Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
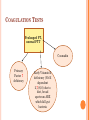
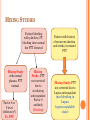
HEME-ONC STEP 3 REVIEW By James K. Rustad, M.D. Copyright © 2009 All Rights Reserved OUTLINE Anemia Iron Deficiency Anemia Beta and Alpha Thalassemia Vitamin B12 deficiency and Schilling test Hemolytic Anemia (Autoimmune, Hereditary Spherocytosis, DrugInduced, and Microangiopathic) G6PD Deficiency Sickle cell Anemia Blood Transfusion Reactions Neutropenia ITP Heparin Induced Thrombocytopenia Coagulation tree, abnormalities, and testing Oncology: Leukemias and Lymphomas Multiple Myeloma ANEMIA ANEMIA What to do? Check CBC and Retic Count If Retic index > 2.5 OR Absolute retic count > 75000 ----- likely Hemolysis or Hemorrhage MEAN CORPUSCULAR VOLUME (MCV) Measure of average RBC volume (size) Microcytic anemia < 80 (Iron Deficiency) Normocytic/Normochromic 80-95: chronic disease, renal failure, hypothyroidism Macrocytic > 95 (B12, Folate deficiency). Remember “hypersegmented (lobed) neutophils” IRON DEFICIENCY ANEMIA VS. ACD Lab Value IRON Deficiency Anemia of Chronic Disease Iron Decreased Decreased TIBC Increased Decreased Ferritin Decreased Normal or Elevated CLINICAL CASE SCENARIO CBC suggests Microcytic Anemia (low MCV), Next step? Iron Studies. If normal, what is next step? Hb electrophoresis. If normal, what is diagnosis? Alpha-Thalassemia Trait, which is a Diagnosis of Exclusion CLINICAL NUGGETS OF KNOWLEDGE Retic Count will increase one week after starting oral iron. Take iron 2 hours before and 4 hours after calcium/antacids (decreases absorption). If not tolerating Ferrous sulfate (abdominal cramps), switch to Ferrous gluconate. BETA THALASSEMIA Normal Minor Major HbA 97-99% 90% 0-10% HbA2 1-3% 4-8% 4-10% HbF 0% 1-5% 90-96% BETA THALASSEMIA MAJOR (COOLEY’S ANEMIA) Homozygous Severe anemia: patient cannot survive without transfusion! To Diagnose? Hb electrophoresis On Peripheral Smear: Target cells (also present in sickle cell, S.C. disease) Basophilic stippling (also present in alcohol abuse, lead poisoning) BETA THALASSEMIA Extramedullary and Intramedullary erythropoiesis Chipmunk facies Hair on end appearance in skull x-ray due to marrow expansion. Treatment: Blood transfusion, allogenic bone marrow transplant. ALPHA THALASSEMIA There are four alpha globin genes. One deleted: silent carrier, normal. Two deleted: Thalassemia minor or Alpha Thalassemia Trait. Low Hct with very low MCV. Three deleted: HbH disease. Severe hemolytic anemia, increased Retic count, Hb Electrophoresis shows HbH. Tx: repeated transfusion. Poor prognosis. Four deleted: Hydrops Fetalis (Hb Electrophoresis shows Hb Bart): Death in utero. SCHILLING TEST To differentiate the cause of Vitamin B12 deficiency: Decreased intake Decreased production of intrinsic factor Decreased absorption Vitamin B12 IM to saturate plasma level then radio-labeled B12 administered orally. ALGORITHM STEP ONE: 24 HOUR URINE TEST If normal (7% of oral dose in urine), cause is decreased intake. If abnormal, give Radio-labeled B12 and Intrinsic Factor. ALGORITHM STEP TWO: RADIO-LABELED B12 AND IF Normal: Intrinsic Factor Deficiency caused by Pernicious anemia or Gastrectomy Abnormal: Cause is decreased absorption of B12 caused by Blind Loop syndrome, Crohn’s or Ileal Resection. SUSPECTED BLIND LOOP SYNDROME Treat with antibiotics and repeat testing. If normal: blind loop syndrome. If still abnormal: caused by Crohn’s or ileal resection. SCHILLING TEST ALGORITHM NML: 7% of oral dose in urine 24 hour urine ABNL: Give B12 and IF NML: I.F. Deficient Pernicious Anemia or Gastrectomy AABNL: Decreased absorption If improved after ABX: Blind loop syndrome. Otherwise: Crohn’s or ileal resection HEMOLYTIC ANEMIA Anemia and jaundice Increased: retic index, LDH, indirect bilirubin Decreased: haptoglobin AUTOIMMUNE HEMOLYTIC ANEMIA Warm antibody induced Cold antibody induced Antibody IgG against Rh antigen IgM against I antigen Causes Idiopathic, drug induced, lymphoma, leukemia Idiopathic, mycoplasma, mono, Waldenstrom’s macroglobulinemia Coomb’s Test + IgG or IgG and C3 - IgG Only