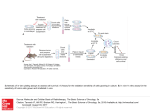
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Supplementary Online Information A.1. Absorption and bioavailability after dermal, inhalatory or oral exposure Bioavailability is defined as the fraction of compound in a certain matrix that reaches the systemic circulation. Three separate processes can be distinguished in bioavailability: (1) release of the compound from its matrix (bioaccessibility), (2) absorption of the released fraction, and (3) metabolism before reaching the systemic circulation (Versantvoort et al. 2005; Oomen et al. 2003). Existing approaches have been summarised in Table A1. Release of the compound from its matrix is required for transport across the dermal, lung or intestinal epithelium and bioavailability of a compound to the body. Therefore, the release depends mainly on the matrix, while absorption and metabolism depend more on the compound specific properties and physiology. In order to predict the bioavailability of a cosmetic ingredient from a cosmetic product, it is therefore important to determine the three different processes involved. However, the bioaccessibility could be used as a measure for the maximal bioavailability. A.1.1. Bioaccessibility models Dermal exposure. The bioaccessibility in vitro model representing the release on skin is a test based on human sweat with the pH of human skin. The preparation of artificial sweat is described in DIN EN ISO 105-E04 (2008). This model could be useful to determine the skin bioaccessibility of a cosmetic ingredient from a cosmetic product. Inhalatory exposure. Artificial lung fluid could be used as in vitro bioaccessibility model for inhalatory exposure. Beeston et al. (2010) described such a model to determine the lung absorption after inhalatory exposure to nanoparticles. In vitro lung bioaccessibility models have also been used to determine exposure after inhalation of soil, man-made fibres and various metals. Inhalatory bioaccessibility model will not be available for pre-validation within the coming few years. However, such a model could be useful to determine the lung bioaccessibility of an ingredient in cosmetics. Oral exposure. In vitro methodologies to study the human oral bioavailability of compounds from consumer products, food (including herbs and spices) and soil have been set-up. Most of the in vitro digestion models simulate in a simplified manner the digestion processes in the mouth, stomach and small intestine, in order to enable investigation the bioaccessibility during transit in the gastrointestinal tract. These models could be used to determine the bioaccessibility of a cosmetic ingredient from a cosmetic product (Daly et al. 2010; Peters et al. 2010; Amiard et al. 2008; Veda et al. 2007; Brandon et al. 2006; Versantvoort et al. 2005; Oomen et al. 2003 and many others). Bacterial metabolism in bioaccessibility. Bacteria (large intestine and skin) could be involved in the metabolism of a cosmetic ingredient. In vitro metabolism studies with skin or intestinal bacteria should be performed if bacterial biotransformation is expected (Platzek et al. 1999; van de Wiele et al. 2005). A.1.2. Absorption models 1 Dermal exposure. Different in silico QSAR models are available for the skin permeability prediction of compounds (Lee et al. 2010; Hansen et al. 2008; Ottaviani et al. 2007). In vitro models that might be useful to study the absorption of cosmetic ingredients from a cosmetic product are artificial membrane PAMPA-skin, the stratum corneum and keratinocytes. Also the spontaneously immortalised human keratinocyte cell line HaCaT has frequently been used as in vitro model to study skin absorption. Furthermore, the cell line has been used in several dermatopharmaceutical studies (Doebis et al. 2002; Boderke et al. 2000; Altenburger and Kissel 1999). However, it is noted that some of these in vitro models have active metabolising enzymes, thus besides absorption, also metabolism may occur (Oesch et al. 2007; Swanson 2004; Baron and Merk 2001). Inhalatory exposure. The lung can be anatomically divided into several parts: trachea, bronchi, bronchioles and alveoli. Absorption of inhaled compounds can be throughout the entire lung. In the upper respiratory airways the absorption is low and is mostly happening in the lower part. With respect to disposition in the airways, CFD (Computational fluid dynamics) could be a useful approach (Kimbell and Subramaniam 2001). No QSAR models predicting lung absorption are known in public literature. In vitro models to study the translocation of compound in the lung are various types of pulmonary epithelial cell lines (e.g. Calu-3 cell line, A549 cell line) and primary lung cells of either human or animal origin (e.g. primary rat type II pneumocytes) (Geys 2009; Geys 2006). The integrity of the cell monolayer was verified by measuring the transepithelial electrical resistance (TEER) and passage of sodium fluorescein. Furthermore, a system of co-cultures of relevant cells (pneumocytes (e.g. A549), macrophages (e.g. THP-1), mast cells (e.g. HMC-1) and endothelial cells (e.g. EAHY926) can be used to study lung absorption (Alfaro-Moreno et al. 2008). These in vitro methods have been used to study the translocation of ultra-fine particles, but could be useful to study the absorption of a cosmetic ingredient after inhalatory exposure. However, the in vitro techniques have active metabolising enzymes to varying extent, thus besides absorption metabolism is investigated (Mansour et al. 2009; Foster et al. 1998). Oral exposure. In silico QSAR-like models can predict specific parameters for an unknown chemical based on structural and physicochemical similarities to various known chemicals. Sophisticated commercial in silico software is available that can predict absorption via the gastrointestinal tract. These in silico tools predict the overall oral absorption of a chemical based on available physicochemical parameters. In vitro experiments (artificial membranes and monolayers of specific cells) can predict the absorption over a single barrier. These in vitro studies are rather standard and could be incorporated in a medium-throughput test strategy. However, cell monolayers (e.g. Caco-2 cells) and artificial membranes lack (completely or to some degree) specific in vivo metabolic and active transport systems. Therefore, these membrane studies can only rank a chemical compared with known reference chemicals and can provide semi-quantitative data. As with in silico models, the validity of these in vitro model predictions for cosmetics remains to be established, because most, if not all, of these models were developed for pharmaceuticals. The ongoing Liintop project provides relevant experimental data on the use of hepatic intestinal in vitro methods to study absorption (http://www.liintop.cnr.it/index.php?PG=project&action=project). A.1.3. Bioavailability models Dermal exposure. OECD guideline 428 (2004)1 describes the in vitro method for measuring dermal absorption of animal and human skin. Three different types of skin preparation may be used: epidermal membranes, consisting of stratum corneum and epidermis, full thickness skin, consisting 1 The EPA guideline on Dermal exposure assessment:Principles and Applications (1992) provides a detailed procedure for in silico calculation of dermal exposure. 2 of stratum corneum, epidermis and dermis (total thickness ca. 1 mm or more) and split thickness skin, consisting of stratum corneum, epidermis and part of the dermis (total thickness ca. 200-400 μm). The last skin preparation is most representative of the in vivo distance between skin surface and dermal blood circulation. The skin preparation may or may not be metabolically active and thus may lead to the determination of absorption alone or the bioavailability of the compound after dermal application (Oesch et al. 2007). Two different types of exposure conditions may be used: finite and infinite dose conditions. Under finite dose conditions, skin load (mg of chemical/cm2) the concentration of dissolved chemicals will change with exposure time, while under infinite conditions they will not, or at least not significantly over the total exposure time. Absorption under finite dose conditions is often expressed as percentage of the applied dose. Its value is specific for the specific exposure time, concentration and skin load employed in the study. Due to this limitation, it is difficult as input parameter for PBPK-models. Absorption under infinite dose conditions is often expressed as a permeation constant, Kp, in cm/h. It is calculated by dividing the steady state flux of the chemical across the skin (mostly expressed in mg/cm2/h) by its concentration in the donor fluid. Unless the chemical investigated or the vehicle in which it has been dissolved alters skin permeability in a concentration dependent fashion, Kp is concentration independent. It can serve as an input parameter for PBPK-models. By multiplying it with the exposure concentration in mg/ml, steady state flux (in mg/cm2/h) can be calculated. The steady state flux across the skin does not scale with body weight, as many other input parameters do. Chemicals may show a significant lag time before reaching steady state flux. This lag time is usually also reported, and can be taken into account when modelling. To conclude, Kp can only be calculated for chemicals in solution or for liquid chemicals. When the chemical is applied as a solid, only (maximum) steady state flux under infinite dose conditions can be calculated. Inhalatory exposure. Currently there are in vitro methods described in public literature concerning lung tissue used to study the bioavailability of compounds after inhalatory exposure (Geys et al 2007). There is a need to investigate whether and how lung preparations of human or animal origin could be used to study the lung bioavailability of a cosmetic ingredient. Oral exposure. The Ussing chamber utilises small sections of intestinal mucosa that are clamped between two chambers containing buffer (Grass et al. 1988; Ussing et al. 1951). This technique enables a fast and reproducible study of intestinal absorption of molecules across the inserted tissue (Gotoh et al. 2005). An additional advantage of the Ussing chamber system is that next to transepithelial drug transport also gut metabolism can be studied (van de Kerkhof et al. 2006). Furthermore, in the everted sac method a segment of the intestine is inverted and placed over a glass rod (Wilson et al. 1954). The mucosal epithelium with villi is turned inside out, while the mucosal membrane forms the inner surface of the everted sac. This system provides information on the mechanism of absorption (Kato et al. 2004). With both in vitro models, the differences in absorption along the length of the gastrointestinal tract can be studied and they can provide quantitative absorption information (absorption over time; ka and Fabs). However, fresh tissue should be used (Bohets et al. 2001; Versantvoort et al. 2000). 