Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
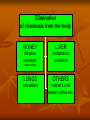
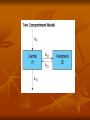
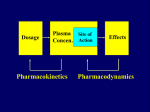
Elimination of chemicals from the body KIDNEY LIVER filtration secretion metabolism excretion (reabsorption) LUNGS OTHERS exhalation mother's milk sweat, saliva etc. 4. Elimination The parameter most commonly used to describe the rate of elimination of a chemical is the half-life. Most toxicokinetic processes are first-order reactions, i.e. the rate at which the process occurs is proportional to the amount of chemical present. High rates (expressed as mass/time) occur at high concentrations and the rate decreases as the concentration decreases; in consequence the decrease is an exponential curve. First order elimination First order elimination kinetics are described by the equation: Ct = C0 * e-kt Taking the natural logarithm of this equation and plotting it semilogarithmically results in a linear graph with a slope of -k, and a y-intercept of ln C0. Again, to determine the half-life, ½ C0 is substituted into the equation to give: ½ C0= C0e-kt1/2 Taking natural logs and solving for t1/2: t1/2= 0.693/k The usual way to analyze exponential changes is to use logarithmically transformed data which converts an exponential into a straight line. The slope of the line is the rate constant (kel) for the process and the half-life for the process is calculated as 0.693/kel. Rate constants and half-lives can be determined for absorption, distribution, and elimination processes. The mechanisms of elimination depend on the chemical characteristics of the compound: volatile chemicals are exhaled, water-soluble chemicals are eliminated in the urine lipid-soluble chemicals are eliminated by metabolism to more water-soluble molecules, which are then eliminated in the urine and/or bile. First-pass metabolism The first-pass effect or presystemic metabolism )is a phenomenon of drug metabolism whereby the concentration of a drug is greatly reduced before it reaches the systemic circulation. It is the fraction of lost drug during the process of absorption which is generally related to the liver and gut wall. Notable drugs that experience a significant first-pass effect are Imipramine ,Propranolol ,and Lidocaine. After a drug is swallowed, it is absorbed by the digestive system and enters the hepatic portal system .It is carried through the portal vein into the liver before it reaches the rest of the body. The liver metabolizes many drugs, sometimes to such an extent that only a small amount of active drug emerges from the liver to the rest of the circulatory system . This first pass through the liver thus greatly reduces the bioavailability of the drug. Elimination by the Kidney Drugs are excreted by the kidneys by 2 processes: 1) Passive: - glomerular filtration - removes molecules up to size of small proteins - therefore, protein bound drugs are poorly filtered - nonsaturable process 2) Active: - tubular reabsorption - saturable process Metabolism in kidneys is a minor elimination route Elimination by the Liver Metabolism - major 1) Phase I and II reactions 2) Function: change a lipid soluble to more water soluble molecule to excrete in kidney 3) Possibility of active metabolites with same or different properties as parent molecule Biliary Secretion EXCRETION BY OTHER ROUTES LUNG - For gases and volatile liquids by diffusion. Excretion rate depends on partial pressure of gas and blood/air partition coefficient. MOTHER’S MILK a) By simple diffusion mostly. Milk has high lipid content and is more acidic than plasma (traps alkaline fat soluble substances). b) Important for 2 reasons: transfer to babies, transfer from animals to humans. OTHER SECRETIONS – sweat, saliva, etc.. minor contribution Clearance (CL) A measure of the ability of the body to eliminate the drug/toxin in ml/min Defined as the rate of drug concentration eliminated from the body in mL/min Can be defined for various organs in the body Sum of all routes of elimination CLtotal = CLliver + CLkidney + CLintestine The Clearance (Cl) of a drug is the volume of plasma from which the drug is completely removed per unit time. The amount eliminated is proportional to the concentration of the drug in the blood. A clearance of 100 mL/minute of a chemical means that 100 mL of blood/plasma is completely cleared of the compound in each minute. The best measure of the ability of the organs of elimination to remove the compound from the body is the clearance (CL): Because the rate of elimination is proportional to the concentration, clearance is a constant for first-order processes and is independent of dose. It can be regarded as the volume of plasma (or blood) cleared of compound within a unit of time (e.g. mL/ min). Renal clearance depends on the extent of protein binding, tubular secretion (active transport) and passive reabsorption in the renal tubule; it can be measured directly from the concentrations present in plasma and urine: CL = rate of elimination/Cp Rate of elimination = Kel * Dose Vd = Dose/Cp Therefore CL = Kel*Vd The total clearance or plasma clearance (which is the sum of all elimination processes, i.e. renal, metabolic, etc.) is possibly the most important toxicokinetic parameter. It is measured from the total amount of compound available for removal (i.e. an intravenous dose) and the total area under the plasma concentration–time curve (AUC) extrapolated to infinity. Plasma clearance reflects the overall ability of the body to remove permanently the chemical from the plasma. Plasma clearance is the