C3 + Cold agglutinin Negative Positive Treatment Steroid, Splenectomy. If treatment refractory: immunosuppressive! Cyclophosphamide or Cyclosporin. No Steroid or Splenectomy. Immunosuppressive treatment!! Cyclophosphamide or Chlorambucil. INCREASED MHC AND SPHEROCYTES MHC: Mean corpuscular hemoglobin Two causes: Autoimmune hemolytic anemia (Coombs test positive) Hereditary Spherocytosis (Coombs test negative) Spherocytes are hyperchromic; they have no area of central pallor (normal red blood cells do!) CLINICAL SCENARIO Child with anemia, icterus, jaundice and splenomegaly on exam. Spherocytes in peripheral smear. Retic count and MCHC increased. Coombs test negative. Most likely diagnosis? Hereditary spherocytosis! HERIDITARY SPHEROCYTOSIS Confirmatory test: Osmotic Fragility Test Treatment: Splenectomy You may also give Folic Acid. DRUG INDUCED HEMOLYTIC ANEMIA AlphaMethyldopa Type Penicillin Type Quinidine Type Direct Coomb’s IgG+ C3+ IgG+ C3+ IgG negative C3+ Indirect Coomb’s Positive without adding Drug Positive ONLY when adding drug Positive ONLY when adding drug CLINICAL CASE 30 year old pregnant patient with anemia. On Methyldopa for Hypertension and started on Penicillin for recent UTI. On exam: pallor. Labs: increased retic count, increased LDH, spherocytes present on smear. Direct Coombs + for IgG and C3. Indirect Coombs positive ONLY when drug added. Most likely cause of anemia? PENICILLIN MICROANGIOPATHIC HEMOLYTIC ANEMIA Fragmented RBC’s Schistocytes (black arrows) Helmet cells (red arrows) Causes: TTP HUS DIC Prosthetic Heart Valve HELLP syndrome in pregnancy THROMBOTIC THROMBOCYTOPENIC PURPURA (TTP) Microangiopathic hemolytic anemia Neuro: change in mental status (confusion waxes and wanes in minutes) May have renal insufficiency PT, PTT normal Cause: unknown Treatment: plasmapheresis HEMOLYTIC UREMIC SYNDROME Causes: unknown OR diarrhea with E. Coli O157: H7, Shigella, Salmonella. G6PD DEFICIENCY X-linked recessive (usually male) Usually in Black Americans G6PD Deficiency can cause hemolytic anemia. Common precipitants include: Infection Drugs such as Dapsone, Quinine, Sulfonamide, Primaquin, Quinidine, Nitrofurantoin Bite cells SICKLE CELL ANEMIA Valine replaces glutamic acid at position 6 of beta chain Sickle Hb forms polymer when dehydrated Hb electrophoresis HbS positive RBC containing HbF does not sickle (fewer crises) Chronic hemolysis: RUQ pain secondary to calcium bilirubinate gallstone Spleen not palpable Poorly healing ulcers of the lower extremity SICKLE CELL ANEMIA (SS) VS. SICKLE CELL TRAIT (SA) Sickle cell anemia (SS): Sickle cell trait (SA) HbS: 75%-95% HbF: 2%-20% HbA2: 2-4% HbS: 40% HbA: 60% Factors precipitating sickling: Hypoxia, dehydration, acidosis, hypothermia Crisis only w/ severe hypoxia: Examples include mountaineering and skiing. Hematuria more frequent than in SS. CLINICAL CASE Patient with sickle cell anemia complains of SOB, weakness. CBC shows hemoglobin of 5 (baseline 9-10) Next study? Retic count to differentiate between Hemolytic Crisis (Retic count high) secondary to splenic sequestration or co-existent G6PD deficiency vs. Aplastic crisis (Retic count low). Treatment of sequestration is splenectomy. On exam of patient with splenic sequestration: spleen palpable whereas it was not palpable on prior exams. CLINICAL CASE Patient with sickle cell complains of weakness. Hematocrit 20 and low Retic index at 0.8. History of febrile illness and skin rash one week ago. Most likely diagnosis? Aplastic crisis secondary to PARVOVIRUS. CLINICAL CASE Patient with sickle cell comes to ER with cough and chest pain worse on inspiration and lying down. CXR shows infiltration and patient started on IV fluids and antibiotics. In 48 hours NO clinical improvement, severe chest pain continues. Diagnosis and next step in management? Acute Chest Pain Syndrome; arrange for Exchange Transfusion BLOOD TRANSFUSION REACTIONS Clinical Case Scenarios CLINICAL CASE SCENARIO Patient with anemia receives blood transfusion and develops chills/fever/chest and flank pain. Most likely diagnosis? ABO incompatibility Treatment: Stop transfusion