3 Table A1. Replacement methods for Absorption and Bioavailability (A). Test name Endpoints measured QSAR dermal exposure Skin permeability Origin of test Area(s) of system (cell applications; line, ex vivo, known primary cells, limitations in silico, in vivo In silico Used mainly for pharmaceuticals Artificial skin (e.g. PAMPAskin) Absorption In vitro Developed for pharmaceuticals Stratum corneum Absorption Ex vivo Developed for pharmaceuticals Status Comments Suitability for specific subcategories of cosmetic ingredients should be evaluated Suitability for cosmetics should be evaluated Not used for cosmetics No validation >2013 necessary, methods have to follow the OECD principles for the validation of QSARs for regulatory purposes Might be useful for >2013 cosmetics as a screening tool Stratum corneum is mainly composed of dead cells and is the first skin barrier. Provides information on the maximal absorption Estimated time to enter prevalidation process <2013 4 Animal or human skin Absorption Ex vivo Developed for pharmaceuticals , the method is in use for cosmetics Frequently used for cosmetics. Primary keratinocytes Bioavailability (absorption + metabolism) In vitro Developed for pharmaceuticals Suitability for cosmetics should be evaluated Immortalised Bioavailability keratinocyte cell (absorption + line metabolism) In vitro Developed for pharmaceuticals Suitability for cosmetics should be evaluated Pig skin resembles <2013 more closely the human skin than that of laboratory animals. Pig skin from slaughter animals can be used. Also human skin from cosmetic surgery (e.g. facelift). An advantage of this model is that phototoxicity can be studied No barrier (stratum >2013 corneum) present, thus bioavailability could be overestimated Absorption and >2013 metabolism are studied, providing information on the bioavailability. No barrier (stratum corneum) present, thus bioavailability could be overestimated 5 Inhalatory bioaccessibility Maximal inhalatory absorption In vitro Currently not frequently used, used to determine the bioaccessibility of metals, soil and man-made fibres Developed for the absorption of ultra-fine particles Model under development Not developed for cosmetics and still under development, but might be useful for cosmetics Primary lung cells Bioavailability (absorption + metabolism) In vitro Pulmonary epithelial cell lines Bioavailability (absorption + metabolism) Co-culture of pulmonary cell lines Bioavailability (absorption + metabolism) >2013 Suitability for cosmetics should be evaluated In vitro Developed for the absorption of ultra-fine particles Suitability for cosmetics should be evaluated In vitro Developed for the absorption of ultra-fine particles Suitability for cosmetics should be evaluated These models have >2013 been shown to be useful to study the absorption of ultra-fine particles. Thus mainly informative of absorption in the alveoli. Animals are needed to obtain primary lung cells These models have >2013 been shown to be useful to study the absorption of ultra-fine particles. Thus mainly informative of absorption in the alveoli Resembles more >2013 closely the lung than lung cell lines and primary lung cells 6 Oral bioaccessibility Maximal oral absorption In vitro QSAR oral exposure Oral absorption prediction In silico Cell monolayers, including transporter transfected (e.g.Caco-2, MDCK) Absorption In vitro Frequently used to determine bioaccessibility of soil, clay and food components. Infrequently used for consumer products Absorption prediction is based on available physicochemica l parameters Absorption over a single layer Some validation for various soil types Not one single method, different models developed which vary. Useful for cosmetics to determine maximal bioavailability >2013 Developed for pharmaceuticals and software is commercially available Might be useful, but remains to be established >2013 Developed for pharmaceutical, transfected cell lines mainly used to identify transporters; problem is that for nonpharmaceutical, low absorption range is more crucial Standard in vitro test >2013 for the pharmaceutical industry. Provides also information on involved transporters (e.g. P-gp, OAT) 7 Artificial intestinal membranes Absorption In vitro Ussing chamber Bioavailability (absorption + metabolism) Ex vivo Everted sac Bioavailability (absorption + metabolism) Ex vivo Absorption. DSPAMPA combines a pH gradient and presence of chemical scavengers to the acceptor to mimic the presence of serum proteins in blood Absorption over parts of the intestine of animal or human origin. Developed for pharmaceuticals Developed for pharmaceuticals as early screening tool Used for early screening. Only information on passive diffusion. A draw-back of this method is the underestimation of the permeability of highly lipophilic drugs >2013 Developed for pharmaceutical, but also used for food components (e.g. vitamins) <2013 Absorption over parts of the intestine of animal or human origin. Developed for pharmaceuticals Developed for pharmaceutical, but also used for food components (e.g. vitamins) The bioavailability depends on intestinal section. Human intestine is available from surgery and pig intestine is available from slaughterhouses. Information on active transport The bioavailability depends on intestinal section. Human intestine is available from surgery and pig intestine is available from slaughterhouse. Information on active transport <2013 8 A.2. Distribution After absorption, the distribution of a compound and its metabolites inside the body is governed by three main factors: (1) the partition of the substance with plasma proteins and (2) between blood and specific tissues; and (2) the permeability of the substance to cross specialized membranes, so called barriers (e.g. blood-brain barrier/BBB, blood-placental barrier/BPB, blood-testis barrier/BTB). Existing approaches have been summarised in Table A2. Concerning QSAR methods, a general review of the application of in silico approaches for predicting ADME properties has been carried out by Cronin et al. (2009) and Madden (2010). Even though a considerable number of substances has been included in the development of these models, most of them considered pharmaceutical compounds – mainly “successful” drugs - and further investigation would be required for the application of these approaches to cosmetic ingredients. In addition, it would be also interesting to have data on drugs with poor pharmacokinetic profile to improve QSAR predictions (Madden, 2010). Furthermore, QSAR methods should comply with the OECD Guidelines for the validation of QSARs for regulatory purposes (OECD, 2004). A.2.1. Estimation of plasma protein binding (PPB) Only the free (unbound) fraction of a compound is available for diffusion and transport across cell membranes. Therefore it is essential to determine the binding of a compound to plasma or serum proteins. The easy availability of human plasma has made it possible to determine the unbound fraction of compounds by performing in vitro incubations directly in human plasma. In vitro approaches. There are three methods generally used for PPB determination: 1) equilibrium dialysis (ED) (Waters et al., 2008); 2) ultrafiltration (UF) (Zhang and Musson, 2006); and 3) ultracentifugation (Nakai et al., 2004). All methods can be automated for high-throughput, are easy to perform and have good precision and reproducibility. The use of combined LC/MS/MS allows high selectivity and sensitivity. Every method has its specific advantages and disadvantages. Actually, ED is regarded as the “gold standard” approach (Waters et al., 2008). However, Skor et al. (2005) argued that similar results may be obtained with UF when compared with ED, but in a higher throughput fashion. Commercial instruments are available and used routinely by pharmaceutical industries, there is no reason why they should not been used for cosmetic ingredients. This test should be requested under present cosmetics practices/regulations. In silico approaches. Two recent reviews on the in silico approaches for the estimation of PPB have been carried out by Wang and Hou (2009); and Mostrag-Szlichtyng and Worth (2010). A general correlation, based on the octanol-water partition coefficient was proposed by de Bruyn and Gobas (2007) after a compilation of literature data and using a broad variety of chemicals, i.e., pesticides, polar organics, polychlorinated biphenyls, dioxins, furans etc. This work points out that with a simple linear estimation, the errors would be in the range of ±2 log units. In addition, the authors found that for compounds with log Pow <2, PPB did not depend on logPow, but a constant average value could be assigned. A2.2. Estimation of blood-tissue partitioning The fate of a compound in the body is determined by partitioning into the human tissues. Therefore, the knowledge of this partitioning is of fundamental importance for the understanding of a compound's kinetic behaviour and toxic potential. The measurement of tissue storage and a molecular understanding of tissue affinity have, historically, not been studied to the same extent as plasma protein binding; however, the knowledge of these partitioning coefficients is essential for 9 the development of PBTK models. Fortunately, a quite large number of approaches have been developed over the last years. In vitro approaches. The available system is the vial-equilibration technique. A spiked sample of organ tissue-buffer homogenate is equilibrated and subsequently, the free (unbound) concentration of the test chemical is determined. The tissue-blood partition coefficient is calculated using results from pure buffer, tissue-buffer and blood-buffer incubations. Tissues can be mixed to obtain average values for e.g. richly perfused tissue groups. Olive oil or octanol are often used instead of adipose tissue. The free (unbound) concentration is typically assessed by one of the following techniques: equilibrium dialysis, ultracentrifugation, headspace analysis (for volatiles) or solid phase (micro-) extraction followed by a classical analysis such as HLPC UV or MS. The purpose of this technique is the prediction of the in vivo tissue blood partitioning and the prediction of an in vivo volume of distribution (Gargas et al., 1989; Artola-Garicano et al., 2000). In silico approaches. A critical review on QSAR estimation of blood-tissue partitioning can be found in Payne and Kenny (2002) who concluded that no single method was able to work in all cases, and that the correlation to apply depended on the species, the tissue and the chemicals lipophilicity. However, in a series of papers Ballard and co-workers (2000, 2003a,b) have carried out a comparison between in vitro and in vivo tissue distribution and have shown that the methodology was viable and comparable results were obtained. Recently, different