Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Linköping University Medical Dissertations
No. 793
From achiral to chiral analysis of citalopram
Björn Carlsson
Department of Medicine and Care, Division of Clinical Pharmacology
University Hospital, SE-581 85 Linköping, Sweden
Linköping 2003
ISBN 91-7373-550-7
ISSN 0345-0082
Printed in Sweden by UniTryck, Linköping 2003
¡A mi amor! María Dolores
Con los años que me quedan,
Yo viviré por darte amor
Borrando cada dolor,
Con besos llenos de pasión
Como te amé por vez primera
(G Estefan & E Estefan Jr)
¿Qué es la vida? Un frenesí.
¿Qué es la vida? Una ilusión,
una sombra, una ficción,
y el mayor bien es pequeño;
que toda la vida es sueño,
y los sueños, sueños son.
Vad är livet? Moln som fara,
rop som inga röster svara.
Intet verkligt är oss givet,
ty en dröm är hela livet,
Själva drömmen drömmar bara
Pedro Calderón de la Barca, 1636-1673
Abstract
Within the field of depression the “monoamine hypothesis” has been the leading theory to explain the biological
basis of depression. This theory proposes that the biological basis of depression is due to a deficiency in one or
more of three key neurotransmitter systems, namely noradrenaline, dopamine and serotonin which are thought to
mediate the therapeutic actions of virtually every known antidepressant agent.
Citalopram is a selective serotonin-reuptake inhibitor (SSRI) used for the treatment of depression and anxiety
disorders. Citalopram is a racemic compound, in other words composed of a 50:50 mixture of two enantiomers
(S-(+)-citalopram and R-(-)-citalopram) and with one of the enantiomers (S-(+)-citalopram) accounting for the
inhibitory effect. At the time of introduction of citalopram the physician needed a therapeutic drug monitoring
service to identify patients with interactions, compliance problems and for handling questions concerning
polymorphic enzymes and drug metabolism. An achiral analytical separation method based on solid-phase
extraction followed by high-performance liquid chromatography (HPLC) was developed for routine therapeutic
drug monitoring (TDM) of citalopram and its two main demethylated metabolites.
As the data available on citalopram were from achiral concentration determinations and to be able to further
investigate citalopram enantiomers effects and distribution, a chiral method for separation of the enantiomers of
citalopram and its demethylated metabolites was established.
The advances within chiral separation techniques have made measurement of the concentrations of the individual
enantiomers in biological fluids possible.
The process behind enantioselective separation is however not fully understood and the mechanism behind the
separation can be further scrutinized by the use of multivariate methods. A study of the optimization and
characterization of the separation of the enantiomers of citalopram, desmethylcitalopram and
didesmethylcitalopram on an acetylated E-cyclodextrin column, by use of two different chemometric programs response surface modelling and sequential optimization was performed. Sequential optimization can be a quicker
mean of optimizing a chromatographic separation; response surface modelling, in addition to enabling
optimization of the chromatographic process, also serves as a tool for learning more about the separation
mechanism.
Studies of the antidepressant effect and pharmacokinetics of citalopram have been performed in adults, but the
effects on children and adolescents have only been studied to a minor extent, despite the increasing use of
citalopram in these age groups.
A study was initiated to investigate adolescents treated for depression, with respect to the steady-state plasma
concentrations of the enantiomers of citalopram and its demethylated metabolites. The ratios between the S- and
R-enantiomers of citalopram and didesmethylcitalopram were in agreement with studies involving older patients.
The concentrations of the S-(+)- and R-(-) enantiomers of citalopram and desmethylcitalopram were also in
agreement with values from earlier studies. The results indicate that the use of oral contraceptives may have
some influence on the metabolism of citalopram. This might be because of an interaction of the contraceptive
hormones with the polymorphic CYP2C19 enzyme.
Even though the SSRIs are considered less toxic compared with older monoamine-active drugs like the
tricyclic/tetracyclic antidepressants, the risk of developing serious side effects such as ECG abnormalities and
convulsions has been seen for citalopram, when larger doses have been ingested. Furthermore, fatal overdoses
have been reported where citalopram alone was the cause of death. Data on the toxicity of each of the
enantiomers in humans have not been reported and no data on blood levels of the enantiomers in cases of
intoxication have been presented.
An investigation was initiated on forensic autopsy cases where citalopram had been found at the routine
screening and these cases were further analysed with enantioselective analysis to determine the blood
concentrations of the enantiomers of citalopram and metabolites. Furthermore the genotyping regarding the
polymorphic enzymes CYP2D6 and CYP2C19 were performed.
In 53 autopsy cases, we found increasing S/R ratios with increasing concentrations of citalopram. We found also
that high citalopram S/R ratio were associated with high parent drug to metabolite ratio and may be an indicator
of recent intake. Only 3.8 % were found to be poor metabolizers regarding CYP2D6 and for CYP2C19 no poor
metabolizer was found.
Enantioselective analysis of citalopram and its metabolites can provide valuable information about the time that
has elapsed between intake and death. Genotyping can be of help in specific cases but the possibility of
pharmacokinetic interactions is apparently a far greater problem than genetic enzyme deficiency.
Publications
This thesis is based on the following publications, referred to in the text by their
designated Roman numerals
I.
Carlsson B, Norlander B. Solid-phase extraction with end-capped C2 columns
for the routine measurement of racemic citalopram and metabolites in plasma by
high- performance liquid chromatography.
J Chromatogr B Biomed Sci Appl 1997;702(1-2):234-9.
II.
Carlsson B, Norlander B. Optimization and Characterization of the Chiral
Separation of Citalopram and its Demethylated Metabolites by Response
Surface Methodology.
Chromatographia 2001;53(5/6):266-272.
III.
Carlsson B., Olsson G., Reis M., Wålinder J., Nordin C., Lundmark J., Scordo
M.G., Dahl M.-L., Bengtsson F. and Ahlner J. Enantioselective Analysis of
Citalopram and Metabolites in Adolescents.
Ther Drug Monit 2001;23(6):658-64.
IV.
Holmgren P., Carlsson B., Zackrisson A.-L., Lindblom B., Dahl M.-L., Scordo
M.G., Druid H., and Ahlner J. Enantioselective analysis of citalopram and its
metabolites in postmortem blood and genotyping for CYP2D6 and CYP2C19
Submitted
The papers are reproduced with the permission of the copyright owners
Contents
List of abbreviations………………………………………………………….. 7
Introduction…………………………………………………………………… 9
Chirality………………………………………………………………………. 10
Drugs and Stereochemistry………………………………………...……… 14
Chirality and Psychopharmacology…………………………………… 14
Chirality and Pharmacodynamics……………………………………. 16
Chirality and Pharmacokinetics…………………………………….... 16
Selective serotonin reuptake inhibitors .........……………..…………………. 18
Cytochrome P450 enzyme system…………………………………………… 20
Citalopram a selective serotonin reuptake inhibitor…………………………. 23
Chemical and physical properties…………………………………… 23
Citalopram and cytochrome P450 superfamily……………………… 23
Pharmacokinetics……………………………………………………. 26
Pharmacodynamics………………………………………………….. 27
Adverse drug reaction – Toxicology………………………………… 28
Chiral bioanalysis…………………………………………………………….. 29
Indirect methods……………………………………………………… 29
Direct methods……………………………………………………….. 30
Mechanistic aspects of enantioseparation……………………………. 30
Chiral stationary phases………………………………………………. 31
Cyclodextrins…………………………………………………………. 33
Fluorescence detection………………………………………………………… 36
Chemometrics…………………………………………………………………. 37
Systematic optimization…………………………………………….... 37
Experimental design………………………………………………….. 39
Design of experiments...........………………………………………… 39
Screening designs……………………………………………………… 40
Full factorial and fractional factorial design…………………………... 40
Optimization design…………………………………………………… 42
Central composite designs………………………………. 42
Modified sequential simplex approach………………… 45
Aims of the present study………………………………………………………. 50
Methods, results and discussion…………………………………………………….
Paper I………………………………………………………………………………
Achiral determination of citalopram
Citalopram extraction version I
Solid-phase extraction
Internal standard
Achiral chromatography
Summary of paper I……………………………………………………….
Paper II……………………………………………………………………………...
Optimization of the chiral separation of citalopram and
its demethylated metabolites
Chiral separation of citalopram
Optimization with Modde
Optimization with Multisimplex
Separation mechanism
Summary of paper II………………………….. ………………………….
Paper III……………………………………………………………………………..
Citalopram and adolescents
Citalopram extraction version II
Results, citalopram and adolescents
Other examples where chiral analysis has been of value
Animal studies
Summary of paper III……………………………………………………....
Paper IV……………………………………………………………………………..
Enantioselective analysis of citalopram in postmortem blood and genotyping
Citalopram extraction version III
Drug levels in post mortem whole blood versus plasma concentrations
from patients with toxicological symptoms
Genotyping
Summary of paper IV………………………………………………………
Conclusions………………………………………………………………………….
Chemometrics-Experimental design
Therapeutic drug monitoring: Achiral or chiral analysis of citalopram
Forensic chemistry: Achiral or chiral analysis of citalopram
Future studies………………………………………………………………….…….
LCMS
On-line extraction and chiral analysis
Free fraction
Chiral switches
Acknowledgements………………………………………………………………….
References…………………………………………………………………………...
Appendix……………………………………………………………………….….…
51
51
56
57
65
66
71
73
75
76
77
79
80
91
List of Abbreviations
5-HT
Į
ĮCT
ĮDCT
ĮDDCT
Į1
Į1
ANOVA
AUC
C2, C8, C18
Cit
DCit
DDCit
S-Cit
R-Cit
S-DCit
R-DCit
S-DDCit
R-DDCit
CitNO
CitAld
CitProp
CCD
CCC
CCF
CD
E-CD Ac
CN
CSP
CYP
EM
PM
UM
HPLC
HSA
IS
LCMS
MAO B
MLR
NA
Pgp
PLS
PRESS
RSM
SSRI
TDM
UV
serotonin, 5-hydroxytryptamine
separation factor, see appendix
separation factor for citalopram enantiomers
separation factor for desmethylcitalopram enantiomers
separation factor for didesmethylcitalopram enantiomers
separation factor 1 between enantiomeric pairs
separation factor 2 between enantiomeric pairs
analysis of variance
area under the concentration time curve
2, 8 or 18 carbon chains bound to silica
racemic citalopram
racemic desmethylcitalopram
racemic didesmethylcitalopram
S-(+)-citalopram (escitalopram)
R-(-)-citalopram
S-(+)-desmethylcitalopram
R-(-)-desmethylcitalopram
S-(+)-didesmethylcitalopram
R-(-)-didesmethylcitalopram
citalopram N-oxide
citalopram aldehyde
citalopram propionic acid
central composite design
central composite circum scribed
central composite face centred
cyclodextrin Į, E and J
acetylated E-cyclodextrin
cyanogroup bound to silica
chiral stationary phase
cytochrome P 450
extensive metabolizer
poor metabolizer
ultrarapid metabolizer
high performance liquid chromatography
human serum albumin
internal standard
liquid chromatography mass spectrometry
monoamine oxidase B
multiple linear regression
noradrenaline, norephinephrine
P-glycoprotein
partial least square
prediction residual sum of squares (SS)
response surface method
selective serotonin reuptake inhibitor
therapeutic drug monitoring
ultra violet
7
Introduction
Stereochemistry (from the Greek “stereos”, meaning solid) refers to chemistry in three
dimensions. Most molecules have a three-dimensional extension in space and if the
chemical compound can not be superimposed on its mirror image it is chiral (greek
“cheir” meaning hand). These molecules related as mirror images are called
enantiomers and they differ only in their spatial arrangement and how they direct plane
polarized light.
Over a century ago Pasteur pointed out the connection between chiral molecules and
living matter. The chiral nature of living systems has evident implications on
biological activity since all of the crucial biopolymers associated with life are
homochiral. The building blocks of life, peptides, proteins and polysaccharides, are
made up of L-amino acids and D-sugars. As a consequence, metabolic and regulatory
processes mediated by biological systems are sensitive to stereochemistry and different
responses can often be observed when comparing the activities of a pair of
enantiomers. The stereochemistry involved in the concept of receptors and other
targets for drugs has been recognized for a long time, however the involvement of
stereochemistry in the process of metabolising drugs has only more recently been
taken into account.
A new tool is provided by the knowledge of the genetics behind polymorphic drug
metabolizing enzymes and why some individuals are poor metabolizers and some
are ultrarapid metabolizers and the consequences for drug effects and metabolism.
Within the field of depression the “monoamine hypothesis” has been the leading
theory to explain the biological basis of depression. This theory proposes that the
biological basis of depression is due to a deficiency in one or more of three key
neurotransmitter systems, namely noradrenaline, dopamine and serotonin, which are
thought to mediate the therapeutic actions of virtually every known antidepressant
agent.
Among the selective serotonin-reuptake inhibitors introduced for treatment of
depression is citalopram. Citalopram is a racemic compound, in other words a mixture
of 50 % of each of two enantiomers and in this case with one of the enantiomers
accounting for the inhibitory effect.
The advances within chiral separation techniques have made the measurement of the
concentrations of the individual enantiomers in biological fluids possible. The
processes behind enantioselective separation are however not fully understood and the
mechanism behind the separation of enantiomers can further be scrutinized by the use
of multivariate methods.
In this thesis analytical separation methods for achiral as well as chiral
analysis of citalopram and its metabolites have been developed. These methods have
further been used as tools in clinical as well as toxicological investigations concerning
citalopram.
9
Chirality
Stereochemistry – chemistry in three dimensions
The term “chirality” is derived from the Greek word for hand ("cheir") and reflects the
fact that the human body is a chiral structure, the right hand, for example, can be said
to be the enantiomer of the left. Molecules that make up living things tend to be chiral:
they have the property of “handedness” and a preference for one kind of mirror-image
isomers or enantiomers (Testa, 1986). Life, in the way we know it, has reached a high
degree of specialization. At the molecular level the macromolecules, for example
proteins and nucleic acid that make up the structures of the body, all contain chiral
structures that use only one of the possible stereoisomers. These biomolecules are
made up of units that have the same configuration of chirality, the proteins are made
up of the L-enantiomers of amino acids whereas most carbohydrates are made up of
sugar monomers with D-configuration. Essential physiological processes are,
therefore, homochiral, they show 100 % stereoselectivty and only involve one of all
the possible stereoisomers of key molecules (Bonner, 2000; Popa, 1997).
We could say that life is homochiral, that it only accepts molecules with the right
configuration. Through this, receptors and enzymes in our body have been obliged to
be able to differentiate between these enantiomers or optical isomers and they can
interact/bind to only one of these molecules. As a result, the structures with which
drugs interact are “handed” and can tell the difference between drug enantiomers, just
as clearly as people can tell if they put their left hands into right-handed gloves.
Discrimination between molecules is therefore at the heart of biology and at the heart
of pharmacology. Therefore, it is not surprising that the enantiomers of the chiral
molecule can have vastly different effects and toxicities (Eliel and Wilen, 1994;
Wainer, 1993).
A good example of this is the interaction of the chiral compound carvone with the
human olfactory receptors. Carvone exists as two enantiomers (R)- and (S)-carvone,
were the (R)-enantiomer is responsible for the distinct taste and smell of oil of
spearmint whereas the S-enantiomer taste and smell of caraway (Allenmark, 1991; Williams
and Wainer, 2002).
The origins of stereochemistry stem from the discovery of plane polarized light and
the discovery by Biot (in 1812) that a quartz plate, cut at right angle to its crystal axis,
rotates the plane polarized light through an angle proportional to the thickness of the
plate. Biot extended his observation to solids and solutions. He recognized the
difference between the rotation produced by quartz and that produced by the organic
substances.
Later on, Pastuer (1848), by recrystallizing racemic sodium ammonium tatrate,
observed that two different crystal forms were formed. He was able to separate these
two forms using a lens and a pair of tweezers and when he separately redissolved the
two kinds of crystals, he found that they rotated plane polarized light differently.
Pastuer postulated that the molecular structures of (+)- and (-)-tartaric acids must be
10
related as an object to its mirror without the knowledge of its molecular structure (Eliel
and Wilen, 1994; Mason, 1986). This method of manual sorting the crystals was
followed by a second method where Pastuer discovered the use of an optically active
alkaloid base to form diastereomeric salt of (+)- and (-)-tartaric acids that have
different solubilities and a third where he found that Penicillium glaucum grown on
racemic tartaric acid, preferentially uses (+)-tartrate as a carbon source (Mason, 1991).
The molecular basis for this observation was solved in 1874 when van´t Hoff and Le
Bel independently and almost at the same time proposed that the four valences of the
carbon atom were not planar, but directed into a three-dimensional space. A tetrahedral
structure, with the four different groups pointing toward the vertices of a regular
tetrahedron, would give two distinctly different nonsuperimposable forms. van´t Hoff
also defined the carbon atom as asymmetric (Wainer, 1993), see figure 1.
In the literature, three main systems of nomenclature are used, each based on a
separate set of principles.
I. Stereoisomers can be defined according to how they rotate the plane of polarized
light in one direction or another. The magnitude of optical rotation is dependent on a
variety of factors, such as analyte concentration, solvent, temperature, wavelength, etc.
Dextrorotatory [(+) or (d-)] denotes a clockwise rotation of the plane of plane
polarized light, while levorotatory [(-) or (l-)] denotes a counter clockwise rotation
(Allenmark, 1991).
II. The second method is derived from knowledge of the absolute structural
configuration of the stereoisomers. To determine the absolute configuration of a chiral
center, the rules proposed by Cahn, Ingold and Prelog (Eliel and Wilen, 1994) are
used. The enantiomers of a chiral compound are termed R (from rectus, Latin for right)
and S (from sinister, Latin for left) based on the sequence of groups around its chiral
center, see figure 2.
III. The final method is based on the structure of the molecule relative to a reference
molecule. A system commonly used in biochemistry where the amino acids and
carbohydrate are termed L- and D- with reference in many cases to the dextrorotary
form of glyceraldehyde (Eliel and Wilen, 1994).
Figure 1.
An asymmetric tetravalent carbon atom and its mirror image.
