Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
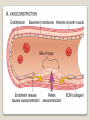
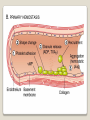
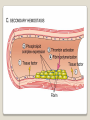
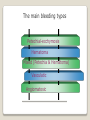
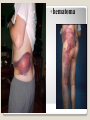
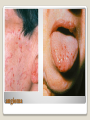
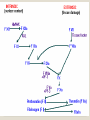
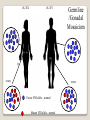
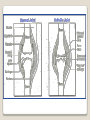
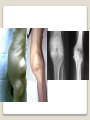
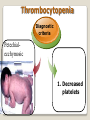
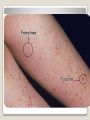
Hemorrhagic diathesis is a disease, characterized by excessive bleeding. According to pathogenesis, it is classified into coagulopathy, platelet disorder (thrombocytopenia and thrombocytopathy) and vasopathy. Each type is subdivided into congenital and acquired. NORMAL BLEEDING DISORDER vascular injury vascular injury vasoconstriction vasoconstriction platelet plug fibrin clot incomplete platelet plug incomplete or delayed fibrin clot The main bleeding types Petechial-ecchymosis Hematoma Mixed (Petechia & Hematoma) Vasculatic Angiomatosic The main bleeding sickness types Hematomic (massive, deep, painful; bleeding may occur anywhere. The most common sites of bleeding are into joints (knees, ankles, elbows), into muscles, from the gastrointestinal tract, cause of the bleeding can be intramuscular injection; characterized by early postoperative & posttraumatic bleeding) hematoma petechia-spotted (macula) macula-hematoma vasculitic purpura angioma The main coagulogram indexes Index Hypo coagulation Normo coagulation Hyper coagulation >5 5-3 <3 <180 180-320 >320 <23 23-44 >45 >120 120-60 <60 Heparin tolerance test, min >11 11-8 <8 Prothrombin index, % <80 80-100 >100 U-factor <80 80-100 >100 Procorventin (VII factor), % <80 80-100 >100 <2 2-4 >4 Bleeding time (by LeeWhite methods), min Platelets number Platelet adhesiveness Time of plasma recalcification, sec Fibrinogen, g/l Screening tests for bleeding disorders Test Abnormality detected Blood count and film Anaemia, leukaemia, disseminated intravascular coagulation Thrombocytopenia Platelet count Activated partial thromboplastin time Deficiency of all coagulation factors except VII, especially follows VIII and IX; heparin Prothrombin time Deficiency of factors I, II, V, VII, and X; warfarin Thrombin time or fibrinogen Hypofibrinogenaemia or dysfibrinogenaemia; heparin; fibrin degradation products Test of platelet-vessel wall interaction Bleeding time Coagulopathy I. Congenital • Deficiency of coagulation factor VIII (hemophilia A) • Deficiency of coagulation factor IX (hemophilia B) • Deficiency of coagulation factor XI (hemophilia C) • Deficiency of other coagulation factors (I, II, V, VII, IX, X and XIII) • Deficiency of XII factor, prekallikrein or kininogen, protein C and S (without excessive bleeding) • von Willebrand’s disease (angiohemophilia) Coagulopathy II. Acquired 1) Hypoprothrombinemia • Deficiency of vitamin K due to acholia of GIT (in cholestatic jaundice) • In overdose of indirect anticoagulant (antagonist of vitamin K) • In liver cirrhosis (due to reduced protein production) 2) Consumption of coagulation factors (II stage of DIC) 3) Heparin overdose 4) Activation of fibrinolytic system • In administration of streptokinase etc. • In trauma, obstetrical or surgical operations • In malignant neoplasms • In shock, sepsis, hematological malignancies, III stage of DIC Platelet disorders Thrombocytopathy I. Congenital (deficiency of platelet membrane glycoprotein) – Glanzmann's Thrombasthenia II. Acquired (normal platelet count in blood and bone marrow but its functions are decreased) • Drugs (after intake of antiplatelet agents: aspirin, Ticlide®); • Immune, toxic, septic processes; • Hematological malignancies and anemias Thrombocytopenia (decrease in the number of platelets) I. Werlhof’s disease - autoimmune thrombocytopenic purpura II. Symptomatic • Immune (heteroimmune, isoimmune,) • Toxic (in phosphorus poisoning etc.) • Methaplastic (in hematological malignancies or myelocarcinosis) • Drug-induced (cytostatics) Thrombocytopenia Thrombocytopenia (decrease in the number