and start IV hydration to prevent Acute Tubular Necrosis! CASE SCENARIO Patient underwent complete compatibility testing, receives blood transfusion and four hours later develops fever, chills. What happened? Leukoagglutinin reaction (not secondary to hemolysis). Treatment? Acetaminophen and Diphenhydramine CASE SCENARIO Patient with lymphoma (immunocompromised) needs blood transfusion. What to do? Transfuse GAMMA Radiated Blood to prevent Graft vs. Host Disease. CASE SCENARIO History of Febrile Reaction with Blood Transfusion. Needs to be transfused again --what to do? Leukocyte depletion filter. CASE SCENARIO A blood transfusion leads to hives and bronchospasm caused by plasma proteins. If another transfusion is necessary, what is the next step? Transfuse WASHED (“like the laundry”) packed RBC’s! NEUTROPENIA 18 y.o. black male, WBC 2000, Polymorphs 38%, asymptomatic Repeat CBC in 3-4 weeks, if WBC same = Benign neutropenia with no risk infection If after 3-4 weeks, WBC increased, repeat again in 3-4 weeks. If WBC goes down, Cyclic Neutropenia. Increased risk of infection and treat with Granulocyte Macrophage Colony Stimulating Factor! IDIOPATHIC THROMBOCYTOPENIA (ITP) Case: Young Female Patient with Platelets 8000. Started on Prednisone 40 mg. After 4 weeks, platelet count 150,000. When Prednisone tapered to 20 mg, platelet count drops below 20,000. Next step? Plan for Splenectomy as patient needs high dose Prednisone to maintain platelet counts. HEPARIN INDUCED THROMBOCYTOPENIA Type I usually mild and self limited; Type II (immune) is of clinical concern. Type II occurs 5-15 days after starting therapy. Increased risk of Thrombosis due to heparin induced platelet aggregation. (HIT) HIT II DIAGNOSIS Diagnosis: 14C serotonin release assay; antiheparin/platelet factor 4 antibody. Treatment: Stop all forms of heparin even the flush for the IV line. HIT II TREATMENT If anticoagulation needed use Danaproid, Lepirudin, Hirudin, or Argatroban. These have no Antidote! Remember, “if you’ve got the poison (Heparin), then I’ve got the remedy (Protamine sulfate).” CLINICAL CASE Patient in hospital received heparin for DVT prophylaxis. Returned home and then after one week came back to ER in severe abdominal pain; on exam abdomen mildly tender in periumbilical area (subjective greater than objective pain). Labs: WBC count and Lactic Acid high, Platelets law. Most likely diagnosis? Acute Mesenteric Ischemia secondary to HIT II. COAGULATION TREE Coagulation Abnormalities and Testing COAGULATION CASCADE Extrinsic (PT) Intrinsic (PTT) TF 12 11 9 8 Factor 10 (changes Prothrombin to Thrombin) 7 COAGULATION CASCADE (CONTINUED) Fibrinogen to Fibrin Monomer via Thrombin Monomer to Polymer Polymer to Cross linked fibrin via Factor 13 VON WILLENBRAND’S DISEASE Deficiency of vWF Most common inherited bleeding disorder. Autosomal dominant. Functions: Bridge for platelet adhesion and aggregation. Carrier for factor 8 (increases half life by five-fold). Prolonged bleeding time despite normal platelet count. Treatment? Desmopressin in mild bleeding. Factor 8 concentrate for severe bleeding or prior to surgery or if patient is still bleeding after desmopressin. COAGULATION TESTS Prolonged PTT and Normal PT No bleeding: Factor 12 deficiency Mild or rare bleeding: Factor 11 deficiency Frequent and severe bleeding: Factor 9 or 8 deficiency!! HEMOPHILIA A VS. HEMOPHILIA B Hemophilia A Hemophilia B Affects only male Affects only male Low factor 8 coagulation activity Low factor 9 coagulant activity Prolonged PTT (normal PTT on mixing study), normal PT Prolonged PTT (normal PTT on mixing study), normal PT Family history Family history Spontaneous bleeding in the joints Spontaneous bleeding in the joints Treatment: Factor 8 concentrate Treatment: Factor 9 concentrate Normal Factor 8 antigen COAGULATION TESTS Prolonged PT, normal PTT Coumadin Primary Factor 7 deficiency Early Vitamin K deficiency (Vit K dependent 2,7,9,10) due to diet, broad spectrum ABX which kill gut bacteria COAGULATION TESTING Both PTT and PT prolonged? Defect in common pathway DIC, Liver Disease, Vitamin K deficiency