11
3
2
F
F
Cl
Br
3
4
Br
H
4
H
Cl
1
2
1
Assign a sequence of priority to the
atoms or groups of atoms attached to the
chiral center. If the four atoms have
different atomic numbers, then priority is
assigned from highest to lowest atomic
number.
Visualise so that the lowest priority atom
is directed away from you. Observe the
arrangement of the remaining ligands.
F
F
Cl
H
Br
Br
H
Cl
If the path goes clockwise, then the
absolute configuration is R. If the path
goes counterclockwise, the absolute
configuration is S.
Figure 2.
The Can Ingold Prelog system.
The chirality rule requires that the model is viewed from the side opposite to that
occupied by the atoms with lowest priority. The remaining three ligands then present a
tripodal array, with the legs extending towards the viewer. If the sequential
arrangement of direction of these three atoms (1,2,3) is clockwise, the configurational
descriptor is R (for Latin rectus meaning right), if it is counter clockwise, it is S (for
Latin sinister meaning left) (Eliel and Wilen, 1994).
12
Basic terminology of stereochemistry
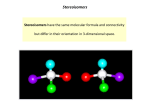
Isomers
Compounds that have the same molecular formula but differ in the way the
constituent atoms are linked together.
Stereoisomers
Compounds having the same molecular formula but with the atoms in a different
three-dimensional arrangement. Stereoisomers can be divided into two distinct
categories, enantiomers and diastereomers.
Enantiomers
Compounds that contain the same atoms linked together in the same way but in a
different three-dimensional arrangement. Enantiomers have identical physical
properties, but rotate the plane of polarised light in opposite directions.
Diastereoisomers
Stereoisomers that are not enantiomeric to each other. They characteristically occur
in molecules with two or more chiral centres. Such compound have 2n
stereoisomers, where n is the number of chiral centers.
Achiral
An entity, such as a molecule, is achiral if it is superposable with its mirror image
Chiral
Not superposable with its mirror image, as applied to molecules, conformations, as
well as macroscopic objects, such as crystals.
Homochiral
Isometric molecules are homochiral if they have the same sense of chirality, that is,
if they are all R or all S.
Chiral centre
Atoms, usually carbon, attached to four different substitutions that could be
swapped to create a new stereoisomer.
Racemate
A mixture of all possible stereoisomers of a compound in equal proportions. It does
not have optical activity.
Stereoselective
Relating primarily to one specific stereoisomer. A biological reaction is
stereospecific if either the substrate or its binding is chiral.
13
Drugs and Stereochemistry
The concept of stereochemistry has been understood by scientists for more than a
hundred years. Many of the drugs used through the years were originally from extract
of plants. In these cases the active moiety taken out was a pure enantiomer. The efforts
by the organic chemists to copy the nature ran into trouble, since the asymmetric
carbon atom with the binding of four different substituents gave rise to a racemic
mixture with two mirror images if there was one asymmetric carbon atom in the
molecule.
For some reason, this knowledge of racemic organic compounds and racemisation
seemed to be forgotten until the question of racemic compounds was raised by Ariens
in the late 1980s (Ariens, 1984; Ariens and Wuis, 1987; Ariens et al., 1988). He asked
the question why we in some cases had to give doses to the patient where half of is
content had no effect or the opposite effect. He also proposed that the active
enantiomer should be named the eutomer and the less active being called the distomer
and that the ratio between the eutomer/distomer (the eudomistic ratio) should be
calculated and could be used as to measure the difference in effect (Ariens, 1986).
After this rediscovery of stereochemistry, the regulatory authorities defined more strict
requirements about drug discovery and chiral compounds (Strong, 1999). A lot of
efforts have, since this regulation, been made in organic chemistry labs to directly
synthesize pure enantiomers or to separate the enantiomers on an industrial scale after
synthesis of a racemic mixture. The possibility of imitating the nature and only
synthesising the desired enantiomer has been possible by the work by the Nobel Prize
winners Knowles, Noyori and Sharpless (http://www.almaz.com/nobel). Single–
enantiomer drug sales show a continuous growth worldwide and many of the topselling drugs are marketed as single enantiomer (Maier et al., 2001; Stinson, 1999).
Chirality and Psychopharmacology
The biosynthetic pathways for noradrenaline (NA) and serotonin (5-HT) are good
examples of stereochemistry within mammalians.
The precursor for NA is L-tyrosine, an aromatic amino acid present in body fluids.
Tyrosine hydroxylase is the enzyme that then converts L-tyrosine to L-dopa (Ldihydroxyphenylalanine), which yields dopamine after enzymatic decarboxylation by
DOPA decarboxylase. DOPA decarboxylase is totally stereospecific, acting only on
the L-enantiomer. L-dopa is used as an antiparkinson agent while D-dopa is
responsible for serious adverse reactions such as agranulocytosis (Scott, 1993;
Williams and Wainer, 2002). Dopamine E-hydoxylase then converts, with a
completely stereospecific enzymatic hydroxylation, dopamine into the chiral
compound L-noradrenaline and is finally changed into adrenaline (epinephrine) by Nmethylation (Allenmark, 1991; Rang et al., 1999).
For 5-HT the biosynthesis follows a pathway similar to that of NA, except that the
precursor amino acid is L-tryptophan instead of L-tyrosine. Tryptophan is converted to 5hydroxytryptophan by tryptophan hydroxylase and then decarboxylated stereospecific
by DOPA decarboxylase to 5-HT. See figure 3.
14
CO2
CO2
NH3
H
NH2
NH3
Tyrosine
hydroxylase
H
DOPA
decarboxylase
HO
OH
HO
OH
L-Tyrosine
OH
L-dihydroxyphenylalanine
(L-dopa)
Dopamine
H NH2
HO
H NHCH3
Phenylethanolamine HO
N-methyl transferase
HO
H
N
H
NH2
HO
OH
OH
L-Noradrenaline
Adrenaline
Tryptophan
hydroxylase
CH2CCOOH
L-Tryptophan
Dopamine
E-hydroxylase
H
N
H
DOPA
decarboxylase
CH2CCOOH
HO
HO
H
N
CH2CH2NH2
NH2
5-hydroxytryptophan
Serotonin (5-hydroxy-tryptamine)
Figure 3.
Stereochemistry involved in the biosynthesis of noradrenaline and serotonin.
15
Chirality and Pharmacodynamics
Among the newer antidepressant drugs, acting on the NA or 5-HT system, many of
them are racemic. For example pharmacodynamic investigations of their action on
receptor subtype show a marked difference between the mirtazapine enantiomers as
well as the fluoxetine enantiomers (Baumann and Eap, 2001; Baumann et al., 2002).
Another example is the selective noradrenaline reuptake inhibitor reboxetine where the
S,S-(+)-enantiomer is approximately 20 times as potent as the R,R-(-)-enantiomer in
inhibiting noradrenaline uptake (Caccia, 1998).
These examples also show that the system proposed by Ariens dealing with eutomer
and distomer have had to be revised and the racemic drugs are better classified as
follows:
I Enantiomer 1 possesses the activity of interest, enantiomer 2 possesses no activity
II Enantiomer 1 possesses the activity of interest, enantiomer 2 possesses some
activity of interest
III Enantiomer 1 possesses the activity of interest, but that which enantiomer 2
possesses is an antagonist of enantiomer 1
IV Enantiomer 1 possesses the activity of interest and enantiomer 2 possesses a
separate activity of interest
V Enantiomer 1 possesses the activity of interest, but enantiomer 2 possesses separate
undesirable activity
Chirality and Pharmacokinetics
Drug absorption, distribution, and excretion are generally processes which do not
differentiate between enantiomers, but when the drugs interact with an enzyme or a
transporter system, a chiral discrimination may be seen and these enantioselective
processes may affect the pharmacokinetics of some drugs.
The pharmacological effect of a drug is directly related to the free fraction rather than
total concentration of the drug in plasma (Simonyi et al., 1986). The major component
of plasma proteins responsible for binding of drugs is human serum albumin (HSA),
which plays a fundamental role in the transport of drugs (mostly acidic compounds),
metabolites, and endogenous ligands. Binding to HSA controls the free, active
concentration of a drug, provides a reservoir for a long duration of action, and
ultimately affects drug absorption, metabolism and excretion. Oxazepam and its
derivates have shown stereoselective binding to HSA (Bertucci and Domenici, 2002;
Kaliszan et al., 1995; Wainer, 1993). Alpha-1-acid glycoprotein, normally present in
plasma at concentrations approximately 100 times lower than HSA, preferably binds
basic drugs, and chiral discrimination between the enantiomers has been shown for
methadone, where R-methadone displays a significantly greater unbound fraction. This
reflects the higher affinity of S-methadone to the main plasma binding protein
(Baumann et al., 2002).
Drug absorption and disposition are regulated, in part, by transport across epithelial
barriers. Transport across specialized capillaries of the blood brain barrier is important
16
for the distribution of drugs to the brain (Evans, 2000; Owens et al., 2001; Schwab et
al., 2002; Tanaka, 1999b). The single enantiomers escitalopram (S-Cit) and Rfluoxetine have been shown to be potent inhibitors of the serotonin transporter protein
(Owens et al., 2001). The transport mechanism for citalopram (Cit) over the bloodbrain barrier is a non-stereoselective, bidirectional and symmetrical carrier-mediated
mechanism without the influence of active efflux mechanisms (Rochat et al., 1999).
The role of P-glycoprotein (Pgp), a drug transporter, in drug disposition includes a
urinary excretion mechanism in the kidney, a biliary excretion mechanism in the liver,
an absorption barrier and determinant of oral bioavailability, and the blood-brain
barrier that limits the accumulation of drugs in the brain. The inhibition of the
transporting function of Pgp can cause clinically significant drug interactions and can
also increase the penetration of drugs into the brain, as well as their accumulation
(Schwab et al., 2002; Tanigawara, 2000). The enantiomers of mefloquine, an
antimalarial agent, interact stereospecifically with Pgp, were the (+)-mefloquine
competitively displaced the Pgp substrate cyclosporine whereas (-)- mefloquine had no
effect on cyclosporine-Pgp binding (Williams and Wainer, 2002).
Chiral discrimination can occur at both the substrate and product levels in drug
metabolism where five different stereochemical conversions may be possible:
I. Prochiral to chiral, a non-chiral compound can become chiral, e.g. the metabolism of
risperidone to the hydroxylated metabolites (+)- and (-)-9-hydroxyrisperidone (YasuiFurukori et al., 2001).
II. Chiral to chiral, numerous of chiral compounds are converted to a chiral metabolite,
e.g. S-fluoxetine to S-norfluoxetine (Caccia, 1998).
III. Chiral to diastereomeric, can occur by conjugation with glucoronic acid, glucose,
glutathione and glutamine which are agents derived from the chiral pools within the
body and of fixed configuration (Caldwell, 1995).
IV. Chiral to non-chiral transformation, chirality may be lost by oxidative metabolism
at a chiral centre, e.g., oxidation of secondary alcohols to yield ketons (Caldwell,
1995).
V. Chiral inversion can be exemplified by racemic ibuprofen when given to humans, a
substantial fraction of the dose of R-(-)-ibuprofen (50 - 60 %) undergoes "metabolic
inversion" to yield S(+)-ibuprofen (Evans, 2001).
17
Selective serotonin reuptake inhibitors
Psychologists and neurobiologists sometimes debate whether it is ego-damaging
experiences and self-deprecating thoughts or biological processes that cause
depression. The mind, however, does not exist without the brain. Considerable
evidence indicates that regardless of the initial triggers, the final common pathways to
depression involve biochemical changes in the brain (Stahl, 2000).
For over 30 years, the leading theory to explain the biological basis of depression has
been the “monoamine hypothesis of depression”. This theory proposes that the
biological basis of depression is due to a deficiency in one or more of three key
neurotransmitter systems, which are thought to mediate the therapeutic actions of
virtually every known antidepressant agent. These systems are NA, dopamine and 5HT.
The development and introduction of selective serotonin-reuptake inhibitors (SSRIs),
including fluoxetine, sertraline, paroxetine, fluvoxamine, and citalopram, represent an
important advance in the pharmacotherapy of psychiatric disorders, not only
depression, but also a wide range of psychiatric disorders from anxiety disorders to
bulimia (Goldstein and Goodnick, 1998; Goodnick and Goldstein, 1998a; Goodnick
and Goldstein, 1998b; Lane et al., 1995; Masand and Gupta, 1999; Murphy et al.,
2000). The SSRIs are chemically unrelated to tricyclic, heterocyclic, and other firstgeneration antidepressants, (Hyttel, 1994) see figure 4. There is also an increasing use
of these new agents for treatment of childhood anxiety disorders (DeVane and Sallee,
1996; Murphy et al., 2000). These five drugs have the predominate effect of inhibiting
the neuronal reuptake of serotonin. SSRIs are the treatment of choice for many
indications, including major depression, dysthymia, panic disorder, obsessivecompulsive disorder, eating disorders, and premenstrual dysphoric disorder, because of
their efficacy, good side-effect profile, tolerability, and safety when overdosed, as well
as with regard to patient compliance (Boerner and Moller, 1999; Pacher et al., 1999).
Pharmacokinetic properties are different due to stereochemistry, metabolism,
interaction/inhibition with cytochrome P450 enzymes (CYP), and participation in drug-drug
interactions. An observable difference between the SSRIs is their different potential
for drug-drug interaction within the CYP enzyme system (Hiemke and Hartter, 2000).
Side effects of SSRIs include gastrointestinal disturbances, headache, sedation,
insomnia, activation, weight gain, impaired memory, excessive perspiration,
paresthesia, and sexual dysfunction (Edwards and Anderson, 1999; Masand and
Gupta, 1999).
SSRIs are prescribed alone and in combination with other psychotropic medications in
the treatment of a variety of psychiatric disorders. Such combinations create the
potential for pharmacokinetic interactions, by affecting the activity of the
drug metabolizing oxidative enzymes (Naranjo et al., 1999; Preskorn, 1998;
Sproule et al., 1997).
18
NC
CH3
O
N CH3
F3C
H
N
O
CH3
Fluoxetine
F
Citalopram
H3C
H
N
NH
O
O
O
F
Paroxetine
Cl
Sertraline
O
N
Cl
NH2
O
CH3
N
Cl
CH2CH2CH2N(CH3)2
F3C
Fluvoxamine
Clomipramine
Figure 4.
The chemical structures of the SSRIs fluoxetine, sertraline, paroxetine, fluvoxamine,
and citalopram and the tricyclic antidepressant clomipramine
Fluoxetine and citalopram are racemic drugs while sertraline and paroxetine have been
launched as single enantiomers. Fluvoxamine is achiral.
19
Cytochrome P450 enzyme system
The reactions catalyzed by drug (xenobiotic)-biotransforming enzymes are generally
divided into two groups, namely phase I and phase II reactions. Phase I reactions
involve hydrolysis, reduction, and oxidation, whereas phase II biotransformation
reactions include glucorinidation, sulfation, acetylation, methylation, conjugation with
gluthatione (mercapturic acid synthesis), and conjugation with amino acids (such as
glycine, taurine, and glutamatic acid). Phase I biotransformation of drugs often
precedes phase II biotransformation and is slower. For this reason, phase I
biotransformation (such as oxidation of drugs by cytochrome P450 enzymes) tends to be
the rate-limiting step in the overall metabolism (Molinoff and Ruddon, 1996).
The CYP superfamily is a group of heme-proteins involved in the metabolism of
exogenous substances such as drugs and chemicals but also of endogenous substances
such as prostaglandins, fatty acids and steroids. The similarities in amino acid
sequence are the base for how these enzymes are divided into families and subfamilies
within this CYP superfamily (van der Weide and Steijns, 1999)
www.imm.ki.se/CYPalleles. The human hepatic CYP system consists of over 30
related isoenzymes with different, sometimes overlapping, substrate specificity.
Approximately 40% of human CYP-dependent drug metabolism is carried out by
polymorphic enzymes, which can cause abolished, quantitatively or qualitatively
altered or enhanced drug metabolism, resulting principally in three types of
phenotypes poor (PM), extensive (EM) and ultrarapid metabolizers (UM) in the
population (Ingelman-Sundberg et al., 1999). All enzymes exhibit major variability in
their activity between different subjects, which is partially caused by genetic factors
(in particular for those enzymes exhibiting genetic polymorphism: such as CYP2D6
and CYP2C19) and by environmental factors (Lin and Lu, 1998; West et al., 1997).
Among enzymes, CYP1A2, CYP2C19, CYP2D6 and CYP3A4 are the most important
enzymes involved in the metabolism of antidepressants or in the occurrence of drug
interactions (Brosen, 1996; Dahl, 2002; Kirchheiner et al., 2001; Meyer et al., 1996;
Nemeroff et al., 1996; Poolsup et al., 2000; Tanaka and Hisawa, 1999).
It is widely accepted that drug metabolism is often responsible for stereoselective
disposition and hence factors modulating the activity of drug-metabolizing enzymes
can modify this effect (Baumann and Rochat, 1995; Eap et al., 2000; Kroemer et al.,
1996).
CYP activities in the brain have also been described where the local cerebral
metabolism of psychotropic drug may have pharmacological and/or toxicological
consequences (Voirol et al., 2000). While the genotype describes the native level of
enzymatic activity, changes in the relative levels and activities of a metabolizing
enzyme can be produced by drug interaction and/or clinical conditions such as disease
progression or malnutrition, and can result in another phenotype (Dahl, 2002; Linder et
al., 1999; Naranjo et al., 1999).
20
CYP1A2
CYP1A2 is expressed at a substantial level in human liver, accounting for
approximately 13 % of the total CYP content (Scordo, 2003). Among the psychotropic
drugs, fluvoxamine is a potent inhibitor of the enzyme (Jeppesen et al., 1996) while
amitriptyline, clomipramine and imipramine undergoes demethylation (Bertilsson and
Dahl, 1996; Coutts and Urichuk, 1999; Nemeroff et al., 1996). Clozapine is known to
be metabolized to a major extent by CYP1A2 (Bertilsson et al., 1994).
Smoking and polycyclic hydrocarbons induce this enzyme. CYP1A2 shows a great
inter-individual variability and it is also polymorphically expressed, with
approximately 50 % of Caucasians being slow or intermediate metabolizers (Landi et
al., 1999). Phenotyping with caffeine has revealed that there is marked inter-individual
variability in the expression of CYP1A2 (Carrillo et al., 2000; Landi et al., 1999).