methodologies have been developed, starting from QSAR, correlations with physicochemical properties, up to mechanistic approaches. Using a mixed approach, Poulin and Theil (2002, 2009) have developed a method to predict tissue partition coefficients based on in vitro data on drug lipophilicity and plasma protein binding as input parameters in a mechanism-based approach. Around 80% of the drugs tested (63 and 123) fall within a twofold error range. The main problem for the generalization of QSAR correlations has been the poor results obtained for charged molecules under physiological conditions and with charged phospholipids. However, in a new mechanistic approach proposed by Schmitt (2008) applied to 59 drug compounds, a deviation less than 3-fold was found for 73% of the 474 experimental partition coefficients. The brain was the organ responsible for most of the errors, but errors were in the conservative side, i.e. higher predicted dose than the measured one. Thus, from a risk assessment perspective, errors are on the safe side. A new surrogate method, biopartitioning micellar chromatography (BMC), that uses micellar mobile phases of Brij35, that structurally resembles the ordered array of the membranous hydrocarbon chains, has demonstrated to be useful in mimicking the drug partitioning process into biological systems (Martín-Biosca et al., 2009). This method combines experimental determination and in silico calculation and offers a new approach to obtain partition coefficients that does not require human or animal tissues. A.2.3. Estimation of substance permeability through Blood-Brain Barrier (BBB) The BBB is a regulatory interface that separates the Central Nervous System (CNS) from systemic blood circulation and may limit or impair the delivery of certain compounds, which makes the brain different from other tissues. There are several mechanisms of transport through the BBB (Mehdipour and Hamidi, 2009): passive diffusion, carrier-mediated transport, receptor-mediated transport, absorptive-mediated transport and active efflux transport. In vitro approaches. Prieto et al. (2004) have reviewed the in vitro models of the BBB and their application to toxicology, whereas the results of a study carried out by Garber et al. (2005) to test the in vitro-in vivo correlation of BBB permeability and several transport mechanisms, using primary bovine and human brain endothelial cells co-cultured with astrocytes, two different immortalized brain endothelial cells and cells not derived from the BBB (ECV/C6, MDCK and Caco-2 cell lines) were unsatisfactory. It was argued by Lai and Kuo (2005) that the absence of 10 pericytes – a CNS cell type - was the main reason for the lack of a clear correlation, but also the absence of close contact between endothelial cells and astrocytes (Cohen-Kashi et al., 2009) and the absence of blood flow that occurs in vivo (Santaguida et al., 2006) were also contributing. New in vitro BBB models have now being developed that integrate pericytes with endothelial cells and astrocytes (Nakagawa et al., 2009) and that allow close contact between endothelial and astrocytes cells (Cohen-Kashi et al., 2009). In both cases, preliminary results confirmed that this new BBB models showed higher barrier integrity and better correlation with in vivo permeability (Pe) and transendothelial electrical resistance (TEER) data. An in vitro BBB model for high throughput screening has been developed by Culot et al. (2008) and used integrated with neuronal cell lines (SH-SY5Y) in neurotoxic assessment of 16 compounds (Hallier-Vanuxeem et al., 2009). Furthermore, MDR-MDCK transfected cell lines have been proposed as one of the standard models to study BBB permeability (Wang et al., 2005, Garberg et al. 2005) In silico approaches. Two recent literature reviews on in silico models for BBB penetration based mainly on the prediction of log BB (log Cbrain/Cblood) has been carried out by Mehdipour and Hamidi (2009) and Mostrag-Szlichtyng and Worth (2010). The first generation of QSAR models tried to predict BBB permeability as a function of several parameters such as lipid solubility, hydrogen bonding, molecular weight and surface area, using generally linear regressions. In the second generation more advanced statistical techniques have been implemented (Mostrag-Szlichtyng and Worth, 2010) and more descriptors have been used: size type (topological indices, molecular weight, surface area, etc.) and polarity descriptors (partial charges, hydrogen bonding, etc.). They concluded that several models exist which are able to provide logBB predictions around 0.35-0.45 log units. However, the data sets used are relatively small and the majority of compounds are drugs. In addition, most of the methods are focused on the prediction of log BB which are an imperfect measure for BBB penetration (Martin, 2004). Furthermore, the vast majority of these approaches do not consider the type of transport mechanism taking place. Recently, some molecular models have been developed to consider the BBB transporters (Allen et al., 2006). However, there is still the need of the 3D structures of these membrane proteins from human cells. A.2.4. Estimation of substance permeability through Blood-Placenta Barrier (BPB) The BPB serves to transport nutrients and waste, and other compounds such as hormones. However, the placenta does not provide a true barrier protection to the fetus from exposure to compounds present in the mother’s systemic circulation, although it might reduce the transport of certain molecules. The transfer across the placenta can occur by four kinds of mechanisms (Myren et al, 2007): passive diffusion, carrier mediated or facilitated diffusion following a concentration gradient, carrier mediated active transport against a gradient and pinocytosis through the formation of a vesicle. In vitro approaches. Experimental methods to study human transplacental exposure to toxic compounds have been reviewed by Vähäkangas and Myllynen (2006). Primary trophoblasts are difficult to cultivate and do not form monolayers, whereas BeWo cells (an immortalized trophoblastic cell line) seem to be the most promising cell lines for transport studies. BeWo has been used to study the placental transport properties by Poulsen et al. (2009) obtaining the same ordering in terms of transfer but much lower rate of transfer when compared with ex vivo human perfused placental experiments. Ex vivo: human perfused placenta cotyledon. This method offers information about transplacental transfer, placental metabolism, storage, acute toxicity and the role of transporters. In addition, it provides an estimation of foetal exposure. The method has recently been reviewed by Pacifici (2006) who showed that all examined antibiotics cross the human placenta including those with a molecular weight greater than 1000 kDa. The ex vivo human perfused placenta cotyledon approach has a minimum number of ethical problems since placentas are normally discarded and incinerated 11 after birth. The only limitations are that the cells remain viable for up to 48 h and that the system may not reflect the placenta during the first months of foetus development (Myren et al., 2007). In vitro models are still under development to obtain data similar to those in ex vivo data. Placental perfusion method is already available. However, the development of a new tiered approach to determine when this test is necessary should be performed for cosmetic ingredients. In Silico approaches. A recent review of QSARs developed for this aspect has been carried out by Hewitt et al. (2007). They found that molecular size, hydrogen bonding and hydrophobicity were the most useful descriptors for passive diffusion. However, the other transport mechanisms and the fact that metabolism can occur in placenta were not addressed. Similar results were found by Giaginis et al. (2009) using 88 structurally diverse drugs. A.2.5. Estimation of substance permeability through Blood-Testis Barrier (BBB) In the testis, the BTB is a physical and physiological barrier which assures functions in hormonal regulation and spermatogenesis (Fawcett et al 1970). BTB is a testis-specific structure composed of side-by-side arranged tight junctions, the basal ectoplasmic specialization, the basal tubulobulbar complexes, the desmosome-like junctions and gap junctions (Siu and Cheng 2008; Xia et al 2005). In vitro approaches. Although there has been a lot of research activity on the exposure of testis to xenobiotics (perhaps due to widely known work on sperm quality), in vitro test systems are still far from being predictive. Actually, few models are presented as in vitro BTB model for toxics studies. Indeed primary Sertoli cells culture are used to study the effects of phorbol ester or cadmium on tightness junctions (Janecki et al 1991; 1992). Culture of Sertoli cells lines on impermeable support has also improved the knowledge about the effects of toxicant on junctional membrane proteins of seminiferous epithelium (Fiorini et al 2004, 2008). Another study was performed combining bicameral chamber and 3D culture with only Sertoli cells and presented another dynamic model of BTB for studying their integrity after bisphenol A exposure (Li et al 2009). To date, no in vitro reprotoxicity test on the male reproduction function allowed the demonstration of the toxic effects both on BTB and spermatogenesis. Only one type of testicular cells (Sertoli or Leydig cells) was tested limitating the interpretation of the observed toxic effects to in vivo extrapolation. Development of an in vitro engineering blood-testis barrier (eBTB) model appears to be the best effective alternative for major step of reprotoxicity test in male: toxicants BTB clearing and further adverse effect on germ cells. Many systems have been tested as organ culture, culture or co-culture of Sertoli cells and germ cells on impermeable support or permeable support as bicameral chamber with or without gel matrix (i.e collagen, matrigel, etc.) (for review see Staub 2008). However these models seem adequate for study dynamics in vitro but is not the exact reflect of in vivo BTB which is composed of a 3D structure with three compartments (interstitial, basal and apical) delimitated by many cell junctions, all contributing to barrier function and germ cells differentiation. A model with a 3D culture in bicameral chamber, the apical and basal compartment has been set up with three types of testicular cells isolated from 18-day-old rat testis (peritubular cells, Sertoli cells and germ cells) (Legendre et al. 2010). In silico approaches.Due to the scarcity of the data sets there are few QSARs for blood-testis transfer (Cronin and Hewitt, 2007) and these are far from being predictive. 