MIXING STUDIES Patient bleeding with platelets, PT, bleeding time normal but PTT elevated Mixing Study with normal plasma: PTT normal. Factor 8 or 9 level (deficiency?) Tx: FFP Mixing Study: PTT not corrected due to circulating anticoagulant Factor 8 antibody (bleeding) Patient with history of recurrent abortion and stroke, increased PTT Mixing Study: PTT not corrected due to Lupus anticoagulant (not bleeding in Lupus: hypercoagulable state) CLINICAL CASE SCENARIO After a dental extraction, patient develops severe bleeding. Normal platelets, bleeding time, PT, PTT. Most likely diagnosis? Factor 13 deficiency (problem with forming cross-linked fibrin) Diagnosis: Clot solubility in 5M urea Treatment: FFP ONCOLOGY Leukemia and Lymphoma ACUTE LEUKEMIA In children: 80% acute leukemia: ALL In adults 80% acute leukemia: AML S/Sx: fatigue, gum bleeding, epistaxis, ecchymosis, purpura, petechiae, bone pain, recurrent infection. On exam: hepatosplenomegaly, lymphadenopathy non tender, firm, rubbery (especially ALL). Labs: decreased platelet, decreased RBC and Hb/Hct, increased WBC with 90% blast cell in peripheral smear (patients have leukopenia but due to blast cells total count increases). Bone marrow: hypercellular with > 30% blast cells ACUTE LEUKEMIA AML AML cells have granules, Auer Rods, histochemical stain suggests myeloid enzyme such as peroxidase. Worst prognosis: Monosomy 5 and 7 Treatment: Cytarabine and Daunorubicin. After remission consider Bone Marrow Transplant. ALL ALL cells have surface marker TdT (terminal dexy nucleotidal transferase). Worst prognosis: t (9:22) and t (4:11) Treatment: Vincristine, Prednisone, Daunorubicin, L-Asperginase – after remission CNS prophylaxis with intrathecal Methotrexate and cranial irradiation; also consider Bone Marrow Transplant. CHRONIC MYELOGENOUS LEUKEMIA Hallmark: WBC > 100,000 Most cells mature (less than 5% blast cells) Low leukocyte Alk. phos. : fatigue, night sweats, low grade fever On exam: splenomegaly, sternal tenderness Drug of choice: Imatinib mesylate (inhibit tyrosine kinase activty of bcl/abl oncogene). Bone marrow transplant (only curative treatment < 60 year old with HLA matched sibling). CML (PHILADELPHIA CHROMOSOME) Philadelphia chromosome negative CML has poorer prognosis than Philadelphia chromosome positive. CHRONIC LYMPHOCYTIC LEUKEMIA Hallmark: lymphocytosis WBC usually greater than 20,000 (75-95% of cells are mature lymphocytes) Usually CD19 is a marker of B and CD5 of T lymphocyte. Usually patient will be >65 yr of age. If Anemia + Thrombocytopenia: IV Fludrabine (IV), Chlorambucil. Refractory CLL: Alemtuzumab HODGKIN’S LYMPHOMA Commonly presents as painless cervical lymphadenopathy in young children. To diagnose? Lymph node biopsy “Reed-Sternberg” cells HODGKIN’S LYMPHOMA Most common: nodular sclerosing type. Staging: I: one lymph node II: two or more lymph nodes on one side of diaphragm III: both sides of diaphragm IV: disseminated disease A: No constitutional symptoms B: fever, night sweats Treatment: IA and IIA: radiation. Others: chemotherapy ABVD: Adriamycin, Bleomycin, Vincristine, Dacarbazine BURKITT’S LYMPHOMA B-cell lymphoma Translocation of protooncogene C-myc from Chromosome 8 to 14 Presentation: abdominal pain/fullness Retroperitoneal or pelvic lymphadenopathy Treatment: low risk – chemotherapy and high risk – autologous stem cell transplant Dx: lymph node biopsy (uniform small cells with scant cytoplasm), “starry sky” MULTIPLE MYELOMA Age > 65 Anemia, bone pain, renal failure, increased calcium, proteinuria Plasma cell malignancy (paraproteins produced) Dx: SPEP, UPEP Bence Jones protein in urine Dipstick (detects albumin, intact globulin) will be negative, urine protein quantitation will have proteinuria Monoclonal spike in beta or gamma globulin region. IgG – 60%, IgA – 25%, light chain – 15% Bone x-ray with lytic lesion Bone marrow infiltrated with plasma cells Treatment: Combination chemo with VAD (Vincristine, Adriamycin, Dexamethasone). If not tolerated: Thalidomide + Dexamethasone < 70 yo: consider autologous stem cell transplant. THANK YOU FOR YOUR ATTENTION Questions?