of platelets) o - Idiopathic thrombocytopenic purpura (ITP) o - Thombotic thrombocytopenic purpura (TTP) o - Heparin-induced thrombocytopenia (HIT) o - Hemolytic-Uremic Syndrome o - Chronic liver disease o Vasopathy I. Congenital • Telangiectasiae (Rendu-Osler-Weber disease) • Aneurysm of fine vessels • Ehlers-Danlos syndrome, Marfan’s syndrome, retinocerebral angiomatosis (von Hippel-Lindau syndrome), encephalotrigeminal angiomatosis (Sturge-Kalisher-Weber syndrome) II. Acquired • Hemorrhagic vasculitis – Henoch-Schönlein purpura • Immune vasculitis • Systemic vasculitis • Avitaminosis of vitamin C THE CLOTTING MECHANISM EXTRINSIC INTRINSIC Collagen XII XI Tissue Thromboplastin VII IX VIII X FIBRINOGEN (I) V PROTHROMBIN (II) THROMBIN (III) FIBRIN Coagulation pathway ◦ Two pathways for fibrin clot formation: Intrinsic ◦ Initiated by negatively charged surface Extrinsic ◦ Initiated on tissue injury ◦ Both pathways converge on a final common pathway Prothrombin Thrombin (Most critical step ) Fibrinogen Fibrin Clot ◦ The pathways are complex and involve many different proteins (called blood clotting factors) Coagulation Cascade - continued Control of coagulation Antithrombins (e.g., antithrombin III) ◦ Proteins C and S ◦ Fibrinolytic cascade Plasminogen plasmin fibrin break down products (*FDP or FSP) – d-dimer is most important of the FDPs *FDP / FSP – Fibrin degradation products / Fibrin split products Bleeding disorders Vascular abnormalities Platelet disorders Clotting factor abnormalities DIC A “Royal Disease” Queen Victoria (1837 to 1901) passed hemophilia on to German, Russian and Spanish royal families. Her son, Leopold, had frequent hemorrhages (British Medical Journal,1868) and died of a brain hemorrhage at 31. His grandson also died of a brain hemorrhage in 1928. www.themegallery.com Haemophilia А factor VIII deficiency В factor IX deficiency С factor XI deficiency Company Logo X-Linked Recessive Inheritance New mutation in germ cell • Affected males (XY): –sons unaffected (no male to male transmission) –daughters obligate carriers • Carrier female (XX): –½ sons affected; ½ daughters carriers • Affected females: very rare. New mutation in maternal or paternal germ cell Carrier female Affected male Normal male 46, XX 46, XY ovary Germline /Gonadal Mosaicism testes Factor VIII allele - normal Mutant VIII allele - normal Forms Haemophilia A - factor VIII deficiency, "classic haemophilia" (X-linked) Haemophilia B - factor IX deficiency, "Christmas disease" (X-linked) Haemophilia C - factor XI deficiency (Ashkenazi Jews, autosomal recessive) The unrelated type 1 and type 2 von Willebrand disease (vWD) Classification (F VIII C level) 2 1 F VIII C level 5-25 % Mild form F VIII C level 1-5 % Moderate form 3 F VIII C level less than 1 % Severe form Company Logo Clinical severity of haemophilia A and B Factor value* Bleeding tendency Less than 0.02 Severe - frequent spontaneous bleeding into joints, muscles, and internal organs 0.02-0.05 Moderate—some “spontaneous” bleeds, bleeding after minor trauma Mild—bleeding only after significant trauma or surgery More than 0.05 * Normal value of factors VIII and IX is 0.5-1.5 Hemophilia Diagnostic criteria Hematomic bleeding sickness types Arhtropathy Family_history_of _coagulation_disor ders: positive APTT:abnormal Mixing_APTT:cor rected Factor_VIII:C_act ivity: abnormal Diagnostic Criteria Condition Vitamin K deficiency or warfarin Disseminated intravascular coagulation von Willebrand disease Hemophilia Aspirin Thrombocytopeni a Uremia Glanzmann's thrombasthenia Bernard-Soulier syndrome Prothrombin time Partial thromboplastin time prolonged normal or mildly unaffected prolonged unaffected prolonged prolonged prolonged decreased unaffected prolonged prolonged unaffected unaffected unaffected prolonged unaffected unaffected prolonged unaffected unaffected unaffected unaffected prolonged decreased unaffected unaffected prolonged unaffected unaffected unaffected prolonged unaffected unaffected unaffected prolonged decreased or unaffected Bleeding time Platelet count Treatment In mild bleeding: 1-desamino-8-D-arginine vasopressin (DDAVP). The level of factor VIII must be maintained at least 30%. In non-life-threatening bleeding or pre-op: factor VIII concentrates. The minimal hemostatic level of factor VIII:C in this case is 50%. Central nervous system hemorrhage and severe bleeding: factor VIII concentrates. The level of factor VIII:C must be maintained at 80% - 100%. Each unit of factor VIII concentrates will raise the level of factor VIII by 2%/kg of body weight. Adjunctive therapy in dental surgery for mild and moderate hemophelia A: epsilon-aminocaproic acid (EACA). Avoidance of aspirin-containing compounds. For monitoring therapy, factor VIII assay is the test of choice. Dosage: the (activity ) units of factor VIII required can be calculated from: 40*BW* (desired factor level-actual factor level)/100 where BW is body weight in kg Treatment The dose of factor VIII concentrate is calculated assuming that one unit of factor VIII is the amount present in 1 mL of plasma. Plasma volume is 40 mL/kg, and the volume of distribution of factor VIII:C is 1.5 times the plasma volume. Thus, to raise the level 100%, the dose should be 40 x 1.5 = 60 units/kg, or approximately 4000 units for a 70-kg individual. To raise the levels to 25% would require 1000 units. The half-life of factor VIII:C is approximately 12 hours. Thus, during major surgery, to achieve an initial level of 100% and maintain it continuously at greater than 50%, a dose of 60 units/kg (approximately 4000 units) initially followed by 30 units/kg (approximately 2000 units) every 12 hours should be adequate. During surgery, initially verify that these doses give the anticipated factor VIII levels. If factor VIII levels fail to rise as expected, an inhibitor should be suspected. Hemophilia B DEFICIENCY OF FACTOR IX Factor IX deficiency X-linked recessive Much less common Clinically= indistinguishable from Hemophilia A with Similar lab findings Diagnosis by factor IX levels Treat with recombinant IX Family_history_of_coagulation_disorders:positive Mixing_APTT:corrected Factor_IX_assay:abnormal APTT:abnormal Diagnostic Criteria Treatment For patients with mild form of factor IX deficiency with non-lifethreatening bleeding, the treatment of choice is FFP. For the severe form of hemophelia B or in life- threatening situation, prothrombin complex concentrates are the treatment of choice. Target levels of factor IX for therapy: Severe hemorrhage: 20% 50% for 3-5 days, then 10% - 20% for the next 10 days. Minor hemorrhage: 20% for 7 days. Surgery: 50% - 70% for 2 days, then 30% - 40% for 3 days, then 20% for 7 days. Dosage: the (activity) units of factor IX required can be calculated from: 80*BW*(desired factor level-actual factor level)/100 where BW is body weight in kg Note that in-vivo recovery of factor IX is about 50%. Hemophilia A Treatment (cryoprecipitate) 1 Mild 15-20 UN/kg 2 3 Moderate Severe . 35-40 UN/kg 70 UN/kg Company Logo von Willebrand's Disease Essentials of Diagnosis Family history with autosomal dominant pattern of inheritance. Prolonged bleeding time, either at baseline or after challenge with aspirin. Reduced levels of factor VIII antigen or ristocetin cofactor. Reduced levels of factor VIII coagulant activity in some patients. Symptoms and Signs von Willebrand's disease is a common disorder affecting both men and women. Most cases are mild. Most bleeding is mucosal (epistaxis, gingival bleeding, menorrhagia), but gastrointestinal bleeding may occur. In most cases, incisional bleeding occurs after surgery or dental extractions. von Willebrand's disease is rarely as severe as hemophilia, and spontaneous hemarthroses do not occur (except in the rare type III). The bleeding tendency is exacerbated by aspirin. Characteristically, bleeding decreases during pregnancy or estrogen use. The main bleeding sickness types Mixed (Petechial & Hematomic) (combined features of both types, but there are some difference: in contrast to hematomic - bleeding are into joints very rare, mostly it is located in subcutaneous, retroperitoneal, mesenteric, subserous intestinal layer or into internal organ);in contrast to petechial-ecchymosic