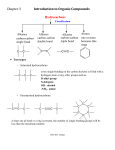
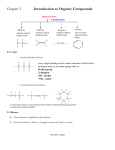
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Gene expression programming wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Minimal genome wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Oncogenomics wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Medical genetics wikipedia , lookup
Y chromosome wikipedia , lookup
Genetic testing wikipedia , lookup
Genetic drift wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Non-coding DNA wikipedia , lookup
Population genetics wikipedia , lookup
Genomic library wikipedia , lookup
Human genome wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
Genome editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genome evolution wikipedia , lookup
Neocentromere wikipedia , lookup
X-inactivation wikipedia , lookup
Genetic engineering wikipedia , lookup
Human genetic variation wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Public health genomics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Microsatellite wikipedia , lookup
Human Reproduction vol.15 no.3 pp.594–598, 2000 Repetitive complete hydatidiform mole can be biparental in origin and either male or female R.A.Fisher1,4, R.Khatoon1, F.J.Paradinas2, A.P.Roberts3 and E.S.Newlands1 1Department of Cancer Medicine, Division of Medicine, Imperial College School of Medicine, Charing Cross Hospital, Fulham Palace Road, London W6 8RF, 2Department of Histopathology, Division of Investigative Sciences, Imperial College School of Medicine, Charing Cross Hospital, Fulham Palace Road, London W6 8RF and 3Directorate of Women’s Services, Newham General Hospital, Glen Road, Plaistow, London E13 8SL, UK 4To whom correspondence should be addressed Complete hydatidiform mole (CHM) is an abnormality in pregnancy due to a diploid conception which is generally androgenetic in origin, i.e. all 46 chromosomes are paternally derived. We have examined the genetic origin of repetitive hydatidiform moles in a patient having three CHM by two different partners, and no normal pregnancies. Using fluorescent microsatellite genotyping, we have shown all three CHM to be biparental, rather than androgenetic, in origin. Examination of informative markers for each homologous pair of chromosomes, in two of the CHM, failed to reveal any evidence of unipaternal disomy, suggesting that the molar phenotype might result from disruption of normal imprinting patterns due to a defect in the maternal genome. It has been suggested that intracytoplasmic sperm injection (ICSI), followed by selection of male embryos, can prevent repetitive CHM; but examination of sex chromosome-specific sequences in the three CHM described here, showed that, while two were female, the first CHM was male. Selection of male embryos is therefore unlikely to prevent repetitive CHM in this patient. Our results suggest that the genetic origin of repetitive CHM should be determined prior to in-vitro fertilization (IVF) and that current strategies for the prevention of repetitive CHM may not be appropriate where the CHM are of biparental origin. Key words: biparental complete mole/genomic imprinting/IVF/ repetitive mole Introduction The premalignant condition, hydatidiform mole (HM), can be classified on the basis of histological examination and genetic origin as complete (CHM) or partial (PHM) (Szulman and Surti, 1978a,b). Genetically CHM are diploid, but abnormal in that they are usually androgenetic, i.e. all 46 chromosomes are paternal in origin (Kajii and Ohama, 1977; Wake et al., 1978). PHM also have two paternal contributions to the nuclear genome but, in contrast to CHM, also have a maternal 594 contribution and are consequently triploid. A patient who has had a molar pregnancy is at an increased risk of a subsequent pregnancy resulting in HM (Bagshawe et al., 1986). Evidence of molar tissue in the uterus of a patient a few months after termination of one molar pregnancy may, therefore, be a second HM. Alternatively, this