12 Table A2. Replacement methods for Distribution (D). Test name Endpoints measured Distribution Bound/unbound fraction of compounds in plasma, specific tissues and organs and permeabilities to cross specialised membranes Estimation of Unbound plasma protein fraction binding blood Origin of test system (cell line, ex vivo, primary cells, in silico, in vivo In silico Human plasma in Area(s) of Status applications; known limitations Comments Estimated time to enter prevalidation process Data available mainly for “successful” drugs but of general applicability to cosmetic ingredients Estimation of partitioning of compounds between blood and plasma and specific tissues more developed that estimation of permeabilities No validation necessary, methods have to follow the OECD principles for the validation of QSARs for regulatory purposes Many tools currently available. Further improvement necessary with focus on data for cosmetics ingredients. Results depend on the validity domain defined for the method Mainly Routinely used pharmaceuticals by but applications pharmaceutical reported for industry. High other chemical throughput classes systems. Optimised; not formally validated, but extensive inhouse evaluation studies. Three methods Formal available with validation commercial necessary instruments: 1) Equilibrium dialysis; 2) Ultrafiltration; 3) ultracentrifugati on not 13 Protocols established, hundreds of compounds tested by a large number of laboratories Estimation of Partition Ex-vivo blood tissue coefficient partitioning between blood and tissues Permeability Blood barrier –brain In vitro, mixed cultures: pericytes with endothelial cells and astrocytes Method produces same results than invivo Research to obtain higher barrier integrity and better correlation with in vivo permeability (Pe) and transendothelial electrical resistance A considerable number of reported applications during PBTK model validation If ex-vivo <2013 animal tissues not accepted then >2013 since alternative approaches are still under development Still research is >2013 needed to develop a more realistic in vitro barrier 14 Permeability Blood – Ex-vivo human placental barrier perfused placenta cotyledon SOP well established and used by several laboratories. Minimum <2013 number of ethical problems since placentas are discarded after birth. Permeability Blood barrier Still research is needed to develop a more realistic in vitro barrier >2013 The only limitations are that the cells remain viable for up to 48 h and that the system may not reflect the placenta during the first months of foetus development –testis In vitro, 3D In vitro systems mixed do not reflect in cultures:peritub vivo BTB ular, Sertoli and germ cells 15 A.3. Metabolism (Biotransformation) Metabolism or biotransformation is the principal elimination route of organic chemicals; roughly 70 to 80 % of pharmaceuticals are partially or practically completely eliminated by metabolism (Zanger et al 2008). Due to a multitude of xenobiotic-metabolizing enzymes possibly acting on a chemical with different metabolic pathways, the first screen should preferably be as comprehensive as possible. Because liver is the principal site of xenobiotic metabolism, the enzyme component in in vitro systems should preferably be liver-derived (Coecke et al 2006; Pelkonen et al 2005; 2008) and of human origin, to avoid species differences (see e.g. Turpeinen et al 2008). There is a generally accepted consensus that metabolically competent human hepatocytes or hepatocyte-like cell lines are the best enzyme source to perform the first primary screening of metabolism (Gomez-Lechon et al 2003, 2008; Houston and Galetin 2008; Riley and Kenna 2004). The two most important end points measured are 1)intrinsic clearance which can be extrapolated into hepatic metabolic clearance and 2) the identification of metabolites (stable, inactive, active or reactive metabolites of concern). At the same time it is possible to have indication about the enzymes participating in the metabolism which allows a number of predictions about physiological and pathological factors affecting the kinetics of a compound of interest. Existing approaches have been summarised in Table A3. A.3.1. Biotransformation, In silico approaches Scientific relevance and purpose. The available systems are of three types: 1) expert systems based on structure-metabolism rules; 2) SAR and QSAR modeling; and 3) systems based on pharmacophore (ligand) or target protein modeling (e.g. CYP2C9 or CYP3A4 molecular models). There are a large number of commercial software available for predicting biotransformation, in various phases of development They provide useful information although none of them allows reliable predictions on the different aspects that should be covered, that is identification of possible metabolites, involved metabolising enzymes, and quantitative kinetics. A recent trend has been to use meta-models (i.e. a combination of models) for partial processes: discussions of various approaches can be found in recent reviews (Testa et al 2004; de Graaf et al 2005; Kulkarni et al 2005; Crivori & Poggesi 2006; Lewis & Ito 2008; Muster et al 2008; Mostrag-Szlichtyng and Worth 2010). Fields of application and limitations. Both knowledge-based algorithms and molecular modelling approaches are currently useful in giving a rapid but only qualitative overview of the potential metabolic fate of a compound. Useful predictions are usually restricted to the class of chemicals which has been used as a training set, but recently promising advances have been achieved (de Groot 2006; Hansch et al 2004; Lewis et al 2008). Standardization of different systems has mainly been in-house of biotechnological enterprises within the pharmaceutical sector. Extrapolation and prediction from in vitro to in vivo human biotransformation from limited in vitro human systems with the help of in silico approaches still represents a major challenge (Pelkonen et al 2009; EPAA 2010). It is worth of stressing that in silico models are usually based on existing human data, so their predictions are directly applicable to humans. Current status and future efforts needed. Although in silico approaches are developing rapidly, they are still inadequate for the production of results which are accepted by regulators and new approaches are needed to predict the major metabolic routes when there are a number of potential metabolic pathways 16 Reliable and good-quality databases (not limited to pharmaceuticals) are of the utmost importance for the development of reliable software for application to a wider assortment of chemicals including cosmetic ingredients and they are still in great need. A.3.2. Biotransformation: Metabolic clearance/stability Scientific relevance and purpose. The metabolic stability test is a relatively simple, fast-toperform, but specialised analytical equipment-based MS study, to find out whether a compound is metabolically stable or labile. It is based on the disappearance of the parent compound over time (with the appropriate analytical technique) when incubated with a metabolically competent tissue preparation (e.g. a human liver preparation, preferably a homogenate added with appropriate cofactors, or human hepatocytes), The rate of parent compound disappearance gives a measure of its metabolic stability and allows for the calculation of intrinsic clearance and extrapolation to hepatic (metabolic) clearance (see e.g. Grime and Riley 2006; Hallifax and Houston 2009; Pelkonen and Turpeinen 2007; Pelkonen et al 2005, 2008; Plant 2004). With the advent of modern MS techniques, the test is suitable also for studying both the detailed qualitative and quantitative metabolic profiles of a compound (Anderson et al 2009; Dalvie et al 2009; Pelkonen et al 2009; Tolonen et al 2009). The use of recombinant enzymes, transfected cells and metabolically competent human (liver-derived) cell lines or subcellular fractions are being very actively employed in pharmaceutical industry and academia. Fields of applications and known limitations. The use of liver based experimental systems should give a fairly reliable view of hepatic intrinsic clearance. However, to be able to predict in vivo clearance, a number of assumptions concerning the substance under study must be made, so an extrapolation model is needed (see e.g. Pelkonen and Turpeinen 2007; RostamiHodjegan and Tucker 2007; Webborn et al 2007). To cover also extrahepatic biotransformation the above described method for metabolic stability can be combined with the use of other tissues. For cosmetic substances, dermal uptake is the most prominent intake pathway and consequently methodologies for skin metabolism would be of considerable significance. Likewise, inhalation (spray applications) is also an important uptake route and metabolism should be taken into consideration in in vitro pulmonary tests. Some efforts would be needed to standardize metabolic stability in skin and pulmonary tissues. Current status and future efforts needed. Although standardization has mainly been made inhouse by pharmaceutical companies and academic groups, reports have been published (e.g. Houston and Galetin 2003; Soars et al 2009; Webborn et al 2007). No formal validation studies are known. However, the stability screening test should be relatively ready for validation after the availability of common procedures and related SOPs. ECVAM has supported a study in which 55 ECVAM/ICCVAM validation substances have been studied in in vitro human and rat liver preparations, to survey metabolic stability and metabolite formation (Pelkonen et al 2009) as well as analytical performance (Rousu et al 2010) As a part of the in vitro testing strategy, the test could replace animal biotransformation studies to a certain extent. It is worth noting that preparations from different species, including man, used under comparable conditions, can provide valuable information on species differences. 