Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Pharmaceutical marketing wikipedia , lookup
Neuropharmacology wikipedia , lookup
NK1 receptor antagonist wikipedia , lookup
Compounding wikipedia , lookup
Drug design wikipedia , lookup
Drug interaction wikipedia , lookup
Pharmacognosy wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Prescription costs wikipedia , lookup
Pharmacokinetics wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Drug discovery wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
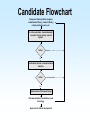
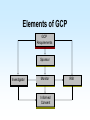
Biomedical Product Development Start with the end in mind Focus on areas of strength US Package Insert Sections • Description • Clinical Pharmacology • Indications and Usage • Contraindications • Warnings • Precautions • Adverse Reactions • Drug Abuse and Dependence • Overdosage • Dosage and Administration • How Supplied • Clinical Studies • References Paul Ehrlich 1854 - 1915 • Immunologist • Tissue staining • 1908 Nobel Prize for Medicine • “Magic Bullet” • Salvarsan Essentials Paul Ehrlich: All who are about to embark on developing a new drug must bring to the task four essentials: – brains – persistence – capital – luck Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/BLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t Research and Development The term includes basic and applied research as well as development activities carried on or supported in the pharmaceutical, biological, chemical, medical, and related sciences, including psychology and psychiatry, if the purpose of such activities is concerned ultimately with the utilization of scientific principles in understanding diseases or in improving health. Discovery The term “Discovery” is used to describe the early phases of the overall biomedical discovery process, that is, the synthesis of or the search for compounds and the screening processes developed to identify “lead” compounds. Idea sources Where do ideas for new products originate? The Process of Discovery Prevalence and Cost of Uncured Disease in the US Approximate Annual Prevalence Cardiovascular 56,000,000 Approximate Economic Cost (billions) $128 Cancer 10,000,000 $104 4,000,000 $100 Diabetes 16,000,000 $ 92 Arthritis 40,000,000 $ 65 Depression 17,400,000 $ 44 3,000,000 $ 30 28,000,000 $ 10 Uncured Diseases Alzheimer’s Stroke Osteoporosis National Institutes of Health • • • • National Cancer Institute National Heart, Lung and Blood Institute National Institute on Aging National Institute of Arthritis and Musculoskeletal and Skin Diseases • National Institute of Diabetes and Digestive and Kidney Diseases Private Organizations • • • • American Cancer Society American Heart Association Howard Hughes Medical Institute Salk Institute for Biological Studies Share of U.S. Ethical Pharmaceutical Market by Product Class, 1995 The Process of Discovery Strategic Plan • Select area of therapeutic or diagnostic interest • Establish long term (5 to 10 year) goals for program • Commit needed resources Program Plan • Short term plans, 1 to 3 years • Identifies areas for discovery research • Allocates resources to carry out the plan – people – space – equipment – money Technology Assessment • Discovery research • Marketing • Clinical research Define the Target • • • • • • • Product Market Proprietary Aspects Technologies used Mechanism of action Regulatory agencies involved Clinical trials Product Definition • • • • Diagnostic Therapeutic Device Combination Know your market • Who will use the product? • What special needs does that group have? • Who will pay for the product? Biomedical Research/Design • • • • Basic Research Feasibility Explore research/design options Lead candidate Biotechnology Discovery Tools • • • • • • • Cloning Protein purification Monoclonal antibodies Carbohydrate technology In vivo genetic modification Transgenic manipulation Cell culture The Screen • Tool to identify new drug candidates • Usually a subcellular component (enzyme, receptor, etc.) removed from a living system and studied in vitro • ACTIVES: agents that stimulate or inhibit normal function Receptors Receptor: any biological macromolecule which can be activated by a drug to cause a biological response or effect. Agonists and Antagonists • Agonist: a drug which binds to a receptor and elicits a biological response • Antagonist: occupies (or blocks) a receptor but does not elicit a response • Intrinsic activity: the measure of a drug’s ability to elicit a response Chemical Candidate Sources • Synthetic program • High through-put screening program • Compound libraries Natural Product Sources • • • • Fermentation/microbial sources Plant/herbal sources Arachnid and amphibian sources Marine sources High Through-put Screening • Tool to identify new drug candidates • Usually a subcellular component (enzyme, receptor, etc.) removed from a living system and studied in vitro Active An active is a substance that causes inhibition or stimulation in a screening model, thereby indicating the substance may have pharmacological effect. Lead Compound A compound that exhibits pharmacological properties which suggest its value as a starting