008; 9(1):54-‐62. Cohen JI. The biology of Epstein-‐Barr virus: lessons learned from the virus and the host. Current Opinion in Immunology 1999; 11:365-‐370. Cyster JG. B cell follicles and antigen encounters of the third kind. Nature Immunology 2010; 11(11):989-‐996. Davey EJ, Greicius G, Thyberg J, Severinson E. STAT6 is required for the regulation of IL-‐4 induced cytoskeletal events in B cells. Int Immunol 2000; 12:995–1003. Davey EJ, Thyberg J, Conrad DH, Severinson E. Regulation of cell morphology in B lymphocytes by interleukin-‐4: evidence for induced cytoskeletal changes. J Immunol 1998; 160:5366–5373. Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. British Journal of Haematology 2010; 149:167-‐180. Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by Epstein-‐Barr virus. Cancer Res 2003; 63(11):2982-‐2989. Dogan I, Bortocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud C-‐A, Weill J-‐C. Multiple layers of B cell memory with different effector functions. Nature Immunology 2009; 10(12):1292-‐ 1299. Dooms H, Abbas AK: Revisiting the role of IL-‐2 in autoimmunity. Eur J Immunol 2010; 40:1538-‐ 1540. Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, Bucher P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem 2001; 276(9):6675-‐6688. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Reviews 2009; 229:152-‐172. Erickson LD, Durell BG, Vogel LA, O’Connor BP, Cascalho M, Yasui T, Kikutani H, Noelle RJ. Short-‐ circuiting long-‐lived humoral immunity by the heightened engagement of CD40. J Clin Invest 2002; 109(5):613-‐620. Faulkner GC, Krajewski AS, Crawford DH. The ins and outs of EBV infection. Trends in Microbiology 2000; 8(4):185-‐189. Feng Q, Cerione RA. Cdc42 and its cellular functions. Handbook of Cell Signaling 2010; 3:1785-‐1794. Flygare J, Rayon Estrada V, Shin C, Gupta S, Lodish HF. HIF1alpha synergizes with glucocorticoids to promote BFU-‐E progenitor self-‐renewal. Blood 2011; 117:3435-‐3444. Gadea G, Sanz-‐Moreno V, Self A, Godi A, Marshall CJ. DOCK10-‐Mediated Cdc42 Activation Is Necessary for Amoeboid Invasion of Melanoma Cells. Current Biology 2008; 18:1456–1465. Gatto D, Brink R. The germinal centre reaction. Mechanisms of allergic diseases 2010; 126:898–907. Gatto D, Brink R. B cell localization: regulation by EBI2 and its oxysterol ligand. Trends in 55 Immunology 2013; 34(7):336-‐341. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-‐coupled receptor EBI2 shapes humoral immune responses. Immunity 2009; 31:259–269. Gerasimcik N, Baptista M, Westerberg L, Severinson E. The guanine nucleotide exchange factor Dock10: expression and function in B lymphocytes. Manuscript 2013. Gerasimcik N, Dahlberg C, Baptista M, Westerberg L, Severinson E. B cells devoid of the Rho GTPase Cdc42 coordinate the actin and microtubule cytoskeleton less effectively and form an extrafollicular antibody response. Manuscript 2013. B cells devoid of the Rho GTPase Cdc42 coordinate the actin and microtubule cytoskeleton less effectively and form an extrafollicular antibody response. Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, Zeidler R, Scheffer B, Ueffing M, Hammerschmidt W. Latent membrane protein 1 of Epstein-‐Barr virus interacts with JAK3 and activates STAT proteins. The EMBO Journal 1999; 18(11):3064-‐3073. Gonzalez SF, Degn SE, Pitcher LA, Woodruff M, Heesters BA, Carroll MC. Trafficking of B cell antigen in lymph nodes. Annu Rev Immunol 2011; 29:215-‐33. Good-‐Jacobson KL, Shlomchik MJ. Plasticity and heterogeneity in the generation of memory B cells and long-‐lived plasma cells: the influence of germinal center interactions and dynamics. The Journal of Immunology 2010; 185:3117-‐3125. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control system and decision making for antibody production. Nature Immunology 2010; 11(8):681-‐688. Goormachtigh G, Ouk T-‐S, Mougel A, Tranchand-‐Bunel D, Masy E, Le Clorennec C, Feuillard J, Bornkamm GW, Auriault C, Manet E, Fafeur V, Adriaenssens E, Coll J. Autoactivation of the Epstein-‐Barr Virus Oncogenic Protein LMP1 during Type II Latency through Opposite Roles of the NF-‐κB and JNK Signaling Pathways. Journal of Virology 2006; 80(15):7382–7393. Graham JP, Arcipowski KM, Bishop GA. Differential B-‐lymphocyte regulation by CD40 and its viral mimic, latent membrane protein 1. Immunol Reviews 2010; 237:226-‐248. Green JA, Suzuki K, Cho B, Willison LD, Palmer D, Allen CDC, Schmidt TH, Xu Y, Proia RL, Coughlin SR, Cyster JG. The sphingosine 1-‐phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nature Immunology 2011; 12(7):672-‐680. Gregory CD, Rowe M, Rickinson AB. Different Epstein-‐Barr virus-‐B cell interactions in phenotypically distinct clones of a Burkitt's lymphoma cell line. Journal of General Virology 1990; 71:1481-‐1495. Greicius G, Tamosiunas T, Severinson E. Assessment of the role of leukocyte function-‐associated antigen-‐1 in homotypic adhesion of activated B lymphocytes. Scand J Immunol 1998; 48:642– 50. Greicius G, Westerberg L, Davey EJ, Buentke E, Scheynius A, Thyberg J, Severinson E. Microvilli structures on B lymphocytes: inducible functional domains? Int Immunol 2003; 16:353-‐364. Gu Yi, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, Kwiatkowski DJ, Williams DA. Hematopoietic cell regulation by Rac1 and Rac2 Guanosine Triphasphatases. Science 2003; 302:445-‐449. 56 Guo F, Velu CS, Grimes L, Zheng Y. Rho GTPase Cdc42 is essential for B-‐lymphocyte development and activation. Blood 2009; 114:2909-‐2916. Guo W and Wu H. Detection of LacZ expression by FACS-‐Gal analysis. Protocol Exchange 2008; DOI:10.1038/nprot.2008.163. Hall, A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans 2005; 33:891-‐895. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol 2001; 19:595-‐621. Harwood NE, Batista FD. Early events in B cell activation. Annu Rev Immunol 2010; 28:185-‐210. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM. Definition of Germinal-‐Center B cell migration in vivo revelas predominant intrazonal circulation patterns. Immunity 2007; 26:655-‐667. Hauser AE, Kerfoot SM, Haberman AM. Cellular choreography in the germinal center: new visions from in vivo imaging. Semin Immunopathol 2010; 32:239-‐255. Hauser AE, Shlomchik MJ, Haberman AM. In vivo imaging studies shed light on germinal-‐centre development. Nat Rev Immunol 2007; 7:499-‐504. Henderson RB, Grys K, Vehlow A, de Bettignies C, Zachacz A, Henley T, Turner M, Batista F, Tybulewicz VLJ. A novel Rac-‐dependent chackpoint in B cell development controls entry into the splenic white pulp and survival. J Exp Med 2010; 207(4):837-‐853. Herzog S, Reth M, Jumaa H. Regulation of B-‐cell proliferation and differentiation by pre-‐B-‐cell receptor signaling. Nat Rev Immunology 2009; 9:195-‐205. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular Responses to Viral Infection in Humans: Lessons from Epstein-‐Barr Virus. Annu Rev Immunol 2007; 25:587–617. Howard M, Farrar J, Hilfiker M, Johnson B, Takatsu K, Hamaoka T, Paul WE. Identification of a T cell-‐ derived B cell growth factor distinct from interleukin 2. J Exp Med 1982; 155:914-‐923. Hömig-‐Hölzel C, Hojer C, Rastelli J, Casola S, Strobl LJ, 1 Müller W, Quintanilla-‐Martinez L, Gewies A, Ruland J, Rajewsky K, Zimber-‐Strobl U. Constitutive CD40 signaling in B cells selectively activates the noncanonical NF-‐κB pathway and promotes lymphomagenesis. J Exp Med 2008; DOI: 10.1084/jem.20080238. Humtsoe JO, Koya E, Pham E, Aramoto T, Zuo J, Ishikawa T, Kramer RH. Transcriptional profiling identifies upregulated genes following induction of epithelial-‐mesenchymal transition in squamous carcinoma cells. Experimental Cell research 2012; 318:379-‐390. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, Rauter I, Benson H, Schneider L, Baxi S, Recher M, Notarangelo L, Wakim R, Dbaibo G, Dasouki M, Al-‐Herz W, Barlan I, Baris S, Kutukculer N, Ochs H, Plebani A, Kanariou M, Lefranc G, Reisli I, Fitzgerald K, Golenbock D, Manis J, Keles S, Ceja R, Chatila T, Geha RS. DOCK8 functions as an adaptor that links Toll-‐like receptor–MyD88 signaling to B cell activation. Nature Immunology 2012; 13(6):612-‐620. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annual Review of Cell and Developmental Biology 2005; 21:247-‐69. Jiang H, Harris MB, Rothman P. IL-‐4/IL-‐13 signaling beyond JAK/STAT. J Allergy Clin Immunol 2000; 57 105:1063-‐1070. Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. The Journal of Immunology 2011; 187:3026-‐3032. Kerfood SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 2011; 34:947-‐960. Khurana Hershey GK. IL-‐13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol 2003; 111(4):677-‐90. Kis LL. The role of the microenvironment on the regulation of Epstein-‐Barr virus latent gene expression. Doctoral Thesis 2009; 1-‐79. Kis LL, Gerasimcik N, Salamon D, Persson EK, Nagy N, Klein G, Severinson E, Klein E. STAT6 signaling pathway activated by the cytokines IL-‐4 and IL-‐13 induces expression of the Epstein-‐Barr virus–encoded protein LMP-‐1 in absence of EBNA-‐2: implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 2011; 117:65-‐174. Kis LL, Nishikawa J, Takahara M, Nagy N, Matskova L, Takada K, Elmberger PG, Ohlsson A, Klein G, KleinE. In vitro EBV-‐infected subline of KMH2, derived from Hodgkin lymphoma, expresses only EBNA-‐1, while CD40 ligand and IL-‐4 induce LMP-‐1 but not EBNA-‐2. Int J Cancer 2005; 113:937–945. Kis LL, Takahara M, Klein G, Klein E. Cytokine mediated induction of the major Epstein Barr virus (EBV)-‐encoded transforming protein, LMP-‐1. Immunol Lett 2006; 104:83-‐88. Kis LL, Takahara M, Nagy N, Klein G, Klein E. IL-‐10 can induce the expression of EBV-‐encoded latent membrane protein-‐1 (LMP-‐1) in the absence of EBNA-‐2 in B lymphocytes and in Burkitt lymphoma–and NK lymphoma–derived cell lines. Blood 2006; 107:2928-‐2935. Kishi Y, Aiba Y, Higuchi T, Furukawa K, Tokuhisa T, Takemori T, Tsubata T. Augmented antibody response with premature germinal center regression in CD40L transgenic mice. The Journal of Immunology 2010; 185: 211-‐219. Klein E, Kis LL, Klein G. Epstein Barr virus infections in humans: from harmless to life endangering virus-‐lymphocyte interactions. Oncogene 2007; 26:1297-‐1305. Klein U, Dalla-‐Favera R. Germinal centres: role in B-‐cell physiology and malignancy. Nat Rev Immunology 2008; 8:22-‐33. Koduru S, Kumar L, Massaad MJ, Ramesh N, Le Bras S, Ozcan E, Oyoshi MK, Kaku M, Fujiwara Y, Kremer L, King S, Fuhlbrigge R, Rodig S, Sage P, Carman C, Alcaide P, Luscinskas FW, Geha RS. Cdc42 interacting protein 4 (CIP4) is essential for integrin-‐dependent T-‐cell trafficking. PNAS 2010; 107(37):16252-‐16256. Kouro T, Takatsu K. IL-‐5-‐ and eosinophil-‐mediated inflammation: from discovery to therapy. Int Immunology 2009; 21(12):1303-‐1309. Kovanen PE, Leonard WJ. Cytokines and immunodeficiency diseases: critical roles of the γc-‐ dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunological Rev. 2004; 202:67-‐83. 