bleeding hemorrhagic syndrome characterized by large bruise) Treatment The bleeding disorder is characteristically mild, and no treatment is routinely given other than avoidance of aspirin. However, patients often need to be prepared for surgical or dental procedures. The bleeding time is probably the best indicator of the likelihood of bleeding, and prophylactic therapy may be reasonably withheld if the procedure is minor and the bleeding time is normal. Desmopressin acetate (DDAVP) is useful for mild type I von Willebrand's disease and should be considered first. The dose is 0.3 mcg/kg, after which vWF levels usually rise two- to threefold in 30– 90 minutes. It can also be given as a nasal spray; levels peak 2 hours after use. The antifibrinolytic agent aminocaproic acid (EACA) is useful as adjunctive therapy during dental procedures. 1. Pl stimulate vasoconstriction of injured vessels 2. Pl form hemostatic plug (platelet adhesion) + platelet aggregation to seal small vessel wall 3. Pl play role in fibrin clot formation Platelet hemostatic activity Bleeding disorders Vascular abnormalities *** Platelet disorders Clotting factor abnormalities DIC Bleeding disorders Platelet disorders ↓production ↑destruction Primary/Idiopathic ITP Acute/Chronic Sequestration Hypersplenism Secondary Drugs, HIV INCREASED DESTRUCTION Immune destruction ◦ Platelets are destroyed by antibodies Platelets with bound antibody are removed by mononuclear phagocytes in the spleen Anti-platlet antibody tests to identify antibodies on platelets are available Fibrin XIIIa Cross-linked fibrin Thrombocytopenia (or -paenia, or thrombopenia in short) is the presence of relatively few platelets in blood. Generally speaking a normal platelet count ranges from 180,000 and 320,000 per mm3. Signs and symptoms Often, low platelet levels do not lead to clinical problems; rather, they are picked up on a routine full blood count. Occasionally, there may be bruising, nosebleeds and/or bleeding gums. It is vital that a full medical history is elicited, to ensure the low platelet count is not due to a secondary process. It is also important to ensure that the other blood cell types red blood cells, and white blood cells, are not also suppressed. Thrombocytopenia Diagnostic criteria Petechialecchymosic 1. Decreased platelets Immune Thrombocytopenic Purpura Cause ◦ Antiplatelet antibodies ◦ Antigen - platelet membrane glycoprotein complexes IIb-IIIa and Ib-IX Morphology ◦ Peripheral Blood thrombocytopenia, abnormally large platelets (megathrombocytes or Giant platelets), ◦ Marrow Normal or Increased magakaryocyte # Diagnosis - by exclusion ◦ Bleeding time - prolonged, but PT & PTT - normal ↓ Marrow magakaryocyte # - your Diagnosis of ITP is ????? Necessary Evaluation • History: Isolated bleeding symptoms consistent with thrombocytopenia without constitutional symptoms (e.g. significant weight loss, bone pain, night sweats). • Physical examination: Bleeding symptoms in the absence of hepatosplenomegaly, lymphadenopathy, or stigmata of congenital conditions. • Complete blood count: Isolated thrombocytopenia (platelet count <100 x 109/L). Anemia only if due to significant bleeding - otherwise normal red cell indices, white blood cell count and differential. • Peripheral blood smear: Identified platelets should be normal to large in size. Red and white blood cell morphology should be normal. • History: Isolated bleeding symptoms consistent with thrombocytopenia without constitutional symptoms (e.g. significant weight loss, bone pain, night sweats). • Physical examination: Bleeding symptoms in the absence of hepatosplenomegaly, lymphadenopathy, or stigmata of congenital conditions. • The presence of abnormalities in the history, physical examination, or the complete blood count and peripheral blood smear should be further investigated, e.g. with a bone marrow examination or other appropriate investigations, before the diagnosis of ITP is made. Bone Marrow Evaluation The main bleeding sickness types Petechia-ecchymosis (is usually localized to superficial sites such as the skin and mucous membranes, wich often combined with