point for drug development. Optimization The process of synthesizing chemical variations, or analogs, of a lead compound, with the goal of creating those compounds with improved pharmacological properties. Neural Discovery From WSJ Jan 27, 2000: Three teams of researchers have discovered a gene for a protein that appears to prevent nerves in the brain and spinal cord from growing back after being damaged by injury or disease. So What? By studying the protein, researchers hope they can design drugs that might help regenerate damaged nerves The Next Steps Scientist are looking for the receptor for the protein. Once it is found, drug companies may be able to design antagonists to block the effect of the protein, allowing damaged nerves to regenerate. Safety Assessment Ames Test In vitro metabolism • microsomes • hepatocytes • liver slices Candidate Flowchart Compound from synthetic program, combinatorial library, chemical library, natural product source, etc. In vitro evaluation - human/mammal receptor/ enzyme assay; reporter system Active No Yes Biochemical, tissue or animal model of function Active Yes Animal model of theraputic target Pharmacokinetics, formulation, acute toxicology Approval for clinical development No Quality Considerations For every experiment the researcher should record: – each item, source, lot number and quantity used – experimental conditions, e.g., times, temperatures, pressures, etc. – all calculations – sampling schedule, results Product Review • • • • • Safety and Efficacy Chemistry/Pharmacy Clinical/Regulatory Marketing/Legal Potential Ups and Downs Pitfalls of Research Stage • Process not well controlled; nonreproducible results • Insufficient experience to adequately predict critical parameters • Process not scaleable “as is” • Documentation incomplete, poorly recorded, poorly organized, or does not support claims Safety Assessment Candidate • Introduction and Summary • Assays • Chemistry • Pharmacy • Patents • Clinical Plan • • • • • • Regulatory Affairs Potential Liabilities Competition Candidate Potential Safety Recommendation Research v. Development Overall objective Corporate Mandate Compounds tested Types of studies Research Development Select a development candidate Broad, Loosely defined Many, diverse Submit an NDA Few Narrow, focused One Many Research v. Development Research Development Regulatory Little or none Extensive Timetable Loose, flexible Innovation Strict, constrained Speed Chaotic Structured Entrepeneurial Interdependent Recognition Culture Workstyle Research v. Development Research Development Quantity µg mg g g kg Safety Ames, P450 1 w 105 w Formulation Capsule Tablet, inject. Metabolism Radiolabeled Assay Budget Departmental Proj. Acct. No. <$100,000 >$1,000,000 Costs Development Tasks 1. Establish raw material specifications 2. Scale-up production processes 3. Establish critical process control parameters 4. Establish final product specifications 5. Validate analytical methods 6. GLP preclinical studies 7. Prepare clinical trial material 8. Initiate stability/reliability studies 9. Establish document systems New Drug Entities New Chemical Entities (NCE) or New Molecular Entities (NME) - active ingredients never before used as drugs Drug Substance or Active Pharmaceutical Ingredient The active ingredient intended to diagnose, treat, cure, or prevent disease or affect the structure or function of the body, excluding other inactive substances used in the drug product. API Requirements • Identity: normally two identity tests required • Strength/potency • Sensitivity • Specificity • Purity: normally 98+% for NCE’s • Stability • Safety and efficacy Drug Product The finished dosage form (tablet, capsule, etc.) that contains a drug substance--generally, but not necessarily, in association with other active or inactive ingredients. Raw Materials Establish specifications and specification testing requirements for: • identity • potency/strength • purity • stability USP/NF United States Pharmacopoeia and National Formulary - designated as the official compendia pursuant to federal and some state statutes, and containing enforceable standards and specifications for strength, quality, purity, packaging, labeling, and where applicable, bioavailability of drugs Nonclinical Laboratory Study Nonclinical laboratory study - in vivo or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety. Good Laboratory Practice (GLP) Regulations established in the U.S. in 1976 to ensure the quality and integrity of bioresearch and animal test data submitted to the FDA Good Laboratory Practice • Regulations on facilities and equipment • Regulations involved in tests and controls • Regulations on personnel and organization GLP Objectives • Verify the quality and integrity of data submitted to FDA • Inspect nonclinical laboratories engaging in safety studies for regulated products • Audit ongoing and completed lab safety studies • Determine degree of compliance with GLP regulations GLP Safety Studies • Toxicology – Acute toxicity – Subacute and chronic toxicity – Reproductive and developmental studies – Mutagenicity • • • • Metabolism Pharmacology Tissue residue Environmental Pharmacokinetics • Study of how the drug is absorbed, distributed throughout the body, metabolized and excreted (ADME) • Determination of the rate constants (kinetics) for ADME Project Team Tasks • Engineering: Determine pilot plant requirements for preparation of clinical trial material • Clinical Affairs: Begin the design of clinical studies to establish efficacy and tolerance of the new drug candidates in human beings • Regulatory Affairs – Prepare IND/IDE – Pre-IND/IDE meeting with the FDA to discuss plans for Phase I clinical trials Development Phase Deliverables • Drug/Biologic/Component Characteristics • Description of actives, excipients, components, and solvents required for formulation or assembly • Analytical test methods • Process or assembly instructions • Processing equipment incompatibilities Development Phase Deliverables Regulatory Affairs • Regulatory status of drug substance and finished product • History or status of communications with FDA • World wide regulatory strategy Development Phase Deliverables Engineering • Equipment/environmental/facility requirements for manufacture • Special handling requirements Pitfalls of Development • The scaled-up version of the product is ineffective or uncharacteristic when compared to the research version • Facilities are inadequate for aseptic handling of product, microbiological testing and/or quality control Development Phase Deliverables • Quality specifications for raw materials, drug substance and processing intermediates • Stability of raw materials • Preliminary product specifications • Storage requirements Development Phase Deliverables Marketing and Medical • Product name • Initial dose levels • Packaging configurations • Projected initial market demand (units per month) Development Phase Deliverables Regulatory Affairs • Regulatory status of drug substance and finished product • History or status of communications with FDA • World wide regulatory strategy Development Phase Deliverables Engineering • Equipment/environmental/facility requirements for manufacture • Special handling requirements Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/PLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t IND/IDE • Investigational New Drug Applications, or Notice of Claimed Exemption for a New Drug Pre-IND Meeting • Brief (1 - 2 hours) conference with FDA to get pre-submission feedback from FDA • Project Manager; FDA staff person that serves as liaison between sponsor and FDA; a Project Manager is assigned to each IND IND Requirements • Preclinical testing:Pharm/tox data including ADME, carcinogenicity and mutagenicity screening • Protection of human subjects IND Contents • • • • • • General Investigational plans Investigator’s Brochure Clinical Protocols Chemistry, Manufacturing and Controls Animal Pharmacology and Toxicology Previous Human Experience IND Requirements • FDA review time: 30 days • Submission size: 4 to 10 (400 page) volumes Human Clinical Trials Clinical Trial Any study in humans intended to – verify effects – identify adverse reactions – determine ADME for an investigational drug GCP Good Clinical Practices • establish procedures to assure the quality and integrity of data obtained during clinical testing • protect the rights and safety of clinical trial subjects Elements of GCP GCP Requirements Sponsor Investigator Monitor Informed Consent IRB Declaration of Helsinki • In research on man, the interest of science and society should never take precedence over considerations related to the well-being of the subject • Biomedical research involving humans must be scientifically sound Principles of GCP • Declaration of Helsinki • Benefits justify the risks • Preserve rights, safety, and well-being of subjects • Adequate information to support trial • Clear, detailed protocol • Prior IRB/IEC approval • Medical care by qualified physician Principles of GCP • • • • • • Qualified personnel Informed consent Record keeping Confidentiality GMP investigational products Quality systems Sponsor An individual, company, institution, or organization that takes responsibility for the initiation, management, and/or financing of a clinical trial. Responsibilities of Sponsor • Trial design • Trial management, Data handling and Recordkeeping – Independent Data Management Committee • Selecting Investigators • Financing Responsibilities of Sponsor • • • • • selecting investigators and monitors informing investigators reviewing ongoing investigations record keeping and record retention ensuring disposition of unused drug supplies Responsibilities of Sponsor • Quality Assurance/Quality Control – SOPs to assure compliance – access to sites and documents – data reliability – contracts with investigators • Contract Research Organization • Medical Expertise Responsibilities of Sponsor • Notification/Submission to Regulatory Authorities • Confirmation of IRB Approval • Investigator’s Brochure • Clinical Trial Material • Ensuring disposition of unused drug supplies • Monitoring Monitoring Clinical Trials Sponsors must monitor trials to • ensure the quality and integrity of the clinical data • ensure that the rights and safety of human subjects involved in the clinical study are