Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Heart failure wikipedia , lookup
Management of acute coronary syndrome wikipedia , lookup
Electrocardiography wikipedia , lookup
Echocardiography wikipedia , lookup
Cardiac contractility modulation wikipedia , lookup
Cardiac surgery wikipedia , lookup
Hypertrophic cardiomyopathy wikipedia , lookup
Arrhythmogenic right ventricular dysplasia wikipedia , lookup
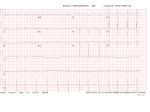
Waldenström macroglobulinemia and amyloid cardiomyopathy—an uncommon association Minghui Zeng, Bingong Li*, Zeqi Zheng, Jian Zhang, Yanhua Liu Department of Cardiology, First Affiliated Hospital, Nanchang University, Nanchang, 330006, China *Corresponding author: Dr. Bingong Li, 17 Yongwaizheng Street, Nanchang, 330006, China. Phone: 86-0791-88695356, [email protected] 1 Abstract Waldenström macroglobulinemia characterized by is a lymphoplasmacytic B-cell lymphoproliferative infiltration disorder of the bone marrow and monoclonal IgM components in the serum. Amyloidosis is a clinical disorder caused by extracellular deposition of insoluble fibrils, due to aggregation of misfolded soluble protein. Cardiac infiltration of amyloid fibril results in progressive cardiomyopathy with a grave prognosis. The coexistence of these two different entities is rare. We herein report the case of a 63-year-old man who presented with amyloid cardiomyopathy that results from waldenström macroglobulinemia. Key words macroglobulinemia; amyloid cardiomyopathy Introduction Waldenström macroglobulinemia (WM) is a B-cell lymphoproliferative disorder characterized by lymphoplasmacytic infiltration of the bone marrow and monoclonal IgM components in the serum1. WM is an uncommon disease. Prominent symptoms include physical weakness, weight loss, hepatomegaly, splenomegaly, lymphadenopathy, purpura, and hemorrhagic manifestations. Amyloidosis is a clinical disorder caused by extracellular deposition of insoluble fibrils, due to aggregation of misfolded soluble protein2. Cardiac infiltration of amyloid fibril results in progressive cardiomyopathy with a grave prognosis. A prominent clinical feature of cardiac amyloidosis is heart failure, characterized by restrictive hemodynamics and 2 progressive deterioration of systolic function. We present an unusual case of cardiac amyloidosis that resulted from WM. Case report A 63-year old Chinese male patient admitted to the hospital presented with a two-year history of slowly progressive bilateral lower extremity edema and weight loss. The patient reported sudden onset of progressive exertional dyspnea four months prior his admission to the hospital. He reported no chest pain, orthopnea, purpura, or paroxysmal nocturnal dyspnea. Subsequent physical examination detected face edema, distended jugular vein, arrhythmia,shortened pulse, pale mucosa, shallow breath, inguinal and cervical lymph nodes enlargement, and edema in the lower extremities. Two years prior hospital admission, the patient experienced symptomatic edema and weight loss. Laboratory results from diagnosis were: erythrocyte sedimentation rate, 134 mm/h; white blood cell count, 3.90×109/L; hemoglobin count, 89 g/L; platelets count, 170×1012/L; activated partial thromboplastin time, 91.6 sec(reference range, 23.7-36.4 sec); serum electrophoresis found alpha 1 level, 3.1%(reference range, 1.4-2.9%); M-PRO level, 61.3% (reference range, 9.0-16.0%); albumin level, 21.1%(reference range, 60.0-71.0%); beta level, 5.1%(reference range, 8.0-13.0%); serum protein level, 89.3 g/d(normal60-85 g/d); albumin level, 15.6 g/d (normal 35-55 g/d), 24-hours urine protein, 8.01 g/day(normal <0.15g/24h ); IgA level, 0.23g/L(normal 0.82-4.53 g/L); IgG level, 1.55 g/L(normal 7.51-15.60 g/L); and IgM level, 95.90 g/L(normal 0.36-3.04 g/L). Bone marrow aspirate morphology showed reduced bone marrow hyperplasia and red blood cells having cotton-like 3 appearance. The CD45/SSC dot-plot showed lymphocytes constituted 11% of nucleated cell and 55.5% of lymphocytes being lymphocytes B, and the presence of CD19, CD20, Ig-kappa and HLA-DR. Immunofixation electrophoresis indicated presence of monoclonal IgM-κ. Based on the aforementioned laboratory results, the patient was diagnosed with WM. The patient subsequently received chemotherapy treatment with a combination of cyclophosphamide, methylprednisolone and thalidomide. The symptoms of bilateral lower extremity edema and weight loss were improved upon this prescribed treatment. IgM concentration was lower than before. At presentation, the patient reported intermittent dyspnea. The patient denied any prior history of coronary heart disease,hypertension, diabetes or pulmonary diseases. Further examination revealed a blood pressure of 99/76 mmHg, a heart rate of 90/min, a body temperature of 36.4˚C and a respiration rate of 20/min. Pitting edema was detected in both lower extremities. The electrocardiography showed low voltage in the limb leads and atrial fibrillation. Echocardiography revealed the septal walls of the left ventricle were thickened by 14 mm, and the posterior wall of left ventricle was thickened 13 mm. Echocardiography also demonstrated atrial enlargement, left ventricular diastolic dysfunction, and left ventricular ejection fraction at 60%. NT-BNP 7202 ng/L(normal<125 ng/L ) , troponin-I was negative. The serum free-light-chain assay revealed