may represent persistent trophoblastic disease from the first HM. In this situation genetic analysis can distinguish between a second, repetitive HM, arising from a new conceptus, or persistent trophoblastic disease from the previous HM (Roberts and Mutter, 1994; Fisher, 1997). In performing genetic analysis of molar tissue in a patient with a second CHM, 5 months after evacuation of a previous CHM, we found that, rather than being androgenetic in origin, both CHM arose from apparently normal, biparental concepti (Fisher and Newlands, 1998). This patient has subsequently had a third molar pregnancy. This report describes genetic studies of the three HM, which confirm that all three are biparental in origin. The implications for patients undergoing in-vitro fertilization (IVF) following repetitive HM are discussed. Materials and methods Patient The patient was a 30 year old Asian woman who, during a 4 year period, had three molar pregnancies terminated by evacuation of the uterus at 16, 6 and 9 weeks gestation respectively. The patient has had no other pregnancies. There was no consanguinity between the patient and her partner and karyotyping did not reveal any chromosomal abnormalities in either the patient or her partner. Molecular genetic studies were carried out to determine the genetic origin of each of the three HM. DNA preparation DNA was prepared from blood samples of the patient and her husband using standard techniques. DNA from each HM was prepared from pathological blocks of formalin-fixed, paraffin-embedded tissue. In each case molar tissue was identified and microdissected from a 5 µm unstained section of tissue with reference to a consecutive section stained with haematoxylin and eosin. DNA was then prepared from this tissue using a modification of a previously described method (Wright and Manos, 1990). Briefly, the tissue was extracted with octane, washed with 100% ethanol and dried under vacuum. When dry, the tissue was incubated in 25 µl of digestion buffer (50 mmol/l Tris pH 8.5, 1 mmol/l EDTA, 0.5 % Tween 20) containing 200 µg/ml of proteinase K. After 3 h at 55°C the tube was incubated for 8 min at 95°C to inactivate the proteinase K. The supernatant (1 µl) was then used as template for polymerase chain reaction (PCR) amplification. PCR amplification In order to determine the sex chromosome complement of each of the three HM, 1 µl DNA from each HM was amplified using both © European Society of Human Reproduction and Embryology Biparental complete mole Table I. Microsatellite polymorphisms identified in DNA from the patient, her partner and the three consecutive CHM. Allele sizes for each polymorphism are given in bp Microsatellite polymorphism Reference Chromosome location Patient CHM1 CHM2 CHM3 Partner Mfd50 D10S89 TH VWA D22S264 Weber et al. (1990) Weber and May (1990) Polymeropoulos et al. (1991) Kimpton et al. (1992) Reed et al. (1994) 7 10 11 12 22 190 146–156 199 138–142 188–198 190–192 150–156 195–199 138–142 188–198 188*–190 144*–146 187*–199 142–146 188–204* 190–192 150–156 195–199 138 188–198 176–192 148–150 195 138–146 188 *Alleles in CHM 2 not present in either the patient or her partner. Table II. Informative microsatellite polymorphisms in the patient, her partner, CHM1 and CHM3. Allele sizes for each polymorphism are given in bp Chromosome Microsatellite Patient CHM1 CHM3 Partner 1 2 3 4 5 6 7 8 9 10 11 12 13 13 14 15 16 17 18 19 20 21 22 X D1S228 D2S165 D3S1262 D4S415 D5S210 D6S290 Mfd50 D8S552 D9S176 D10S192 D11S925 D12S97 D13S158 D13S170 D14S74 D15S207 D16S516 D17S807 D18S61 D19S220 D20S120 D21S270 IL2RB DXS996 121–123 141–151 108–116 191–193 219 260 191 169–173 254–260 239–259 269–280 263–273 110–116 137–151 296–298 163–168 282–284 121–123 220–222 277 212 211–217 133 153–159 123–125 151–155 116–118 169–191 217–219 256–260 191–193 173–176 256–260 239–255 269–284 267–273 110–118 ND 298–306 163–170 280–284 119–123 218–222 264–277 212–230 215–217 133–141 159 123–125 151–155 108–118 169–193 219–225 256–260 191–193 173–176 252–254 239–255 269–284 263–267 NI 137–147 294–298 163–164 280–282 119–121 222–224 264–277 212–239 215–217 133–135 128–159 125 155–159 106–118 169 217–225 252–256 177–193 176 252–256 249 –255 284–286 259–267 116–118 147 294–306 164–170 280–288 119 218–224 264–270 230–239 211–215 135–141 128 NI ⫽ not informative; ND ⫽ not done. X and Y chromosome-specific primers (Witt and Erickson, 1989). 