58 Kracker S, Gardes P, Mazerolles F, Durandy A. Immunoglobulin class switch recombination deficiencies. Clinical Immunology 2010; 135:193-‐203. Kucik DF. Rearrangement of integrins in avidity regulation by leukocytes. Immunol Research 2002; 26(1-‐3):199-‐206. Kurosaki T. B-‐lymphocyte biology. Immunological Reviews 2010; 237: 5-‐9. Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunology 2010; 28:21-‐55. Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, Küppers R. EBV-‐infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity 2000; 13:485-‐495. Küppers R. Mechanisms of B-‐cell lymphoma pathogenesis. Nat Rev Cancer 2005; 5:251-‐262. Küppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-‐cell lymphomas. N Engl J Med 1999; 11:1520-‐1529. Linternam MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. IL-‐21 acts directly on B cells to regulate Bcl-‐6 expression and germinal center responses. J Exp Med 2010; DOI: 10.1084/jem.20091738. Linternam MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, Fagarasan S, Liston A, Smith KGS, Vinuesa CG. Foxp3+ follicular regulatory T cells control the germinal center response. Nature Medicine 2011; 17(8):975-‐982. Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-‐derived signals. J Exp Med 1999; 190(1):75-‐89. Lundgren M, Larsson C, Femino A, Xu M, Stavnezer J, Severinson E. Activation of the Ig Germ-‐Line gamma 1 promoter. Involvement of C/enhancer-‐binding protein transcription factor and their possible interaction with an NF-‐IL-‐4 site. The Journal of Immunology 1994; 153:2983-‐ 2995. Luzuriaga K, Suvillan JL. Infectious mononucleosis. NEJM 2010; 362(21):1993-‐2000. Mackay F, Figget WA, Saulep D, Lepage M, Hibbs ML. B-‐cell stage and context-‐dependent requirements for survival signals from BAFF and the B-‐cell receptor. Immunol Reviews 2010; 237:205-‐225. MacLennan ICM. Germinal centers. Annu Rev Immunol 1994; 12:117–139. Malek TR. The biology of Interleukin-‐2. Annu Rev Immun 2008; 26:453-‐79. Malet-‐Engra G, Viaud J, Ysebaert L, Farce M, Lafouresse F, Laurent G, Gaits-‐Iacovoni F, Scita G, Dupre L. CIP4 controls CCL19-‐driven cell steering and chemotaxis in chronic lymphocytic leukemia. Cancer research 2013; 73(11):3412-‐3424. McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Disease Markers 2010; 29:151-‐156. McHeyzer-‐Williams M, Okitsu S, Wang N, McHeyzer-‐Williams L. Molecular programming of B cell memory. Nat Rev Immunology 2012; 12:24-‐34. 59 Melendez J, Grogg M, Zheng Yi. Signaling role of Cdc42 in regulating mammalian physiology. J Biol Chem 2011; 286(4):2375-‐2381. Meller N, Irani-‐Tehrani M, Ratnikov BI, Paschal BM, Schwartz MA. The novel Cdc42 guanine nucleotide exchange factor, Zizimin1, dimerizes via the Cdc42-‐binding CZH2 domain. J Biol Chem 2004; 279(36):37470-‐37476. Meller N, Merlot S, Guda C. CZH proteins: a new family of Rho-‐GEFs. Journal of Cell Science 2005; 118:4937-‐4946. Meyer-‐Hermann M, Mohr E, Pelletier N, Zhang Y, Victora GD, Toellner K-‐M. A theory of germinal center selection, division, and exit. Cell Reports 2012; 2:162-‐174. Miyamoto Y, Yamauchi J. Cellular signaling of Dock family proteins in neural function. Cellular Signalling 2010; 22:175-‐182. Montecino-‐Rodriguez E, Dorshkind K. B-‐1 B cell development in the fetus and adult. Immunity 2012; 36:13-‐21. Mulloy JC, Cancelas JA, Filippi MD, Kalfa TA, Guo F, Zheng Y. Rho GTPases in hematopoiesis and hemopaties. Blood 2010; 115:936-‐947. Muppidi JR, Arnon TI, Bronevetsky Y, Veerapen N, Tanaka M, Besra GS, Cyster JG. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J Exp Med 2011; 208(10):1941-‐1948. Murray PJ. The JAK-‐STAT Signaling Pathway: Input and Output Integration. The Journal of Immunology 2007; 178:2623–2629. Natkunam Y. The biology of the germinal center. Hematology 2007; 210-‐215. Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-‐4 Receptor: Signaling Mechanisms and Biologic Functions. Ann Rev Immunol 1999; 17:701-‐738. Nelson BH. Interleukin-‐2 signaling and the maintenance of self-‐tolerance. Curr Dir Autoimmun. 2002; 5:92-‐112. Nishikimi A, Meller N, Uekawa N, Isobe K-‐I, Schwartz MA, Maruyama M. Zizimin2: a novel, DOCK180-‐related Cdc42 guanine nucleotide exchange factor expressed predominantly in lymphocytes. FEBS Letters 2005; 579:1039-‐1046. Oracki SA, Walker JA, Hibbs ML, Corcoran LM, Tarlinton DM. Plasma cell development and survival. Immunological Reviews 2010; 237:140-‐159. Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011; 331:1203-‐1207. Paulie S, Ehlin-‐Henriksson B, Mellstedt H, Koho H, Ben-‐Aissa, Perlmann P. A p50 surface antigen restricted to human urinary bladder carcinomas and B lymphocytes. Cancer Immunol Immunother 1985; 20(1):23-‐8. Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-‐cell migration in the early phases of T-‐ dependent antibody responses. Int Immunol 2010; 22(6):413-‐419. 60 Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature 2009; 460(27):1122-‐1127. Pieper K, Grimbacher B, Eibel H. B-‐cell biology and development. J Allergy Clin Immunol 2013; 131:959-‐71. Pillai S Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunology 2009; 9:767-‐777. Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, Comerma L, Chorny A, Shan M, Xu W, Magri G, Knowles DM, Tam W, Chiu A, Bussel JB, Serrano S, Lorente JA, Bellosillo B, Lloreta J, Juanpere N, Alameda F, Baro T, de Heredia CD, Toran N, Catala A, Torrebadell M, Fortuny C, Cusi V, Carreras C, Diaz GA, Blander JM, Farber CM, Silvestri G, Cunnigham-‐Rundles C, Calvillo M, Dufour C, Notarangelo LD, Lougaris V, Plebani A, Casanova JL, Ganal SC, Diefenbach A, Arostegui JI, Juan M, Yague J, Mahalaoui N, Donadieu J, Chen K, Cerutti A. B cell-‐helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nature Immunology 2012; 13(2):170-‐80. Puls A, Eliopolos AG, Nobes CD, Bridges T, Young LS, Hall A. Activation of the small GTPase Cdc42 by the inflammatory cytokines TNFα and IL-‐1, and by the Epstein-‐Barr virus transforming protein LMP1. J Cell Science 1999; 112:2983-‐2992. Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS. Memory B cells, but not long-‐lived plasma cells, posses antigen specificities for viral escape mutants. J Exp Med 2011; 208(13):2599-‐2606. Qin J, Vinogradova O, Plow EF. Integrin Bidirectional Signaling: A Molecular View. PLoS Biology 2004; 2(6):0726-‐0729. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KGC, Dörner T, Hiepe F. Competence and competition: the challenge of becoming a long-‐lived plasma cell. Nat Rev Immunology 2006; 6:741-‐750. Reth M. B cell antigen receptors. Curr Opin Immunol 1994; 6(1):3-‐8. Reth M, Wienands J. Initiation and processing of signals from the B cell antigen receptor. Annu Rev Immun 1997; 15:453-‐79. Revenu C, Athman R, Robine S, Louvard D. The co-‐workers of actin filaments: from cell structures to signals. Nat Rev Mol Cell Biol 2004; 5:1-‐12. Richnau N, Aspenström P. RICH, a Rho GTPase-‐activating protein domain-‐containing protein involved in signaing by Cdc42 and Rac1. J Biol Chem 2001; 276(37):35060-‐35070. Rossman KL, Der CJ, Sondek J. GEF means go:turning on Rho GTPases with guanine nucleotide-‐ exchange factors. Nat Rev Mol Cell Bio 2005; 6:167-‐180. Rowe M, Rowe DT, Gregory CD, Young LS, Farell PJ, Rupani H, Rickinson AB. Differences in B cell growth phenotype reflect novel patterns of Epstein-‐Barr virus latent gene expression in Burkitt's lymphoma cells. The EMBO Journal 1987; 6(9):2743-‐2751. Roy B, Shukla S, Lyszkiewicz M, Krey M, Viegas N, Düber S, Weiss S. Somatic hypermutation in peritoneal B1b cells. Molecular Immunology 2009; 46:1613-‐1619. Sakabe I, Asai A, Iijima J, Maruyama M. Age-‐related guanine nucleotide exchange factor, mouse Zizimin2, induces filopodia in bone marrow-‐derived dendritic cells. Immunity & Ageing 2012; 61 9(2):1-‐9. Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, Tybulewicz VLJ. The BAFF receptor transduces survival signals by co-‐opting the B cell receptor signalling pathway. Immunity 2013; 38:475-‐488. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-‐Vilbois MH, Dustin ML, Nussenzweig MC. In vivo imaging of germinal centres reveals a dynamic open structure. Nature 2007; 446:83-‐87. Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, Dustin ML, Nussenzweig MC. A dynamic T cell-‐limited checkpoint regulates affinity-‐dependent B cell entry into the germinal center. J Exp Med 2011; 10.1084/jem.20102477. Severinson E, Westerberg L. Regulation of adhesion and motility in B lymphocytes. Scand J Immunol 2003; 58:139-‐144. Shapiro-‐Shelef M, Calame K. Regulation of plasma-‐cell development. Nat Rev Immunology 2005; 5:230-‐242. Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunological Reviews 2012; 247:52-‐63. Skinnider BF, Elia AJ, Gascoyne RD, et al. Signal transducer and activator of transcription 6 is frequently activated in Hodgkin and Reed-‐Sternberg cells of Hodgkin lymphoma. Blood 2002; 99(2):618-‐626. Smith KGC, Hewitson TD, Nossal GJV, Tarlinton DM. The phenotype and fate of antibody-‐forming cells of the splenic foci. Eur J Immunol 1996; 26:444-‐448. Snapper CM, Finkelman FD, Paul WE. Differential regulation of IgG1 and IgE synthesis by interleukin 4. J Exp Med 1988; 167:183-‐196. Spolski R, Leonard WJ. Interleukin-‐21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008; 26:57-‐79. Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and Regulation of Class Switch Recombination. Annu Rev Immunol 2008; 26:261–292. Sumoza-‐Toledo A, Santos-‐Argumedo L. The spreading of B lymphocytes induced by CD44 cross-‐ linking requires actin, tubulin, and vimentin rearrangements. J Leukoc Biol 2004; 75:233-‐239. Sverremark E, Fernandez C. Germinal center formation following immunization with the polysaccharide dextran B512 is substantially increased by cholera toxin. International Immunology 1998; 10(7):851-‐859. Sverremark E, Fernandez C. Role of T cells and germinal center formation in the generation of immune responses to the thymus-‐independent carbohydrate Dextran B512. The Journal of Immunology 1998; 161:4646-‐4651. Takatsu K, Kouro T, Nagai Y. Interleukin 5 in the link between the innate and acquired immune response. Adv Immunol 2009; 101:191-‐236. Takenawa T, Suetsugu S. The WASP-‐WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 2007; 8:37-‐48. 62 Taylor JJ, Pape KA Jenkins MK. A germinal center-‐independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 2012; 209(3):597-‐606. Taylor JJ, Jenkins MK, Pape KA. Heterogeneity in the differentiation and function of memory B cells. Trends in Immunology 2012; 33(12):590-‐597. Teitel M, Pandolfi PP. Lymphoid malignancies. Mouse Models of Human Cancer 2004; 237-‐259. Thaunat O, Granja AG, Barral P, Filby A, Montaner B, Collinson L, Martinez-‐Martin N, Harwood NE, Bruckbauer A, Batista FD. Assymetric segregation of polarized antigen on B cell division shapes presentation capacity. Science 2012; 335:475-‐479. Thorley-‐Lawson DA. Epstein-‐Barr virus: exploiting the immune system. Nat Rev Immunol 2001; 1:75-‐82. Thorley-‐Lawson DA. EBV the prototypical human tumor virus-‐just how bad is it? J Allergy Clin Immunol 2005; 116(2):251-‐261. Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol 2010; 10:182-‐ 192. Tian L, Nelson DL, Stewart DM. Cdc42-‐interacting protein 4 mediates binding of the Wiskott-‐ Aldrich syndrome protein to microtubules. J Biol Chem 2000; 275(11):7854-‐7861. Tybulewicz VLJ, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immun 2009; 9:630-‐644. Uchida J, Yasui T, Takaoka-‐Shichijo Y, Muraoka M, Kulwichit W, Raab-‐Traub N, Kikutani H. Mimicry of CD40 signals by Epstein-‐Barr virus LMP-‐1 in B lymphocyte responses. Science 1999, 289:300-‐303. van Kooyk Y, Figdor CG Avidity regulation of integrins: the driving force in leukocyte adhesion. Current Opinion in Cell Biology 2000; 12:542–547. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol 2012; 30:429-‐57. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-‐Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivable fluorescent reporter. Cell 2010; 143:592-‐605. Vinuesa CG, Cook MC, Ball J, Drew M, Sunners Y, Cascalho M, Wabl M, Klaus GGB, MacLennan IMC. Germinal centers without T cells. J Exp Med 2000; 191(3):485-‐493. Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. T cells and follicular dendritic cells in germinal center B-‐cell formation and selection. Immunological Reviews 2010; 237:72-‐89. Vitetta WS, Ohara J, Myers CD, Layton JE, Krammer PH, Paul WE. Serological, biochemical, and functional identity of cell-‐stimulatory factor 1 and B cell differentiation factor for IgG1. J Exp Med 1985; 162:1726-‐1731. Vossenkämper A, Spenser Jo. Transitional B cells: how well are the checkpoints for specificity understood? Arch Immunol Ther Exp 2011; 59:379-‐384. Walmsley MJ, Ooi SKT, Reynolds LF, Harless Smith S, Ruf S, Mathiot A, Vanes L, Williams DA, Cancro MP, Tybulewicz VLJ. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science 2003; 302:459-‐462. 63 Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, Cyster JG. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med 2011; 208(12):2497-‐2510. Westerberg L, Greicius G, Snapper SB, Aspenström P, Severinson E. Cdc42, Rac1, and the Wiskott-‐ Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood 2001; 98:1086-‐1094. Westerberg L, Larsson M, Hardy SJ, Fernandez C, Thrasher AJ, Severinson E. Wiskott-‐Aldrich syndrome protein deficiency leads to reduced B-‐cell adhesion, migration, and homing, and a delayed humoral immune response. Blood 2005; 105:1144-‐1152. Williams DA, Zheng Yi, Cancelas JA. Rho GTPases and regulation of hematopoietic stem cell localization. Methods in Enzymology 2008; 439:365-‐393. Wills-‐Karp M, Finkelman FD. Untangling the complex web of IL-‐4-‐ and IL-‐13-‐mediated signaling pathways. Science Signaling 2008; 1(51):1-‐4. Wollenberg I, Agua-‐Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, Graca L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol 2011; 187:4553-‐ 4560. Wong P, Hattangadi SM, Cheng AW, Frampton GM, Young RA, Lodish HF. Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 2011; 118:128-‐138. Yelo E, Bernardo MV, Gimeno L, Alcaraz-‐Garcia MJ, Majado MJ, Parrado A. Dock10, a novel CZH protein selectively induced by interleukin-‐4 in human B lymphocytes. Molecular Immunology 2008; 45:3411–3418. Yoshida T, Mei H, Dörner T, Hiepe F, Radbruch A, Fillatreau S, Hoyer BF. Memory B and memory plasma cells. Immunol Rev 2010; 237:117-‐139. Young LS, Dawson CW, Eliopoulos AG. The expression and function of Epstein-‐Barr virus encoded latent genes. Mol Pathol 2000; 53:238-‐247. Young LS, Rickinson AB. Epstein-‐Barr virus: 40 years on. Nat Rev Cancer 2004; 4(10):757-‐768; Zotos D, Coquert JM, Zhang Y, Light A, D’Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, Nutt SL, Tarlinton DM. IL-‐21 regulates germinal center B cell differentiation and proliferation through a B cell-‐intrinsic mechanism. J Exp Med. 2010; 207(2):365-‐378. 64