CYP2C19
CYP2C19 accounts for about 3 % of the total CYP content in the liver (Scordo, 2003).
Psychotropic drugs such as hexobarbital, diazepam, citalopram, imipramine,
clomipramine and amitriptyline are metabolized via CYP2C19 (Poolsup et al., 2000).
Fluvoxamine and fluoxetine are moderate inhibitors (Jeppesen et al., 1996) and the
antiepileptics carbamazepine and phenytoin induce CYP2C19 (Scordo and Spina,
2002). Deficiencies of these enzymes are inherited as autosomal recessive traits, which
result from a variety of mutations. About 20 % of Japanese subjects are poor
metabolizers of S-mephenytoin (an anticonvulsant metabolized by CYP2C19),
whereas less than 3 % of Caucasians are affected. In Caucasians, a single base pair
mutation (m1) is the major defect responsible for the PM phenotype, where the m2
mutation is more important in Orientals (de Morais et al., 1994a; de Morais et al.,
1994b).
CYP2D6
CYP2D6 accounts for about 2 % of the total CYP content in the liver (Scordo, 2003).
It is quantitatively one of the less prominent hepatic enzymes. CYP2D6 metabolizes a
number of antidepressants, antipsychotics, beta-adrenoreceptor blockers, and
antiarrhythmic drugs (Dahl and Sjöqvist, 2000; Otani and Aoshima, 2000; Poolsup et
al., 2000). Deficiencies of these enzymes are inherited as autosomal recessive traits,
which result from a variety of mutations leading to about 7 % of the Caucausians not
having a functional enzyme (PM). In addition to the defective CYP2D6 alleles, several
genes cause impaired enzyme activity where a subgroup of 10-15% of Caucasians are
termed phenotypical intermediate metabolizers of drug substrates of CYP2D6
(Raimundo et al., 2000). On the other hand, there are some individuals that have
unusually high levels of enzyme activity, presumably because of gene duplication,
named ultrarapid metabolizers (UM) (Bernal et al., 1999; Dahl et al., 1995; Lundqvist
et al., 1999). This high capability of metabolization is explained by a duplication or
multiduplication of a functional CYP2D6*2 gene (Lundqvist et al., 1999).
21
Several antidepressants are inhibitors of the CYP2D6 enzyme, although the individual
SSRIs differ in potency of this effect. Co-pharmacy should be considered as a
possibility for pharmacokinetic drug interactions leading to increased plasma drug
concentrations (Kirchheiner et al., 2001; Lam et al., 2002).
CYP3A4
CYP3A4 is the predominant CYP enzyme in the liver, accounting for approximately 30 %
of the total P-450 content, and the enzyme is also expressed in gut mucosa (Scordo,
2003). Unlike other human CYPs (e.g. CYP2D6 and CYP2C19), there is no evidence
that CYP3A4 exhibits genetic polymorphism. There are genetic variations in the
flanking, intronic end exonic regions of the gene that may influence the level of
functional CYP3A4 protein, however full length mRNA has been detected in all adults
studied to date (Lamba et al., 2002). The new antidepressant nefazodone is
metabolized by this enzyme (Spina and Scordo, 2002). Most of the SSRIs with the
exception of norfluoxetine do not inhibit this enzyme, and interactions between SSRIs
and CYP3A4 would not appear to be significant (Brosen, 1998).
Drugs can not only be substrates for CYP3A4 or an inhibitor. They can also be
inducers of a CYP3A4 and thereby increase the activity of the enzyme. One example
of this is carbamazepine that is both a substrate and inducer of CYP3A4 (Levy, 1995;
Tanaka, 1999a).
22
Citalopram a selective serotonin reuptake
inhibitor
Overview
Citalopram is a selective serotonin reuptake inhibitor with no, or only minimal, effect on
NA and dopamine reuptake (Sanchez and Hyttel, 1999). The ability of Cit to
potentiate serotonergic activity in the central nervous system via inhibition of the
neuronal reuptake of 5-HT is thought to be responsible for its antidepressant action
(Joubert et al., 2000).
It has been shown that only one of the enantiomers, S-Cit, stands for the reuptake
inhibitor effect (Hyttel et al., 1992). S-Cit has recently been introduced onto the
market under the name of escitalopram (Cipralex®, Swedish trade name)
(Montgomery et al., 2001).
Chemical and physical properties
Cit with the molecular formula of the racemate (RS)-1-[3-(dimethylamino)propyl]-1(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitril has one asymmetric carbon
atom in the 1 position in the isobenzofuran ring, leading to a racemic formulation, of
the S(+)-enantiomer (S-Cit) and the R-(-)-enantiomer (R-Cit). The molecular weight
of the hydrobromide is 405.31 g/mol and the white to off-white crystals of the
racemate have a melting point of 185-188 qC. The hydrobromide salt is sparingly
soluble in water and it is soluble and stable in ethanol for up to one year at +4 qC. The
pKa for the base (molecular weight 324.39 g/mol) of Cit is 9.5. The structures of the
enantiomers are chemically unrelated to other SSRIs and antidepressants (se figure 4
on page 19).
Citalopram and the cytochrome P450 superfamily
Fenotyping data obtained by using sparteine and mephenytoin showed that CYP2D6
and CYP2C19 partially contributed to the metabolism of Cit to desmethylcitalopram
(DCit) and that the further metabolism of DCit to didesmethylcitalopram (DDCit)
appeared to a large extent to be mediated via CYP2D6 (Sindrup et al., 1993). Further
studies in human liver microsomes found that even CYP3A4, but not CYP1A2, was
involved in the N-demethylation of Cit, (Kobayashi et al., 1997; Rochat et al., 1997)
but the relative contribution of CYP2D6, CYP2C19 and CYP3A4 were not in
accordance. Other studies in human liver microsomes have indicated that, at
therapeutic concentrations, CYP3A4 was responsible for 40-50 % of the formation of
DCit, while the contribution of CYP2C19 increased and that of CYP2D6 tended to
decrease with increasing drug concentration (Olesen and Linnet, 1999; von Moltke et
al., 2001). CYP2D6 exclusively mediated the second demethylation step, and
citalopram N-oxide (Cit-NO) was also exclusively formed by CYP2D6.
23
As Cit is a racemic compound, the stereoselective metabolism has also been studied
showing that the three enzymes CYP2D6, CYP2C19 and CYP3A4 favour a more
rapid demethylation of the S-Cit over the R-Cit. Further metabolism of DCit to DDCit
is also stereoselective but here the R-DCit is favoured over the S-DCit (Olesen and
Linnet, 1999). Another metabolite that is not formed by these P450 enzymes is a
propionic metabolite formed by deamination of Cit, DCit and DDCit via monoamine
oxidase B (MAO B). This process is also stereoselective where the production of S-Cit
propionic acid was 5.6 times higher than R-Cit propionic (Kosel et al., 2001; Rochat
et al., 1998). In figure 5, the metabolic pathways are summarized.
These in vitro experiments have been followed up by in vivo studies on healthy
volunteers, showing that the AUC of S-, but not R-Cit is significantly higher in PM of
CYP2C19 (Herrlin, 2001). Steady-state pharmacokinetics of the enantiomers of Cit,
DCit and DDCit in healthy human subjects who were extensive metabolizers by
CYP2D6 and CYP2C19, showed, after multiple doses of Cit, for 21 consecutive days
(40 mg per day), that the (S)(+)-enantiomers were eliminated faster than their
antipodes (Sidhu et al., 1997).
A single dose of ketozonazole, a potent inhibitor of CYP3A4, does not have any effect
on Cit pharmacokinetics (Gutierrez and Abramowitz, 2001). Cit and concomitant
treatment with fluvoxamine, a CYP2C19 inhibitor, increased the average plasma levels
of S-Cit and R-Cit (Bondolfi et al., 1996).
Carbamazepine, an inducer of CYP3A4 gave a considerable decrease in Cit plasma
concentrations (Leinonen et al., 1996; Leinonen et al., 1991; Steinacher et al., 2002)
but in these studies, concentration determination of Cit enantiomers has not been performed.
24
NC
CH3
O
N CH3
CYP2D6
NC
NC
CH3
O
O
N CH3
CH2OH
O
Citalopram (Cit)
CitNO
MAO-A
MAO-B
F
CYP2C19
CYP3A4
CYP2D6
CH3
NC
O
CitOH
F
F
CYP? other
enzymes
NC
N H
25
MAO-A
MAO-B
NC
O
O
C H
Aldehyde
oxidase
O
COOH
DCit
CitProp
CitAld
F
F
CYP2D6
?
H
NC
O
CYP?
unknown metabolite(s)
N H
MAO-A
MAO-B
DDCit
F
F
Figure 5.
Summary of the metabolic pathways for Cit
and its metabolites.
Pharmacokinetics
The absorption of Cit is not affected by food, and its oral bioavailability is reported to
be approximately 80 % (Joffe et al., 1998). Peak plasma levels occur at about 4 hours
(range 1 to 6 hours) after single or multiple doses and the plasma concentrations of the
metabolites are much less than the plasma concentration of Cit (Baumann and Larsen,
1995).
Cit is bound to plasma protein to 80 %, while the protein binding of the demethylated
metabolite is lower, 74 %. It is widely distributed among peripheral tissues, with the
volume of distribution estimated to 14 L/kg (Fredricson Overo, 1982; Joffe et al.,
1998; Kragh-Sorensen et al., 1981).
The single- and multiple dose pharmacokinetics of Cit are linear and dose proportional
over a range of 10-60 mg/day. Steady-state plasma levels are achieved in patients in 12 weeks. At a daily dose of 40 mg, the average plasma concentration is about 83 ng/ml
(255 nmol/L) (n=114) with a range from 20-200 ng/ml (62-620 nmol/L) (Fredricson
Overo, 1982). However, no clear correlation has been established between plasma
concentration and clinical response due to, among other things, that Cit is a racemic
compound.
The elimination half-life of Cit is approximately 33 hours (range 23-45 hours) (KraghSorensen et al., 1981) which allows recommendation for once-daily dosing. The
systemic Cit plasma clearance is 0.33 L/min. Cit is eliminated primarily via the liver
(85 %) and the remainder via the kidneys, approximately 12 % (range 6-21 %) of the
daily dose is excreted in urine as unchanged Cit (Kragh-Sorensen et al., 1981), see
table 1.
In the elderly, clearance after oral administration was reduced to 25-75 % of the values
in younger volunteers and the half-life was longer. Steady state plasma concentrations
of Cit were higher than expected but there were no signs or symptoms of adverse
effects in patients receiving therapy for 3 weeks (Fredericson Overo et al., 1985;
Gutierrez and Abramowitz, 2000). In patients with a mild to moderate renal
dysfunction, the clearance after oral administration of Cit was decreased by 17 %,
T1/2 was moderately increased and peak plasma concentration was unaffected. Impaired
hepatic function reduced the oral clearance of Cit by 37 % and doubled the T1/2
(Joffe et al., 1998).
26
Parameter
Bioavailability (F)
Plasma protein binding
Time to peak plasma level (Tmax)
Mean peak plasma concentration Cit (Cmax)
Volume of distribution (Vd)
Metabolizing enzymes
Metabolites
Value
80 %
80 %
2-4 h
311 nmol/L (40 mg/day)
12-16 L/kg
CYP2C19, 3A4, 2D6, MAO-B
Desmethylcitalopram
Didesmethylcitalopram
Propionic acid derivates
Citalopram-N-oxide
Steady-state trough plasma concentration (mean±SD):
Citalopram
Desmethylcitalopram
Didesmethylcitalopram
Systemic Clearance (Cl)
Elimination half-life (T1/2):
Citalopram
Desmethylcitalopram
Didesmethylcitalopram
Renal clearance (ClR)
Urinary extraction of intact parent compound after oral
dose
130±70 nmol/L (20 mg)
<50 % of parent
<10 % of parent
26 (23-38) L/h
33 (23-45) h
50 h
100 h
2.8 to 3.3 L/h
12 (6-23) %
Table 1.
Summary of the pharmacokinetic parameters for racemic citalopram (Baumann and
Larsen, 1995; Bezchlibnyk-Butler et al., 2000; Fredricson Overo, 1982; KraghSorensen et al., 1981; Noble and Benfield, 1997; Rochat et al., 1997; Rochat et al.,
1998; Joffe et al., 1998).
Pharmacodynamics
The pharmacological activity resides in the S-Cit. The metabolite S-DCit, although,
less potent than the parent compound, is still a selective inhibitor of 5-HT reuptake. SCit increases the amount of 5-HT in the synapse, thus prolonging its activity at
postsynaptic receptor sites. Inhibition of reuptake leads to a reduced serotonin turnover
(Hyttel et al., 1992). That Cit is a very selective 5-HT reuptake inhibitor has been
shown both in vitro and in vivo, see table 2. In vitro brain studies showed Cit to be 300
times more selective in inhibiting 5-HT reuptake than clomipramine, the TCA with
highest affinity for 5-HT inhibition (Hyttel, 1994; Sanchez and Hyttel, 1999). The
metabolites are less liphohilic than the parent compound, leading to them entering the
brain less readily than Cit. Cit has only weak affinity to a series of receptors. Receptor
binding studies indicate that Cit has no significant activity for muscarinic, histamine
H1, serotonin 5-HT1A, 5-HT1B, 5-HT2, dopamine, Į1, Į2, E-adrenergic, and Jaminobutyric acid (GABA).
27
Racemate/Enantiomer
Cit
(S)-Cit
(R)-Cit
Inhibitory constant,
5-HT (nM)a
1,8
1,5
250
S/R activity
eudismic ratiob
167
DCit
(S)-DCit
(R)-DCit
14
9,9
65
6,6
Inhibitory constant for inhibition of the synaptosomal uptake of serotonin as IC50, a
higher concentration denotes a less effective inhibitor.
b
Ratio between the inhibitory constant for the (S)-enantiomer (eutomer) divided by the
(R)-value (distomer).
a
Table 2.
Pharmacological effects of citalopram enantiomers (Hyttel et al., 1992).
Adverse drug reactions-Toxicology
Cases of serotonin syndrome with fatal outcome, a rare but serious event that can
occur when medications that act to increase 5-HT at the synaptic junction are
coadministrated, have been documented for Cit in combination with moclobemide and
buspirone (Brosen and Naranjo, 2001; Neuvonen et al., 1993). When taken alone in
overdose, Cit appears to have a relatively wide margin of safety (Muldoon, 1996;
Personne et al., 1997). SSRIs are believed to have a more benign cardiovascular safety
profile than do the tricyclic antidepressants (Pacher et al., 1999). The effects of Cit on
cardiac conduction and repolarization have been extensively evaluated, both in
prospective studies in volunteers and patients, showing that the only effect of Cit on
ECG findings is a small reduction in heart rate (Rasmussen et al., 1999). Although
these new SSRIs seem to be a better alternative regarding adverse drug reactions and
interaction with other drugs, they are found among autopsy cases (Rogde et al., 1999).
Overdose by an antidepressant was the probable cause of death in 2.1 % of the men and
7.9 % of the women (Isacsson et al., 1999). Cit alone has been found to be the probable
cause of death in some autopsy cases (Anastos et al., 2002; Ostrom et al., 1996; Worm
et al., 1998). Postmortem concentrations of Cit in blood, and different organs have
been investigated but no study on the distribution of the Cit enantiomers has been
performed (Anastos et al., 2002; Jenkins and Gubanich, 2002; Levine et al., 1996;
Worm et al., 1998).
28
Chiral bioanalysis
Background
Within the field of chromatographic chiral separation, the basic mechanism behind
chiral recognition and other fundamental problems concerning separation of
enantiomers of drugs, pesticides, pheromones and other bioactive substances have
been solved during the last two decades. For pheromones and more volatile chiral
substances, gas chromatography is still the method of choice. In the field of drug
analysis, high performance liquid chromatography (HPLC) and capillary
electrophoresis have been established as the major enantioseparation techniques within
the last decade (Maier et al., 2001; Scriba, 2002). Thus, chiral separation has become
one of the most active areas of analytical chemistry.
There are two main approaches used to separate enantiomers, direct and indirect. The
indirect method is based on the formation of a pair of diastereoisomers of the racemic
mixtures with a chiral reagent. The direct approach utilizes chiral discrimination
achieved by a chiral selector. The chiral selector may be a mobile-phase additive or the
stationary phase in the chromatographic column.
Indirect methods
The indirect methods are divided into two categories: one is to derivatize the
enantiomers using an achiral derivatizing reagent and to separate the derivatives using
a chiral stationary phase (CSP). The other is to derivatize the enantiomers using a
homochiral derivatizing reagent and to separate the derivatives using an achiral
stationary phase (Haginaka, 2002). The crucial step for this is to convert the
enantiomers of a compound into diastereoisomers which make them suitable for
normal reversed or straight phase chromatography on a standard column such as C18.
It is also possible to achieve temporary diastereoisomers by adding an enantiopure
counter ion (acid or base) to the mobile phase. It is essential that the chiral
derivatization reaction proceeds to completion since enantiomers may display different
kinetics during reaction with another chiral molecule. There is always a risk that the
transformation of enantiomers to diastereomers will lead to racemization of the
compounds leading in turn to dubious results (Gorog and Gazdag, 1994; Subramanian,
1994). It is also important that the derivatization reagent is pure otherwise unwanted
reaction products will be formed, see scheme 1.
[(+)-A+(-)-A] + [(+)-B+(-)-B] o
Enantiomers (50:50 %)
Reagents (98:2 %)
(+)-A-(+)-B + (-)-A-(+)-B + (+)-A-(-)-B + (-)-A-(-)-B
I (49 %)
II (49 %)
III (1 %)
IV (1 %)
Reaction products
Scheme 1.
Derivatization of enantiomers with an impure chiral reagent.