17 A.3.3. Biotransformation: Bioactivation assays Scientific relevance and purpose. The formation of reactive metabolites by biotransformation seems to be the cause of deleterious effects for a large number of compounds (Park et al 2006; Williams 2006). Even though mechanistic details of relationships between toxicities and reactive metabolites are still somewhat unclear, there is ample indirect, albeit circumstantial, evidence for their associations (Baillie 2006; 2008; Tang and Lu 2010). Nevertheless, it has been extensively demonstrated that reactive metabolites form covalent adducts with nucleophiles on cell proteins or DNA, or initiate free radical chain reactions within cells which then may lead to drug-induced toxicity, cell damage or other adverse reactions. There are direct and indirect methods to test the potential formation of reactive metabolites. Most direct assays use trapping agents (i.e. glutathione or its derivatives, semicarbazide, methoxylamine or potassium cyanide), that are able to trap both soft and hard electrophiles: conjugates are then analytically measured. Recently, a simple and efficient LC/TOFMS-based screening method for reactive drug metabolites was presented, utilizing several stable isotope labeled trapping agents (Rousu et al 2009). The Ames test is a prime example of an indirect method for the bioactivation assay making use of metabolically competent enzyme system (which could be human-derived, if needed) and properly engineered bacteria to detect reactive, DNA-bound metabolites. It has been in use since 1970s and extensively validated. Another indirect method to detect the role of reactive intermediates in toxicity is based on employing metabolically competent cells in culture (primary hepatocytes, genetically engineered cell lines, etc.) and non-metabolising cells (cell lines are often used). By comparing the concentration–toxicity curves (or IC50 data) of the compound in both models, it is possible to know whether a bioactivation of the xenobiotic is required to elicit the toxic effects. An additional possibility consists in the addition of inhibitors of metabolising enzymes to the hepatocyte culture medium: the reduction of toxicity indicate that it is associated to the bioactivation of test compound. Fields of application and limitations. Subcellular screening tests are simple, robust and can be employed in high throughput screening conditions. The most important limitation is that the most convincing evidence about an association between reactive metabolites and toxicity come from limited sets of compounds, which are mostly drugs and hepatotoxicants. Due to the species differences in metabolism, there is a great need for the application of these methods to human-based in vitro models that offer better predictions of potential hazard to humans. Current status and future efforts needed. A number of in vitro systems are available for studying the production of reactive metabolites or biotransformation-mediated effects. However, apart from the Ames test, no in vitro methods for bioactivation have been properly validated. A.3.4. Biotransformation: Induction assays Scientific relevance and purpose. Since induction has a complex underlying mechanism, it is a good indicator for high quality metabolic competent systems that can be used for long-term purposes (Coecke et al 1999; Pelkonen et al 2008): that is why developments are ongoing to assess CYP induction in bioreactor-based systems. Obviously, the most relevant intake routes 18 for cosmetics (dermal, inhalation) should be considered when in vitro test systems are developed. Fields of application and limitations. A large number of test systems ranging from nuclear receptor binding assays to induction-competent cell lines is currently available All these test systems are widely used in pharmaceutical industry to help early drug development and are designed to detect induction of CYP enzymes relevant for the pharmaceutical area. This can represent a potential limitation since for cosmetics, other CYP forms might play an additional or more prominent role (e.g. CYP 2E1, 1A1). Thus, further progress is needed to cover this potential gap. Current status and future efforts needed. For two human hepatic metabolic competent test systems ECVAM is carrying out a multi-study validation trial for cytochrome P450 induction providing a reliable human metabolic competent standard model using the human cryopreserved HepaRG® (CryoHepaRG®) cell line and human cryohepatocytes (Cryohep). Two detailed standard operating procedures (SOPs) have been established for the CYP induction test methods for both cells (Kanebratt and Andersson, 2008; Antherieu et al 2010; Richert et al 2010; Lubberstedt et al 2010; Stephenne et al 2010). However, again probe substrate are pharmaceuticals, and it would be useful to investigate its use with a wider assortment of chemicals of interest, e.g. typical cosmetic ingredients with known toxicokinetic properties. A.3.5. Biotransformation: Inhibition assays Scientific relevance and purpose. Due to the broad substrate specificity of metabolizing enzymes, there is always a possibility that compounds would interfere with each other’s biotransformation. Inhibition of biotransformation leads to higher concentrations and delayed clearance and may cause adverse effects. At the site of entry (i.e. gi-tract, skin, lung) inhibition of the first-pass metabolism would increase the blood concentration of the parent compound. Fields of application and limitations. There are currently available a large number of test systems ranging from recombinant expressed enzymes (principally CYP and UGT enzymes, but increasingly also other xenobiotic-metabolizing enzymes) to primary cells (hepatocytes) and permanent cell lines (Li 2008; Farkas et al 2008). All these test systems are widely used in pharmaceutical industry and can be judged to be validated at least for pharmaceuticals. This can represent a potential limitation since for cosmetics, other CYP forms might be relevant. In addition some cosmetics contain complex plant-derived mixtures and it is not elucidated to what extent current inhibition assays would be applicable. Thus, further progress should cover chemical and compositional peculiarities characteristic for cosmetics field. Current status and future efforts needed. There is an extensive literature available about the current situation (see e.g. Pelkonen et al 1998, 2008) and both FDA and EMA have published revised draft guidelines for testing interactions, including inhibition, of drugs under development. Because inhibition tests have been used principally for drugs and within pharmaceutical companies, it would be useful to apply them with a wider assortment of chemicals of interest, e.g. typical cosmetic ingredients with known toxicokinetic properties. 19 Table A3. Replacement methods for Metabolism (M). Test name Endpoints measured Biotransformation / in silico approaches Metabolite profile, principal metabolites, metabolizing enzymes Metabolic stability / metabolic clearance / metabolite formation Disappearance of parent compound and/or appearance of (major) metabolites Metabolite profile Origin of test system (cell line, ex vivo, primary cells, in silico, in vivo Expert systems QSAR Pharmacophore or molecular protein modelling Human (cryopreserved) hepatocytes, permanent hepatocytederived cell lines, Subcellular organelles Recombinant enzymes Area(s) of applications; known limitations Status Comments Estimated time to enter prevalidation process Developed mostly for pharmaceuticals (drug development tools) Quantitative predictions not reliable Mainly pharmaceuticals but applications reported for other chemical classes Used during early drug development for screening and “negative” selection Predictive capability heavily dependent on selected parameters and model compounds No validation necessary, methods have to follow the OECD principles for the validation of QSARs for regulatory purposes Optimised; not formally validated, but extensive inhouse evaluation studies, Protocols adequately established, hundreds of compounds tested; a large number of laboratories With suitable analytical techniques, covers also elucidation of primary metabolite profile ready prevalidation to 20 Metabolic activation Adducts with trapping agents Suicide inhibition Production of oxidative stress Metabolismdependent toxicity As above Mainly pharmaceuticals but applications reported for other chemical classes Some in-house evaluation; mostly in development phase; no consensus about pros and cons of different methods Correlations between activation and toxic outcomes not well established the Ames test is validated Other approaches >2013 Induction metabolism of CYP induction (function and/or expression of mRNA) Cultured human hepatocytes; permanent cell lines (e.g. HepaRG) Mainly pharmaceuticals; a number of other compounds studied; in principle any chemicals CryoHepaRG and cryohepatocytes are under validation at ECVAM Suitable also for the study of inhibitory interactions with probe substrates <2013 Inhibition metabolism of CYP inhibition (function) Human liver microsomes and other preparations; Recombinant enzymes; cultured human hepatocytes or permanent cell lines Mainly pharmaceuticals; a number of other compounds studied; in principle any chemicals Optimised; not formally validated, but extensive inhouse evaluation studies, Protocols adequately established, hundreds of compounds tested; a large number of laboratories High-troughput screening and cocktail methods established practically ready to prevalidation 21 A.4 Excretion Predicting major excretion pathways of compounds is important in relation to their kinetic behaviour and the relationship to pharmacological/toxicological effects. The kidneys and the hepatobiliary system have the capacity to excrete either as the parent compound or as metabolites and are important routes for elimination of xenobiotics and their metabolites. Excretory processes seem to be the least developed area in the context of in vitro toxicokinetic methods (see 5.6.). Consequently, some background on the current knowledge of excretory processes is given and thereafter only a few examples of approaches are described, which may point to definite advances for the field. Background on renal excretion Excretion by the kidney encompasses three different mechanisms: firstly, glomerular filtration of the unbound fraction, secondly, secretion by transport mechanisms at the tubular site and thirdly, tubular reabsorption. Whereas glomerular filtration and tubular secretion are mechanisms by which the xenobiotic substance is excreted from the body, tubular reabsorption is a process by which the substance is taken back from the intratubular fluid into the renal blood flow and hence, into the body. The amount excreted by the urine is resulting from all three processes. Tubular secretion of a variety of organic anionic drugs and polar metabolites is mediated by a family of multispecific organic anion transporters (OAT genes) that are part of the SLC22 family of solute carriers. Different OATs localize to the apical (OAT2, OAT4, and RST/URAT) or basolateral (OAT1/NKT and OAT3) membranes of the renal proximal tubule; the net transport of organic anions from blood to urine is believed to require both apical and basolateral OATs. The extent of functional redundancy among OATs remains uncertain, but closely related OAT genes are tightly linked in the genome. Hence, a better understanding of human variation in organic anionic xenobiotic excretion may be obtained by studying OAT genes in combination rather than individually (Xu et al. 2005) and co-expressing them in specific cell lines. There are also transporters for cationic substances. The process of reabsorption can be also mediated by transporters. For endogenous substances like sugar, Tmax and Vmax have been characterised by in vivo studies in humans. Lipophilic, not-charged substances are diffusing back from the primary urine during the transport through the tubular system of the kidney. This process is very efficient so that for many drugs