preserved Monitoring The act of overseeing the progress of a clinical trial, and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, standard operating procedures (SOP's), GCP, and the applicable regulatory requirement(s). Monitoring Responsibilities • • • • • • selection of a monitor written monitoring procedures preinvestigation site visits periodic site visits review of subject records record of on-site visits Investigator A person responsible for the conduct of the clinical trial at a trial site. If a trial is conducted by a team of individuals at a trial site, the investigator is the responsible leader of the team and may be called the principal investigator Investigators’ Responsibilities • • • • • control of drug record keeping and record retention investigator reports assurance of IRB review handling of controlled substances Investigators’ Responsibilities • • • • • • • Provide adequate resources Medical care of Trial Subjects Communication with the IRB Compliance with the protocol Control of investigational product Informed Consent Records and reports Institutional Review Board (IRB) An independent body constituted of medical, scientific, and nonscientific members, whose responsibility it is to ensure the protection of the rights, safety, and well-being of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trials, of protocols and amendments, and of the methods and material to be used in obtaining and documenting informed consent of the trial subjects. IRB/IEC Responsibilities • Institutional Review Board/Independent Ethics Committee • Minimize risk to subjects • Risk v. Benefit must be reasonable • Subject selection must be equitable • Informed consent • Adequate monitoring for safety • Subject Privacy Informed Consent A process by which a subject voluntarily confirms his or her willingness to participate in a particular trial, after having been informed of all aspects of the trial that are relevant to the subject's decision to participate. Informed consent is documented by means of a written, signed, and dated informed consent form. Clinical Definitions • Adverse Drug Reaction (ADR): all noxious and unintended responses to a medicinal product related to any dose • Adverse Event (AE): any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and that does not necessarily have a causal relationship with this treatment. IND Safety Reports Unexpected, Fatal or Life Threatening and Associated with the Use of the Drug Serious, Unexpected and Associated With the Use of the Drug Serious, Expected and Nonserious To FDA by Telephone Within 3 Working Days To FDA and all Participating Investigators in a Written Report Within 10 Working Days To FDA in Next IND Annual Progress Report Written Report Within 10 Working Days Written Report to all Participating Investigators Phase I Clinical Trials Number of Subjects: Length: 20 to 80 Purpose: Primarily safety Several months Phase I Subject Selection FDA General Considerations for the Clinical Evaluation of Drugs • normal volunteers • generally, no concomitant drug therapy • generally excludes women of childbearing potential and children • pretreatment physical exams and follow-up studies Clinical Protocols • • • • • • objectives of study investigator; IRB approval patient selection/exclusion study designs dosing schedules description of observations and measurements • clinical procedures and lab tests Phase I Data • • • • • metabolism pharmacology toxicology dose ranging side effects Recording Phase I Data Case Report Form • Baseline information on patient’s existing medical condition and personal characteristics • drug related changes e.g., blood pressure • adverse events (side effects) • patient feedback Phase I Wrap-up • • • • Evaluation of Phase I data for safety Prepare Phase II Protocol Update Investigator’s Brochure Recruit Phase II clinical sites Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/PLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t Developmental Pathways • • • • • Clinical trials Formulation development/Stability Chemical process development Metabolism/ Pharmacokinetics Toxicology Phase 1 Assay Development • • • • • Well characterized drug substance Broad specifications Stability indicating assays developed Start assay validations Stability studies Dosage Form Development • • • • • • Preformulation Formulation Preliminary stability Package selection Formal stability Manufacture and Packaging for clinical supplies • Technology transfer Metabolism -1• Pharmacokinetics - ADME • Duration of effect v. drug blood levels • Bioequivalency/bioavailability of alternate dose forms • Biotransformation of the drug Metabolism -2• Studies in special populations – Age – Gender – Hepatic/renal impaired – Metabolic interaction – Ethnic groups Toxicology • • • • • • • Acute studies Rangefinding studies Subacute studies Genetic Toxicity Reproductive Toxicity Chronic Toxicity (6 months, 1 year) Carcinogenicity Studies (2 year dosing) Phase 2 Clinical Trial Number of patients: Length: Purpose: 100 to 300 patient volunteers 2 years Initial trials in patients to determine efficacy Phase 2 Clinical Trial • Dose ranging to determine optimal effect • Strength • Frequency