excessive increase in kappa free light chain, resulting in high kappa-to-lambda ratio, with free kappa (κ) light chain concentration at 7170 mg/dL(normal 629-1350 mg/dL), and free lambda (λ) light chain concentration at 149.00 mg/L(normal 313-723 mg/dL). The gingival biopsies detected presence of 4 amyloid deposits. From these test results, the patient was diagnosed with primary (AL) cardiac amyloidosis, with manifestations of heart failure. The patient was initially treated with loop diuretics by intravenous torasemide injection at 20 mg/day, which improved dyspnea symptoms and lower leg edema. The patient was also treated with a combination chemotherapy consisted of fludarabine, cyclophosphamide and mesna. Upon the treatment described above, the patient showed improvement in dyspnea, and an alleviation of lower extremities edema, and improvement in overall physical shape. At present, he still was followed up. Discussion WM is a rare disease, with overall incidence of approximately 3 per million persons per year. The pathogenesis of WM is poorly understood. The disorder has two main characteristics: lymphoplasmacytic infiltration of the bone marrow and serum monoclonal IgM3. The patient presents initial symptoms of edema and weight loss. Diagnosis of WM was confirmed from serum electrophoresis, immunohistochemistry, immunofixation, bone marrow aspirate morphology analyses. Two years following WM diagnosis, exertional dyspnea emerges. Amyloid cardiomyopathy is diagnosed upon gingival biopsies. Cardiac amyloidosis that results from WM is rare. Amyloidosis is rare systemic disorder that derived from tissue deposition of amyloid protein. Amyloidosis includes primary AL amyloidosis and secondary AA amyloidosis. AL amyloidosis has an estimated incidence of 9 cases per million in developed countries, with 65 being the average age of diagnosed patients; less than 10% of patients are under 504. Amyloidosis is particularly challenging to clinicians. There is 5 some relation between WM and amyloidosis. It is reported immunoglobulin M clones can give rise to both AL and AA amyloidosis, with 4% to 7% of cases being AL amyloidosis. The Mayo Clinic Group reported 2% of smoldering WM develop AL amyloidosis. The heart is the frequently affected organ in IgM-AL amyloidosis5. Amyloid cardiomyopathy is especially devastating for WM patients, especially primary light chain (AL) amyloidosis. Cardiac amyloidosis is characterized by progressive diastolic and systolic dysfunction and arrhythmia. Cardiac amyloidosis symptom is similar to restrictive cardiomyopathy. The symptom is not apparent at the early stage, when cardiac complication becomes clinically evident however, irreversible damage has often already occurred. B-type natriuretic peptide and troponins are significantly in IgM-AL amyloidosis, which suggest cardiac dysfunction6. Cardiac amyloidosis is diagnosed and evaluated by specialized tests-low QRS voltages with poor R wave progression in the chest leads are detected by electrocardiography7; thickening of the right ventricule, normal/near normal ejection fraction and valvular thickening are found by from echocardiography; myocardial tissue is characterized by cardiac magnetic resonance; And early stage damage is detected by radionuclide imaging. Endomyocardial biopsy however, is the gold standard for demonstrating cardiac amyloid deposition. While cardiac amyloidosis has a poor prognosis, the treatment may be classified as follows: supportive therapy, chemotherapy, and cardiac transplantation8. Owing to the catastrophic nature of cardiac amyloidosis, physician should diagnose and implement treatment for the disorder early. Conventional treatment for amyloidosis entails 6 chemotherapy and symptomatic therapy. The combination of chemotherapy drugs includes fludarabine, cyclophosphamide, ituximab, bortezomib, thalidomidea and dexamethasone4. WM accounts for approximately 2% of all hematologic malignancies. The corresponding therapy of WM, by consensus, entails alkylating agents, nucleoside analogs and monoclonal antibody rituximab, with the monoclonal antibody rituximab being the choice of first-line therapy for WM9. Conclusions While WM-induced amyloid cardiomyopathy is not uncommon, the relevant treatment is poorly understood. This study advocates for cooperative treatment from both hematological and cardiovascular approaches. References 1. Leleu X, Roccaro AM, Moreau AS, et al. Waldenstrom macroglobulinemia. Cancer Lett. 2008; 270: 95-107. 2. Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011; 29: 1924-1933. 3. Stone MJ, Pascual V. Pathophysiology of waldenström’s macroglobulinemia. Haematologica. 2010; 95: 359-364. 4. Desport E, Bridoux F, Sirac C, et al. AL amyloidosis. Haematology. 2013; 41: 299-301. 7 5. Palladini G, Merlini G. Diagnostic challenges of amyloidosis in waldenström macroglobulinemia clinical lymphoma, Clin Lymphoma Myeloma Leuk. 2013; 13: 244-246. 6. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol. 2007; 50: 2101-2110. 7. Gallucci G, Guariglia R, Cangiano R, Mansueto G, Martorelli MC, Musto P. Modification of QRS pattern in a patient with AL amyloidosis. Int J Cardiol. 2013; 164: 9-12. 8. Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012; 1: e000364. 9. Gertz MA, Merlini G, Treon SP. Amyloidosis and waldenström macroglobulinemia. Hematology Am Soc Hematol Educ Program. 257-282. 8