29
Direct methods
A variety of CSPs are now available for the separation of enantiomers by highperformance liquid chromatography, and they have been shown to be very
useful in the chromatographic resolution of racemic mixtures. The basic principle for
the separation of the enantiomers is the temporary diasteromeric complexes that are
formed on the column. The fundamental mechanism for chiral recognition is the “three
point rule” (Allenmark, 1991). Chiral recognition requires a minimum of three
simultaneous interactions between the chiral selector and one of the enantiomers in the
racemate to obtain separation. At least one interaction must depend on the
stereochemistry at the chiral center of the chiral selector and the enantiomer (Han,
1997). Forces such as electrostatic, hydrogen bonding, repulsive/attractive van der
Waal, S-S or dipolar interactions and inclusion phenomena, contribute to the
recognition process (Maier et al., 2001). In the separation of enantiomers by
chromatography, the separation factor, Į, is determined by the difference between the
free energy of adsorption of each enantiomer. In HPLC, the enantioseparation is
dominated by enthalpic contributions in most cases, because the experiment is
commonly performed at comparatively low temperatures (Okamoto, 2002).
Mechanistic aspects of enantioseparation
Understanding how and where chiral recognition by chiral selector molecules occurs
may provide valuable information of the qualitative magnitude of enantioseparation,
types of analytes separable on a given selector, predictability of elution order, and
appropriate chromatographic conditions. Compared with the number of chiral selectors
available, relatively few detailed studies on enantioseparation mechanisms are
available (Maier et al., 2001).
The most popular strategy to establish chiral recognition models for a given selector
involves the collection of a representative body of chromatographic enantioseparation
data with a series of analytes displaying incremental structural information. A more
sophisticated strategy for development is chemometrically driven prediction of
retention and enantioselectivity by the construction of quantitative structure activity
relationships (QSAR). For macromolecules and polymeric selectors these studies are
more problematic and elucidation of the chiral recognition mechanism of protein type
selectors may be even more challenging. The “three-point-interaction model” applies
mainly for the Pirkel-type and other brush-type CSPs (Lipkowitz, 2001).
30
Chiral stationary phases
Chiral selectors can be obtained from natural sources or can be generated from natural
or synthetic building blocks. A large number of CSPs developed are made of natural
material such as protein, and cyclodextrin, seminatural synthetic of amylase or pure
synthetic products.
Chiral selectors can be classified in many different ways. Wainer has suggested a
classification scheme for high-performance liquid chromatography CSPs based on the
mode of formation of the solute-CSP complex divided in to five categories (Wainer,
1993).
Type I. Where the analyte-CSP complexes are formed by attractive interactions like
hydrogen bonding, S-S interactions and dipole stacking as represented by Pirkle-like
CSPs.
Type II. Where the analyte-CSP complexes are formed by attractive interactions and
through the inclusion into a chiral cavity or ravine as represented by some cellulosebased CSPs.
Type III. Where the primary mechanism involves the formation of inclusion
complexes, as represented by cyclodextrins.
Type IV. Where the analyte is part of a diastereomeric metal complex as in chiral
ligand exchange chromatography.
Type V. Where the CSPs are a protein and the analyte-CSP complexes are based on
combinations of hydrophobic and polar interactions.
Another way to classify these different CSP would be according to their origin (Maier
et al., 2001), see table 3.
31
Source
Type
Chiral selector
Natural
Proteins
Serum albumin
Orosomucoid
(Į1-acid glycoprotein)
Ovomucoid
Cellobiohydrolase I
Avidine
Chymotrypsine
Ovantranferrin
Oligosaccharides
Į-E-and J-Cyclodextrin
Antibiotics
Vancomycin
Teicoplanin
Ristocetin
Avoparcin
Amino acids
Cholic acids/bile acids
Alkaloids
Tartaric acid
Low Mw molecules
Semisynthetic
Synthetic
Modified oligosaccharides
Derivatized cyclodextrins
Cyclodextrin polymers
Modified Polysaccaride
Polysaccharide carbamates
Polysaccharide esters
Modified low Mw molecules
Ion exchange selectors
Synthetic low Mw molecules
Pirkle type selectors
Receptor molecules
LEC selectors a
Crown ethers
Proline derivate
Helical synthetic polymers
Polyacrylamides
Polyacrylates
Crosslinked tartaramides
MIPs b
Table 3.
Main groups of chiral selectors used for analytical HPLC, arranged according to their
origin. Adapted from Maier et al (Maier et al., 2001). Mw=molecular weight
a
LEC= Ligand Exchange Chromatography, b MIPs= molecular imprinted polymers
32
Cyclodextrins
Cyclodextrins (CDs) are cyclic oligosaccharides that form cavities. They are produced
by partial degradation of starch and enzymatic coupling of cleaved units into
crystalline, homogenous toroidal structures of different molecular weight. The three
most characterized CDs, denoted Į, E and J, contain six, seven and eight glucose units
respectively. The different number of glucose units leads to different internal
diameters and size of the cavities, see table 4. A Į-CD has a size suitable for
complexing a single six-membered aromatic ring, a E-CD can easily accommodate a
molecule with the size of a biphenyl or naphthalene and J-CD can contain molecules
as large as substituted pyrenes (Han, 1997). Cyclodextrins are chiral structures and, for
example, E-cyclodextrin has 35 stereogenic centres.
Cyclodextrin
Į
E
J
Number of
glucose
units
6
7
8
Molecular
weight
g/mol
972
1135
1297
Cavity
diameter
0,57 nm
0,78 nm
0,95 nm
Cavity
volume
Å3
174
262
427
Water
solubility
(g/100 mL)
14,5
1,85
23,2
Table 4.
Physical properties of Į-, E- and J-cyclodextrin (Schneiderman and Stalcup, 2000).
The seven Į-D-glucose units in E-CD are linked through the 1,4 position (Į-1,4linked) adopting a chair conformation, forming a rigid torus-shaped molecule with a
central cavity. The toroidal structure has a hydrophilic surface resulting from
secondary 2-, 3-hydroxyl groups and primary 6-hydroxyl groups, making the
cyclodextrin water soluble. All the hydroxyl groups of the glucose building blocks are
oriented to the exterior of the molecule, with the primary hydroxyl groups located at
the narrow opening of the torus and the secondary hydroxyl groups on the wide
opening. The 2´-OHs are pointed in clockwise mode, while the 3´-OHs are counter
clockwise. One or two of the 6´-OHs are used to attach the cyclodextrin through a
spacer arm to the silica (Hinze et al., 1985). The cavity is composed of the skeletal
carbons, ether oxygen atoms and methylene hydrogens giving it an apolar character.
As a consequence, cyclodextrins can include other apolar molecules of appropriate
dimensions and bind them through dipole-dipole interactions, hydrogen bonding or
van der Waals forces (Armstrong and Li, 1987; Bressolle et al., 1996; Hinze et al.,
1985), see figure 6.
The high density of secondary hydroxyl groups at the opening of the toroid acts as an
energy barrier for polar molecules attempting to form complexes, and instead
hydrogen bonding occurs. Amines and carboxyl groups interact strongly with these
hydroxyl groups as a function of the pKa of the analyte and pH of the aqueous system.
This relationship is important in understanding how to design mobile phases for chiral
separations on bonded native cyclodextrins.
33
Inclusion complexation are considered to be the driving force to obtain
enantioselectivity in the reversed phase mode but in most cases the cylindrical binding
cavity is itself too symmetrical to induce large enantioselectivity. The hydroxyl groups
in the 2- and 3-position situated on the rim of the toroidal structure make it possible for
potential interactions between these hydroxyl groups and substituent(s) present in the
guest enantiomer molecule (Hinze et al., 1985). The selectivity, resolution and
retention times are, in this mode, dependent on the type and amount of organic
modifier, buffer, flow rate, temperature and choice of CD. The competition between
analyte and the organic modifier for the CD cavity controls the retention, as well as the
chiral discrimination process, which in this instance depends on the relative
hydrophobicity of the solvent and analyte (Armstrong and Zhang, 2001).
H
OH
H
O
O
OH HO
H
H
OH
H
O
H
H
O HO
H
H
OH
H
H
HO
OH
O
H
O
OH
H
H
HO
H H
O
H
H
OH
HO
H
OH
H
H
H
H
H
H
O H
H
OH
OH
O
HO
H
O
OH
O
OH
OH
H
H
H
H
H
O
H
OH
O
O
H
HO
X
X
C
+
Y
Z
Figure 6.
E-CD and the inclusion complex mechanism between the toroidal structure and guest
molecule.
34
C
Z
Y
The CD with the broadest applicability is the E form. It has demonstrated the greatest
suitability for small analytes of general interest in the pharmaceutical, chemical and
environmental areas. Several derivatives of the native CDs have been made to change
the physical and chemical properties and to incorporate various special characteristics.
This provides specific interactions with certain functional groups and produces highly
selective separations for a vast number of analytes. The CD rim hydroxyls have been
acetylated or derivatized with R,S-hydroxypropylether, S-naphtylethylcarbamate and
R-naphtylethylcarbamate etc. In table 5, the currently available underivatized and
derivatized CD-CSPs are listed.
Material
E-CB
E-CB acetylated
E-CB S-hydroxypropyl ether
E-CB R,S-hydroxyproyl ether
E-CB S-naphtylethyl carbamate
E-CB R-naphtylethyl carbamate
E-CB 3,5-dimethylphenyl carbamate
E-CB 3,5-dimethylcarbamate
J-CB
J-CB acetylated
Į-CB
Į-CB acetylated
Trade name
Cyclobond I 2000
Cyclobond I 2000 Ac
Cyclobond I 2000 SP
Cyclobond I 2000 RSP
Cyclobond I 2000 SN
Cyclobond I 2000 RN
Cyclobond I 2000 DMP
Cyclobond I 2000 DM
Cyclobond II
Cyclobond II Ac
Cyclobond III
Cyclobond III Ac
Company
Asctec
E-CB
Į-CB permethylated
E-CB permethylated
J-CB permethylated
Nucleodex E-OH
Nucleodex Į-PM
Nucleodex E-PM
Nucleodex J-PM
MachereyNagel
E-CB
J-CB
ChiraDex
ChiraDex Gamma
VWR
Table 5.
Currently available CD columns (Armstrong and Zhang, 2001).
In HPLC these columns are mainly used in reversed phase modes but normal phase
and polar organic mode have also been used. In normal phase mode the derivatized
CDs are more useful than native CDs with chiral recognition and retention
mechanisms arising from S-S interactions with the derivative group. In the polar
organic mode, the mobile phase contains acetonitrile and small portions of methanol,
triethylamine and glacial acetic acid, which act as hydrogen bonding modifiers. As in
normal phase mode, the inclusion mechanisms do not seem to play an important role,
instead hydrogen bonding with the secondary and primary hydroxyl groups of the CD
cavity and the analyte act more like a lid on top of the cavity (Armstrong, 1998).
35
Fluorescence detection
When certain molecules or atoms are exposed to high-intensity light, the molecules or
atoms absorb the energy and enter an excited state. As a molecule moves from this
excited state back to its normal state, some of the energy is released in the form of
fluorescence. Fluorescence is a type of luminescence. The atoms and molecules in
different compounds emit different levels of fluorescence when exposed to the same
energy levels (Kemp, 1975).
The process of fluorescence detection involves several components and processes: an
excitation source, filtering the source light, excitation of the analyte with a selected
wavelength, collecting and filtering the emitted fluorescence, measuring the emitted
fluorescence and amplifying the emitted signal, see figure 7.
The typical energy source used for fluorescence detection is a lamp that provides an
intense, stable spectrum of light in the UV and visible range. The fluorescence
intensity is directly related to the intensity of the excitation spectrum, so highsensitivity detectors use the most intense excitation source available (Lindsay, 1992).
Common excitation light sources include the following. Vapour lamps: mercury,
cadmium, or zinc. Arc lamps: deuterium or xenon.
Of these light sources, the vapour lamps provide high intensity, narrow-band outputs
in the UV and visible ranges. The arc lamps provide a wider, lower intensity spectrum.
Among the arc lamps, the xenon lamp provides the widest spectrum, making it the
excitation source of choice for general-purpose fluorescence detectors.
Radiation
Slit
Filter
Slit
Filter
Flow
cell
Photomultiplier
detector
Figure 7.
Schematic layout of a fluorescence detector (Lindsay, 1992).
36
Chemometrics
Chemometrics can be broadly defined as the application of mathematical and
statistical methods to chemistry (Deming, 1986). In pharmaceutical and biomedical
analysis, we are often confronted with several variables at one time and hitherto the
approach has been to scrutinize one variable (factor) at a time. There are however a lot
of problems associated with this approach, as the following list demonstrates:
Does not lead to optimum
Inefficient
Unnecessarily many runs
No information about what happens when the factors are varied
simultaneously
No information about interactions between factors
The information of the variability in the response is less
Chemometrics, on the contrary, works with the variables multivariately, analysing
everything together, giving information about which factors have a real influence on
the response, settings of the factors to achieve optimal conditions and the possibility of
predicting values for the responses when the factors are varied within the model
(Berridge et al., 1991; Deming, 1986; Wold, 1991).
The application of chemometrics is greatly motivated by the fact that chemical and
biological data that are produced in studies have to be used both efficiently and
economically. Applications within pharmaceutical research and development areas
range from analysis of drug-interactions, culture media and to how to optimize the
process of the final product (Gabrielsson et al., 2002; Lapinsh et al., 2001; Walters,
1999). Chemometrics can work both with data analysis, utilizing inherent information
in chemical and biological data in the best way or for planning and performing
experiments in a way that the data contain maximum information about stated
questions (Wold, 1991).
Systematic optimization
Systematic optimization are carried out in the following sequence:
Choice of objective function
Selection of important factors
Optimization
In order to find the most suitable factor combination, we can distinguish between
simultaneous and sequential optimization approaches.
With simultaneous strategies, the relationship between responses and factors is studied
by running experimental designs, constructing a mathematical model, and
investigating the relationship by use of so-called response surface methods (RSM).
Very often RSMs aim to judge this relationship graphically and the consequences are
drawn from those plots.
37
Sequential strategies of optimization are based on an initial experimental design
followed by a sequence of further measurement in the direction of the steepest ascent
or descent. That is, no quantitative relationship between factors and responses is
evaluated but the response surface is searched along an optimal path (invisible path)
(Otto, 1999). The two strategies are exemplified in figure 8.
2
4
Simplex 2
Simplex 1
1
Simplex 3
5
3
Figure 8.
With response surface methods, the response is described by a mathematical model
(dotted contour lines of the response surface). By use of sequential strategy methods
the response is measured along a search path, here along a Simplex path (See section
Multisimplex) beginning with trials 1, 2 and 3 and followed by the next calculated trial
4.
38
Experimental design
The objective of experimental design is to plan and conduct experiments in order to
extract the maximum amount of information from collected data in the presence of
noise from the measured values, with the fewest number of experimental runs. The
basic idea is to vary all relevant factors simultaneously, over a set of planned
experiments, and then connect the results by means of a mathematical model. This
model is then used for interpretation, prediction and optimization (Gabrielsson et al.,
2002; Otto, 1999).
Experimental design can be used in many areas within analytical chemistry. For the
optimisation of drug separation with high-performance liquid chromatography, the
designs employed include the central composites design (Do et al., 2001), full factorial
design (Karlsson and Aspegren, 2000), Plackett-Burman (Altria et al., 1995; Bempong
and Honigberg, 1996), simplex (Carlsson and Norlander, 2001; Fell et al., 1988) and
D-optimal design (Hermann et al., 2002). Analytical variability can also be estimated
for an analytical procedure with experimental design (Tsang et al., 1998; Wieling et
al., 1996). Another area is robustness testing which is performed at the end of method
development to test the susceptibility of an analytical procedure to small changes in
the experimental conditions (Eriksson et al., 2000; Hund et al., 2000; Hund et al.,
2002; Small et al., 1995).
The three primary experimental objectives can be divided into screening, optimization
and robustness testing. It can be concluded that the use of experimental designs and
statistical data evaluation are, in conjunction, of great benefit in the optimisation and
robustness evaluation of HPLC methods.
Design of experiments
Prior to the experiments, one has to specify some input conditions about the purpose
and objectives of the investigation/experiment such as identifying the number of
factors, ranges for the factors and types of factors (quantitative or qualitative) and
number of responses.
Then the experimental design is created, and its experiments are carried out, either in
parallel, or one after another. Each experiment gives some results, i.e., values of the
response variables. Thereafter, these data are analyzed by regression analysis. This
gives a model relating the changes in factors to changes in the responses. The model
will indicate which factors are important, and how they combine in influencing the
responses.
During an investigation, one needs answers to the following questions:
I
Which factors have a real influence on the responses (results)?
II
Which factors have significant interactions (synergy or antagonism)?
III
What settings of the factors are required to achieve optimal conditions for
the best performance of a process or a system or a product?
IV
What are the predicted values of the responses (results) for given settings
of the factors? (Eriksson et al., 2000; Otto, 1999)
39
Screening designs
Screening is the first stage of an investigation where the goal is simply to identify the
important factors:
I
Which factors are most influential?
II
What are their appropriate ranges?
An important factor is a factor that causes substantial changes (effects) to the response
when it varies.
In the screening stage, one uses simple models (linear or linear with interactions), and
experimental designs that allow the identification of the factors with largest effects in
the fewest possible number of experimental runs. The model represents the
relationship between the response y and the factors x1, x2 etc. Using the models, the
data are separated into two parts; the systematic part explained by the model, and the
“noise”, the remaining unmodelled part of data. Data (y) = Model [factor values (x),
parameters (E)] + Noise (İ) (Wold, 1991).
Linear model: y=E0+E1x1+E2x2+…+İ
Interaction model: y=E0+E1x1+E1x2+E12x1x2+…+İ
After some initial experiments have been performed, a common approach is to identify
an interesting standard reference experiment and then to perform new, representative
experiments around it. The experiments can be illustrated graphically as points in a coordinate system defined by the x-axes. With three (factors) variables x1, x2 and x3 the
experiments will be distributed as in table 6 and figure 9.
All variables are changed simultaneously to cover the entire area of interest. Another
reason for this is that it makes it possible to examine interaction effects. A center
point, the middle value for each variable interval, is also included to detect curvature
in the experimental design. More center points will give a statistical measure of noise.
The size and sign of the scaled and centered regression coefficients indicate the
influence of variables on the response.
Full factorial and fractional factorial design
Whether the objective is screening of process factors or optimization of a chemical
reaction, the variables that influence the responses are the center of attention
(Gabrielsson et al., 2002; Lundstedt et al., 1998).