of administration • Acceptable level of side effects • Initial determination of risk-to-benefit ratio Phase 2 Product Development • Pilot scale to larger scale-up batches • Start process validation • Assay Development – Set or tighten specifications as needed – Continue assay development – Continue stability studies • shelf-life • storage requirements • primary packing materials End-of-Phase 2 Meetings • Determine the safety of proceeding to Phase 3 • Evaluate Phase 3 plan and protocols • Identify any additional information necessary to support marketing application Phase 3 Clinical Trials Patients: Length: Purpose: 1,000 to 3,000 patient volunteers 3 years PIVOTAL Verify safety and effectiveness Monitor adverse reactions from longterm use Phase 3 Differences • Larger patient pool • Genetic, lifestyle and physiologic diversity • Concomitant therapies and conditions permitted • Out-patient population, less rigorously monitored Pivotal Studies At least two pivotal (adequate and well controlled) studies are required to provide “substantial evidence” supporting claims of effectiveness for new drugs and antibiotics. Criteria for Pivotal Studies • adequate size • must be a controlled trial • must have a blinded design (when practical and ethical) • must be randomized Adequate Size Determinants • Degree of response sought • Desired assurance against false positive • Acceptable risk of failure to demonstrate response Study Controls • Clear statement of objectives • Design that permits valid comparison with control • Method of subject selection that provides adequate assurance that subjects have the disease or condition being studied Study Design • Adequate measures to minimize bias by subjects, observers and analysts of the data • Well-defined and reliable methods of assessment of subject’s responses • Adequate analysis of the study results to assess the effects of the drug. Discontinuance of a Study • Dangerous adverse effect is found • Drug lacks significant effects or has an effect less advantageous than that of an existing therapy • Drug has a significant effect, but that effect does not justify the risks associated with its use • Drug shows clear evidence of being safe and effective Additional Phase 3 Components • • • • • Pharmacology/Toxicology Completion Prescribing Information Registration/Pricing Sales Formulation Marketing plan/support trials Success Rate • 70% of INDs successfully complete Phase I Clinical Trials • 33% of INDs successfully complete Phase 2 Clinical Trials • 27% of INDs successfully complete Phase 3 Clinical Trials and continue to NDA Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/PLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t Establishment of Commercial Scale Manufacturing Technology Transfer NDA Phase 2 Early Development Preformulation Report Phase 3 Full Development Process Optimization Process Validation Biobatch (>10% Full Scale Process Validation Batches (3) Development Report Biobatch Qualification Validation Activities FMI, including packaging Provisional Scale-up Preliminary Release Notification Specifications Full Scale Process Optimization Provisional Transfer Document Pilot scale stability Provisional Scale-up Report Full Scale Packaging Preliminary process parameters Validation Protocol Full Scale Stability Specifications for dose form Launch Production Experience Final Validation Report Production Acceptance of the Process (10 batches or 1 year) Final Transfer Report Pre-approval Inspection Assessment of Bio/Validation Batch Equivalence Establish Technology Transfer Team 15 -23 Months 10 - 16 Months 10 Months 15 Months Early Development • • • • • • • Preformulation Report Development Report FMI, including packaging Specifications Pilot Scale stability Preliminary process parameters Specifications for dose form Process Optimization • • • • • • Biobatch (>10% Full Scale) Biobatch Qualification Provisional Scale up Full Scale Process Optimization Provisional Scale up Report Validation Protocol Process Validation • • • • • • • Process Validation Batches (3) Validation Activities Preliminary Release Notification Provisional Transfer Document Full Scale Packaging Full Scale Stability Assessment of Bio-/Validation Batch Equivalence Production Experience • • • • • Final Validation Report Production Acceptance of the Process (10 Batches or 1 Year) Final Transfer Report Pre-Approval Inspection Validation • The defining and testing of processes, specifications and/or equipment used, and to prove the capability and suitability of achieving required results consistently • A requirement of GMP’s for drugs and devices • “Organized, documented common sense” Validation Protocol A written plan stating how validation will be conducted, including test parameters, product characteristics. production equipment, and decision points on what constitutes acceptable test results. Installation Qualification • • • • Suitability of building Services Materials of construction Suitability, positioning, accuracy and calibration of instruments Operation Qualification • • • • • Consistent operation System failsafes Maintenance program Equipment records Equipment and system service records