Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Biochemical switches in the cell cycle wikipedia , lookup
Tissue engineering wikipedia , lookup
Extracellular matrix wikipedia , lookup
Cell encapsulation wikipedia , lookup
Signal transduction wikipedia , lookup
Programmed cell death wikipedia , lookup
Cytokinesis wikipedia , lookup
Cell growth wikipedia , lookup
Cell culture wikipedia , lookup
Cellular differentiation wikipedia , lookup
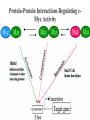
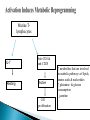
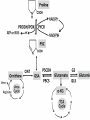
The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation Zeba Shahnaz 09-arid-1567 PhD Zoology The Myc family genes i.e., c-Myc, MYCN and L-Myc genes encode transcription factors that play essential roles in cell proliferation, cell growth, differentiation, and apoptosis The Myc protein consists of two regions 1- N-terminal transactivation domain (TAD) (required for transcriptional activation) 2- C-terminal domain (CTD) (critical for DNA binding and protein Interactions) Additionally, Myc has four conserved regions known as Myc boxes (MB) that are essential for different functions. MBI MBII MBIII MBIV The CTD encompasses a basic region required for binding to the consensus CACGTG E-box and a helix-loop-helix leucine zipper (HLHZip) domain, necessary for dimerization with Max. The function of c-Myc is modulated by the availability of its heterodimeric binding partner Max and the concentrations of competing Max/Max homodimers and Max/Mad heterodimers, all of which bind to a hexameric DNA sequence termed the E box. Myc/Max heterodimers bound to DNA recruit protein complexes (TRRAP) associated with histone acetylase activity that modify chromatin and activate transcription, whereas Max/Mad heterodimers recruit inhibitory complexes (Sin3/NCoR) associated with histone deacetylase activity. Proliferating animal cells consume considerable amounts of energy and require de novo synthesis of macromolecules to support their growth and proliferation One of the fundamental problems in animal cells is coordination of the metabolic program with cell growth- and proliferation-associated bioenergetic demand. Prokaryotes and unicellular eukaryotes rely primarily on homeostatic regulation of cell metabolism, while metabolic programs in multicellular eukaryotes have evolved to be actively controlled by extracellular signals (Deberardinis et al., 2008). Antigenic stimulation T- cell T-cell expansion +differentiation Immune response Accumulation of cell biomass during the initial growth and rapid proliferation during the expansion phase is associated with dramatically increased bioenergetic and biosynthetic demand. So, T cell activation provides a unique physiological cellular model to help us understand how animal cells coordinate their metabolic program with cell growth and proliferation in response to extracellular fate-decisive signals Polyclonal mitogen (concanavalin A & phytohaemagglutinin ) ↑glycolysis & Glutamine oxidation (In thymocyte & Mature lymphocytes) The uptake of glutamine and glucose and the consumption of the latter, mainly through glycolysis, are substantially up regulated upon stimulation with anti-CD3 and anti-CD28 Signaling via ERK and AKT pathways are used in the uptake of glutamine and glucose, respectively mammalian target of rapamycin (mTOR), an essential component of PI3KAkt pathway, used in regulating fatty acid metabolism in memory T cells However, the robustness of metabolic changes after T cell activation suggests the presence of additional regulatory steps in the T cell metabolic program, and the molecular mechanisms behind these remain to be explored. The metabolic changes associated with T cell activation are reminiscent of the characteristic metabolic activities of tumor cells and may represent a general metabolic reprogramming during cell growth and proliferation . Murine Tlymphocytes IL-7 Resting Anti-CD4 & anti-CD28 Active Cell proliferation ↑ metabolites that are involved in anabolic pathway s of lipids, amino acids & nucleotides ↑ glutamine & glucose consumption ↓carnitne Activation of T cells dramatically up regulated glycolysis In contrast, both mitochondria-dependent fatty acid βoxidation (FAO) and pyruvate oxidation through the tricarboxylic acid cycle (TCA cycle) were markedly down regulated upon activation. Glutamine and glucose consumption through