50 ng of DNA from the patient and her partner were amplified with the same primers as controls. To determine the genetic origin of each HM, 50 ng DNA from the patient and her partner and 1 µl DNA from each of the HM was amplified using five pairs of primers which flank polymorphic microsatellite repeat (two tetranucleotide and three dinucleotide) sequences on different chromosomes (Table I). One of each pair of primers was labelled with the fluorescent dye FAM (blue) or TAMRA (yellow) Applied Biosystems, Warrington, UK. Following amplification, 5 µl of each PCR reaction product was analysed by electrophoresis in a 1% agarose gel to assess the yield of product. PCR products were diluted as appropriate and subsequently resolved by capillary electrophoresis using an ABI PRISM 310 Genetic Analyser (Applied Biosystems Ltd). Analysis and sizing of the microsatellite polymorphisms was performed using ABI PRISM GeneScan software (Applied Biosystems Ltd). Further analysis of CHM1 and CHM3 was carried out by amplification of DNA from the maternal, paternal and molar DNA for at least one polymorphic marker on each of the 22 autosomes and the X chromosome (Reed et al., 1994) (Table II). Where a marker was uninformative with respect to the origin of the molar DNA, a further marker for that chromosome was examined. Primers were labelled with FAM (blue), TAMRA (yellow), HEX (green) or TET (green) and analysis of fluorescent products performed using ABI PRISM GeneScan software. Results All three hydatidiform moles showed cistern formation, excess circumferential trophoblast, karyorrhexis and polyploid villi characteristic of CHM in early gestation (Paradinas, 1994; Paradinas et al., 1996) (Figure 1). The allele sizes of five microsatellite polymorphisms on different chromosomes are shown in Table I for the patient, her partner and the three HM. Analysis of DNA from CHM1 and CHM3 showed that in each case one allele for the microsatellite polymorphisms Mfd50, D10S89 and TH was maternal in origin while the other allele was inherited from her partner demonstrating that these concepti were biparental in origin (Figure 2). For the markers VWA and D22S264, 595 R.A.Fisher et al. Figure 1. Microscopic appearance of complete hydatifiorm mole (CHM3), evacuated at 9 weeks gestation, showing the branching villi with excess trophoblast characteristic of complete hydatidiform mole (CHM) in early gestation. Haematoxylin and eosin staining. Bar ⫽ 500 µm; V ⫽ branching villi; T ⫽ excess trophoblast (arrows). alleles found in the CHM, while not fully informative, were compatible with a contribution from both parents. Examination of the alleles found in CHM2, identified several alleles which were not present in either parent (Figure 2). However, for each marker, the results were compatible with one of the two alleles being maternal in origin showing that CHM2 was also of biparental origin but arose from a conception with a different partner to the first and third CHM. In all three HM the relative heights of the peaks representing each of the alleles in the molar tissue were consistent with disomy, i.e. a single maternally derived and a single paternally derived allele. No evidence of trisomy was observed for any of the polymorphisms examined. Analysis of products following amplification with X and Y chromosome-specific primers showed that tissue from CHM1 was male (Y-positive), while CHM2 and CHM3 were female (Y-negative) (Figure 3). Further informative microsatellite markers examined in the patient, her partner, CHM1 and CHM3 are shown in Table II. For each marker examined, CHM1 and CHM3 were shown to be