Process Qualification • Demonstrate control of process within defined limits • Demonstrate consistent performance • Demonstrate consistent results New Drug Application FDA reviewers must determine • Whether drug is safe and effective for intended use • Whether benefits outweigh risks • Whether proposed labeling is appropriate • Whether manufacturing methods and controls are adequate NDA Contents -1• • • • Application Form FDA-356h Index Summary Pharmacologic Class, Scientific Rationale • Proposed Label • Foreign Marketing History NDA Contents -2• Chemistry, Manufacturing and Controls • Human Pharmacokinetics and Bioavailability • Nonclinical, Pharmacology, Toxicology • Microbiology (anti-infective) NDA Contents -3Clinical Data Summary • List of Investigators • Background/Overview of Clinical Investigators • Clinical Pharmacology • Controlled Clinical Trials • Uncontrolled Clinical Trials NDA Contents -4Clinical Data Summary • Integrated Summary of Effectiveness • Integrated Summary of Safety • Drug Abuse and Overdose • Benefits/Risks NDA Contents -5Clinical Data Summary • Review of Literature for Analogs • Bibliography for Compound • Statistical Section • Case Report Forms and Tabulations NDA Submission • 150 volumes • 50,000 pages • 12 to 48 months of review and negotiations • record: 42 days Pre-approval Inspection Prior to NDA approval, the FDA will inspect the proposed manufacturing facility to assure that the conditions presented in the NDA do exist and have been adequately documented. Records for PAI • CMC Section of NDA • Master Formula – specific manufacturing instructions for full scale commercial lots – in process specifications – product specifications • History section of NDA Records for PAI • • • • • Raw materials Laboratory Equipment qualification Cleaning validation SOPs Records for PAI • Batch record for first full scale production run • Validation protocols and reports • History of production • Failure investigation reports • All complaints • Microbiological data PAI Documentation • Report to pilot plant detailing R&D formulation development • Report covering pilot plant experiences during scale-up • Report setting product specification US Package Insert Sections • Description • Clinical Pharmacology • Indications and Usage • Contraindications • Warnings • Precautions • Adverse Reactions • Drug Abuse and Dependence • Overdosage • Dosage and Administration • How Supplied • Clinical Studies • References Description • • • • • proprietary name and established name dosage form and route of administration quantitative ingredient information pharmacological or therapeutic class chemical name and structural formula Clinical Pharmacology • Actions of the drug in humans • Pharmacokinetic data (ADME) • Clinical Trial Results Adverse Reactions • • • • Clinical Adverse Experiences Concomitant Therapy Laboratory Abnormalities Hypersensitivity Reactions Dosage And Administration • General Recommendations • Dosage in Patients with Renal Insufficiency • National Drug Code FDA Post Dug Approval Activities • Post-marketing surveillance • Prescription drug advertising and promotional labeling • Pharmaceutical industry surveillance • Drug shortages • Therapeutic inequivalence reporting • Medication errors Post Market Surveillance • Phase 4 Clinical Trials • General Reporting Requirements – Field Alert Reports – Annual Reports – Adverse Drug Reaction Reporting (ADR) – Special reports • cGMP Requirements Phase 4 Clinical Trials • Satisfy pre-approval FDA request • Evaluate safety and effectiveness in general use • Cost/benefit analysis • Augmentation of original indication • Marketing implications • Expansion of claim structure Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/PLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t Success Rate • 70% of INDs successfully complete Phase I Clinical Trials • 33% of INDs successfully complete Phase 2 Clinical Trials • 27% of INDs successfully complete Phase 3 Clinical Trials and continue to NDA New Drug Application FDA reviewers must determine • Whether drug is safe and effective for intended use • Whether benefits outweigh risks • Whether proposed labeling is appropriate • Whether manufacturing methods and controls are adequate Advisory Committee Advisory Committee - a panel of outside experts convened periodically to advise FDA on safety and efficacy issues about drugs and other FDAregulated products. FDA isn’t bound to take committee recommendations, but usually does. Preparing for the Panel • Learn as much as possible about the committee members • Fully understand the issues the review division presents • Understand how the committee functions • Don’t over do the presentation; keep it short FDA Verdict • • • Approval letter Approval letter Not approvable letter Success Rate 20% of IND’s result in successful NDA’s Development Time Line Stage Discovery The Screen The Lead Development Safety Assess Cand Patents Dosage Form Dev IND/IDE Clinical Trials Phase 1 Review Safety Data Phase 2 End Phase 2 Clin Rev Phase 3 NDA/PLA/PMA FDA Review Approval Product Launch Year 1 Year 2 Year 3 Year 4 Year 5 Year 6 Year 7 Year 8 Year 9 Year 10 Year 11 Year 12 t t R R t t t t t t t t t