parameter that is altered by factors such as enzyme induction, liver disease, kidney disease, inter-individual or interspecies differences in hepatic enzymes or in some cases organ blood flow. Once the chemical is in the general circulation, the same volume of plasma will be cleared of chemical per minute (i.e. the clearance value) applies irrespective of the route of delivery of chemical into the circulation. However, the bioavailability (F) will determine the proportion of the dose reaching the general circulation. Therefore, bioavailability has to be taken into account if clearance is calculated from data from a nonintravenous route (e.g. oral): The overall rate of elimination, as indicated by the terminal halflife (t ), is dependent on two physiologically related and independent variables: CL=Vd*Kel where CL is the ability to extract and remove irreversibly the compound from the general circulation, and V the extent to which the compound has left the general circulation in a reversible equilibrium with tissues. Chemicals that are extremely lipid-soluble and are sequestered in adipose tissue are eliminated slowly. Lipid soluble organochlorine compounds, which are not substrates for P450 oxidation, due to the blocking of possible sites of oxidation by chloro-substituents, are eliminated extremely slowly: for example, the half-life of 2,3,7,8tetrachlorodibenzodioxin (TCDD) is about 8 years in humans. Classical Toxicokinetics If we assume that the concentration of a chemical in blood or plasma is in some describable dynamic equilibrium with its concentrations in tissues, then changes in plasma toxicant concentration should reflect changes in tissue toxicant concentrations and relatively simple kinetic models can adequately describe the behavior of that toxicant in the body system. Classic toxicokinetic models typically consist of a central compartment representing blood and tissues that the toxicant has ready access and equilibration is achieved almost immediately following its introduction, along with one or more peripheral compartments that represent tissues in slow equilibration with the toxicant in blood. One-Compartment Model The most straightforward toxicokinetic assessment entails quantification of the blood or more commonly plasma concentrations of a toxicant at several time points after a bolus intravenous (iv) injection. Often, the data obtained fall on a straight line when they are plotted as the logarithms of plasma concentrations versus time; the kinetics of the toxicant is said to conform to a one-compartment model. Mathematically, this means that the decline in plasma concentration over time profile follows a simple exponential pattern as represented by the following mathematical expressions: Ct = C0 × e−kel*t First order elemination First order elimination kinetics are described by the equation: Ct = C0 * e-kt Taking the natural logarithm of this equation and plotting it semilogarithmically results in a linear graph with a slope of -k, and a y-intercept of ln C0. Again, to determine the half-life, ½ C0 is substituted into the equation to give: ½ C0= C0e-kt1/2 Taking natural logs and solving for t1/2: t1/2= 0.693/k Two compartments model Two-Compartment Model After rapid iv administration of some toxicants, the semilogarithmic plot of plasma concentration versus time does not yield a straight line but a curve that implies more than one dispositional phase. In these instances, it takes some time for the toxicant to be taken up into certain tissue groupings, and to then reach an equilibration with the concentration in plasma; hence, a multi-compartmental model is needed for the description of its kinetics in the body. The concept of tissue groupings with distinct uptake and equilibration rates of toxicant becomes apparent when we consider the factors that govern the uptake of a lipid-soluble, organic toxicant. Plasma concentration-time profile of a toxicant that exhibits multi-compartmental kinetics can be characterized by multiexponential equations. For example, a two-compartment model can be represented by the following bi-exponential equation. C = A × e−α×t + B × e−β×t where A and B are coefficients in units of toxicant concentration, and α and β are the respective exponential constants for the initial and terminal phases in units of reciprocal time. The initial (α) phase is often referred to as the distribution phase, and terminal (β) phase as the post-distributional or elimination phase. Zero order elimination A constant amount of drug is absorbed regardless of dose. A plot of this equation is linear with a slope, k, and a y-intercept, C0. The elimination halflife may be calculated from this equation for a drug which exhibits zero order elimination. This occurs when Ct = ½ Co and t = t1/2 Loading Doses The loading dose is one or a series of doses that are administered at the beginning of therapy. The objective is to reach the target concentration rapidly. The loading dose can be estimated with the following formula: Loading Dose = Target Cp x Vss/F where Cp = Concentration in plasma Vss= Volume of Distribution at steady state F = Fractional bioavailability of the dose Dosing Rate In the majority of clinical situations, drugs are administered as a series of repeated doses or as a continuous infusion in order to maintain a steady state concentration. Therefore, a maintenance dose must be calculated such that the rate of input is equal to the rate of drug loss. This may be determined using the following formula: Dosing Rate = Target concentration × CL/F where CL= Clearance F= Fractional bioavailability of the dose