biparental in origin. In CHM1 a single polymorphism of maternal origin was identified in the molar tissue for markers on the X chromosome, compatible with a male genotype. CHM3 had two X-specific alleles, one maternal and one paternal in origin, confirming a female genotype. No evidence of unipaternal disomy was found for any of the autosomes or the sex chromosomes. Discussion CHM are generally diploid (Szulman and Surti, 1978a) and androgenetic in origin (Kajii and Ohama, 1977; Wake et al., 1978), all 46 chromosomes being derived from the father. They may be monospermic, arising by fertilization of an enucleate egg by a single spermatozoon which then doubles to 596 Figure 2. Fluorescently labelled polymerase chain reaction (PCR) products identified following amplification of the microsatellite TH in the patient, her partner, and the three hydatidiform moles (HM) (CHM1, CHM2, CHM3). The patient was homozygous for the 199 bp allele while her partner was homozygous for the 195 bp allele. The maternal 199 bp allele (shaded) was present in all three HM. While CHM1 and CHM3 each had the 195 bp allele identified in the partner, in CHM2 the second, 187 bp, allele was not present in either the patient or her partner. Figure 3. Polymerase chain reaction (PCR) products produced following amplification of DNA with X and Y-chromosome specific primers in the patient, her partner, and the three hydatidiform mole, (HM) (CHM1, CHM2, CHM3). A 130 bp, X-specific product was present in all samples. A 170 bp, Y-specific product was present only in the partner and CHM1. provide a diploid chromosome complement, or dispermic, arising from fertilization of an enucleate egg by two spermatozoa (Ohama et al., 1981). PHM are usually triploid (Szulman and Surti, 1978a) and generally arise by dispermy (Jacobs et al., 1982; Lawler et al., 1982). In both CHM and Biparental complete mole PHM, the trophoblastic hyperplasia, characteristic of a molar pregnancy, is associated with the presence of two paternal genomes and thus involves imprinted genes, that is genes which are normally only expressed from the maternally or paternally derived allele. Further evidence that the trophoblastic hyperplasia typical of molar pregnancies results from increased expression of paternally derived genes is provided by studies of the development of reconstituted mouse eggs. While those embryos which received a male and a female pronuclei developed to term following implantation (Surani et al., 1984), androgenetic embryos with two male pronuclei showed much greater trophoblastic development than those with two female pronuclei (Barton et al., 1984) in which fetal development was favoured. HM is therefore an imprinted condition in that the pathology is dependent on the parental origin of the genome. Although the majority of CHM are androgenetic in origin, occasionally CHM have been shown to be biparental in origin (Vejerslev et al., 1987; Ko et al., 1991; Kovaks et al., 1991; Sunde et al., 1993; Fisher et al., 1997). These unusual CHM have only one chromosome complement from the father, the second set of chromosomes being inherited from the mother as in a normal pregnancy. The rarity of these cases makes it difficult to estimate their true frequency. However, a recent study of two families in which several sisters had one or more CHM, found that all CHM examined were biparental in origin (Moglabey et al., 1999) suggesting that familial repetitive HM is of biparental origin. In one family in particular there was a high degree of consanguinity. The case described here represents an unrelated couple who had three CHM and no normal pregnancies. This case is of particular interest in that the second of the three CHM was conceived with a different partner to the first and third CHM. In androgenetic CHM, where the whole paternal chromosome complement is over-represented, it is difficult to specifically identify those genes that contribute to the abnormal development. Since biparental CHM are pathologically indistinguishable