The number of experiments in an experimental design grows rapidly with increasing
number of variables. For the purpose of screening, a full factorial design is not a
realistic option. A reduced design, called a fractional factorial design, is then a more
appropriate choice. For a fractional factorial design, experiments are chosen to give the
maximum amount of variation with fewer experimental runs. The drawback is lost
information caused by confounding of main effects with interaction effects if the
resolution is lower than four (Gabrielsson et al., 2002).
40
Experiment
Factor
x1
-1
+1
+1
-1
-1
+1
+1
-1
1
2*
3
4*
5*
6
7*
8
Factor
x2
-1
-1
+1
+1
-1
-1
+1
+1
Factor
x3
-1
-1
-1
-1
-1
+1
+1
+1
Response
y1
y2
y3
y4
y5
y6
y7
y8
Table 6.
Full factorial design at two levels, 23 design, see also figure 9. The star labelled
experiments represent the half-fraction factorial design of figure 9.
+1
+1
x3
x3
+1
x2
-1
-1
x1
+1
+1
x2
-1
-1
-1
Full factorial design
x1
+1
-1
Fractional factorial design
Figure 9.
Using fractional design, the number of experiments is reduced by a number p
according to a 2k - p . For p=1, so-called half-fraction design result.
41
Optimization designs
After screening, the goal of an investigation is usually to understand in more detail
how the factors influence the responses and create a valid map of the experimental
domain.
To make predictions, optimize or find a region of operability the use of more complex
models is needed. Quadric polynomial models are used and, due to the increased
complexity, more experiments are required.
Quadric polynomial model: y=E0+E1x1+E2x2+E11x12+E22x22+E12x1x2+…+İ
How can we find the optimum? Is there a unique optimum, or is a compromise
necessary to meet conflicting demands on the responses?
Different types of response surface modelling (RSM) designs can be used in this
optimization process: three-level full factorial, central composite (CCD), Box Behnken
and D-optimal.
Presented here are two strategies for optimization: response surface modelling and
simplex optimization. Both these designs support the estimation of quadratic terms
(Gabrielsson et al., 2002).
Central composite designs
Central composite circumscribed (CCC) and central composite face centered (CCF)
are the two most frequently used composite designs (Eriksson et al., 2000). The CCC
(stare design) design prescribes experiments, the axial points, whose factor values are
located outside the low and high settings of the factor definition whereas in the CCF
design, the axial points are centered on the face of the cube, see figure 10.
42
x3
x3
x2
x2
x1
x1
Central composite circumscribed
(CCC)
Central composite face centered
(CCF)
Figure 10.
To the left central composite circumscribed (CCC) design and, to the right, central
composite face centered design (CCF) with three factor/variables are graphically
illustrated. Central composite design for three factors consists of a full factorial two
level design and a star design. The experimental points are symmetrically distributed
around a center-point.
Analysing the resulting experimental data
Analysing the resulting experimental data, that have been laid out according to a
statistical design, usually consist of three stages (Eriksson et al., 2000).
I. In the first stage, the raw data are evaluated. To see whether the data are normally
distributed and to check that the data replicate error is not larger than the total
variation in the design points.
II. Thereafter, in the second step calculation of models that link the design data to the
recorded responses, and the interpretation of these models. The data collected by the
experimental design are used to estimate the coefficients of the model. The data
obtained are analysed by means of multiple regression (MLR) or generalisations
thereof such as partial least squares projection to latent structures (PLS) and will show
which factors are important, and how they combine together to influence the results.
III. The third stage is to use the obtained regression model. This involves prediction of
optimum conditions for verifying experiments or finding settings for an optimization
design by e.g. response surface plots.
To determine an optimum in a response surface plot, it is necessary for the polynomial
function to contain quadratic terms (Gabrielsson et al., 2002).
The most interesting variables with regards to the optimum are plotted against the
response in three dimensions. This graphically illustrates the relationship between
43
different experimental variables and the responses and enables identification of the
region of optimum.
Revision of the fitted model
All results of model fitting, by MLR or PLS are displayed, in the same way,
graphically and in lists. Both MLR and PLS compute regression coefficients from each
response. Thus y is expressed as a function of the x´s according to the selected model
(i.e. linear, linear plus interactions or quadratic).
Goodness of fit statistics, and information about model adequacy are obtained by,
calculating the predictive power Q2 of the model (according to cross validation) and
the percent of the variation of the response R2 explained by the model.
In these calculations Q2=1-PRESS/SS, R2=(1-SSResid/SS) and PRESS the prediction
residual sum of squares. Small deviations between the actual residuals and the
predicted ones will render a low PRESS and high values of predicted variation (Q2).
SS is the sum of squares of the total variation of a selected response, corrected for the
mean. The total SS consists of two parts, one part resulting from the regression model
(SSRegr) and another due to the residuals (SSResid). Small residuals will render a high
degree of explained variation (R2).
Large Q2, 0.7 or lager, indicates that the model has good predictive ability and will
have small predictive errors.
Examination of the regression coefficients plotted in a bar chart will indicate how the
factors influence the response(s). This is normally used to detect strong interactions,
while response contour plots are used to interpret their meaning.
The analysis of variance (ANOVA) for selected responses, partitions the total variance
of the response (SS) into a component due to the regression model and a component
due to residuals. If replicate observations are available the residual sum of squares is
further partitioned into pure error and lack of fit.
Model adequacy can be further assessed by normal probability plots of residuals
and by checking for optimal transformation of the response through the use of BoxCox plots. For PLS, a summary of the fit by component and PLS score and loading
plot’s are available. Effect plots are available for screening designs.
MLR
With Multiple Linear Regression, the coefficients of the model are computed to
minimize the sum of the squares of the residuals, i.e. the sum of squared deviations
between the observed and fitted values of each response. The least squares regression
method yields small variances from the coefficients and small prediction errors. It is
important to note that MLR separately fits one response at a time and hence assumes
them to be independent (Eriksson et al., 2000).
44
PLS
When several responses (3 or more) have been measured, it is important to develop a
model representing the relationship of all the responses to the factors. PLS takes MLR
one step further, as it deals with many responses simultaneously, taking their
covariances into account. This provides you with an overview of how all the factors
affect all the responses. PLS is one of the most common methods for analyzing
multivariate data, where a quantitative relationship between a descriptor matrix x and
a response matrix y is sought. The best possible correlation between x and y is
calculated using the least squares technique. Hence, the resulting PLS model is not
always the best description of x and y, but rather of the relationship between them
(Gabrielsson et al., 2002).
Modified sequential simplex approach
The simplex method of optimization of chromatographic resolution selectively directs
the chromatographer through the response surface towards an optimum (Berridge,
1982). The simplex algorithm can handle only one response at a time, but there are
usually many response variables to optimize simultaneously. A method of forming a
joint response measure, from the individual response variables, is therefore needed.
Zadhe introduced the concept “fuzzy sets” (Otto, 1988; Zadeh, 1965) that provides
flexible and efficient techniques for handling different and conflicting optimization
criteria.
The modified sequential simplex algorithm differs from the standard method by the
addition of the concept of contraction and extension (Berridge, 1982; Berridge et al.,
1991). The enhancement seeks to direct the algorithm selectively towards optimum,
the zone of data covered becoming progressively focused in the process.
45
Simplex optimization
The simplex method is based on an initial design of k+1 trials, where k is the number
of variables. A k+1 geometric figure in a k-dimensional space is called a simplex. The
corners of this figure are called vertices. A simplex is a geometric figure having one
more vertex than its number of factors. Therefore a simplex in one dimension is a line,
in two dimensions a triangle, in three dimensions a tetrahedron, see figure 11, and in
multiple dimensions a hyper-tetrahedron. A geometric interpretation is difficult with
more variables, but the basic mathematical approach outlined below can handle the
search for optimum conditions (Otto, 1999).
Figure 11.
Geometric interpretation of lower dimensional simplexes. The shapes of the simplex in
a one, a two and a three variable search space, are a line, a triangle or a tetrahedron.
The basic simplex method
The basic simplex method is easy to understand and apply. The optimization begins
with the initial trials. The trial conditions are spread out efficiently. These initial trials
form the first simplex. The number of initial trials is equal to the number of control
variables plus one. With two variables, the first simple design is based on three trials
for three variables it is four trials, etc. This number of trials is also the minimum for
defining a direction of improvement. Therefore, it is a time saving and economical
way to start an optimization project. After the initial trials, the simplex process is
sequential, with the addition and evaluation of one new trial at a time. The simplex
searches systematically for the best levels of the control variables. The optimization
process ends when the optimization objective is reached or when the responses cannot
be further improved, see figure 12.
46
Start
Make first
simplex
Rank trials in
order:
B, Nw & W
Make
reflection R
Re-test
retained trial
Stop
Yes
Yes
Let Nw be W
Rank the
other trials:
B & Nw
No
Reevaluation
rule activated
?
No
Objectives
fulfilled
?
Replace
W for R
Figure 12.
Basic simplex rules.
The calculations in the basic simplex algorithm are outlined in the flow chart. For each
simplex, the following labels are used: W for the least favourable trial or the trial being
rejected, B for the most favourable trial and Nw for the second least favourable trial
(i.e. next to worst).
The first rule is to reject the trial with the least favourable response value in the current
simplex. A new set of control variable levels is calculated, by reflection into the
control variable space opposite the undesirable result. This new trial (R) replaces the
least favourable trial in the next simplex. This leads to a new least favourable response
in the simplex that, in turn, leads to another new trial, and so on. Through this, the
simplex will move steadily towards more favourable conditions away from the least
favourable conditions
These steps are repeated until the simplex begins to rotate around the optimum or the
response satisfies the experimenters´ needs (1996; Otto, 1999), se figure 13.
The fixed step width of the fixed size simplex might lead to problems if the step width
chosen is either too large or too small. In the first case, the optimum might be missed
and in the latter the number of experiments required becomes very large. These
disadvantages can be circumvented if the step width is tuneable, as with in the
variable-size simplex or modified simplex method (Otto, 1999).
47
R
Nw
B
W
Figure 13.
Illustration of a simplex path with two variables. After three initial trials the corners of
the triangle are classified as W for the least favourable trial or the trial being rejected,
B for the most favourable trial and Nw for the second least favourable trial (i.e. next to
worst). Then the next trial 4 (R) is calculated and performed.
The modified simplex method
The modified simplex method has much in common with the basic method, but can
adjust its shape and size depending on the response in each step. Several new rules are
added to the basic simplex rules. These rules make the simplex expand, in a direction
of more favourable conditions, or contract if a move was taken in a direction of less
favourable conditions.
The procedure for expansion and contraction enable the modified simplex both to
accelerate along a successful track of improvement and to home in on the optimum
conditions. Therefore the modified simplex will usually reach the optimum region
quicker than the basic method will and will pinpoint the optimum levels more closely.
Moves in a typical optimization sequence are easy to show for control variables (the
response is shown separately).
The degree of contraction depends on how unfavourable the new response is.
With the modified simplex, the step width is changed by expansion (E) and contraction
(C- and C+) of the reflected vertices, see figures 14 and 15.
48
E
R
C
W
C-
Figure 14.
Illustration of the different moves with the modified simplex method, adding the
expansion and contraction of the reflected vertices.
Control variable 2
Control variable 1
Figure 15.
An example of a typical optimization sequence with the modified simplex method.
Change in the levels for two control variables.
49
Aims of the present study
With the introduction of citalopram (Cipramil®) in Sweden, a lot of questions were
raised about its effect due to the fact it being a racemic compound. All plasma
concentration data for citalopram and its metabolites, used to correlate the clinical data
and pharmacological effect, were all based on determination of the total concentration
(sum of the two enantiomers) with achiral chromatography. The rediscovery of
chirality and its implication on clinical pharmacology and a more rigorous view from
the regulatory authorities, together with the awareness that racemic drugs have to be
further elucidated, formed the grounds of this thesis.
Specific aims:
Development of a routine therapeutic drug monitoring (TDM) method for
determination of racemic citalopram and its two main demethylated metabolites.
Development of an enantioselective method for determination of the
enantiomers of citalopram and the demethylated metabolites that are also
enantiomers.
Studies of the distribution of the enantiomers in patients treated with citalopram.
Study of the distribution of the enantiomers in whole blood from autopsy cases.
50
Methods, results and discussion
Introduction
At the time of introduction of citalopram the physician needed a therapeutic drug
monitoring service to identify patients with interactions, compliance problems and to
provide answers to questions concerning polymorphic enzymes and drug metabolism.
Anyone engaged in routine therapeutic drug monitoring must keep in mind that the
plasma concentration achieved and maintained is a direct consequence of a wide
variety of processes in the body. These processes include drug absorption, distribution,
metabolism and excretion in combination with the physiological status of the patient.
When long term oral therapy is initiated, the drug will accumulate within the body
until the time when the rate of elimination equals the rate of administration. It requires
5 half-lives of drug administration before a steady state concentration is reached and
stabilized. It is important to note that the principles that govern the gradual
accumulation of a drug to a steady state also apply when the drug is discontinued. For
any changes of dosage, after the steady state has been achieved, these principles
regulate the time to reach a new steady state.
Paper I
Achiral determination of citalopram
Citalopram extraction version I
Solid-phase extraction
Solid-phase extraction (SPE) has become one of the most common sample clean-up
and concentration techniques used by analytical chemists. Over the last two decades,
the use of SPE has gained in popularity and is today considered as being the technique
of choice for sample work-up. SPE offers several benefits over liquid-liquid
extraction, such as improved recoveries, less solvent consumption, smaller sample
volumes, and increased sample throughput. A large choice of sorbents such as highly
cross-linked copolymers, functionalized copolymers, graphitized carbons or some
specific n-alkylsilicas are now available for trapping analytes over a wide range of
polarities.
The method development is based on prediction from liquid chromatographic retention
data or solvation parameters in order to determine the main parameters for extraction.
These parameters are type and amount of sorbent, sample volume, which can be
applied without loss of recovery, composition and volume of the clean-up solution,
and composition and volume of the elution solution. Obtaining extracts free from
matrix interferences in a few steps or even in one step when possible, has now been
included in the development of the SPE procedure. New selective phases such as
51
mixed-mode and restricted access matrix sorbents or phases such as immunosorbents
or molecularly imprinted polymers are also available. Additional selectivity is
obtained by combinination of non-polar sorbents with ion-exchange or ion-pair
sorbents. SPE is relatively easy to use and the technique is often incorporated into
dedicated fully automatized sample preparation systems of various formats, with online coupling to liquid chromatography followed by various detection modes (Boos
and Rudolphi, 1998a; Boos and Rudolphi, 1998b; Fritz and Masso, 2001; Hennion,
1999; Rouan et al., 2001; Takeuchi and Haginaka, 1999).
Conditioning
Solid phases must be conditioned, solvated, or activated before the sample is applied.
Failure to activate the column sufficiently results in poor retention of the molecules
because sample capacity is reduced on a dry column. The conditioning of the column
with an organic solvent, for example methanol or acetonitrile forces the bonded carbon
stationary phase to stand up from the silica so they are ready to accept the molecules.
A C2 column, with two-carbon chains bound to the silica, is used in this method.
Because of the very short chain length of the bound group, leaving the silica surface
somewhat exposed, C2 is a fairly polar sorbent with its polarity slightly greater than
CN. C2 is often used as a replacement for C18 and C8 when analytes are too strongly
retained by the latter.
In this extraction method, version I the C2 column is conditioned with acetonitrile
because methanol has the disadvantage that small amounts will remain on the column,
after the flush step, due to hydrogen bonding to the silica surface. Acetonitrile is the
preferred choice if secondary silanol interactions are to be utilized for sample clean up.
The organic solvent that has passed through the column may interfere with subsequent
hydrophobic binding of the analyte and must be flushed away, in this case with 25
mmol/L phosphate buffer pH 11.5.
Buffering/sample pretreatment
The purpose of buffering the sorbent is to ensure optimal pH conditions for extraction.
For hydrophobic extraction, retention is promoted when the functional groups of the
analytes are not charged. For optimal retention of a drug it must be in a hydrophobic
form, therefore the buffer must be at a pH at which the molecules are uncharged. The
pKa of the analyte will determine the pH for buffering. Addition of an organic solvent,
like methanol or acetonitrile, in low concentrations, to the matrix may also be useful
for breaking the protein binding of the drug.
The pKa of Cit is 9.5 and, by using a phosphate buffer of 11.5, it will be uncharged.
The buffered plasma sample is passed through the column followed by two washing
steps. See figure 16.
Washing
The washing and elution stages of any extraction are extremely important and require
knowledge of polarity of the analyte and relative polarity of solvent or solvent
mixtures. Incorrect selection of a washing solution or elution solvent will result in low
recovery, poor reproducibility, and/or dirty extracts.
52
In this method two washing steps are used, the first with water and the second with
acetonitrile. The water will wash out the polar matrix and gradually change the pH
from 11.5 (phosphate buffer) to approximately 6.5 (micro milieu at silanol level). The
Cit molecules have now become positively charged and, after drying the column with
air, they will be retained on the column by ion interaction with negatively charged
secondary silanols. Uncharged non-polar molecules can now be removed by washing
with acetonitrile. This step is called phase switching because there is a switch from
interaction between the sorbent and water to an interaction between sorbent and a
water miscible organic solvent. See figure 16.
Elution
Ideal elution solvents selectively remove analytes and leave remaining interferences on
the column. Hydrophobic bonds are broken by solvent elution, while ionic interactions
are broken by high or low pH aqueous elution. The final elution solvent must
simultaneously disrupt both these interactions, that is, a high (basic) or low (acidic) pH
modified organic solvent.
For the elution of Cit, DCit, DDCit and internal standard (IS-Cit) methanol with 1 %
acetic acid was used to break hydrophobic and ionic interaction and effectively eluate
the analytes. See figure 16.
