Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
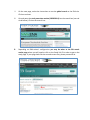
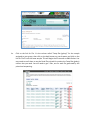
MappingRNAsequencedata Part1:RNA-RocketRNAseqpipeline ThegoalofthisexerciseistoretrieveanRNA-seqdatasetinFASTQformatandrunitthrough an RNA-sequence analysis pipeline. We will be using Pathogen Portal’s RNA-Rocket which includesaworkflowformappingRNA-Seqreadstoareferencegenome,usingthismappingto assembletranscripts,mappingtranscriptstoexistingannotations,anddeterminingexpression levels.Themappingworkflowusestwoalgorithms,TopHatforaligningreadsandCufflinksfor transcriptpredictionandcalculatingexpressionlevels.TheinputrequiredisFASTQfilesandthe outputsarereadalignments(BAMFiles),tabdelimitedassemblyandexpressionfilesforknown genes,isoformsandnoveltranscripts. 1. CreateanaccountonRNARocket a. Go tohttp://rnaseq.pathogenportal.org/ b. Click on Create an Account and fill in the required information. Clickheretocreateanaccount orlogintoyourexisting account 2. UploadtheRNAsequencingreadstoyourRNARocketlaunchpad.RNARocketallowsyou todirectlyretrieveFASTQfilesofthesequencingreadsusingSRAaccessionnumbers. a. Background: This exercise will rely on data deposited in the sequence read archive (SRA). ThedataisbasedontranscriptomicanalysisofthreedevelopmentalstagesofPlasmodium falciparum: 1.Salivaryglandsporozoites 2.Culturedsporozoites,and 3.Culturedasexualstages. EachdevelopmentalstagewasassayedbyRNAsequencing(2replicatespersample).Thestudy accession number for this data on SRA is SRP033414 and additional information about this experimentmaybeobtainedfromGEO: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52867 Examining the information available in GEO and under the SRA accession numbers you will noticethatthisdataispairedend.Soforeachsamplethereshouldbetwofilesoneforeachof thepairs.Moreinformationforeachsequencingruncanbefoundat: Salivaryglandsporozoitessample1: http://www.ncbi.nlm.nih.gov/sra/SRX385640 Salivaryglandsporozoitessample2: http://www.ncbi.nlm.nih.gov/sra/SRX385641 Culturedsporozoitessample1: http://www.ncbi.nlm.nih.gov/sra/SRX385642 Culturedsporozoitessample2: http://www.ncbi.nlm.nih.gov/sra/SRX385643 Asexualstageparasitessample1: http://www.ncbi.nlm.nih.gov/sra/SRX385644 Asexualstageparasitessample2: http://www.ncbi.nlm.nih.gov/sra/SRX385645 TherequiredinputfileforRNARocket’sanalysispipelineisaFASTQfile,atextfile(similarto FASTA)thatincludessequencequalityinformationanddetailsinadditiontothesequence(ie. name,qualityscores,sequencingmachineID,lanenumberetc.).FASTQfilesarelargeandasa result not all sequencing repositories will store this format. However, tools are available to convert, for example, NCBI’s SRA format to FASTQ. Sequence data is housed in three repositoriesthataresynchronizedonaregularbasis. ▪ ▪ ▪ ThesequencereadarchiveatGenBank TheEuropeanNucleotideArchiveatEMBL TheDNAdatabankofJapan b. UploaddataintoyourLaunchpad. Note:DuringthisexerciseyouwillNOTdownloadanydatatoyourcomputer.Insteadyouwill beprovidinginformationtoenabletransferringdatafromENA/SRAtoRNA-Rocket. i. Clickonthe“LaunchPad”linkintheGalaxymenubar.Thenselect“FromENA/SRA”. ii. Onthenextpage,noticetheinstructionstousetheglobalsearchontheENAsite. Clickoncontinue. iii. Cutandpastethestudyaccessionnumber(SRP033414)intothesearchbox(seered circlebelow).Clickonthesearchicon. iii. Depending on RNA-rocket’s configuration you may be taken to the EBI search resultspagewhereyouwillneedtoclickontheStudylinkIDinordertogettothe studypage.Ifyourpagelookslikethesecondscreenshot,pleaseproceedtoiv. iv. Click on the link for File 1 in the column called “Fastq files (galaxy)” for the sample assignedtoyourgroup,thenclickonthebackbuttononyourbrowserandclickonthe linkforFile2fromthesamesample.ThiswillbeginthefiletransfertoRNA-Rocket.You mayneedtoscrolldowntoseetheReadFilestabwhichcontainstheFastqfiles(galaxy) columnthatyouneed.Youwillneedtoget2files,oneforeachfilegeneratedbythe pairedendsequencing. Youshouldnowseeawindowthatlookssimilartothis: Toviewtheprogressofyourupload,clickon“ProjectView”(redsquareinimageabove). Youcaninspectthecontentsofcompletedtasks(likeuploadedfiles)byclickingonthe eyeiconnexttothenameofthefile(arrowinaboveimage).InspectingaFASTQfile shouldlooklikethis: c. ConfigureandinitiatetheRNAsequenceanalysispipeline. i. Background: Pathogen portal uses two algorithms for mapping (TopHat) and transcript prediction and expression value calculation (Cufflinks). Note that there aremanyalgorithmsandmethodsforRNA-seqmappingandanalysiseachwithits advantages and disadvantages. You are encouraged to learn more about the algorithmyouareusing. o TopHat: o Cufflinks: http://tophat.cbcb.umd.edu/ http://cufflinks.cbcb.umd.edu/index.html ii. Navigatetotheworkflow.Clickonthe“LaunchPad”linkintheuppermenubar.On the next page, scroll down to the “RNA-Seq Analysis” section and click on “Map Reads&AssembleTranscripts”. iii. SelectAnalysisType.Onthenextpage,scrolldownandchooseEukaryoticPairedEnd Analysis under Select Analysis Type. We are analyzing a paired end eukaryotic sample. iv. Selectthetargetprojectfromthedropdownmenu.Youshouldonlyhaveoneor two projects one of which will contain both FASTQ files you uploaded (probably called“UploadedFiles”).Onceyouselectthecorrectprojectyoushouldseethetwo FASTQfilescontainedwithinit.Nextclickoncontinue. v. Configurethepipeline.Thepipelineconsistsof7steps. Step1:Inputdataset–Selecttheupstreamreadfile(endsin_1)andclickonthearrowto moveittothe“Selected”window. Step2:Inputdataset–Selectthedownstreamreadfile(endsin_2)andclickonthearrow tomoveittothe“Selected”window. Step3: TopHat2 – Under Select a reference genome choose Plasmodium falciparum 3D7. There are a number of optionsthatmaybemodified,however, for the purposes of this exercise the defaultparametersmaybeused. Step4:Cufflinks– Set the Maximum Intron Length (-I): 5000. The reference annotation should be automatically selected:Plasmodiumfalciparum3D7 Select how to use the provided annotation: AssembleNovel+annotatedtranscripts. Once again there are a number of options to modify but we only need to change the maximumIntronLength. Step5:BAMtoBigWig–Nochangeneeded Step6:BAMtoBigWig–Nochangeneeded Step7:CreateaBedGraphofgenomecoverage–Nochangeneeded ClickontheRunWorkflowbutton. After you start the workflow you should get a confirmation window listing all the steps that have been added to the queue. The progress of your workflow can be viewed to the right. Completedtasksareingreen,runningtasksareinyellowandtaskswaitinginthequeuearein grey. The workflow will run overnight and we will view the results and calculate differential expressioninasubsequentexercise.