Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
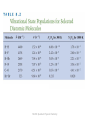
Ch 8. The Vibrational and Rotational Spectroscopy of Diatomic Molecules - light interacts with molecules to induce transitions between states. - Absorption of electromagnetic radiation in the infrared and microwave regions of the spectrum. - Transitions between eigenstates of vibrational and rotational energy induced by light MS310 Quantum Physical Chemistry 8.1 An introduction to spectroscopy What is Spectroscopy? : see the ‘bond’ → a lot of information about the character of chemical bond and reactivity Our focus in this chapter : rotation and vibration (Atomic spectroscopy : Ch 11, electron spectroscopy : Ch 15, NMR : Ch 18) Bond length : rotational spectroscopy Frequency of characteristic oscillation with chemical bond : vibrational spectroscopy Discrete energy level in Q.M → absorption and emission spectrum make individual peaks Transition energy is given by h | E1 E2 | MS310 Quantum Physical Chemistry Energy, wavelength of light(photon) and name of wave Visible light : very small range MS310 Quantum Physical Chemistry Used wave in spectroscopy → from microwave to X-ray region : nine order of magnitude 1 ~ In spectroscopy, we use the wave number Therefore, energy is given by | E2 E1 | h hc~ Experimentally, only a few transition occur(not arbitrary chosen state) : selection rule MS310 Quantum Physical Chemistry Electric and magnetic fields associated with a traveling light wave. → consider the dipolar diatomic molecule in time-dependent electric field MS310 Quantum Physical Chemistry Interaction between classical harmonic oscillator in electric field. (arrow : direction of mass) Oscillator absorbs energy in both the stretching and compression half cycles. Real Q.M oscillator : similar Electric field effects 2 way : permanent and dynamic Ex) polar HCl molecule has permanent dipole moment, and dynamic dipole moment can be generated. MS310 Quantum Physical Chemistry 8.2 Absorption, Spontaneous emission, and Stimulated emission Photon-assisted transition Absorption : photon induces a transition to higher level Spontaneous emission : excited state relaxes to lower level Stimulated emission : photon induces a transition from excited state to lower level MS310 Quantum Physical Chemistry Spontaneous emission : random event, related to lifetime of excited state Absorption, Stimulated emission : related to radiation density ρ(ν) In equilibrium, overall transition 1 to 2 and 2 to 1 must be same. System is described by B12 ( ) N1 B21 ( ) N 2 A21 N 2 Use blackbody spectral density function, we can obtain A21 16 2 3 B12 B21 , B21 c3 MS310 Quantum Physical Chemistry Ex) 8.1 Derive the result by the (1) the overall rate of transition is zero at equilibrium, (2) ratio of N2 to N1 is governed by the Boltzmann distribution Sol) overall transition rate is zero : B12 ( ) N1 B21 ( ) N 2 A21 N 2 N 2 g2 h / kT e e h / kT ( g1 g2 ) Boltzmann distribution is given by N 1 g1 ( ) A21 B12e h / kT 8h 3 1 B21 c 3 e h / kT 1 A21 8h 3 16 2 3 Two expression must be same → B12 B21 , 3 B21 c c3 Lightbulb : incoherent photon source(random direction) Laser : coherent photon source(all photons are in phase and same direction) MS310 Quantum Physical Chemistry 8.3 An introduction to vibrational spectroscopy Vibrational frequency → depends on the 2 vibrating atoms at the end of bond other atoms affect much less degree → group frequency : characteristic frequency of bond Can caculate the # of first excited state(N1) and # of ground state(N0) at 300K and 1000K by the Boltzmann distribution Except