from the more common androgenetic CHM the underlying mechanism giving rise to these CHM is also likely to be an over-expression of paternally transcribed genes. These rare biparental CHM are, therefore, potentially valuable for identifying the imprinted genes involved in molar development, since much smaller regions of the genome are likely to be abnormal in these cases. There are a number of human genetic disorders involving imprinted genes such as Angelman syndrome and Beckwith–Wiedemann syndrome (BWS) which result from a number of different mechanisms including duplication of a single paternal chromosome with loss of the corresponding maternal chromosome (Hall, 1997). In order to investigate whether biparental CHM might result from unipaternal disomy of a specific chromosome, at least one informative microsatellite polymorphism was examined for each pair of autosomes in CHM1 and CHM3. No evidence of unipaternal disomy was found in either CHM. This suggests that the molar pathology in these cases results from uniparental disomy of only a small region of the paternal genome. Alternatively two active copies of the genes involved in molar development might result from expression of maternal genes which are normally imprinted and therefore not transcribed. While some cases of BWS are due to the presence of two copies of paternal genes, others have a normal genotype and result instead from loss or relaxation of imprinting of the maternally inherited gene (Hall, 1997). That the defect may be in the maternal, rather than paternal, genome in diploid, biparental molar pregnancies is suggested by the fact that two different partners were involved in the three molar pregnancies in this study. This is supported by a recent report (Moglabey et al., 1999) of two families in which several sisters have repetitive HM. The repetitive HM in both the families described by Moglabey et al. were also shown to be biparental in origin. Linkage and homozygosity analysis suggested that, in their families, there is a defective gene located on chromosome, 19q13.3–13.4. This region of the genome has recently been shown to be the location of at least two imprinted genes, PEG3, a paternally expressed gene (Kim et al., 1997) involved in maternal behaviour and offspring growth in mice (Li et al., 1999) and Zim1, a Kruppel-type zinc-finger gene which is maternally expressed (Kim et al., 1999). These observations suggest the presence of an imprinted domain in human chromosome 19q13.4 in which other imprinted genes involved in molar development might be located. Cases of familial HM, for further linkage analysis, are extremely rare. However, a number of cases of recurrent HM are available. Although some cases of repetitive HM are androgenetic (Roberts and Mutter, 1994; Fisher, 1997), others are clearly biparental in origin. A pathological and genetic review of patients with repetitive CHM may identify additional patients with biparental HM for further investigation and identification of the genes involved in trophoblastic development. Identification of the genetic origin of repetitive HM is also important for patients considering IVF to avoid repetitive CHM. It has been suggested that in patients with repetitive HM, intracytoplasmic sperm injection (ICSI) followed by preimplantation genetic diagnosis (PGD) can be used to prevent recurrent HM (Reubinoff et al., 1997). ICSI of oocytes ensures that only a single spermatozoon enters the egg, thus preventing CHM, or PHM, which arise by dispermy. Following fertilization, the sex of the embryo can be determined from one or two blastomeres. Although CHM which arise by dispermy may be male or female (Fisher and Lawler, 1984; Wake et al., 1984) the 75% of CHM (Fisher et al., 1989) which arise following fertilization of an anucleate egg by a haploid spermatozoon are always female with a 46,XX karyotype. A 46,YY karyotype is presumed to be non-viable. Subsequent rejection of 46,XX embryos in favour of 46,XY embryos will therefore eliminate CHM