menorrhagia, nose bleeding, gumms bleeding; rare – with gastrointestinal bleeding or brain hemorrhage; it is small, painless, provoked by simple action like skin cleaning, measure of blood pressure etc; pinch test is positive) Diagnostic Criteria: Plt_count: abnormal (low) Peripheral blood smear:no increase in schistocytes Bone_marrow:megakaryocytosis Immune Thrombocytopenic Purpura Feature Age / Sex Onset Predisposing Factors Duration Pathogenesis Peripheral smear Bone marrow Acute Children Abrupt Viral infection/ vaccine <2 months - Chronic Adult/Female Gradual - >6 mnoths Ig G against Platelet GP Thrombocytopenia & Same Giant PLTS Normal or Same ↑Megakaryocytes ITP Feature Acute Tests Prolonged BT & Same Normal PT & PTT Intracranial Same bleed Complication (most dangerous) Clinical course Treatment PLT. Transfusion Splenectomy Chronic Spontaneous remission No If <20,000 No If <50,000 Yes (refractory cases) Condition Partial Prothrombin thromboplast Bleeding time Platelet count time in time prolonged normal or mildly prolonged unaffected unaffected Disseminated intravascular prolonged coagulation prolonged prolonged decreased von Willebrand disease unaffected prolonged prolonged unaffected Hemophilia unaffected prolonged unaffected unaffected Aspirin unaffected unaffected prolonged unaffected Vitamin K deficiency or warfarin Thrombocytopenia unaffected unaffected prolonged decreased Uremia Glanzmann's thrombasthenia Bernard-Soulier syndrome unaffected unaffected prolonged unaffected unaffected unaffected prolonged unaffected unaffected unaffected prolonged decreased or unaffected Initial Management of ITP Assessment of Disease Status: • What bleeding is the patient experiencing? • Determine the timing, location, and severity of bleeding symptoms. • Does this patient have any additional risk factors for bleeding such as use of antithrombotic agents or high-risk occupation? • Is a surgical procedure anticipated? • Is this patient likely to comply with recommended treatments? • Is the bleeding experienced by this patient interfering with his or her daily activities or causing significant anxiety? • The majority of patients with no bleeding or mild bleeding (defined here as skin manifestations only, such as petechia and bruising) can be treated with observation alone regardless of platelet count. • First-line treatment includes observation, corticosteroids, IV Ig, or anti-D immunoglobulin (anti-D). • Anti-D should be used with caution given recent FDA warnings of severe hemolysis. It is therefore not advised in patients with bleeding causing a decline in hemoglobin, or those with evidence of autoimmune hemolysis. General Considerations for Initial Management Drug induced thrombocytopenia Heparin induced thrombocytopenia (HIT) Seen in 3-5% of patients treated with unfractionated heparin thrombocytopenic after 1-2 weeks of Rx Caused by IgG antibodies against platelet factor 4/heparin complexes on platelet surfaces Exacerbates thrombosis, both arterial and venous (in setting of severe thrombocytopenia) ◦ Antibody binding results in platelet activation and aggregation. Rx - cessation of heparin Other drugs??? Platelet functional disorders Bleeding disorders Vascular abnormalities Platelet disorders Clotting factor abnormalities DIC Causes Infections Meningococcemia, Rickettsioses, Infective endocarditis Drug reactions Hereditary hemorrhagic telangiectasia Autosomal dominant Cushing syndrome Henoch - Schönlein Purpura systemic hypersensitivity disease of unknown cause polyarthralgia, and acute Glomerulonephritis Palpable purpuric rash, colicky abdominal pain ◦Scurvy and the Ehlers-Danlos syndrome ◦Amyloid infiltration of blood vessels Vascular abnormalities The main bleeding sickness types Vasculatic (hemorrhage due to inflammatory changes of small vessels, the main cause are immune disorders or infectious agent) The main bleeding sickness types Angiomatosic (hemorrhage due to vascular dysplasia, teleangiectasia; the main clinical criteria is relapsing bleeding without hemorrhage in skin, subcutaneous and other tissue; nose bleeding are most often, dangerous and massive)