the Br2, N1<<N0 acceptable even 1000K → absorption of light at characteristic frequency occur at n=0 Experimental result in Q.M harmonic oscillator : ∆n = nfinal – ninitial = +1 MS310 Quantum Physical Chemistry MS310 Quantum Physical Chemistry MS310 Quantum Physical Chemistry Use more higher sensitive instrument : ∆n = +2, +3, … can measure, but much weaker than ∆n = +1 → selection rule ∆n = +1 is not rigorously for anharmonic potential Model of anharmonic potential : Morse potential V ( R) De [1 e ( R Re ) ]2 De : dissociati on energy relative to the bottom of potential, k / 2 De d 2V force constant k ( 2 ) x xe , dx k : also valid for harmonic oscillator Bond energy D0 : respect to lowest allowed level 1 ( h )2 1 2 E h ( n ) ( n ) Energy level is given by n 2 4 De 2 : anharmonic correlation MS310 Quantum Physical Chemistry MS310 Quantum Physical Chemistry Material-dependent parameter is given by this table. B : rotational constant, r : bond length MS310 Quantum Physical Chemistry 8.4 The origin of selection rule Transition probability is not zero → transition dipole moment follow this condition xmn m* ( x ) x ( x ) n ( x )d 0 In real case, position depends on time. We can take Taylor expansion at the x=0(equilibrium position) x ( x(t )) 0 x x(t )( d x ) x 0 ... dx First term : permanent dipole moment Second term : dynamic dipole moment(time-dependent) We can think absorption occurs state of n=0 : reasonable MS310 Quantum Physical Chemistry We can calculate the transition dipole moment m0 x Am A0 0 x H m ( 1/ 2 x )H 0 ( 1/ 2 x )e x 2 dx d x 1/ 2 1/ 2 x 2 Am A0 [( ) x x0 ] H m ( x )xH 0 ( x )e dx dx First integral : 0 by orthogonality, We only see the second integral m=even, Hm(α1/2x) : even, m=odd, Hm(α1/2x) : odd Therefore, if m is even, overall integral becomes integral of odd function and it becomes zero. → value of integral may not zero if the transition of n=0 → m=2b+1, b=0,1,2,… MS310 Quantum Physical Chemistry Figure of m=1,3,5 Except the m=1 case, area of red and blue region is same. xm 0 0 Therefore, only m=1 transition allowed. → ∆n=1 : selection rule for absorption ∆n=-1 : selection rule for emission Vibrational excitation : only dμx/dx≠0 H2, N2, O2 : μx0 =0, dμx/dx=0 → 99.93% of atmosphere doesn’t absorb the IR radiation IR radiation only absorb by CO2, NOx, and hydrocarbon : they occur greenhouse effect MS310 Quantum Physical Chemistry 8.5 Infrared absorption spectroscopy IR spectroscopy follows the Beer-Lambert Law Incident light I0(λ) through the distance dl Absorption related to dl, intensity, and concentration(also it related to # of molecule), ε(λ) : molar absorption coeffieicnt dI ( ) ( ) MI ( )dl dI ( ) I ( ) ( ) Mdl , e ( ) Ml I ( ) I 0 ( ) MS310 Quantum Physical Chemistry Ex) 8.4 ε(λ) of ethane : 40 (cm bar)-1 at 12μm Calculate the I(λ)/ I0(λ) when 10cm absorption cell length and 2.0ppm. Also, how long the cell length if I(λ)/ I0(λ)=0.90? Sol) I ( ) e ( ) Ml exp(40(cm bar) -1 (2.0 10-6 bar)(10cm) ) I 0 ( ) 0.9992 1.0 Therefore, it is difficult to measure. Rearrange the equation, we can obtain 1 I ( ) 1 3 l ln ln 0 . 90 1 . 