excretion by urine is practically zero. Even if there are examples how the involved transporters can be identified, it is difficult to use the findings to feed into a physiological model of renal excretion which includes tubular secretion and tubular reabsorption. A general review on transporters is given by Klaassen and Lu (2008). Background on biliary excretion In humans, biliary excretion does not play an important role for most of the substances. To undergo biliary excretion, a compound present in the portal venous or hepatic portal blood must first pass through the sinusoidal membrane or be taken up by transporters and enter the hepatocytes. Inside the hepatocytes the compound can be metabolised by phase 1 and phase 2 biotransformation enzymes, bind non-specifically, efflux back into the blood across the sinusoidal membrane or to be transported across the canalicular membrane to be excreted into the bile. As a result biliary excretion represents at least one active process involving transporters embedded in the canicular membrane of hepatocytes. These transporters belong to the ATP binding cassette (ABC) superfamily of transporters. The ABC-transporter 22 intercepts compounds at the level of the plasma membrane and effluxes them before they are able to reach their intracellular target structures (Chiba et al., 2006). The major transporters involved in the biliary excretion of xenobiotics include ABCB11 (P-glycoprotein, MDR1, Pgp), ABCC2 (multi-drug resistance associated protein 2 (MRP2)), and ABCG2 (breast cancer resistance protein (CRP). ABCB4 (MDR3) and ABCB11 (bile salt export pump (BSEP)) are two other ABC transporters present in the canalicular membrane, although they play a minor role in biliary excretion. It is assumed that the molecular weight also plays a role (> 600 ) and that the transporters specifically transport charged molecules. The recent review of Klaassen and Aleksunes (2010) provides a general survey of hepatic transporters. Examples of current approaches and future efforts needed. There have been a few attempts for developing expert systems or computational approaches to predict renal excretion from some basic molecular and physicochemical properties. Based on the intravenous dose for 160 drugs, Manga et al. (2003) developed a decision tree model to predict unchanged drug excreted in the urine as percentage of the i.v. dose. The data were categorized into classes and the classes were assigned correctly in 90% of the cases using a test set of 40 compounds. The dependence of the of properties such as the distribution coefficient at pH 6.5, the H-bond donors, counts of OH and COOH groups, etc. shows that ionization has a prominent role in the modeling of this process. Kusama and co-workers (2010) demonstrated on 141 approved drugs that only 4 physicochemical parameters (charge, molecular weight, lipophilicity, and protein unbound fraction in plasma) are required to predict major excretion pathways. For those compounds major clearance pathways were determined to be metabolism by CYP3A4, CYP2C9 and CYP2D6, or renal excretion in unchanged form. Efforts have been undertaken to use collagen-sandwich cultures of hepatocytes as an in vitro test system. Using this system bilirubin conjugation and canalicular versus sinusoidal disposition of bilirubin glucuronides has been studied. Biliary excretion index was estimated by measuring disposition of bilirubin glucuronides into standard and Ca(2+), Mg(2+)-free medium. From the results the authors concluded that sandwich-cultured primary hepatocytes might provide a useful in vitro method to differentiate between sinusoidal and canalicular disposition of bilirubin glucuronides (Lengyel et al., 2005). Based on published data in silico modeling attempts are being made to evaluate the molecular weight dependence of biliary excretion and to develop quantitative structurepharmacokineticc relationships to predict biliary excretion (Yang et al., 2009). Due to the fact that progress in the field is very recent, no real global efforts have been systematically undertaken to standardize the above mentioned approaches. Standardization has mainly been in-house. Some pharmaceutical companies as well as academic groups have published reports on their experiences. No formal validation studies are known. An understanding of mechanisms that determine these processes is required for the prediction of renal and biliary excretion. Physiologically based in-vitro/in-silico/in-vivo methods could potentially be useful for predicting renal and biliary clearance. Whereas for biliary excretion some advances have been made with in vitro models (i.e. sandwich-cultured hepatocytes), no reports could be indentified in the literature on in vitro models of renal excretion nor were reports available on in silico methods. 23 References. Alfaro-Moreno E, Nawrot TS, Vanaudenaerde BM, Hoylaerts MF, Vanoirbeek JA, Nemery B, Hoet PHM (2008) Co-cultures of multiple cell types mimic pulmonary cell communication in response to urban PM10. The European Respiratory Journal 32: 1184–1194. Allen DD, Geldenhuys WJ (2006) Molecular Modelling of blood-barrier nutrient transporters: in silico basis for evaluation of potential drug delivery to the central nervous system. Life Sciences 78: 1029-1033. Altenburger R, Kissel T (1999) The human keratinocyte cell line HaCaT: an in vitro cell culture model for keratinocyte testosterone metabolism. Pharmaceutical Research 16:766-771. Amiard JC, Amiard-Triquet C, Charbonnier L, Mesnil A, Rainbow PS, Wang WX (2008) Bioaccessibility of essential and non-essential metals in commercial shellfish from Western Europe and Asia. Food and Chemical Toxicology 46:2010-2022. Anderson S, Luffer-Atlas D, Knadler MP (2009) Predicting Circulating Human Metabolites: How Good Are We? Chemical Research in Toxicology 22(2):23-56. Anthérieu S, Chesné C, Li R, Camus S, Lahoz A, Picazo L, Turpeinen M, Tolonen A, Uusitalo J, Guguen-Guillouzo C, Guillouzo A (2010) Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG® cells Drug Metabolism Disposition 38:516-525. Artola-Garicano E, Vaes, WH, Hermens JL (2000) Validation of negligible depletion solidphase microextraction as a tool to determine tissue/blood partition coefficients for semivolatile and nonvolatile organic chemicals. Toxicology and Applied Pharmacology 166:138-144. Baillie TA (2006) Future of toxicology-metabolic activation and drug design: challenges and opportunities in chemical toxicology. Chemical Research in Toxicology 19: 889-893. Baillie TA (2008) Metabolism and toxicity of drugs. Two decades of progress in industrial drug metabolism Chemical Research in Toxicology 2008; 21: 129-137. Ballard P, Leahy DE, Rowland M (2000) Prediction of In Vivo Tissue Distribution from In Vitro Data 1. Experiments with Markers of Aqueous Spaces. Pharmaceutical Research 17:660-663. Ballard P, Arundel PA, Leahy DE, Rowland M (2003) Prediction of In Vivo Tissue Distribution from In Vitro Data 2. Influence of Albumin Diffusion from Tissue Pieces During an in Vitro Incubation of Estimated Tissue-to-Unbound Plasma Partition Coefficients (Kpu). Pharmaceutical Research 20:857-863 Ballard P, Leahy DE, Rowland M (2003) Prediction of In Vivo Tissue Distribution from In Vitro Data 2. Correlation Between in vitro and in vivo tissue distribution of a homologous series of nine 5-n-Alkyl-5-Ethyl Barbituric Acids. Pharmaceutical Research 20:864-872 24 Baron JM, Merk HF (2001) Drug metabolism in the skin. Current Opinion in Allergy and Clinical Immunology 1:287-291. Beeston MP, van Elteren JT, Selih VS, Fairhurst R (2010) Characterization of artificially generated PbS aerosols and their use within a respiratory bioaccessibility test. Analyst 135:351-357. Boderke P, Schittkowski K, Wolf M, Merkle HP (2000) Modeling of diffusion and concurrent metabolism in cutaneous tissue. Journal of Theoretical Biology 204:393-407. Bohets H, Annaert P, Mannens G (2001) Strategies for absorption screening in drug discovery and development. Current Topics in Medicinal Chemistry 1:367-383. Brandon EF, Oomen AG, Rompelberg CJ, Versantvoort CH, van Engelen JG, Sips AJ (2006) Consumer product in vitro digestion model: Bioaccessibility of contaminants and its application in risk assessment. Regulatory Toxicology and Pharmacology 44:161-171. Chiba P, Mihalek I, Ecker GF, Kopp S, Lichtarge O (2006) Role of transmembrane domain/transmembrane domain interfaces of P-glycoprotein (ABCB1) in solute transport. Convergent information from photoaffinity labeling, site directed mutagenesis and in silico importance prediction. Curr Med Chem. 13, 793-805. Coecke S, Rogiers V, Bayliss M, Castell J, Doehmer J, Fabre G, Fry J, Kern A, Westmoreland C (1999a) The use of long-term hepatocyte cultures for detection induction of drug metabolising enzymes: the current status. ATLA 27:579-638. Coecke S, Ahr H, Blaauboer BJ Bremer S, Casati S, Castell J, Combes R, Corvi R, Crespi CL, Cunningham ML, Elaut G, Eletti B, Freidig A, Gennari A, Ghersi-Egea JF, Guillouzo A, Hartung T, Hoet P, Ingelman-Sundberg M, Munn S, Janssens W, Ladstetter B, Leahy D, Long A, Meneguz A, Monshouwer M, Morath S, Nagelkerke F, Pelkonen O, Ponti J, Prieto P, Richert L, Sabbioni E, Schaack B, Steiling W, Testai E, Vericat JA, Worth A (2006)Metabolism: a bottleneck in in vitro toxicological test development. The report and recommendations of ECVAM workshop 54. ATLA 34(1):49-84. Cohen-Kashi Malina K, Cooper I, Teichberg VI (2009) Closing the gap between in-vivo and in-vitro blood-brain barrier tighness. Brain Research 1284:12-21. Crivori P, Poggesi I (2006) Computational approaches for predicting CYP-related metabolism properties in the screening of new drugs. European Journal of Medicinal Chemistry 41(7):795-808. Cronin MT, Bajot F, Enoch SJ, Madden JC, Roberts DW, Schwöbel J (2009) The in chemicoin silico interface: challenges for integrating experimental and computational chemistry to identify toxicity. Alternatives to Laboratory Animals 37:513-521. Cronin MTD, Hewitt M (2007) In silico models to predict passage through the skin and other barriers. In B Testa, H van de Waterbeemd (eds) Comprehensive Medicinal Chemistry II, Volume 5, ADME-Tox Approaches. Elsevier, Oxford, pp 725-744. 