oxidative catabolism and pentose phosphate pathway (PPP) were significantly elevated in activated T cells. Therefore, activation rapidly switches T cell metabolism from FAO and pyruvate oxidation via the TCA cycle to aerobic glycolysis, PPP, and glutamine oxidation. To explore the impact of metabolic reprogramming on T cell growth and proliferation, T cells were stimulated in either complete or nutrient-free media. Deprivation of glutamine but not glucose led to an impairment of activation-induced cell growth associated with a reduction of lipid and protein biosynthesis . However, both conditions resulted in impaired activationinduced cell proliferation 2DG (I) HK DHEA(I) G6PD Blocked T-cell proliferation DON (I) GLs In order to test the dependency of T cell proliferation on glucose and glutamine catabolism in vivo, 2DG and DON were applied in an adoptive transfer model by using ovalbumin (OVA)-specific, TCR-transgenic OT-II T cells. Both 2DG and DON partially inhibited the proliferation of antigen-specific active T cells after OVA immunization in vivo . These results show that both glucose and glutamine catabolic pathways are required for activation-induced T cell proliferation in vitro and in vivo. Two transcription factors, hypoxia inducible factor 1, alpha subunit (HIF1α), and myelocytomatosis oncogene (Myc) were used to know the regulation of metabolic reprogramming in activated T cells . Both HIF1 α and Myc are recently suggested to regulate cell metabolism in transformed cells , and were highly induced at both the transcription and protein levels upon T cell activation . The induction of these two transcription factors caused the up regulation of glycolysis and glutaminolysis A mouse model was generated carrying a conditional HIF1a allele (Hif1aflox/flox) and a tamoxifen-inducible Cre recombinase (CreERT2) transgene to delete HIF1a flox alleles in an acute manner. T cells Cultured With 4Hydroxytamoxif en (4OH) activated with anti-CD3 and antiCD28 (active) Deletion of HIF1 α IL-7 Cultured Without 4Hydroxytamoxif en (4OH) No/less effect on glycolysis, glutaminolysi s & FAO activated with anti-CD3 and antiCD28 (active) Another mouse model was generated carrying a conditional Myc allele (Myc flox/flox) and a tamoxifen-inducible Cre recombinase (CreERTam) to delete Myc flox alleles in an acute manner. T cells Cultured With 4Hydroxytamoxif en (4OH) activated with anti-CD3 and antiCD28 (active) Deletion of Myc IL-7 Cultured Without 4Hydroxytamoxif en (4OH) cell proliferation stopped,Low accumulation of lipid, a.a, nucleotides activated with anti-CD3 and antiCD28 (active) The role of Myc in regulating T cell proliferation and growth in vivo was observed by using the staphylococcal enterotoxin B (SEB) which acts as an antigen and activate the naive Vb8+ T cells. The Myc deletion blocked SEB-induced Vb8+ T cell proliferation and growth ( consistent with in vitro results) Deletion of Myc significantly impaired activation-induced glycolytic flux at both early and late time points. Myc deficiency led to a reduction of HK2 and pyruvate kinase muscle isoform 2 (PKM2) at the protein level. In vivo studies ( effects of deletion of Myc in regulating T cell metabolism in response to SEB) also showed that Myc is required for the induction of enhanced glycolytic activity and metabolic gene expression in SEB-responsive T cells in in vivo. . As glycolysis interconnects with other metabolic pathways, such as the PPP, the TCA cycle, and FAO, the metabolic flux through these pathways was investigated. So, Acute deletion of Myc moderately inhibited activationinduced upregulation of metabolic activity through the PPP The induction of two key metabolic enzymes in the PPP, Tkt and G6PDx, was compromised in Myc-deficient T cells upon activation. There are a large number of Myc target genes that are activated by an increase in the level of Myc. Many of these Myc targets have an E-box element containing the 5′-CACGTG-3′ sequence. In quiescent G0 or differentiated cells, the many Myc target genes are repressed by Max/Mad or Max/Mnt dimers, together with the SIN3/HDAC transcriptional co-repressor complex. The HDACs deacetylate chromatin, making it inaccessible to transcription. Myc activates transcription by replacing the Mad/Mnt, which removes the repressor complex. The Myc/Max dimer bind to the E-box and provides a complex to bring in various co activators. The