3 10 cm 1 6 ( ) M I 0 ( ) (40 cm bar ) (2.0 10 bar ) This order of length can make by the mirror and it uses to the gas detection. MS310 Quantum Physical Chemistry How does ε(λ) depend on the wavelength? Consider the ketone molecule Vibration frequency of carbonyl group : determined by the force constant of C=O bond. Force constant depends on the chemical bond between C and O and other alkyl groups affect much less! Vibrational modes : depends on the degree of freedom total degree of freedom : 3n translational freedom : 3 rotational freedom : 3(nonlinear), 2(linear) vibrational freedom : 3n-6(nonlinear), 3n-5(linear) Benzene : 30 vibrational modes, but only 20 distinct vibrational frequencies because of the degeneracy MS310 Quantum Physical Chemistry MS310 Quantum Physical Chemistry See the IR spectroscopy of CH4 and CO Case of CH4, 9 vibrational frequency expected. However, there are only 2 peaks and a lot of additional frequencies. Also, broadening in CO peak. Why? → rotational spectra MS310 Quantum Physical Chemistry There are many rotational states with only 1 vibrational transition, n=0 → n=1. We can analyze the rotational spectra if we use the highresolution instrument. In CH4 spectra, there are only 2 peaks instead of 9. Why? Case of CH4, there are only 2 peaks 1306 cm-1 : 3 degenerate C-H bending modes 3020 cm-1 : 3 degenerate C-H stretching modes Where are other 3 modes? Use group theory, these modes are symmetric and doesn’t satisfy the condition dμx/dx≠0. Therefore, these modes are infrared inactive. However, all modes of CH4 and CO are raman active.(section 8.8) MS310 Quantum Physical Chemistry Consider the CO2 case. 2 C=O bonds are equivalent and expected there are only 1 peaks. However, there are 2 peaks by the experiment. Why? → symmetric and antisymmetric stretching MS310 Quantum Physical Chemistry Symmetric stretch : only depends on k1 , symmetric 1 2 k1 Asymmetric stretch : C moves → opposite direction of each oscillator, F=-(k1+2k2)x : 1 k1 2k 2 antisymmet ric 2 Symmetric and antisymmetric O-H stretching modes MS310 Quantum Physical Chemistry 8.6 Rotational spectroscopy Rotational selection rule : ∆J=Jfinal – Jinitial=±1 We see the Example 8.1 Ex) 8.1 Use these eigenfunctions of rigid rotor, J=0 →J=1 transition is allowed and J=0 → J=2 is forbidden. 1 12 Y ( , ) ( ) 4 3 12 0 Y1 ( , ) ( ) cos 4 1 5 Y20 ( , ) ( ) 2 ( 3 cos 2 1) 16 0 0 MS310 Quantum Physical Chemistry Sol) assume the electromagnetic field : z direction → μz=μcosθ 2 0 0 zJ 0 d YJ0 ( , )(cos )Y00 ( , ) sin d J=0 → J=1 2 3 cos 3 3 d cos sin d [ ] 0 0 4 0 0 2 3 3 3 10 z 2 J=0 → J=2 z20 5 8 2 0 0 2 d ( 3 cos 1) cos sind 3 cos 4 cos 2 5 1 1 [ ] 0 [ ] 0 8 4 2 8 4 4 5 Therefore, J=0 → J=1 transition is allowed, J=0 → J=2 transition is forbidden. Also, μzJ0 is zero unless ∆MJ=0. MS310 Quantum Physical Chemistry Consider the charged two particles in rotational motion. Rotational absorption : nonzero ‘permanent’ dipole moment! (by contrast, in vibrational absorption, nonzero dynamic dipole moment) We use the angular quantum number J instead l.(l used for orbital angular momentum) Energy is given by the 2 h2 E J ( J 1) J ( J 1) hcBJ ( J 1) 2r02 8 2 r02 B : rotational constant MS310 Quantum Physical Chemistry Simulated rotational spectroscopy MS310 Quantum Physical Chemistry calculate the energy change of the transition ∆J=1 and ∆J=-1 E E ( J final ) E ( J initial ) 2 2 J ( J 1) 2hcB( J 1) ( J 1)( J 2) E 2 r02 2 r02 2 2 J ( J 1) 2hcBJ ( J 1)J E 2 r02 2 r02 | ∆E+ | ≠ | ∆E- | : energy level not spaced equally Rotational energy not depends on mJ : 2J+1 fold degenerate Difference between ∆ν, but ∆(∆ν) is same and its value is 2cB MS310 Quantum Physical Chemistry In Real case : rotational and vibrational change simultaneously ∆Erotational << kT : many rotational peaks observe Calculate the ratio of ∆Erot and ∆Evib E rot 2 / r02 2 Evib k / r0 k This ratio is 0.028(H2) and 0.00034(I2). moment of inertia, force constant large → smaller ratio MS310 Quantum Physical Chemistry Relative ratio of given J state nJ gJ ( J 0 ) / kT 2 J ( J 1) / 2 IkT e ( 2J 1)e n0 g0 Degeneracy (2J+1) dominant