which arise by doubling of a haploid spermatozoon (Reubinoff et al., 1997). However, selection of male embryos would not prevent a further CHM in cases of repetitive CHM of biparental origin which we have shown may be either male or female. Thus ICSI followed by PGD may prevent the recurrence of triploid PHM and androgenetic CHM but not repetitive biparental CHM. Patients with recurrent HM have been reported in which some HM are CHM and others PHM (Rice et al., 1989); we are unaware of any cases of repetitive CHM in which patients have both androgenetic and biparental CHM. 597 R.A.Fisher et al. Studies of fertilization, syngamy and cleavage in eggs from patients with a history of repetitive CHM have revealed a variety of abnormal pronuclei (Edwards et al., 1992; Edwards, 1994). In one case a small number of eggs were observed to have only a single pronucleus. These cells which later cleaved into normal 2-cell embryos and continued to divide, were interpreted as androgenetic diploid embryos. Observation of pronuclear development would also identify any embryo with three polar bodies that might represent a triploid PHM. It would obviously be of interest to examine pronuclear development in cases of biparental repetitive CHM which might be expected to have two pronuclei. Further genetic studies of repetitive HM are therefore important in the clinical management of patients with repetitive HM and for a better understanding of the role of imprinted genes in the development of trophoblast. Acknowledgements Facilities for 310 analysis were provided through funds from the Wellcome Trust and the Trustees of Charing Cross Hospital. This work was supported by grants from the Cancer Treatment and Research Trust. Note added at proof Since submitting this manuscript we have examined the genetic origin of the HM in two further cases of repetitive HM in which the patient had 3 or more CHM. In both cases all CHM were shown to be biparental in origin. References Bagshawe, K.D., Dent, J. and Webb, J. (1986) Hydatidiform mole in England and Wales, 1973–83. Lancet, ii, 673–677. Barton, S.C., Surani, M.A.H. and Norris, M.L. (1984) Role of maternal and paternal genomes in mouse development. Nature, 311, 374–376. Edwards, R.G. (1994) Hydatidiform moles. Hum. Reprod., 9, 1783–1785. Edwards, R.G., Crow, J., Dale, S. et al. (1992) Pronuclear, cleavage and blastocyst histories in the attempted preimplantation diagnosis of the human hydatidiform mole. Hum. Reprod., 7, 994–998. Fisher, R.A. (1997) Genetics of gestational trophoblastic disease. In Hancock, B.W., Newlands, E.S. and Berkowitz, R.S. (eds), Gestational Trophoblastic Disease. Chapman and Hall, London, UK, p. 15. Fisher, R.A. and Lawler, S.D. (1984) Heterozygous complete hydatidiform moles: do they have a worse prognosis than homozygous complete moles? Lancet, ii, 51. Fisher, R.A. and Newlands, E.S. (1998) Molecular and genetic studies of gestational trophoblastic disease. J. Reprod. Med., 43, 53–59. Fisher, R.A., Povey, S., Jeffreys, A.J. et al. (1989) Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Hum. Genet., 82, 259–263. Fisher, R.A., Paradinas, F.J., Soteriou, B.A. et al. (1997).Diploid hydatidiform moles with fetal red blood cells in molar villi: 2—Genetics. J. Pathol., 181, 189–195. Hall, J.G. (1997) Genomic imprinting: nature and clinical relevance. Ann. Rev. Med., 48, 35–44. Jacobs, P.A., Szulman, A.E., Funkhouser, J. et al. (1982) Human triploidy: relationship between parental origin of the additional haploid complement and development of partial hydatidiform mole. Ann. Hum. Genet., 46, 223–231. Kajii, T. and Ohama, K. (1977) Androgenetic origin of hydatidiform mole. Nature, 268, 633–634. Kim, J., Ashworth, L., Branscomb, E. et al. (1997) The human homolog of a mouse imprinted gene, Peg3, maps to a zinc finger gene-rich region of human chromosome, 19q13.4. Genome Res., 7, 532–540. Kim, J., Lu, X. and Stubbs, L. (1999) Zim1, a maternally expressed mouse Kruppel-type zinc-finger gene located in proximal chromosome 7. Hum. Mol. Genet., 8, 847–854. 