53
CH3
H3C
N
NC
CH3
O
N
CH3
O
NC
CH3
F
CH3
HO
OH
F
CH3
CH3
CH3
O
O
O
O
O
Si
O Si O
Si
O Si O
Si
O Si O
Si
O
O
O
O
O
O
O
SiH3
SiH3
O
Citalopram, IS F=Cl
CH3
SiH3 H3Si
O
SiH3 H3Si
HO
SiH3 H3Si
O Si OH
Washing with water
NC
NC
CH3
O
+
N
CH3
H
F
O
F
CH3
HO
H3C
+
N
CH3
O
O-
Si
O Si O
O
O
SiH3 H3Si
HCH3
CH3
CH3
-
O
O
O
O
O
Si
O Si O
Si
O Si O
Si
O
O
SiH3 H3Si
O
O
SiH3 H3Si
CH3
CH3
CH3
O
O
O Si OH
O
SiH3
HO
Si
O
O
CH3
OH
O Si O
O
SiH3 H3Si
SiH3
Acetonitrile washing ”phase switching”
CH3
CH3
O
O
O
Si
O Si O
Si
O Si O
O
SiH3 H3Si
O
O
HO
O
O
CH3
CH3
O
SiH3 H3Si
Si
O Si OH
O
O
SiH3
SiH3
Elution with methanol/acetic acid
Figure 16.
Extraction procedure for citalopram and metabolites on a C2 extraction column,
showing the mechanisms involved.
54
Internal standard
When working with biological matrices, the addition of an internal standard (IS) will
compensate for errors caused by inaccuracy in volume transfers in the extraction step,
as well as in the injection step. One important criteria for the IS is that binding strength
of the analytes and the IS to proteins and other components of the matrix should be
similar. The physiochemical properties of the IS should be comparable with those of
the analyte(s) so it behaves in a similar way to the analyte(s) during sample pretreatment. Yet the properties must be sufficiently unique to allow the IS to be clearly
distinguished from the other analyte in the HPLC separation. The IS can not be
another drug on the market because there is always a risk that that drug has also been
prescribed to the patient. In such a case, a quantitative determination will be
impossible. In this method, the internal standard IS-Cit is the chlorinated version of
Cit, see figure 16.
Protein binding of analytes may be a problem and should be considered if recoveries
differ greatly when comparing extractions from plasma and water.
Achiral chromatography
After evaporating the eluate, the samples were dissolved in the mobile phase for the
achiral chromatography and injected.
For many older chromatographic C18 columns, organic additives in the mobile phase
are used to reduce secondary silanol interactions so as to obtain good performance.
With newer generations of analytical reversed phase material however, this problem
has been considerably reduced and amine additives are very seldom necessary
(Tournel et al., 2001). At the time as this method was developed, secondary
interactions were avoided by the use of a phenyl-based stationary phase. After the
publication of this method a few improvements have been made. The dimension of the
column has been changed from 8*100 mm to 3.9*100 mm, protected by a precolumn
of 3.9*20 mm, making it possible to reduce injection volume from 20 µl to 10 µl while
maintaining the same sensitivity.
55
As this method has been our routine method for several years, both internal and
external controls can in addition to the validation presented in paper I, further
demonstrate the accuracy and stability of the method. In figure 17, the spiked values
versus found concentrations of Cit external controls, followed for 5 years and the
internal controls for Cit, DCit and DDCit during the last two years can be seen.
External control for citalopram
septem ber 1998 to decem ber 2002
Citalopram internal controls from 2001 to
2002
700
y = 0,924x + 4,686
R2 = 0,9892
600
600
Cit high
CV=4.0 %
500
Cit low
CV=5.5 %
400
DCit High
CV=5.4 %
300
DCit low
CV=10.7 %
200
DDCit High
CV=5.7 %
100
DDCit low
CV=7.6 %
400
nmol/L
Found nmol/L
500
300
200
100
0
0
200
400
600
0
1 6 11 16 21 26 31 36 41 46
Num ber of controls
Spiked value nm ol/L
Figure 17.
External control for citalopram for a 5 years period analysed every quarter and low and
high internal controls for Cit, DCit and DDCit during the last two years. New
standardisation was carried out on every analysis batch.
Summary of paper I
A routine TDM method has been established for achiral concentration monitoring of
Cit, DCit and DDCit.
The introduction of solid–phase sample preparation utilizing “phase-switching”
technique allowed a more effective washing procedure.
The use of a phenyl column made it possible to use a mobile phase without any amine
additives.
56
Paper II
Optimization of the chiral separation of
citalopram and its demethylated
metabolites
Chiral separation of citalopram
With the enantioselective chromatography column/phases available today, the
possibility of finding a column that can solve the problem of separating the
enantiomers of the parent drug is good. However, in bioanalysis of a drug there is
always the possibility of finding metabolites of that drug in the sample and these may
either be of interest to measure or they may interfere with the analysis of the parent
drug.
Among the different E-CD columns available, the acetylated and 3,5dimethylcarbamate derivates of E-CD have been used for chiral separation of Cit (Carlsson
and Norlander, 2001; Rochat et al., 1995a; Sidhu et al., 1997). A cellulose-based CSP
(Chiracel OD) column has also been used for chiral separation of Cit, after
derivatization to minimize retention times (Rochat et al., 1995b). The enantiomers of
the propionic metabolite of citalopram (Cit-Prop) could also be separated, after
derivatization, on the same column but with a different mobile phase. With the
column, chirobiotic V, where vancomycin is the chiral selector used in the polar
organic mode with a mobile phase of methanol, acetic acid and triethyl amine the
eluation order of the enantiomers is reversed as compared to E-CD, the R- enantiomers
eluates before the S-enantiomers (Kosel et al., 1998; Zheng et al., 2000). The
separation of S-Cit and R-Cit have also been performed on a protein type Į1-acid
glycoprotein column (Chiral-AGP), with 3.0 mM N-dodecyl-N,N- dimethylammonio3-propanesulfonate and 10 mM hexanoic acid in phosphate buffer pH 6.5 as the
mobile phase. On the Į1-acid glycoprotein column it was not possible to separate the
demethylated metabolites (Haupt, 1996). Detection principles used are either UV or
fluorescence.
In the initial attempts to separate citalopram and its demethylated metabolites a
screening was performed on different columns. With the AGP column it was not
possible to obtain any separation of the two metabolites, which is why it was excluded.
A batch of a column with vancomycin as chiral selector was also tried early on but the
time to obtain a stabile base line could take a whole weak, so that it was impossible to
use. The native form of E-CD could separate the enantiomeric pairs of Cit, DCit and
DDCit, but one problem was not solved, i.e. the overlapping of different enantiomer
pairs.
Modde, in both screening and optimization design (CCF), was used in these attempts
to find the optimum for separation on these columns but they were unsuccessful. The
57
derivatized forms of E-CD offer additional opportunities for chiral separation and
among them the acetylated form of E-CD (E-CD Ac). The E-CD Ac, where the
hydroxyl groups on the rim of the E-CD cone are modified to acetoxy groups, leading
to a somewhat extended cone, gave promising results.
This study had two main objectives:
I. To further scrutinize and understand the separation mechanism within enantiomeric
pairs, as well as between enantiomeric pairs of Cit, DCit and DDCit.
II. To evaluate the use of response-surface methodology as an optimization tool for
enantioselective analysis, and to compare response surface modelling versus sequential
simplex optimization.
Optimization with Modde
With the use of Modde, an initial screening design resulted in four factors i.e. pH (4,56,5), buffer concentration (5-145 mM), methanol content (45-65 %) and temperature
(5-35 qC) to be used in a full factorial optimization design (CCF). Responses were the
adjusted retention time (tR´) of the different enantiomers, separation factors within
enantiomeric pairs (ĮCT, ĮDCT and ĮDDCT) and between enantiomeric pairs (Į1 and
Į2). With a CCF design this resulted in 27 experiments including 3 replicates at the
center point.
Evaluation of the model with linear, quadratic, and cross terms showed a good fit for
all terms except for Į2 where the predictive ability Q2 was low, illustrated in graphic
form in figure 18. Acceptable values for chemical data are R2 0.8 and Q2 0.5 and
excellent when Q2 > 0.8 (Lundstedt et al., 1998). The low predictive ability for Į2 is
also shown in the plot “lack of fit”, as the model also suffers from a significant lack of
fit for Į2, as shown in figure 19 comparing ĮCT and Į2. Suspected outliers among the
experiments can be found in a residual plot. In figure 20, the residual plots for ĮCT
and ĮDCT show that experiment number 5 could be an outlier. Subsequently, the non
significant quadratic and cross terms were removed to try to improve Q2 for Į2. In
figure 21, an overview of factors and responses with A, all terms included and in B,
non-significant terms removed. Every response can also be presented with main
effects, cross and quadratic terms and confidence interval, se figure 22.
The very low value of Q2 was not improved by modifying the model and as other Q2´s,
for the other responses did not improve, the default model was used to further
investigate the separation mechanism. Interpretation of the model by response surface
plots gave that the optimum for separating all the six enantiomers has to be done with
a mobile phase of 55:45 methanol:10 mM citric acid-triethylamine buffer pH 6,3 at 30
qC, see figure 23 and paper II, page 269.
58
Investigation: Citalopram (PLS, Comp=5)
Summary of Fit
1,0
2
R2
Q2
2
R &Q
0,8
0,6
Modde
4.0
0,4
0,2
0,0
Log tR´
R-CT
DCT
DDCT
DDDCT
D1
D2
N=27 CondNo=6.1592
DF=12 Y-miss=0
Figure 18.
Summary of fit: the levels of explained (R2) and predicted (Q2) variations were good
for all responses except for Į2.
Investigation: Citalopram (PLS, Comp=5)
Investigation: Citalopram (PLS, Comp=5)
Lack of Fit for ĮCT
Lack of Fit for Į2
0,014
0,05
0,012
0,04
0,010
0,008
0,03
0,006
0,02
0,004
0,01
0,002
0,00
0,000
SD-LoF
N=27
DF=12
SD-pe
R2=0,9587
Q2=0,7920
SD-LoF
SD-pe * sqrt(Fcrit)
R2Adj=0,9104
RSD=0,0084
AlphaLev=0,05
N=27
DF=12
SD-pe
R2=0,7075
Q2=0,1317
SD-pe * sqrt(Fcrit)
R2Adj=0,3661
RSD=0,0485
AlphaLev=0,05
Figure 19.
ANOVA: Plot of lack of fit for ĮCT and Į2. If the standard deviation of lack of fit
(SD-LoF) is smaller than the standard deviation of the pure error multiplied by the
square rot of the critical F (SD-pe*sqrt(Fcrit) there is no lack of fit in the model. For
ĮCT there is no lack of fit but for Į2 there is a lack of fit.
59
Investigation: Citalopram (PLS, Comp=5)
ĮCT
99,9
N-Probability
99,5
99
98
95
90
80
70
60
50
40
30
20
10
5
2
1
0,5
12
3
113
0
16
9
11
1
19
24
525
27 18
21
1720
8
23
26 14
6
2215
4
7
Modde 4.0
2
-1,0
-0,5
0,0
0,5
1,0
1,5
Standardized Residuals
R2=0,9587 R2Adj=0,9104
Q2=0,7920 RSD=0,0084
N=27
DF=12
Investigation: Citalopram (PLS, Comp=5)
ĮDCT
99,9
N-Probability
99,5
99
98
95
90
80
70
60
50
40
30
20
10
5
2
1
0,5
7
10
21
8
1923
1
22
26
182
24
16
13
15
17
143
25
4 27
11 20
9
12
6
Modde 4.0
5
-1,5
N=27
DF=12
-1,0
-0,5
0,0
0,5
1,0
Standardized Residuals
R2=0,9650 R2Adj=0,9242
Q2=0 7719 RSD=0 0068
Figure 20.
Residual N-plot for ĮCT and ĮDCT, showing the distribution of the residuals. For
ĮDCT, experiment number 5 can be considered as an outlier.
60
Investigation: Citalopram (PLS, Comp=5)
Normalized Coefficients
pH
Ko
Me
Te
pH*pH
Ko*Ko
Me*Me
Te*Te
pH*Ko
pH*Me
pH*Te
Ko*Me
Ko*Te
Me*Te
0,4
0,2
0,0
-0,2
-0,4
-0,6
Modde 4.0
-0,8
Log tR´
R-CT DCT
DDCT
DDDCT
D1
D2
A N=27 CondNo=6.1592
DF=12 Y-miss=0
Investigation: Citalopram (PLS, Comp=3)
Normalized Coefficients
pH
Ko
Me
Te
pH*Ko
pH*Me
Ko*Me
0,4
0,2
0,0
-0,2
Modde 4.0
-0,4
-0,6
-0,8
Log tR´
R-CT DCT
B
DDCT
DDDCT
D1
D2
N=27 CondNo=1.1331
DF=19 Y-miss=0
Figure 21.
A. Normalized coefficient with all interaction and quadratic terms included.
B. Normalized coefficient with non-significant quadratic and cross terms removed.
61
Investigation: Citalopram980210RT (PLS, Comp=5)
Scaled & Centered Coefficients for ĮCT
Modde 4.0
0,01
0,00
pH*Te
Ko*Me
Ko*Te
Me*Te
pH*Te
Ko*Me
Ko*Te
Me*Te
pH*Me
pH*Ko
Te*Te
Ko*Ko
pH*pH
Te
Me
Ko
pH
-0,02
Me*Me
-0,01
N=27 R2=0,9585 R2Adj=0,9101
DF=12 Q2=0,8232 RSD=0,0084 ConfLev=0,95
Investigation: Citalopram980210RT (PLS, Comp=5)
Scaled & Centered Coefficients for Į2
0,04
Modde 4.0
0,02
0,00
-0,02
pH*Me
pH*Ko
Te*Te
Me*Me
Ko*Ko
pH*pH
Te
Me
Ko
pH
-0,04
N=27 R2=0,7076 R2Adj=0,3665
DF=12 Q2=0,1552 RSD=0,0485 ConfLev=0,95
Figure 22.
Coefficient plot. Plot of scaled and centred coefficients for the responses ĮCT and Į2.
All factors for the response are displayed in bars with their confidence intervals.
Factors that are within the boundaries of the confidence interval have no significance.
62
Figure 23
D CT
tR´ R-CT
tR´
D DCT
D
D
1,19
1,20
1,19
40
1,18
1,18
1,17
1,17
1,16
1,16
20
4,5
5,0
1,15
6,5
6,0
20
40
1,14
140 120
100
5,5
60
80
100
Concentration
pH
5,0
120
140
80
60
Concentration
4,5
D DDCT
20
140
1,14 120 100
pH
6,0
40
5,5
80
60
Concentration
6,5
D
pH
6,0
40
20
6,5
D
D
D
D
4,5
5,0
1,15
5,5
1,10
1,05
1,160
1,155
1,00
1,150
0,95
1,05
1,00
1,145
4,5
5,0
1,140
140 120
100
0,90
5,5
80
60
Concentration
6,0
40
20
6,5
6,0
20
pH
6,5
40
60
80
100
Concentration
20
pH
5,0
120
140
6,5
6,0
0,95
5,5
40
5,5
60
80
Concentration
4,5
100
pH
5,0
120
140
4,5
A. Response surface for the retention time of the last-eluated enantiomer of citalopram (RCT) and for the separation factors for DCT, DDCT, DDDCT, D1 and D2.Methanol content
and temperature were held at their centre points, 55 % and 20 qC, respectively; the other
factors were varied between their minimum and maximum values.
tR´ R-CT
DDCT
D CT
tR´
D
D
1,17
1,17
120
1,16
100
1,15
80
1,14
1,16
1,15
1,14
1,13
60
1,13
1,12
40
5
10
50
15
45
20
5
10
55
20
25
Temperature
60
30
35
Methanol
65
D DDCT
D
50
15
1,11
45
1,10
1,12
5
10
55
20
25
Temperature
60
30
35
Methanol
65
D
D
50
15
45
1,11
55
20
25
Temperature
60
30
35
Methanol
65
D
D
1,12
1,10
1,16
1,11
1,15
1,08
1,10
Alfa1
1,14
1,09
1,06
1,13
1,08
1,04
1,12
5
1,11
45
10
50
15
55
20
25
Temperature
60
30
35
65
Methanol
1,07
35
30
1,02
65
60
25
20
55
15
Temperature
50
10
5
45
Methanol
5
1,06
45
10
50
15
55
20
25
Temperature
60
30
35
Methanol
65
B. Response surface for the retention time of the last-eluated enantiomer of citalopram (RCT) and for the separation factors for DCT, DDCT, DDDCT, D1 and D2. pH and buffer
concentration were 6,3 and 10 mM, respectively; the other factors were varied between
their minimum and maximum values.
63
Optimization with Multisimplex
The control variables pH, buffer concentration, methanol content and temperature
were the same as the factors in the Modde optimization and likewise the response
variables were separation factors within enantiomeric pairs (ĮCT, ĮDCT and ĮDDCT)
and between enantiomeric pairs (Į1 and Į2). The adjusted retention time was not used
in this optimization process. The reference values from which the five initial trials
were calculated were the same as the center point in the Modde optimization i.e.
methanol content 55%, pH 5.5, temperature 20 qC and buffer concentration 75 mM.
For the Multisimplex optimization 12 experiments were performed to obtain an
adequate separation within and between enantiomer pairs. The resulting mobile phase
was 57:43 methanol: 22 mM citric acid-triethylamine pH 6.3 at 13 qC. Comparing the
results obtained with Modde 55:45 methanol: 10 mM citric acid-triethylamine pH 6.3
at 30 qC. The divergence between the results of these two optimization programs is to
be found in the obtained optimal column temperature. One possible explanation, which
has come forward after this study, is that the reduction of the separation factors (ĮCT,
ĮDCT, ĮDDCT, Į1 and Į2) is caused by aging of the column, and can be compensated
for by lowering the temperature of the column. As the optimization trials with the two
programs were done on different occasions, with several months inbetween, a certain
degree of aging may have occurred. The storage media for this type of column was
methanol but today the manufacturer recommends 2-propanol, as it has less effect on
the stationary phase.
Separation factors (Į) have been used as responses in Modde and response variables in
Multisimplex. Instead of separation factors, Kaiser peak separation index or resolution
can be used. The Kaiser peak separation index is defined as the average valley depth
expressed as a ratio to the average peak height of the two adjacent peaks (Owens et al.,
1996), see appendix. In these chromatograms, the use of resolution would require that in
every experiment, the single enantiomers have had to be injected to obtain the parameters for
the calculations.