for small J and large T nJ/n0 decrease to 0 rapidly J increase Moment of inertia increase : upper value of J increase HD case : 4 for 300K, 7 for 700K CO case : 13 for 100K, 23 for 300K, 33 for 700K MS310 Quantum Physical Chemistry Higher frequency(∆J=+1) : R branch Lower frequency(∆J=-1) : P branch Center of spectrum : ∆J=0 : forbidden transition For raman spectroscopy, selection rule becomes to ∆J=0, ±2 MS310 Quantum Physical Chemistry Higher resolution IR spectra of CO molecule MS310 Quantum Physical Chemistry 8.7 Fourier transform infrared spectroscopy FTIR : one pulse → same as many single-wavelength experiment(multiplex advantage) → short time! Instrument : Michelson interferometer MS310 Quantum Physical Chemistry At first, we consider the one incident wave Incident light : amplitude A0ei(kx-ωt), intensity I0 Through the beam splitter S : 50% of light transmitted, and other 50% of light reflected and go to M2 Transmitted light : reflect by M1 and 50% is reflected by S Reflected light : reflect by M2 and 50% is transmitted through S These 2 waves make interference. A A A( t ) 0 [e i ( kyD t ) e i ( k [ y D d ]t ) ] 0 (1 e i ( t ) )e i ( kyD t ) 2 2 δ(t) : phase difference from the path difference, Δd 2 2 (t ) ( 2 SM1 2 SM 2 ) d ( t ) intensity I : related to A*(t)A(t) I0 I0 2d ( t ) I ( t ) (1 cos (t )) (1 cos ), I 0 A02 2 2 MS310 Quantum Physical Chemistry Δd=nλ : constructive, measured Δd=(2n+1)λ/2 : destructive, not measured Measured signal : interferogram, single sine wave MS310 Quantum Physical Chemistry After, consider the many incident waves(different frequencies) Amplitude is given by A( t ) j Aj 2 (1 e i 2d ( t ) j i( )e Measured intensity is given by I ( t ) 2 j y D j t ) 1 2v I ( )( 1 cos( t )) j j 2 j c FTIR gives all frequency data simultaneously → use for determine of fraction of air MS310 Quantum Physical Chemistry 8.8 Raman spectroscopy Consider the oscillating electric field E E0 cos 2 t and characteristic frequency of molecule is νvib Electric field induce the induced magnetic moment, μinduced and it related to polarizability induced (t ) E0 cos 2 t Polarizability related to bond length xe+x(t) and we can expand by the Taylor-Mclaurin series ( x ) ( xe ) x ( d ) x xe ... dx Consider the vibration of molecule x(t ) xmax cos 2 vibt MS310 Quantum Physical Chemistry We can calculate induced dipole moment by these results d induce ( t ) E E 0 cos 2 t[ ( xe ) ( ) x xe xmax cos 2 vib t ] dx 1 d ( xe ) E 0 cos 2 t ( ) x xe xmax E 0 [cos(2 2 vib )t cos(2 2 vib )t ] 2 dx Therefore, allowed frequency is ν, ν-νvib, ν+νvib ν : Rayleigh frequency ν-νvib : Stokes frequency ν+νvib : anti-Stokes frequency Unless the dα/dx≠0, stokes and anti-stokes peak : zero. It means raman active bond satisfies dα/dx≠0. However, it is not related to dμx/dx≠0 → can be raman active although IR inactive! MS310 Quantum Physical Chemistry Stokes : n=0 to n=1 Anti-stokes : n=1 to n=1 Intensity of stokes and antistokes peak same? I anti stokes nexcited e 3 h / 2 kT h / 2 kT e h / kT I stokes nground e Range of 1000cm-1 to 3000cm-1 , ratio is 8x10-3 to 5x10-7 at 300K Therefore, 2 peaks are quite different. Raman and IR spectroscopy are complementary. MS310 Quantum Physical Chemistry 8.9 How does the transition rate between states depend on frequency? Solution of time-independent Schrödinger equation : constant energy → it cannot describe transition state Consider the 2-level system and E2>E1, wavefunction is written by iE t iE t 1 2 ( x , t ) a1 1 ( x )e a2 2 ( x )e a11 a2 2 At t=0, system in ground state : a1=1, a2=0 Write the initial hamiltonian as H0 Light turns