Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
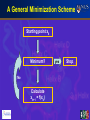
LSM2104/CZ2251 Essential Bioinformatics and Biocomputing Protein Structure and Visualization (3) Chen Yu Zong [email protected] 6874-6877 LSM2104/CZ2251 Essential Bioinformatics and Biocomputing Lecture 11 Receptor Ligand Binding, Energy Minimization and docking concepts Structural Modelling 1. Receptor-Ligand Interactions 2. Energy description of structures 3. Structure optimization by energy minimization 4. Receptor-ligand docking Protein-Protein Interaction Protein-Protein interaction: Surface contact, shape complementarity Intermolecular forces: Van der Waals, hydrogen bonding, electrostatic force Hydrogen Bond Types of Hydrogen Bond: N-H … O N-H … N O-H … N O-H … O V r Protein-DNA Interaction Protein-DNA interaction: • DNA recognition by proteins is primarily mediated by certain classes of DNA binding domains and motifs Protein-RNA Interaction Protein-RNA interaction: • RNA recognition by proteins is primarily mediated by certain classes of RNA binding domains and motifs Protein-Ligand Interaction Ligand Binding: A small molecule ligand normally binds to a cavity of a protein. Why? Effect of Binding: Activate, inhibit, being metabolized or transported by, the protein Protein-Ligand Interaction Ligand Binding: A small molecule ligand normally binds to a cavity of a protein. Why? Effect of Binding: Activate, inhibit, being metabolized or transported by, the protein Protein-Ligand Interaction Ligand Binding: A small molecule ligand normally binds to a cavity of a protein. Why? Effect of Binding: Activate, inhibit, being metabolized or transported by, the protein Protein-Drug Interaction Mechanism of Drug Action: A drug interferes with the function of a disease protein by binding to it. This interference stops the disease process Drug Design: Structure of disease protein is very useful Protein-Drug Interaction Mechanism of Drug Action: A drug interferes with the function of a disease protein by binding to it. This interference stops the disease process Drug Design: Structure of disease protein is very useful Example of Binding Induced Shape Change Example 2: Induced Fit of Hexokinase (blue) Upon Binding of Glucose (red). Note that the active site is a pocket within the enzyme. Energy Description Energy is needed to make things or objects change: Movement, Chemical reaction, Binding, Dissociation, Structural Change, Conformational change etc. Why Energy Description for molecular structure? • Structure determination (“evolution” of a structural-template into the correct structure) • Binding induced shape change (binding sometimes induces shape change, one of the mechanisms for the interference of the function of a molecule by another) • Protein motions (proteins undergo internal motions that have implications such as the switch between active and in-active state) Energy Description Kinetic energy -- motional energy Kinetic energy is related to the speed and mass of a moving object. The higher the speed and the heavier the object is, the bigger work it can do. Potential Energy -- "positional" energy. Water falls from higher ground to lower ground. In physics such a phenomenon is modeled by potential energy description: Objects move from higher potential energy place to lower potential energy place. Potential Energy Description of Protein Structure “Evolution” • A molecule changes from higher potential energy form to lower potential energy form. • Potential energy is determined by inter-molecular, intra-molecular, and environmental forces • Protein structural “evolution” can be performed by systematic variation of the atom positions towards the lower energy directions. This procedure is called “structure optimization” or “energy minimization” Energy Minimization for Structural Optimization • Protein structure “evolution” can be performed by systematical variation of the atom positions towards the lower energy directions. This procedure is called “structure optimization” or “energy minimization” Potential Energy Surface (PES) A force field defines for each molecule a unique PES. Each point on the PES represents a molecular conformation characterized by its structure and energy. Energy is a function of the coordinates. (Next) Coordinates are function of the energy. CH 3 energy CH3 CH 3 coordinates Goal of Energy Minimization A system of N atoms is defined by 3N Cartesian coordinates or 3N6 internal coordinates. These define a multi-dimensional potential energy surface (PES). • Minima (stable conformations) • Maxima • Saddle points (transition states) energy A PES is characterized by stationary points: Goal of Energy Minimization • Finding the stable conformations coordinates Classification of Stationary Points 1st Derivative 0 0 0 Type Minimum Maximum Saddle point 2nd Derivative* positive negative negative 20.0 16.0 energy transition state 12.0 8.0 local minimum 4.0 global minimum 0.0 0 90 180 270 360 coordinate * Refers to the eigenvalues of the second derivatives (Hessian) matrix Minimization Definitions Given a function: f f ( x1 , x2 , x3 x3 N ) Find values for the variables for which f is a minimum: f 0 xi 2 f 0 2 xi Functions • Quantum mechanics energy • Molecular mechanics energy Variables • Cartesian (molecular mechanics) • Internal (quantum mechanics) Minimization algorithms • Derivatives-based • Non derivatives-based A Schematic Representation Starting geometry Easy to implement; useful for well defined structures Depends strongly on starting geometry Population of Minima Active Structure Most populated minimum Global minimum Most minimization method can only go downhill and so locate the closest (downhill sense) minimum. No minimization method can guarantee the location of the global energy minimum. No method has proven the best for all problems. A General Minimization Scheme Starting point x0 Minimum? No Calculate xk+1 = f(xk) yes Stop Two Questions Where to go (direction)? How far to go (magnitude)? f(x,y) This is where we want to go How Far To Go? Until the Minimum Line search in one dimension • Find 3 points that bracket the minimum (e.g., by moving along the lines and recording function values). • Fit a quadratic function to the points. • Find the function’s minimum through differentiation. • Improved iteratively. Arbitrary Step Real function Cycle 1: 1, 2, 3 Cycle 2: 1, 2, 4 3 1 2 • xk+1 = xk + lksk, lk = step size. • Increase l as long as energy reduces. • Decrease l when energy increases. 4 5 Steepest Descent Where to go? • Parallel to the force (straight downhill): Sk = -gk How far to go? • Line search • Arbitrary Step Steepest Descent: Example f ( x, y) x 2 y 2 2x g 4y 2 Sk g k Starting point: (9, 9) 15 Cycle 1: Step direction: (-18, -36) Line search equation: y 2 x 9 Minimum: (4, -1) 10 Cycle 2: Step direction: (-8, 4) Line search equation: -5 5 0 -10 y 0.5 x 1 Minimum: (2/3, 2/3) -15 -15 -10 -5 0 5 10 15 Steepest Descent:Overshooting SD is forced to make 90º turns between subsequent steps (the scalar product between the (-18,-36) and the (-8,4) vector is 0 indicating orthogonality) and so is slow to converge. Why Ligand-Protein Docking? Molecular recognition is a central phenomenon in biology • Enzymes Substrates • Receptors Signal inducing ligands • Antibodies Antigens Classifying docking problems in biology • Protein-ligand docking – Rigid-body docking – Flexible docking • Protein-protein docking • Protein-DNA docking • DNA-ligand docking Ligand-Protein Docking • Proteins Drugs • Proteins Natural Small Molecule Substrates The Molecular Docking Problem Given two molecules with 3D conformations in atomic details: • Do the molecules bind to each other? If yes: • How does the molecule-molecule complex looks like? • How strong is the binding affinity? Structures of protein-ligand complexes • X-ray (PDB: 30,179 entries from X-ray crystallography, NMR and neutron diffraction) • NMR Importance of the protein 3D structures • Resolution < 2.5Å • Homology modeling problematic Basic Principles The association of molecules is based on interactions • H-bonds, salt bridges, hydrophobic contacts, electrostatic • Very strong repulsive (VdW) interactions on short distances. Association interactions are weak and short ranged. • Strong binding implies surface complementarity. Most molecules are flexible. Docking Concept Representation of a Cavity HIV-1 Protease Generation of Cavity Model X-ray structure of HIV protease Molecular surface model at active site Active site filled with spheres. Sphere centers become potential locations for ligand atoms. Ligand-protein docking concept Ligand-protein .docking concept Ligand-Protein Docking Concept Checking Chemical Complementarity in Ligand-Protein Docking Potential Energy Between Ligand and Protein: • A ligand with sufficiently low ligand-protein potential energy is considered as a drug candidate • Chemical database can be searched to find which chemical molecules can be docked to a disease protein with sufficiently low ligand-protein energy Summary Receptor-ligand binding Energy minimization for structural optimization Receptor-ligand docking concept