598 Kimpton, C.P., Walton, A. and Gill, P. (1992) A further tetranucleotide repeat polymorphism in the VWF gene. Hum. Mol. Genet., 1, 287. Ko, T.-M., Hsieh, C.-Y., Ho, H.-N. et al. (1991) Restriction fragment length polymorphism analysis to study the genetic origin of hydatidiform mole. Am. J. Obstet. Gynecol., 164, 901–906. Kovaks, B.W., Shahbahrami, B., Tast, D.E. et al. (1991) Molecular genetic analysis of complete hydatidiform moles. Cancer Genet. Cytogenet., 54, 143–152. Lawler, S.D., Fisher, R.A., Pickthall, V.J. et al. (1982) Genetic studies on hydatidiform moles. I. The origin of partial moles. Cancer Genet. Cytogenet., 5, 309–320. Li, L.-L., Keverne, E.B., Aparicio, S.A., et al. (1999) Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science, 284, 330–333. Moglabey, Y.B., Kircheisen, R., Seoud, M. et al. (1999) Genetic mapping of a maternal locus responsible for familial hydatidiform moles. Hum. Mol. Genet., 8, 667–671. Ohama, K., Kajii, T., Okamoto, E. et al. (1981) Dispermic origin of XY hydatidiform moles. Nature, 29, 551–552. Paradinas, F.J. (1994) The histological diagnosis of hydatidiform moles. Curr. Diagn. Pathol., 1, 24–31. Paradinas, F.J., Browne, P., Fisher, R.A. et al. (1996) A clinical, histopathological and flow cytometric study of 149 complete moles, 146 partial moles and 107 non-molar hydropic abortions. Histopathology, 28, 101–109. Polymeropoulos, M.H., Xiao, H., Rath, D.S. et al. (1991) Tetranucleotide repeat polymorphism at the human tyrosine hydroxylase gene (TH). Nucleic Acids Res., 19, 3753. Reed, P.W., Davies, J.L., Copeman, J.B. et al. (1994) Chromosome-specific microsatellite sets for fluorescence-based, semi-automated genome mapping. Nature Genet., 7, 390–395. Reubinoff, B.E., Lewin, A., Verner, M. et al. (1997) Intracytoplasmic sperm injection combined with preimplantation genetic diagnosis for the prevention of recurrent gestational trophoblastic disease. Hum. Reprod., 12, 805–808. Rice, L.W., Lage, J.M., Berkowitz, R.S. et al. (1989) Repetitive complete and partial hydatidiform mole. Obstet. Gynecol., 74, 217–219. Roberts, D.J. and Mutter, G.L. (1994) Advances in the molecular biology of gestational trophoblastic disease. J. Reprod. Med., 39, 201–208. Sunde, L., Vejerslev, L.O., Jensen, M.P. et al. (1993) Genetic analysis of repeated, biparental, diploid, hydatidiform moles. Cancer Genet. Cytogenet., 66, 16–22. Surani, M.A.H., Barton, S.C. and Norris, M.L. (1984) Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature, 308, 548–550. Szulman, A.E. and Surti, U. (1978a) The syndromes of hydatidiform mole. I. Cytogenetic and morphologic correlations. Am. J. Obstet. Gynecol., 131, 665–671. Szulman, A.E. and Surti, U. (1978b) The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. Am. J. Obstet. Gynecol., 132, 20–27. Vejerslev, L.O., Fisher, R.A., Surti, U. et al. (1987) Hydatidiform mole: Cytogenetically unusual cases and their implications for the present classification. Am. J. Obstet. Gynecol., 157, 180–184. Wake, N., Takagi, N. and Sasaki, M. (1978) Androgenesis as a cause of hydatidiform mole. J. Natl Cancer Inst., 60, 51–57. Wake, N., Seki, T., Fujita, H. et al. (1984) Malignant potential of homozygous and heterozygous complete moles. Cancer Res., 44, 1226–1230. Weber, J.L. and May, P.E. (1990) Dinucleotide repeat polymorphism at the D10S89 locus. Nucleic Acids Res., 18, 4637. Weber, J.L., Kwitek, A.E. and May, P.E. (1990) Dinucleotide repeat polymorphisms at the D7S435 and D7S440 loci. Nucleic Acids Res., 18, 4039. Witt, M. and Erickson, R.P. (1989) A rapid method for detection of Ychromosome DNA from dried blood specimens by the polymerase chain reaction. Hum. Genet., 82, 271–274. Wright, D.K. and Manos, M.M. (1990) Sample preparation from paraffinembedded tissues. In Innis M.A., Gelfand, D.H., Sninsky, J.J., and White, T.J. (eds), PCR Protocols. A Guide to Methods and Applications. Academic Press, London, UK, pp. 153–158. Received on July 20, 1999; accepted on November 10, 1999