histone acetyltransferases (HATs), such as CBP/p300, CGN5 and TIP60, acetylate histones to open up the chromatin to make it accessible so that the Myc target genes can be transcribed. These target genes contribute to cell growth and activation of the cell cycle. Module 4: Figure Myc as a gene activator Cell Signalling Biology - Michael J. Berridge - www.cellsignallingbiology.org - 2012 Myc drives the expression of Gfpt1, Cad, Ppat, and Oat , which not only are required for glutaminolysis but also control critical steps in the hexosamine, pyrimidine, purine, and polyamine biosynthetic pathways . Difluoromethylornithine (DFMO), a potent inhibitor of ornithine decarboxylase (ODC) and polyamine biosynthesis, inhibited activation-induced T cell proliferation in vitro and in vivo , and the addition of exogenous polyamines completely restored proliferation in the presence of DFMO in vitro Addition of either hypoxanthine and thymidine (HT) or polyamines (PAs) alone was insufficient to permit cell proliferation in the absence of glutamine. Because a-KG is required to maintain cell viability and minimal cell proliferation in the absence of glutamine, addition of a-KG alone was sufficient to drive only one cell division in the absence of glutamine. However, the combination of a-KG with either HT or with PAs promoted additional cell divisions and growth . While in Myc deficient T cells even the addition of a-KG with HT and PAs failed to promote cell proliferation in such Mycdeficient T cells. This indicates that Myc-driven glutaminolysis coordinates multiple biosynthetic pathways to support activation-induced cell proliferation and growth. Although ornithine is produced largely from arginine via the action of arginase as part of the urea cycle , urea cycle enzymes including arginase were not induced upon activation nor were any defects in proliferation observed in T cells lacking arginase I, the dominant isoform in the thymus, these results indicate that arginine is not the only precursor for ornthine in T cells. Previous studies in bovine endothelial and intestinal epithelial cells suggested the possible presence of a noncanonical metabolic pathway, which generates ornithine from glutamine . Intriguingly, the metabolic enzymes Aldh18A1, Prodh, and OAT that link glutamine and proline to ornithine synthesis were induced in a Myc dependent manner upon T cell activation. Therefore, U-13C-isotope labeled glutamine (U-13C-glutamine) flux were followed and the incorporation of 13C into all five carbons of ornithine (U-13C-ornithine) were observed, which accounted for up to 30% of total ornithine in active T cells but only 1% in resting T cells. Finally, deletion of Myc partially abolished the incorporation of glutamine-derived carbons into both ornithine and putrescine . These results indicate that a Myc-dependent noncanonical ornithine biosynthetic pathway is coupling glutaminolysis to ornithine and polyamine biosynthesis in activated T cells . To fulfill the bioenergetic and biosynthetic demand of proliferation, T cells reprogram their metabolic pathways from fatty acid b-oxidation and pyruvate oxidation via the TCA cycle to the glycolytic, pentose-phosphate, and glutaminolytic pathways. HIF1a and Myc, transcription factors were induced upon T cell activation, but only the acute deletion of Myc markedly inhibited activation induced glycolysis and glutaminolysis in T cells. Glutamine as an important source for biosynthetic precursors in active T cells. A Myc-dependent metabolic pathway link glutaminolysis to the biosynthesis of polyamines. Therefore, drives metabolic reprogramming in activated, primary T lymphocytes. This may represent a general mechanism for metabolic reprogramming under patho-physiological conditions. Cantor JR, Sabatini DM. 2012. Cancer cell metabolism: One hallmark, many faces. Cancer Discov 2: 881–898. Chi V. Dang.2013. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harbor Laboratory Press Dang CV. 2011. Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb Symp Quant Biol 76: 369–374. DeBerardinis, R.J., Lum, J.J., Hatzivassiliou, G., and Thompson, C.B. (2008).The biology of cancer: metabolic reprogramming fuels cell growth and proliferation.Cell Metab. 7, 11–20. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormic k LL, Fitzgerald P,Chi H, Munger J, et al. 2011. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871882.