Separation mechanism
With the E-CD Ac, the separation was improved, as compared to E-CD. The
acetylation of sterically fixed hydroxyl groups changes the nature of the interaction
from a diastereomeric complex formed by simple hydrogen bonding to sites with
hydrogen donor and acceptor mechanisms which are a much stronger type of
interaction. The size of the CD cone and the possibility of a stronger type of
diastereomeric interaction between the analytes and the chiral phase make the
separation between enantiomers of the parent drug and metabolites possible.
The temporary complex formation between the different citalopram enantiomers and
the cyclodextrin cone (which is proposed in reversed phase mode) can not fully base
line separate these six enantiomers. To obtain a complete baseline separation between
the enantiomer pairs, pH has to be 4 (limit for the column), and a buffer concentration
around 100 mM has to be used at low temperature. This will result in overlapping
peaks from parent drug and metabolites and, to be able to quantify the enantiomers, a
64
mass spectrometric detector is needed. The mobile phase, however, in this application
is not compatible with the mass spectrometric detector.
Summary of paper II
Modde vs Multisimplex
Both programs use response surfaces to determine an optimum. To be able to
determine an optimum it is necessary that the polynomial function contains quadric
terms (Lundstedt et al., 1998). Modde gives more information about the mechanism
behind a chiral chromatographic separation, which has been shown in many articles.
Multisimplex is a more straightforward way of finding an optimum, but a disadvantage
is that there is also the risk that a local optimum has been found. Multisimplex and
Modde complement each other. Chromatographers might find the optimum faster with
Multisimplex but they are not gaining as much information about the separation
mechanism as they do with Modde.
65
Paper III
Citalopram and adolescents
Citalopram extraction version II
The extraction procedure from paper I and the optimized chromatographic procedure
for the chiral chromatography from paper II were intended to be used for
determination of the enantiomers of Cit and its metabolites.
At the very first analytical test, it turned out that the mix of the extraction methodology
and the chiral separation were not compatible with regard to the resulting
chromatography, see figure 24.
Standard ethanol solutions of Cit, DCit and DCCit diluted in the mobile phases were
used during the optimization study. The conclusion was drawn that the cause had to be
found in the extraction procedure. An investigation was performed to find out how to
avoid these disturbances. An unwanted residue of acetic acid after the evaporating step
could lower the pH of the injection solution, leading to distortion in the chromatogram.
This could be solved by using 2.5 % ammonia in acetonitrile as the eluating agent, a
solvent that was discarded during the evaluation of the achiral chromatography due to
losses in the evaporation step. The losses could however be avoided by the addition of
100 µl 10 mM citric acid to the tube used for collecting the eluate, but to obtain good
extraction recovery 2*2 ml of 2.5 % ammonia in acetonitrile had to be used. The
problem was not solved totally by these measures. It seems also that the phosphate
buffer used contributed to the chromatographic problem and as a consequence this was
substituted for by 30 mM NaOH. The change from methanol to acetonitrile improved
the extraction in another way as methanol usually gives more dirty extracts than
acetonitrile.
66
A
B
Figure 24.
A. Injection of a solution of Cit, DCit and DDCit in mobile phase. B. Injection of a
plasma extract using extraction method I and re-dissolved in the mobile phase for the
chiral separation of Cit, DCit and DDCit.
Results, citalopram and adolescents
As no investigations had been done regarding Cit enantiomers and adolescents, such a
study was initiated. Together with the Child and Adolescent Department at Uppsala
University Hospital, adolescent patients on Cit medication were recruited. The data on
the concentration of the enantiomers were found to be in agreement with earlier
studies on adults. However in these other studies, see table 7, additional CNS drugs
were prescribed. In our study of adolescent patients, there were a few females using
contraceptive hormones. In earlier studies a correlation between the ratio of the S/R
Cit and S/R DCit had been found. In this study however an interesting finding was that
the correlations were good for females not using contraceptive hormones but not for
females using contraceptive hormones, se figure 25 and 26. A possible explanation
may be an interaction of contraceptive hormones with the metabolism of citalopram.
Several studies have shown that contraceptive hormones may interact with the
polymorphic enzyme CYP2C19 (Hägg et al., 2001; Laine et al., 2000; Tamminga et
al., 1999; Tanaka, 1999b).
67
20
20
min
-10
0,0
5,0
10,0
15,0
20,0
25,0
30,0
R-IS-CIT - 21,493
R-CIT - 16,113
Int_Chan_1
S-IS-CIT - 19,358
40
R-DCIT - 12,844
S-IS-CIT - 19,043
R-IS-CIT - 21,059
S-DDCIT - 9,827
40
S-CIT - 14,654
60
R-CIT - 15,874
60
R-DCIT - 12,662
80
R-DDCIT - 10,747
S-DCIT - 11,580
80
Patient number 18
S-CIT - 14,922
100 Citalopram enantiomers #30
mV
Int_Chan_1
S-DCIT - 11,789
Standard I
S-DDCIT - 10,015
R-DDCIT - 10,853
100 Citalopram enantiomers #1
mV
min
-10
0,0
5,0
10,0
15,0
20,0
25,0
30,0
Figure 25.
Standard I with 3.5 nmol/L of S- and R-DDCit, 7.5 nmol/L of S- and R-DCit, 15
nmol/L of S- and R-Cit and a patient with 4 S- DDCit, 7 R-DDCit, 27 S-DCit, 38 RDCit, 40 S-Cit and 90 nmol/L R-Cit.
Correlation S/R ratio Without contraceptive pill
1,2
1,2
1
1
S/R Desmethylcitalopram
S/R Desmethylcitalopram
Correlation S/R ratio With contraceptive pill
0,8
0,6
y=0.416*x+0.581
r2=0.163
r=0.403
p=0.2480
0,4
0,2
0,8
0,6
y=1.355 *x+0.095
r2=0.926
r=0.962
p=0.0005
0,4
0,2
0
0
0
0,2
0,4
0,6
0,8
1
0
S/R Citalopram
0,2
0,4
0,6
0,8
1
S/R Citalopram
Figure 26.
Correlation between the S/R ratio for Cit and S/R ratio for DCit for females using and
those not using oral contraceptives.
68
Comparison with data from adults
Four studies have hitherto been published were the concentration of the enantiomers
have been measured in patients on Cit medication. In table 7 a summary of the four
studies is presented with the intention of giving an indication of what concentrations of
S-Cit are to be expected.
Genotyping
In this study, samples for determination of the enantiomers of Cit and for genotyping
of the polymorphic enzymes CYP2D6 and CYP2C19 were drawn from every patient.
With the frequencies of 7 % for PM and 1-3 % for UM of 2D6 and 3 % of 2C19 PM
the study had to have a design of at least 100 patients, if it were to be possible to draw
any conclusions regarding these polymorphic enzymes. The genotyping results could
however be a tool to explain possible outliers found among the patients in the
enantioselective analysis.
Other examples where chiral analysis has been of value
During the journey from achiral to chiral analysis of Cit, other problems/questions
have emerged, such as inquiries from clinicians about Cit metabolism or interesting
findings obtained with the achiral method, leading to a further analysis with the chiral
method. The first example is from a patient with Crohn´s disease who was followed
using plasma samples during a 24 hours period. In figure 27, the plasma concentration
and ratio of the different enantiomers versus time is shown. These led to the dose
given to the patient being increased to 120 mg daily. This plasma concentration profile
could be compared with the study by Sidhu et al (Sidhu et al., 1997) on healthy human
subjects, who were extensive metabolizers of CYP2D6 and CYP2C19 and who took
multiple doses (40 mg per day) of Cit for 21 consecutive days. The results of this study
showed the same stereoselectivity in the disposition of Cit, DCit and DDCit.
69
Concentration of the enantiomers
Ratio between S och R enantiomers
250
R-Cit
S-Cit
150
Ratio
S/R
nmol/L
200
R-DCit
S-DCit
100
R-DDCit
S-DDCit
50
0
0
10
20
30
1,00
0,90
0,80
0,70
0,60
0,50
0,40
0,30
0,20
0,10
0,00
S/R-DCit
S/R-DDCit
S/R-Cit
0
Hours after dose
10
20
30
Hours after dose
Figure 27.
Concentration- and ratio-time curves on a patient suffering from Crohn´s disease on a
Cit dose of 60 mg daily.
Carbamazepine, an inducer of CYP3A4, has been shown to affect the metabolism of
Cit (Leinonen et al., 1996; Leinonen et al., 1991). Phenytoin is also an inducer of
CYP3A4, but in contrast to carbamazepine, it is not itself metabolized via CYP3A4
(Tanaka, 1999a). Phenytoin is mainly metabolized by CYP2C19 and CYP2C9. In this
example a sample from a patient on 20 mg of Cit and 100 mg of phenytoin daily was
analysed by the enantioselective method with the following concentrations found
[nmol/L] 17 S-DDCit, 41 R-DDCit, 124 S-DCit, 183 R-DCit, 114 S-Cit and 141 R-Cit.
S/R ratios were S/R DDCit 0.41, S/R DCit 0.68 and S/R Cit 0.81, see figure 28. In contrast
to the normal parent/metabolite ratio (Cit/DCit) of between 0.3-0.5, this patient
showed a ratio of 0.83 indicating an induction of CYP3A4 metabolism. The ratio
between the different enantiomers did not differ as compared to earlier studies.
70
150
100
50
-10
0,0
R-IS-CIT - 20,501
R-CIT - 15,784
S-DDCIT - 8,975
R-DDCIT - 9,333
200
S-CIT - 14,849
250
Int_Chan_1
S-IS-CIT - 18,928
CIPRALEX03033 #11 [modified by Administratör]
mV
S-DCIT - 11,081
R-DCIT - 11,677
300
min
5,0
10,0
15,0
25,0
20,0
30,0
Figur 28.
Chromatogram of a serum sample from a patient at steady state, before dose, receiving
20 mg Cit and 100 mg phenytoin daily with the following concentrations found [nmol/L]
17 S-DDCit, 41 R-DDCit, 124 S-DCit, 183 R-DCit, 114 S-Cit and 141 R-Cit.
Animal studies
The chiral method has also been used in studies on rats. Enantioselective analysis of
Cit and its metabolites in blood and brain has been used in a study on open-field
behaviour in rats (Wikell et al., 1999). To further scrutinize the enantiomer-selective
distribution of Cit beyond clinical doses, an investigation concerning the toxicological
effects of Cit has been carried out (Kugelberg et al., 2001) on rats with a further
extension as a model for investigating drug distribution postmortem.
Summary of paper III
With this study on adolescents, the distribution of Cit enantiomers have been
investigated and found to be in agreement with studies on adult patients.
As the polymorphic enzyme CYP2C19 is involved in the metabolism of contraceptive
hormones as well as of citalopram a possible interaction has been shown.
71
Carlsson B et al (2001) Rochat B et al (1995)
72
Racemic drug dose in mg/day 10-30 (30r14)
(meanrS.D.)
Diagnosis
Major depressive
disorder (dysthymia)
Number
19
Female
16
Male
3
Age year (mean)
15-20(18)
Steady state
>14 days
Trough value (hours
10-30
after dose)
Other drugs
No (contraceptives)
Citalopram nmol/L
S (mean rS.D.)
62r53
S/R (mean rS.D.)
0,59r0,13
DCit nmol/L
S (mean rS.D.)
26r17
S/R (mean rS.D.)
0,82r0,13
DDCit nmol/L
S (mean rS.D.)
4r1
S/R (mean rS.D.)
0,46r0,14
Zheng Z et al (2000)
20-60 (44r15)
20-40 (22r7)
Foglia JP et al (1997) Normalized to
20 mg/day
20
Depressive patients
29
13
16
20-65
TDM
12-24
Stroke-induced
depression
16
13
3
61-88
One week
Trough
Dementia + behavioral
disturbances
8
4
4
76-90 (77,2)
15-90
Two weeks
SS
12-15
Trough value
CNS drugs
CNS, somatic
+ lorazepam
103r66
0,56r0,14
88r40
0,7r0,17
100r76
0,65r0,19
54r41
50r26
0,72r0,2
37r36
0,95r0,2
50r13
1,08r0,3
25r15
?
?
?
?
Table 7.
Data comparing the four investigations carried out using concentration determination of citalopram’s enantiomers. Recalculated
to 20 mg daily of Cit. Data from (Carlsson et al., 2001; Foglia et al., 1997; Rochat et al., 1995a; Zheng et al., 2000).
Paper IV
Enantioselective analysis of citalopram in
postmortem blood and genotyping
The enantioselective analysis on this material was carried out before the clinical study
in paper III. Genotyping for the polymorphic enzymes CYP2D6 and CYP2C19 has
prolonged the process of publication of paper IV, thus explaining its later publication.
Citalopram extraction version III
Extraction of whole blood was a new challenge. To be able to use solid phase
extraction, and to extract the analytes with sufficient recovery the femoral whole blood
samples had to be diluted, sonicated (treated by ultrasonic bath) and centrifuged before
extraction on solid-phase columns (Verweij et al., 1996).
Blood should be handled much like serum or plasma. The difference is the presence of
whole red blood cells and, in this special case, that many postmortem samples are
clotted.
Buffering/sample pretreatment
To optimize the extraction for whole blood samples, the sample pretreatment step was
modified. As the concentrations in these samples could be assumed to be higher than
in the TDM service 0.1-0.5 g blood (corresponding approximately to 0.1-0.5 ml) were
used and the standard curves were prepared in the concentration range 0.01 to 0.16
µg/g (a30-500 nmol/L) for S- and R-DDCit, 0.05 to 0.8 µg/g (a150-2500 nmol/L) for
S- and R-DCit and 0.1 to 1.6 µg/g (a300-5000 nmol/L) for S- and R-Cit.
IS-Cit was added to the sample, followed by dilution with 3 ml 5:95 acetonitrile:0.025
M phosphate buffer. After vortex mixing, sonication and centrifugation the supernatant
was poured on the conditioned solid phase column and the extraction procedures given
in paper I were followed.
As the determination of the enantiomers was performed before study III, a minor
problem was seen regarding skew ness of the peaks when the samples from the whole
blood extraction were re-dissolved in the mobile phase for the chiral chromatography
(55:45 methanol:10 mM citric acid-triethylamine).
This problem was solved by re-dissolving the samples in a “mobile phase” with higher
content of buffer, 55:45 methanol:100 mM citric acid-triethylamine pH 6.3, which
brought the pH to 6.3.
In table 8, the extraction recoveries for the three extraction procedures are
summarised.
73
Extraction
method
Cit
DCit
DDCit
IS
I
Achiral method
plasma
98
102
96
89
II
Chiral method
plasma
89
108
102
75
III
Chiral method
whole blood
91
93
94
85
Table 8.
Extraction recovery for the three extraction procedures.
Drug levels in postmortem whole blood versus plasma
concentrations from patients with toxicological symptoms
The findings that a S/R–Cit ratio close to 1.0 in postmortem whole blood is associated
with an intake of the drug only a few hours or less before death can further be
confirmed by some examples from routine toxicology. In table 9 six patients found to
have toxic symptoms were analysed regarding the racemic plasma concentrations and
later followed up by enantioselective analysis.
Sample
number
1
2
3
4
5
6
DCit
S/R Ratio
DDCit
S/R Ratio
Cit
S/R Ratio
Total
Total
Total
concentration
concentration
concentration
nmol/L (µg/g)
nmol/L (µg/g)
nmol/L (µg/g)
2472 (0.80)
1.03
318 (0.097)
1.37
43 (0.013)
981 (0,32)
0.94
198 (0.061)
0.92
<2
2.07
1591 (0.52)
0.99
239 (0.074)
1.88
23 (0.007)
640 (0.21)
0.90
115 (0.036)
0.69
<2
1188 (0.39)
0.89
191 (0.059)
0.84
15 (0.004)
1.14
2484 (0.81)
1.14
172 (0.053)
1.00
<2
Table 9.
Plasma concentration and S/R ratio from six patients with toxic symptoms.
In case number 8 in paper IV, a higher concentration of DCit than Cit was found.
Phenytoin was also found together with Cit in this case. Interaction with phenytoin, as
in the example shown from a patient on citalopram and phenytoin, gives further
support to that CYP3A4 being induced by phenytoin.
74
Genotyping
In this study, the patients were also genotyped for the polymorphic enzymes CYP2D6
and CYP2C19. With the frequencies of 7 % for PM and 1-3 % for UM of 2D6 and
3 % of 2C19 PM, the study had to have a design of at least 100 patients, if it were to be
possible to draw any conclusions regarding these polymorphic enzymes. The low
frequency approximately 3.5 % of PM in this study is however in agreement with an
earlier study (Druid et al., 1999). In this paper pyrosequencing has been used for the
genotyping of CYP2D6 but in the earlier paper the technique by Heim and Mayer
(Heim and Meyer, 1990) was used.
Summary of paper IV
Investigation of these autopsy cases showed that pharmacokinetic interactions have a
greater impact on the cause of death than metabolic deficiency regarding the
polymorphic enzymes CYP2D6 and CYP2C19. The enantioselective analysis of Cit
and the S/R ratio can give additional information when interpreting the results,
especially with regard to the time elapsed between intake and death.
75
Conclusions
Chemometrics – Experimental design
Experimental design has been shown to be an effective way of planning and
performing chromatographic optimization systematically. Optimization with response
surface methodology using central composite or modified simplex design led to a
refined chiral chromatographic process for the analysis of citalopram and its
demethylated metabolites. Simplex design was a quicker way to obtain the optimum
while the central composite design gave additional information about the separation
mechanism on the acetylated E-cyclodextrin column.
Therapeutic drug monitoring:
Achiral or chiral analysis of citalopram
An achiral method for TDM may still have a function in the daily routine to identify
problems e.g. with compliance. With the introduction of S-Cit (escitalopram) the
possibility of finding a concentration-effect relationship may be possible and if so the
chiral method could support clinicians in optimization of the effect. As racemic
citalopram is still on the market, the chiral method can support the switch from
racemic to pure enantiomer or vice versa. Patients may by mistake take the racemic
form instead of the pure enantiomer or vice versa, where the chiral method may unveil
the presence or not of R-Cit in the blood sample.
