Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
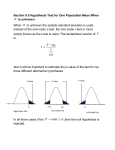
Bioinformatics II Fall 2006 Homework 4 Due Oct. 19, 2006 email to: [email protected] Email the R code and hand in the output. All plots should have titles We are going to do a few statistical tests using the Golub data, which is already loaded into the Biobase package. library(Biobase) data(golubMergeSub) To extract the data about the samples: SampleInfo=pData(golubMergeSub) To extract the probeset summaries (in log2) golubExpr=log2(exprs(golubMergeSub)) The normalization used produces both negative and zero expression summaries, so that taking log2, results in NaN ("not a number") and –Inf, which we will have to remove. Print the first few lines of SampleInfo and of golubExpr to see what you have. (Do not turn this in.) A. Testing differential expression when the samples are independent. We will use probeset 4. (Row 4 of golubExpr) It is a bit inconvenient to show the differences between the methods using all 72 patients, so I have chosen 12 patients samp=c(13, 42, 6 ,37, 15, 51, 45, 68 ,62, 27, 34, 71) gene4=golubExpr[4,samp] ALtype=SampleInfo[samp,2] # saves the patient numbers # save the 12 gene expression values # saves ALL or AML for each patient We will also look at what happens when the data have outliers. y1=gene4 y1[1]=14 y2=y1 y2[12]=3 Bioinformatics II Fall 2006 1. Get boxplots of gene4, y1 and y2 by cancer type. Note: If x is a factor and y is a vector of numbers, boxplot(y~x) produces a boxplot for each level of x. 2. a) Do a two-sample t-test for differential expression using gene4, y1, y2. t.test(y~x) # does a 2-sample t-test of the equality of the mean of y broken into 2 # groups by the values of x. b) Notice that the p-value decreased as single points were moved to more extreme separation of the ALL and AML samples. But even an extremely high ALL value and an extremely low AML value did not lead to statistical significance. Why is this? (Look at the formula for the t-test, or have a look at the 95% confidence interval for the difference in means that is printed by R.) 3. a) Do a two-sample Wilcoxon test for differential expression using gene4, y1, y2. wilcox.test b) Notice that the p-value for gene4 and y1 are identical. Why is this? (Look at how the Wilcoxon test statistic is computed.) 4. a) 1:12 generates the numbers 1,2,3,...,12. What does samp(1:12,12) do? b) We will do a permutation test (just for gene4) by computing 100 permutations of the cancer types, and extracting the t-value. To start, you need to find the component of the t.test output that holds the t-value. t.out=t.test(y~x) unclass(t.out) # saves the output # prints the output object to the screen without formatting. t.out$compname #extracts the data from component "compname" What is the name of the component that holds the computed t value? Bioinformatics II Fall 2006 c) Here is a function you can write that will do the permutation test. perm.t = function(y,x,nperms){ out=numeric(nperms) for (i in 1:nperms){ ptype=sample(x,12) t.out=t.test(y~ptype) out[i]=t.out$compname } out } #create an empty vector to store the t-values #permute the cancer types #replace with the right component d) Run your function 100 times using gene4 for y and obtain an histogram of t-values. This is an estimate of the null distribution of the t-test. e) Use the 100 t-values to estimate the percentage of samples for which |t| is greater than the value you obtained from gene4. f) In perm.t we have permuted the values of the disease classification. Suppose what we permuted the data instead – would this have the same effect? 5. a) 1:12 generates the numbers 1,2,3,...,12. What does samp(1:12,12,replace=T) do? b) We will do a bootstrap test (just for gene4) by computing 100 samples from the combined ALL and AML data, and extracting the t-value. Here is a function you can write that will do the permutation test. boot.t = function(y,x,nboot){ out=numeric(nboot) #create an empty vector to store the t-values for (i in 1:nboot){ yboot=sample(y,12,replace=T) #sample from the combined sample t.out=t.test(yboot~x) out[i]=t.out$compname #replace with the right component } out } c) Run your function 100 times using gene4 for y and obtain an histogram of t-values. This is an estimate of the null distribution of the t-test. d) Use the 100 t-values to estimate the percentage of samples for which |t| is greater than the value you obtained from gene4. Bioinformatics II Fall 2006 B. Testing differential expression when the samples are NOT independent. We will look at the difference in gene expression for genes 1 and 4 in the AML patients. The patients with valid data for both genes are: AML=c(21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 32, 33, 34, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72) AMLG1=GolubExpr[1,AML] AMLG4=GolubExpr[4,AML] M=AMLG4 – AMLG1 6. Obtain a boxplot of AMLG1 and AMLG4 and compute the correlation in expression. boxplot(list(AMLG1,AMLG4), names=c("Gene 1","Gene 4")) cor cor.test #computes the correlation #tests if the correlation is 0 The correlation is not high, but because we are measuring gene expression on genes in the same individuals, we should handle the data as dependent. 7. Do a one-sample t-test to test if the mean of M is 0 and a paired t-test to determine if the difference between the mean of AMLG1 is the same as the mean of AMLG2. ?t.test will give the options. If you pick, the option: paired=T you should get the same value of the test statistic. Now try omitting paired=T. The p-value may be bigger or smaller than the value from the paired test, depending on the correlation. 7. Permutation test: We do not want to break the pairing. The simplest way to do the test is to save abs(M) and sign(M). Then permute the signs, andcompute the t-tests on the product of abs(M) and the permuted signs. Write a function to do this, and turn in a histogram of 100 t-values. 8. Bootstrap test: Under the null hypothesis of no difference, M and –M are equally likely. Save the vector c(M,-M) and pick samples of size M with replacement from this vector. Write a function to do this and turn in a histogram of 100 t-values.