on : electric field applied → permanent or dynamic dipole moment generate Assume the electric field along the x axis, time-dependent potential energy is given by E Hˆ int E x E0 cos 2 t x 0 (e 2it e 2it ) 2 : dipole approximation MS310 Quantum Physical Chemistry We must solve time-dependent Schrödinger equation ( x, t ) ( Hˆ 0 Hˆ int )( x, t ) i , ( x, t ) a1 (t )1 a2 (t )2 t ˆ E ,H ˆ E : trivial H 0 1 1 1 0 2 2 2 Therefore, equation changes to da ( t ) da ( t ) a1 ( t ) Hˆ int 1 a2 (t ) Hˆ int 2 i1 1 i2 2 dt dt Multiply Ψ2* left side and integration da1 (t ) da2 (t ) * ˆ * * ˆ a1 (t ) H int 1dx a2 (t ) 2 H int 2dx i dx i 2 1 2 2 dx dt dt * 2 Ψ1 and Ψ2 are orthonormal, equation can be simply i da2 (t ) a1 (t ) 2* Hˆ int 1dx a2 (t ) 2* Hˆ int 2dx dt MS310 Quantum Physical Chemistry Assume a1(t) and a2(t) are small change(it means a1=1, a2=0 on the right side, but not left side!) Equation is change by i ( E 2 E1 )t da 2 ( t ) * * i 2 Ĥ int 1dx e 2 ( x ) H int 1 ( x )dx dt E0 i ( E 2 E1 )t 2it e (e e 2it ) 2* ( x ) x 1 ( x )dx 2 i ( E 2 E 1 h ) t E0 i ( E 2 E1 h )t (e e ) 2* ( x ) x 1 ( x )dx 2 x21 i ( E 2 E 1 h ) t E0 i ( E 2 E1 h )t (e e ) 2 MS310 Quantum Physical Chemistry We use dummy variable t’ and integrate it. t i i ( E 2 E 1 h ) t ' ( E 2 E 1 h ) t ' i 21 E0 a2 x (e e )dt' 2 0 21 x i ( E 2 E 1 h ) t i ( E 2 E 1 h ) t E0 1 e 1 e ( ) 2 E 2 E 1 h E 2 E 1 h MS310 Quantum Physical Chemistry Unless the μx21 ≠ 0, a2(t) must be zero. First term of a2(t) : stimulated emission However, our focus is absorption : second term period of oscillation becomes zero when E2 - E1 → hν(we assume E2 > E1, E1 - E2 → hν can neglect) Use L’Hôpital’s rule f(x) f' ( x ) lim lim x 0 g( x ) x 0 g' ( x ) i ( E 2 E 1 h ) t i ( E 2 E 1 h ) t 1 e d( 1 e ) / d ( E 2 E 1 h ) lim a 2 ( t ) lim lim E 2 E 1 h 0 E 2 E 1 h 0 E E h E 2 E 1 h 0 d ( E E h ) / d ( E E h ) 2 1 2 1 2 1 it i ( E 2 E1 h )t it lim e E 2 E 1 h 0 Resonance condition : E1 - E2 = hν In this case, a2(t) increase linearly. MS310 Quantum Physical Chemistry E1 - E2 is slightly different to hν : do not resonance → transition probability almost zero : no transition Therefore, transition occurs only the hν is equal or extremely close to E1 - E2. MS310 Quantum Physical Chemistry Final goal : find the transition probability, a2*(t)a2(t) Neglect the first term, we can calculate easily 2 sin [( E 2 E1 h )t / 2] a2* ( t )a2 ( t ) E02 [ x21 ]2 ( E 2 E1 h )2 → when resonance condition, a2*(t)a2(t) increases as t2 Plot a2*(t)a2(t) when 40ps, 120ps, and 400ps time increase → height increases as t2 and width decreases as 1/t. Therefore, there are no transition without resonance, E1 - E2 = hν. Why height of peak decrease when time increase? → uncertainty principle MS310 Quantum Physical Chemistry a2*(t)a2(t) : closely related to observed in an absorption spectra Then, what is measured by the ‘real’ instrument? Intrinsic linewidth of vibrational spectra : less than 10-3 cm-1 Resolution of instrument : ~ 0.1cm In this case, measurement peak broaden and no information about the intrinsic linewidth by the measurement → ‘inhomogeneous broadening’ MS310 Quantum Physical Chemistry Summary -Light interacts with molecules to induce transitions between states and molecular spectroscopy were described. - It was discussed the absorption of electromagnetic radiation in the infrared and microwave regions of the spectrum. - Light of these wavelengths induces transitions between eigenstates of vibrational and rotational energy. - The frequency at which energy is absorbed or emitted is related to the energy levels. MS310 Quantum Physical Chemistry