25 Culot M, Lundquist S, Vanuxeem D, Nion S, Landry C, Delplace Y, Dehouck M-P, Berezowski V, Fenart L, Cecchelli R (2008) An in vitro blood-brain barrier model for high throughput (HTS) toxicological screening. Toxicology in Vitro 22:799-811. Dalvie D, Obach RS, Kang P, Prakash C, Loi CM, Hurst S, Nedderman A, Goulet L, Smith E, Bu HZ, Smith DA (2009) Assessment of Three Human in Vitro Systems in the Generation of Major Human Excretory and Circulating Metabolites. Chemical Research in Toxicology 22(2):357-68. Daly T, Jiwan MA, O'Brien NM, Aherne SA (2010) Carotenoid content of commonly consumed herbs and assessment of their bioaccessibility using an in vitro digestion model. Plant Foods for Human Nutrition 65:164-169. De Bruyn AMH, Gobas FAPC (2007) The sorptive capacity of animal protein. Environmental Toxicology and Chemistry 2007; 26, 1803-1808. de Graaf C, Vermeulen NP, Feenstra KA (2005) Cytochrome P450 in silico: an integrative modeling approach. Journal of Medicinal Chemistry 48(8):2725-2755. de Groot MJ (2006) Designing better drugs: predicting cytochrome P450 metabolism. Drug Discovery Today 11(13-14):601-606. DIN EN ISO 105-E04:2008 (2009). Textiles - Tests for colour fastness - Part E04: Colour fastness to perspiration. Doebis C, Ritter T, Brandt C, Schönberger B, Volk HD, Seifert M (2002) Efficient in vitro transduction of epithelial cells and keratinocytes with improved adenoviral gene transfer for the application in skin tissue engineering. Transplant Immunology 9:323-329. FDA Guidance (2006): Drug Interaction Studies – Study Design, Data Analysis, and Implications for Dosing and Labeling. Farkas D, Shader RI, von Moltke LL, Greenblatt DJ (2008) Mechanisms and consequences of drug-drug interactions. In: Preclinical Development Handbook: ADME and Biopharmaceutical Properties (ed. Shayne Cox Gad), John Wiley & Sons, chapter 25, pp. 879-918. Fawcett DW, Leak, LV, Heidger, PM Jr (1970) Electron microscopic observations on the structural components of the blood-testis barrier. Journal of Reproduction and Fertility Supplement 10:105-22. Fiorini C, Gilleron J, Carette D, Valette A, Tilloy A, Chevalier S, Segretain D, Pointis G (2008) Accelerated internalization of junctional membrane proteins (connexin 43, N-cadherin and ZO-1) within endocytic vacuoles: an early event of DDT carcinogenicity. Biochimica et Biophysica Acta 1778(1):56-67. Fiorini C, Tilloy-Ellul A, Chevalier S, Charuel C, Pointis G. (2004) Sertoli cell junctional proteins as early targets for different classes of reproductive toxicants. Reproductive Toxicology 18(3):413-21. 26 Foster KA, Oster CG, Mayer MM, Avery ML, Audus KL (1998) Characterization of the A549 cell line as a type ii pulmonary epithelial cell model for drug metabolism. Experimental Cell Research 243:359–366. Garberg P, Ball M, Borg N, Cecchelli R, Fenart L, Hurst RD, Lindmark T, Mabondzo A, Nilsson JE, Raub TJ, Stanimirovic D, Terasaki T, Öberg J-O, Österberg T (2005) In vitro models for the blood-brain barrier. Toxicology in vitro 19:299-334. Gargas ML, Burgess RJ, Voisard DE, Cason GH, Andersen ME (1989) Partition coefficients of low-molecular-weight volatile chemicals in various liquids and tissues. Toxicology and Applied Pharmacology 98:87-99. Geys J, de Vos R, Nemery B, Hoet PHM (2009) In vitro translocation of quantum dots and influence of oxidative stress. American Journal of Physiology Lung Cellular and Molecular Physiology 297:L903–L911. Geys J, Coenegrachts L, Vercammen J, Engelborghs Y, Nemmar A, Nemery B, Hoet PHM (2006) In vitro study of the pulmonary translocation of nanoparticles. A preliminary study Toxicology Letters 160:218–226 Geys J, Nemery B, Hoet PH (2007) Optimisation of culture conditions to develop an in vitro pulmonary permeability model. Toxicol In Vitro 21:1215-1219. Giaginis C, Zira A, Tsantili-Kakoulidou A, Theocheris S (2009) Application of quantitative structure-activity relationships (QSARs) for modelling drug and chemical transport across the human placenta barrier: A multivariate data analysis approach. Toxicology Letters 189S, S258. Gómez-Lechón MJ, Castell JV, Donato MT (2008) An update on metabolism studies using human hepatocytes in primary culture. Expert Opinion on Drug Metabolism and Toxicololy 4(7):837-54. Gómez-Lechón MJ, Donato MT, Castell JV, Jover R (2003) Human hepatocytes as a tool for studying toxicity and drug metabolism. Current Drug Metabolism 4(4):292-312. Gotoh Y, Kamada N, Momose D (2005) The advantages of the Ussing chamber in drug absorption studies. Journal of Biomolecular Screening 10:517-523. Grass GM, Sweetana SA (1998) In vitro measurement of gastrointestinal tissue permeability using a new diffusion cell. Pharmaceutical Research 5:372-376. Grime K, Riley RJ (2006) The impact of in vitro binding on in vitro-in vivo extrapolations, projections of metabolic clearance and clinical drug-drug interactions. Current Drug Metabolism 7(3):251-64. Hansch C, Mekapati SB, Kurup A, Verma RP (2004) QSAR of cytochrome P450. Drug Metabolism Reviews 36(1):105-156. 27 Hallier-Vanuxeem D, Prieto P, Culot M, Diallo H, Landry C, Tähti H, Cecchelli R (2009) New strategy for alerting central nervous system toxicity: Integration of blood-barrier toxicity and permeability in neurotoxicity assessment. Toxicology in Vitro 23:447-453. Hallifax D, Houston JB (2009) Methodological uncertainty in quantitative prediction of human hepatic clearance from in vitro experimental systems. Current Drug Metabolism 10(3):307-321. Hansen S, Henning A, Naegel A, Heisig M, Wittum G, Neumann D, Kostka KH, Zbytovska J, Lehr CM, Schaefer UF (2008) In-silico model of skin penetration based on experimentally determined input parameters. Part I: experimental determination of partition and diffusion coefficients. European Journal of Pharmaceuticals and Biopharmaceuticals 68:352-367. Hewitt M, Madden JC, Rowe PH, Cronin MTD (2007) Structure-based modelling in reproductive toxicology:(Q)SARs for the placental barrier. SAR and QSAR in Environmental Research 18:57-76. Houston JB, Galetin A (2003) Progress towards prediction of human pharmacokinetic parameters from in vitro technologies. Drug Metabolism Reviews 35(4):393-415. Houston JB, Galetin A (2008) Methods for predicting in vivo pharmacokinetics using data from in vitro assays. Current Drug Metabolism 9(9):940-51. Janecki A, Jakubowiak A, Steinberger A (1992) Effect of cadmium chloride on transepithelial electrical resistance of sertoli cell monolayers in two-compartment cultures. A new model for toxicological investigations of the blood-testis barrier in vitro. Toxicology and Applied Pharmacology 112:51-7. Janecki A, Jakubowiak A, Steinberger A (1991) Effects of cyclic AMP and phorbol ester on transepithelial electrical resistance of Sertoli cell monolayers in two-compartment culture. Molecular and Cellular Endocrinology 82(1):61-9. Kanebratt KP, Andersson TB (2008) HepaRG® cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metabolism Disposition 36:137–145. Kato T, Hayashi Y, Inoue K, Yuasa H (2004) Functional characterization of the carrier-mediated transport system for glycerol in everted sacs of the rat small intestine. Biological and Pharmaceutical Bulletin 27:1826-30. Kimbell JS, Subramaniam RP. (2001) Use of computational fluid dynamics models for dosimetry of inhaled gases in the nasal passages. Inhalat Toxicol 13; 325-334. Klaassen CD, Lu H (2008) Xenobiotic transporters: ascribing function from gene knockout and mutation studies. Toxicological Sciences 101(2):186-196. Klaassen CD, Aleksunes LM (2010) Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacological Reviews 62(1):1-96. Kulkarni SA, Zhu J, Blechinger S (2005) In silico techniques for the study and prediction of xenobiotic metabolism: a review. Xenobiotica 35(10-11):955-973. 28 Kusama M, Toshimoto K, Maeda K, Hirai Y, Imai S, Chiba K, Akiyama Y, Sugiyama Y (2010) In Silico Classification of Major Clearance Pathways of Drugs with their Physiochemical Parameters. Drug Metabolism Disposistion [Epub ahead of print] Lai C-H, Kuo K-H (2005) The critical component to establish in vitro BBB model: Pericyte. Brain Research Reviews 50:258-265. Lee PH, Conradi R, Shanmugasundaram V (2010) Development of an in silico model for human skin permeation based on a Franz cell skin permeability assay. Bioorganical and Medicine Chemical Letters 20:69-73. Legendre A, Froment P, Desmots S, Lecomte A, Habert R, Lemazurier E (2010) An engineered 3D blood-testis barrier model for the assessment of reproductive toxicity potential, Biomaterials 31:4492-4505. Lengyel G, Veres Z, Szabó P, Vereczkey L, Jemnitz K (2005) Canalicular and sinusoidal disposition of bilirubin mono- and diglucuronides in sandwich-cultured human and rat primary hepatocytes. Drug Metabolism Disposition 33(9):1355-1360. Lewis DF, Ito Y (2008) Human cytochromes P450 in the metabolism of drugs: new molecular models of enzyme-substrate interactions. Experts Opinion Drug Metabolism and Toxicology 4(9):1181-1186. Lewis DF, Ito Y, Goldfarb PS (2006) Structural modelling of the human drug-metabolizing cytochromes P450. Current Medical Chemistry 13(22):2645-2652. Li AP (2008). In vitro evaluation of metabolic drug-drug interactions: scientific concepts and practical considerations. In: Preclinical Development Handbook: ADME and Biopharmaceutical Properties (ed. Shayne Cox Gad), John Wiley & Sons, chapter 24, pp. 853-878. Li MW, Mruk DD, Lee WM, Cheng CY (2009) Disruption of the blood-testis barrier integrity by bisphenol A in vitro: Is this a suitable model for studying blood-testis barrier dynamics? The international journal of biochemistry & cell biology 41(11):2302-14. Lübberstedt M, Müller-Vieira U, Mayer M, Biemel KM, Knöspel F, Knobeloch D, Nüssler AK, Gerlach JC, Zeilinger K (2010) HepaRG Human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. Journal of Pharmacology and Toxicolology Methods [Epub ahead of print]. Madden JC (2010) In silico approaches for predicting ADME properties. In T Puzyn, J Leszczynski, MTD Cronin (eds) Recent Advances in QSAR Studies – Methods and Applications. Springer, Dordrecht, pp 283-304. Manga N, Duffy JC, Rowe PH, Cronin MTD (2003) A hierarchical QSAR Model for Urinary Ecretion o Drugs in Humans as a Predictive Tool for Biotransformation. QSAR and Combinatorial Science 22, 263-273. 29 Mansour HM, Rhee YS, Wu X (2009) Nanomedicine in pulmonary delivery. International Journal of Nanomedicine 4:299-319. Martin I (2004) Prediction of blood-brain barrier penetration: are we missing the point? Drug Discovery Today 9:161-162. Martín-BioscaY, Torres-Cartas S, Villanueva-Camañas, Sagrado S, Medina-Hernández MJ (2009) Biopartitioning micellar chromatography to predict blood to lung, blood to liver, blood to fat and blood to skin partitioning coefficients of drugs. Analytica Chimica Acta 632:296303. Mehdipour AR , Hamidi M (2009) Brain drug targeting: a computational approach for overcoming blood-brain barrier. Drug Discovery Today 14:1030-1036. Mostrag-Szlichtyng A, Worth A (2010) Review of QSAR Models and Software Tools for predicting Biokinetic Properties. EUR 24377 EN. Luxembourg, European Commission. Myren M, Mose T, Mathisen L, Knudsen LE (2007) The human placenta- An alternative to foetal exposure. Toxicology in Vitro 21:1332-1340. Muster W, Breidenbach A, Fischer H, Kirchner S, Muller L, Pahler A (2008) Computational toxicology in drug development. Drug Discovery Today 13(7-8):303-310. Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M (2009) A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochemistry International 54:253-263. Nakai D, Kumamoto K, Sakikawa Ch, Kosaka T, Tokui T (2004) Evaluation of protein binding ratio of drugs by a micro-scale uktracentrifugation method. Journal of Pharmaceutical Sciences 93:847-851. OECD guideline 428 (2004). Skin Absorption: in vitro Method. Available via http://www.oecdilibrary.org/docserver/download/fulltext/9742801e.pdf?expires=1275558971 &id=0000&accname=Dutch+Government&checksum=4396D617A72B24A90D1DE616728 D8258. OECD (2004). OECD Series on Testing and Assessment Number 49. Report from the expert group on (Quantitative) Structure-activity relationships [(Q)SARs] on the principles for the validation of (Q)SARs. Oesch F, Fabian E, Oesch-Bartlomowicz B, Werner C, Landsiedel R (2007) Drug-metabolizing enzymes in the skin of man, rat, and pig. Drug Metabolism Reviews 39:659-698. Oomen AG, Rompelberg CJ, Bruil MA, Dobbe CJ, Pereboom DP, Sips AJ (2003) Development of an in vitro digestion model for estimating the bioaccessibility of soil contaminants. Archives of Environmental Contamination and Toxicology44:281-287. 30 Ottaviani G, Martel S, Carrupt PA (2007) In silico and in vitro filters for the fast estimation of skin permeation and distribution of new chemical entities. Journal of Medicinal Chemistry 50:742-748. Pacifici GM (2006) Placental transfer of antibiotics administered to the mother: a review. Int. Journal of Clinical Pharmacology and Therapeutics 44:57-63. Park KB, Dalton-Brown E, Hirst C, Williams DP (2006) Selection of new chemical entities with decreased potential for adverse drug reactions. European Journal of Pharmacology 2006; 549: 1-8. Payne M.P. and Kenny, L.C (2002) Comparison of models for the estimation of biological partition coefficients. Journal of Toxicology and Environmental Health Part A 65:897-931. Pelkonen O, Mäenpää J, Taavitsainen P, Rautio A, Raunio H (1998) Inhibition and induction of human cytochrome P450 enzymes. Xenobiotica 28:1203-1253. Pelkonen O, Tolonen A, Turpeinen M, Uusitalo J (2008). In vitro metabolism in preclinical drug development. In: Preclinical Development Handbook: ADME and Biopharmaceutical Properties (ed. Shayne Cox Gad), John Wiley & Sons chapter 21, pp. 743-774. Pelkonen O, Tolonen A, Korjamo T, Turpeinen M, Raunio H (2009) From known knowns to known unknowns: predicting in vivo drug metabolites. Bioanalysis 1:393-414. Pelkonen O, Tolonen A, Rousu T, Tursas L, Turpeinen M, Hokkanen J, Uusitalo J, Bouvier d'Yvoire M, Coecke S (2009) Comparison of metabolic stability and metabolite identification of 55 ECVAM/ICCVAM validation compounds between human and rat liver homogenates and microsomes - a preliminary analysis. ALTEX, Alternativen zu Tierexperimenten 26:214222. Pelkonen O, Turpeinen M (2007) In vitro–in vivo extrapolation of hepatic clearance: Biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 37:1066–1089. Pelkonen O, Turpeinen M, Uusitalo J, Rautio A, Raunio H (2005) Prediction of drug metabolism and interactions on the basis of in vitro investigations. Basic Clin.Pharmacol.Toxicol 96(3):167-175. Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H (2008) Inhibition and induction of human cytochrome P450 enzymes – current status. Archives of Toxicology 82:667-715. Tolonen A, Turpeinen M, Pelkonen O (2009) Liquid chromatography-mass spectrometry in in vitro drug metabolite screening. Drug Discovery Today (DDT) 14:120-133. Peters CM, Green RJ, Janle EM, Ferruzzi MG (2010) Formulation with ascorbic acid and sucrose modulates catechin bioavailability from green tea. Food Research International 43:95102. 31 Plant N. Strategies for using in vitro screens in drug metabolism (2004) Drug Discov Today 9:328-336. Platzek T, Lang C, Grohmann G, Gi US, Baltes W (1999) Formation of a carcinogenic aromatic amine from an azo dye by human skin bacteria in vitro. Human and Experimental Toxicology 18:552-559. Poulsen MS, Rytting E, Mose T, Knudsen LE (2009) Modeling placental transport: Correlation of in vitro BeWo cell permeability and ex vivo human placental perfusion. Toxicology in Vitro 23:1380-1386. Poulin P, Theil FP (2002) Prediction of Pharmacokinetics Prior to In Vivo Studies. 1. Mechanism-Based Prediction of Volume of Distribution. Journal of Pharmaceutical Sciences 91:129-156. Poulin P, Theil FP (2009) Development of a Novel Method for Predicting Human Volume Distribution at Steady-State of Basic Drugs and Comparative Assessment with Existing Methods. Journal of Pharmaceutical Sciences 98:4941-4961. Prieto P, Blaauboer BJ, de Boer AG, Boveri M, Cecchelli R, Clemedson C, Coecke S, Forsby A, Galla HJ, Garberg P, Greenwood J, Price A, Tähti H (2004) Blood–Brain Barrier In Vitro Models and Their Application in Toxicology. ATLA 32:37-50. Riley RJ, Kenna JG (2004) Cellular models for ADMET predictions and evaluation of drugdrug interactions. Current Opinion on Drug Discovery and Development 7(1):86-99. Richert L, Abadie C, Bonet A, Heyd B, Mantion G, Alexandre E, Bachellier P, Jaeck D, Kingston S, Pattenden C, Illouz S, Dennison A, Hoffmann S, Coecke S (2010) Interlaboratory evaluation of the response of primary human hepatocyte cultures to model CYP inducers – A European Centre for Validation of Alternative Methods (ECVAM) - funded validation study. Toxicology In Vitro ; 24:335-345. Rostami-Hodjegan A, Tucker GT (2007) Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nature Reviews Drug Discovery 6:140148. Rousu T, Hokkanen J, Pelkonen O, Tolonen A (2010) Applicability of generic assays based on liquid chromatography-electrospray mass spectrometry to study in vitro metabolism of 55 structurally diverse compounds. Frontiers in Pharmacology – Drug Metabolism and Transporters, doi: 10.3389/fphar.2010.00010 Rousu T, Pelkonen O, Tolonen A (2009) Rapid detection and characterization of reactive drug metabolites in vitro using several isotope-labeled trapping agents and ultra-performance liquid chromatography/time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 23: 843855. Santaguida S, Janigro D, Hossain M, Oby E, Rapp E, Cucullo L (2006) Side by side comparison between dynamic versus statics models of blood-brain barrier in vitro: A permeability study. Brain Research 1109:1-13. 32 Schmitt W (2008) General approach for the calculation of tissue to plasma partition coefficients. Toxicology in Vitro 22:457-467. Siu MKY, Cheng CY (2008) Extracellular matrix and its role in spermatogenesis. In: Molecular mechanisms in Spermatogenesis, Vol 636, Cheng CY (Ed.) (Chapter 5). Skor H, Sahly Y, Rahavendran S (2005) Evaluation of a 96 Well Plate Ultrafiltration Method compared to a Validated 96 Well Equilibrium Dialysis Method for Plasma Protein Binding Determination in a Drug Discovery Setting. The APPS Journal (Annual Meeting Abstracts) 7(S2):1579. Soars MG, Webborn PJ, Riley RJ (2009) Impact of hepatic uptake transporters on pharmacokinetics and drug-drug interactions: use of assays and models for decision making in the pharmaceutical industry. Molecular Pharmaceuticals 6(6):1662-77. Staub C (2001) A century of research on mammalian male germ cell meiotic differentiation in vitro. Journal of Andrology 22(6):911-26. Stéphenne X, Najimi M, Sokal EM (2010) Hepatocyte cryopreservation: is it time to change the strategy? World Journal of Gastroenterology 16(1):1-14. Review. Swanson HI (2004) Cytochrome P450 expression in human keratinocytes: an aryl hydrocarbon receptor perspective. Chemico-Biological Interactions 149:69-79. Tang W, Lu AYH (2010) Metabolic bioactivation and drug-related adverse effects: current status and future directions from a pharmaceutical research perspective. Drug Metab Rev 42:225-249. Testa B, Balmat AL, Long A (2004) Predicting drug metabolism: concepts and challenges. Pure Appilied Chemistry 76(5):907-914. Turpeinen M, Ghiciuc C, Orpitoui M, Kankainen L, Pelkonen O, Pasanen M (2007) Predictive value of animal models for human cytochrome P450 (CYP)-mediated enzymes: A comparative study in vitro. Xenobiotica 37:1367-1377. Ussing HH, Zerahn K (1925) Active transport of sodium as the source of electric current in the short-circuited isolated frog skin. Acta Physiol Scand 23:110-27. Vähäkangas K, Myllynen P (2006) Experimental methods to study human transplacental exposure to genotoxic agents. Mutation Research 608:129-135. van de Kerkhof EG, Ungell AL, Sjöberg AK, de Jager MH, Hilgendorf C, de Graaf IA, Groothuis GM (2006) Innovative methods to study human intestinal drug metabolism in vitro: preci-sion-cut slices compared with Ussing chamber preparations. Drug Metabolism Disposition 11:1893-1902. van de Wiele T, Vanhaecke L, Boeckaert C, Peru K, Headley J, Verstraete W, Siciliano S (2005) Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environmental and health Perspectives 113:6-10. 33 Veda S, Platel K, Srinivasan K (2007) Varietal differences in the bioaccessibility of beta-carotene from mango (Mangifera indica) and papaya (Carica papaya) fruits. Journal of Agriculture and Food Chemistry 55:7931-7935. Versantvoort CH, Oomen AG, van de Kamp E, Rompelberg CJ, Sips AJ (2005) Applicability of an in vitro digestion model in assessing the bioaccessibility of mycotoxins from food. Food and Chemical Toxicology 43:31-40. Versantvoort CHM, Rompelberg CJM, Sips AJAM (2000) Methodologies to study human intesti-nal absorption. RIVM Report 63003001. Wang J, Hou T (2009) Recent advances on in silico ADME modeling, Annual Reports in Computational Chemistry 5:101-127. Elsevier Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ (2005) Evaluation of the MDR-MDCK cell line as a permeability screen for the BBB. International Journal of Pharmaceutics 288:349-359. Waters NJ, Jones R, Williams G, Sohal B (2008) Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. Journal of Pharmaceutical Sciences 97:4586-4595. Webborn PJ, Parker AJ, Denton RL, Riley RJ (2007) In vitro-in vivo extrapolation of hepatic clearance involving active uptake: theoretical and experimental aspects. Xenobiotica 37: 1090-1099. Williams DP (2006) Toxicophores: investigations in drug safety. Toxicology 2006; 226: 1-11. Wilson TH, Wiseman G (1954) The use of sacs of everted small intestine for the study of the transference of substances from the mucosal to the serosal surface. Journal of Physiology 123:116-125. Xia W, Mruk DD, Lee WM, Cheng CY (2005) Cytokines and junction restructuring during spermatogenesis--a lesson to learn from the testis. Cytokine Growth Factor Review 16(45):469-93. Xu G, Bhatnagar V, Wen, G, Hamilton B, A Eraly, S, Knigam S (2005) Analyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)] Kidney International 68, 1491–1499 Yang X, Gandhi YA, Duignan DB, Morris ME (2009) Prediction of biliary excretion in rats and humans using molecular weight and quantitative structure-pharmacokinetic relationships. AAPS Journal 11(3):511-525. Zanger UM, Turpeinen M, Klein K, Schwab M (2008) Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 392(6):1093-108. Zhang J, Musson DG (2006) Investigation of high-throughput ultrafiltration for the determination of an unbound compound in human plasma using liquid chromatography and 34 tandem mass spectrometry with electrospray ionization, Journal of Chromatography B 843,47-56 35