Forensic chemistry:
Achiral or chiral analysis of citalopram
The use of enantioselective analysis within forensic chemistry can serve as a
complement to the achiral routine. In the case of Cit, the introduction of escitalopram
and the concomitant prescribing of racemic generic products of Cit, a statement from
the forensic laboratory including enantioselective analysis may give certain additional
information about the cause of death. Furthermore citalopram is not, or will not be, the
only drug that will go from racemic to single enantiomer (chiral switches) (Agranat et
al., 2002; Baumann and Eap, 2001).
76
Future studies
LCMS
It is possible to further improve the chiral method for Cit analysis by changing from
fluorescence detection to mass spectrometric detection. The mobile phase used
however has to be replaced because triethyl amine and to some extent citric acid is not
suitable. Maybe another type of column has to be used instead of acetylated Ecyclodextrin with a mobile phase that is more compatible with a mass spectrometric
detector. There is also the possibility of using the mass spectrometer, as it examines
different masses, in order to measure different enantiomers in overlapping peaks if this
is the only way to obtain baseline separation between the enantiomers.
On-line extraction and chiral analysis
A more advanced method construction is that of automatizing the solid phase
extraction followed by direct injection and separation on a chiral column. Another
combination is on-line extraction with alkyl-diol silica (Öhman et al., 2001) followed
by mass spectrometric detection.
Free fraction
A question to be solved is if the total concentration, the sum of free fraction and drug
bound to plasma proteins, or free fraction only best reflect the effect/concentration in
the brain.
Chiral switches
Today most of the new drugs reaching the market are single enantiomers, rather than
the racemic mixtures that dominated up to ten years ago. Many of the new singleenantiomer drugs were developed as such, but there are also important examples of
new single-enantiomer drugs derived from “chiral switches” of established racemates
with examples such as, omeprazole to esomeprazole or bupivacaine to ropivacaine the
first local anaesthetic marketed as pure S-(-) enantiomer (Agranat et al., 2002; Agranat
and Caner, 1999; Baker and Prior, 2002; Caldwell, 2001; Ekatodramis and Borgeat,
2001; Tucker, 2000; Williams and Wainer, 2002).
Examples of “chiral switches” within the field of psychopharmacology are the switch
from citalopram to escitalopram (Baumann et al., 2002; Montgomery et al., 2001; von
Moltke et al., 2001) and that the enantiomer S-fluoxetine is in the clinical trial phase
for migraine headaches, as well as R-fluoxetine as an antidepressant (Baumann
and Eap, 2001).
As patents for the racemic drugs are expiring and generic products will be on the
market at the same time as the “chiral switches” there will be a need for
77
enantioselective analysis both within TDM and forensic medicine. It should not be
forgotten that metabolic processes can give unwanted effects, e.g. chiral inversion as
exemplified by thalidomide, which was withdrawn due to teratogenicity and
neuropathy (Reist et al., 1998), where the non teratogenic form is converted to the
teratogenic form in the body.
78
Acknowledgements
To my supervisor, Professor, Johan Ahlner. After many persuasive attempts from you
and a trip to Barcelona to listen to the controversial speech by E.J. Ariens about
racemic drugs, I finally gave up and began a somewhat slow journey with citalopram
towards this thesis.
To Björn Norlander, my co-supervisor, co-author and roommate at work. For making
sense of my fragmental or overheadish lines and for twenty years of fruitful scientific
discussions, friendship and how to win the pools.
To Professor Curt Peterson. Head of the Division of Clinical Pharmacology. For
giving me the time to complete my work
To my co-authors, specially Gunilla Olsson, Margareta Reis and Per Holmgren
To Eva Ekefeldt-Hultin and Karin Walldén. For not complaining about my absence
during the last six months and for their help with analysis of citalopram.
To the Citalopram mafia Fredrik Kugelberg, Daniel Öhman and Finn Bengtsson.
To the rest of the staff and PhD students at the Division of Clinical Pharmacology
To the pensioners/veterans at the Division of Clinical Pharmacology, with special
thanks to Åke Bertler for giving me the opportunity to begin my thesis and studies in
the Spanish languish.
To my children Mario and Paloma. Although the have complained about the mess in
the study room (Paloma) and about me not having time for soccer or ice hockey
(Mario).
To my mother Stina and in remembrance of my father Bertil.
To my sister Eva, and my brothers Mats and Jan for support and always offering a
helping hand when needed.
A Pepe Chermá y Dolores Yeste porque me dejaron traerme a su Fallera a Suecia. I por
las peladillas, jamónes, chorizos y vinos.
To Lions, the Faculty of Health Sciences, Linköping University, the County
Council of Östergötland, Anna-Lisa and Anders Olofssons’ fund for the
investigation of mental health, H. Lundbeck (Copenhagen, Denmark), the National
Board of Forensic Medicine and Laboratory Medicine Östergötland for financial
support.
79
References
Agranat, I., Caner, H., and Caldwell, J. (2002). Putting chirality to work: the strategy of chiral switches. Nat Rev
Drug Discov 1, 753-768.
Agranat, I. I., and Caner, H. (1999). Intellectual property and chirality of drugs. Drug Discov Today 4, 313-321.
Allenmark, S., ed. (1991). Chromatographic enantioseparation methods and applications, second edn
(Chichester, Ellis Horwood Limited).
Altria, K. D., Clark, B. J., Filbey, S. D., Kelly, M. A., and Rudd, D. R. (1995). Application of chemometric
experimental designs in capillary electrophoresis: a review. Electrophoresis 16, 2143-2148.
Anastos, N., McIntyre, I. M., Lynch, M. J., and Drummer, O. H. (2002). Postmortem concentrations of
citalopram. J Forensic Sci 47, 882-884.
Ariens, E. J. (1984). Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical
pharmacology. Eur J Clin Pharmacol 26, 663-668.
Ariens, E. J. (1986). Chirality in bioactive agents and its pitfalls. Trends Pharmacol Sci 7, 200-205.
Ariens, E. J., and Wuis, E. W. (1987). Bias in pharmacokinetics and clinical pharmacology. Clin Pharmacol Ther
42, 361-363.
Ariens, E. J., Wuis, E. W., and Veringa, E. J. (1988). Stereoselectivity of bioactive xenobiotics. A pre-Pasteur
attitude in medicinal chemistry, pharmacokinetics and clinical pharmacology. Biochem Pharmacol 37, 9-18.
Armstrong, D., and Li, W. (1987). Optimization of liquid chromatography separations on cyclodextrin-bonded
phases. Chromatography 2, 43-48.
Armstrong, D. W. (1998). The evolution of chiral stationary phases for liquid chromatographaphy. LC GC
International April, 23-31.
Armstrong, D. W., and Zhang, B. (2001). Chiral stationary phases for HPLC. Anal Chem 73, 557A-561A.
Baker, G. B., and Prior, T. I. (2002). Stereochemistry and drug efficacy and development: relevance of chirality
to antidepressant and antipsychotic drugs. Ann Med 34, 537-543.
Baumann, P., and Eap, C. B. (2001). Enantiomeric antidepressant drugs should be considered on individual
merit. Hum Psychopharmacol 16, S85-S92.
Baumann, P., and Larsen, F. (1995). The pharmacokinetics of citalopram. Rev Contemp Pharmacother 6, 287295.
Baumann, P., and Rochat, B. (1995). Comparative pharmacokinetics of selective serotonin reuptake inhibitors: a
look behind the mirror. Int Clin Psychopharmacol 10 Suppl 1, 15-21.
Baumann, P., Zullino, D. F., and Eap, C. B. (2002). Enantiomers' potential in psychopharmacology--a critical
analysis with special emphasis on the antidepressant escitalopram. Eur Neuropsychopharmacol 12, 433-444.
Bempong, D. K., and Honigberg, I. L. (1996). Multivariate analysis of capillary electrophoresis separation
conditions for Z-E isomers of clomiphene. J Pharm Biomed Anal 15, 233-239.
Bergström, T., and Öberg, T. (1996). User´s Guide to Multisimplex TM 1.0, 1.0 edn (Karlskrona, Bergström &
Öberg).
Bernal, M. L., Sinues, B., Johansson, I., McLellan, R. A., Wennerholm, A., Dahl, M. L., Ingelman-Sundberg,
M., and Bertilsson, L. (1999). Ten percent of North Spanish individuals carry duplicated or triplicated CYP2D6
genes associated with ultrarapid metabolism of debrisoquine. Pharmacogenetics 9, 657-660.
Berridge, J. C. (1982). Unattended optimisation of reversed-phase high-performance liquid chromatographic
separations using the modified simplex algorithm. J Chromatogr 244, 1-14.
80
Berridge, J. C., Jones, P., and Roberts-McIntosh, A. S. (1991). Chemometrics in pharmaceutical analysis. J
Pharm Biomed Anal 9, 597-604.
Bertilsson, L., Carrillo, J. A., Dahl, M. L., Llerena, A., Alm, C., Bondesson, U., Lindstrom, L., Rodriguez de la
Rubia, I., Ramos, S., and Benitez, J. (1994). Clozapine disposition covaries with CYP1A2 activity determined by
a caffeine test. Br J Clin Pharmacol 38, 471-473.
Bertilsson, L., and Dahl, M. L. (1996). Polymorphic drug oxidation. Relevance to the treatment of psychiatric
disorders. CNS Drugs 5, 200-223.
Bertucci, C., and Domenici, E. (2002). Reversible and covalent binding of drugs to human serum albumin:
methodological approaches and physiological relevance. Curr Med Chem 9, 1463-1481.
Bezchlibnyk-Butler, K., Aleksic, I., and Kennedy, S. H. (2000). Citalopram--a review of pharmacological and
clinical effects. J Psychiatry Neurosci 25, 241-254.
Boerner, R. J., and Moller, H. J. (1999). The importance of new antidepressants in the treatment of
anxiety/depressive disorders. Pharmacopsychiatry 32, 119-126.
Bondolfi, G., Chautems, C., Rochat, B., Bertschy, G., and Baumann, P. (1996). Non-response to citalopram in
depressive patients: pharmacokinetic and clinical consequences of a fluvoxamine augmentation.
Psychopharmacology (Berl) 128, 421-425.
Bonner, W. A. (2000). Parity violation and the evolution of biomolecular homochirality. Chirality 12, 114-126.
Boos, K. S., and Rudolphi, A. (1998a). The use of novel restricted-access media in HPLC, part I - classification
and review. LC-GC International February, 84-95.
Boos, K. S., and Rudolphi, A. (1998b). The use of novel restricted-access media in HPLC, part II - applications.
LC-GC International April, 224-233.
Bressolle, F., Audran, M., Pham, T. N., and Vallon, J. J. (1996). Cyclodextrins and enantiomeric separations of
drugs by liquid chromatography and capillary electrophoresis: basic principles and new developments. J
Chromatogr B Biomed Appl 687, 303-336.
Brosen, K. (1996). Drug-metabolizing enzymes and therapeutic drug monitoring in psychiatry. Ther Drug Monit
18, 393-396.
Brosen, K. (1998). Differences in interactions of SSRIs. Int Clin Psychopharmacol 13 Suppl 5, S45-47.
Brosen, K., and Naranjo, C. A. (2001). Review of pharmacokinetic and pharmacodynamic interaction studies
with citalopram. Eur Neuropsychopharmacol 11, 275-283.
Caccia, S. (1998). Metabolism of the newer antidepressants. An overview of the pharmacological and
pharmacokinetic implications. Clin Pharmacokinet 34, 281-302.
Caldwell, J. (1995). Stereochemical determinants of the nature and consequences of drug metabolism. J
Chromatogr A 694, 39-48.
Caldwell, J. (2001). Do single enantiomers have something special to offer? Hum Psychopharmacol 16, S67S71.
Carlsson, B., and Norlander, B. (2001). Optimization and Characterization of the Chiral Separation of
Citalopram and its Demethylated Metabolites by Response Surface Methodology. Chromatographia 53, 266-272.
Carlsson, B., Olsson, G., Reis, M., Walinder, J., Nordin, C., Lundmark, J., Scordo, M. G., Dahl, M. L.,
Bengtsson, F., and Ahlner, J. (2001). Enantioselective analysis of citalopram and metabolites in adolescents.
Ther Drug Monit 23, 658-664.
Carrillo, J. A., Christensen, M., Ramos, S. I., Alm, C., Dahl, M. L., Benitez, J., and Bertilsson, L. (2000).
Evaluation of caffeine as an in vivo probe for CYP1A2 using measurements in plasma, saliva, and urine. Ther
Drug Monit 22, 409-417.
81
Coutts, R. T., and Urichuk, L. J. (1999). Polymorphic cytochromes P450 and drugs used in psychiatry. Cell Mol
Neurobiol 19, 325-354.
Dahl, M. L. (2002). Cytochrome p450 phenotyping/genotyping in patients receiving antipsychotics: useful aid to
prescribing? Clin Pharmacokinet 41, 453-470.
Dahl, M. L., Johansson, I., Bertilsson, L., Ingelman-Sundberg, M., and Sjoqvist, F. (1995). Ultrarapid
hydroxylation of debrisoquine in a Swedish population. Analysis of the molecular genetic basis. J Pharmacol
Exp Ther 274, 516-520.
Dahl, M. L., and Sjoqvist, F. (2000). Pharmacogenetic methods as a complement to therapeutic monitoring of
antidepressants and neuroleptics. Ther Drug Monit 22, 114-117.
de Morais, S. M., Wilkinson, G. R., Blaisdell, J., Meyer, U. A., Nakamura, K., and Goldstein, J. A. (1994a).
Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in
Japanese. Mol Pharmacol 46, 594-598.
de Morais, S. M., Wilkinson, G. R., Blaisdell, J., Nakamura, K., Meyer, U. A., and Goldstein, J. A. (1994b). The
major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem
269, 15419-15422.
Deming, S. N. (1986). Chemometrics: an overview. Clin Chem 32, 1702-1706.
DeVane, C. L., and Sallee, F. R. (1996). Serotonin selective reuptake inhibitors in child and adolescent
psychopharmacology: a review of published experience. J Clin Psychiatry 57, 55-66.
Do, B., Robinet, S., Pradeau, D., and Guyon, F. (2001). Speciation of arsenic and selenium compounds by ionpair reversed-phase chromatography with electrothermic atomic absorption spectrometry. Application of
experimental design for chromatographic optimisation. J Chromatogr A 918, 87-98.
Druid, H., Holmgren, P., Carlsson, B., and Ahlner, J. (1999). Cytochrome P450 2D6 (CYP2D6) genotyping on
postmortem blood as a supplementary tool for interpretation of forensic toxicological results. Forensic Sci Int 99,
25-34.
Eap, C. B., Bender, S., Gastpar, M., Fischer, W., Haarmann, C., Powell, K., Jonzier-Perey, M., Cochard, N., and
Baumann, P. (2000). Steady state plasma levels of the enantiomers of trimipramine and of its metabolites in
CYP2D6-, CYP2C19- and CYP3A4/5-phenotyped patients. Ther Drug Monit 22, 209-214.
Edwards, J. G., and Anderson, I. (1999). Systematic review and guide to selection of selective serotonin reuptake
inhibitors. Drugs 57, 507-533.
Ekatodramis, G., and Borgeat, A. (2001). The enantiomers: revolution or evolution. Curr Top Med Chem 1, 205206.
Eliel, E. L., and Wilen, S. H., eds. (1994). Stereochemistry of organic compound, first edn (New York, John
Wiley&Sons, Inc).
Eriksson, L., Johansson, E., Kettaneh-Wold, N., Wikstrom, C., and Wold, S. (2000). Design of experiments.
Principles and applications (Stockholm, Learnways AB).
Evans, A. M. (2000). Influence of dietary components on the gastrointestinal metabolism and transport of drugs.
Ther Drug Monit 22, 131-136.
Evans, A. M. (2001). Comparative pharmacology of S(+)-ibuprofen and (RS)-ibuprofen. Clin Rheumatol 20, S914.
Fell, A. F., Noctor, T. A., Mama, J. E., and Clark, B. J. (1988). Computer-aided optimisation of drug enantiomer
separation in chiral high-performance liquid chromatography. J Chromatogr 434, 377-384.
Foglia, J. P., Pollock, B. G., Kirshner, M. A., Rosen, J., Sweet, R., and Mulsant, B. (1997). Plasma levels of
citalopram enantiomers and metabolites in elderly patients. Psychopharmacol Bull 33, 109-112.
Fredericson Overo, K., Toft, B., Christophersen, L., and Gylding-Sabroe, J. P. (1985). Kinetics of citalopram in
elderly patients. Psychopharmacology (Berl) 86, 253-257.
82
Fredricson Overo, K. (1982). Kinetics of citalopram in man; plasma levels in patients. Prog
Neuropsychopharmacol Biol Psychiatry 6, 311-318.
Fritz, J. S., and Masso, J. J. (2001). Miniaturized solid-phase extraction with resin disks. J Chromatogr A 909,
79-85.
Gabrielsson, J., Lindberg, N., and Lundstedt, T. (2002). Review. Multivariate methods in pharmaceutical
applications. J of Chemometrics 16, 141-160.
Goldstein, B. J., and Goodnick, P. J. (1998). Selective serotonin reuptake inhibitors in the treatment of affective
disorders--III. Tolerability, safety and pharmacoeconomics. J Psychopharmacol 12, S55-87.
Goodnick, P. J., and Goldstein, B. J. (1998a). Selective serotonin reuptake inhibitors in affective disorders--I.
Basic pharmacology. J Psychopharmacol 12, S5-20.
Goodnick, P. J., and Goldstein, B. J. (1998b). Selective serotonin reuptake inhibitors in affective disorders--II.
Efficacy and quality of life. J Psychopharmacol 12, S21-54.
Gorog, S., and Gazdag, M. (1994). Enantiomeric derivatization for biomedical chromatography. J Chromatogr B
Biomed Appl 659, 51-84.
Gutierrez, M., and Abramowitz, W. (2000). Steady-state pharmacokinetics of citalopram in young and elderly
subjects. Pharmacotherapy 20, 1441-1447.
Gutierrez, M., and Abramowitz, W. (2001). Lack of effect of a single dose of ketoconazole on the
pharmacokinetics of citalopram. Pharmacotherapy 21, 163-168.
Hagg, S., Spigset, O., and Dahlqvist, R. (2001). Influence of gender and oral contraceptives on CYP2D6 and
CYP2C19 activity in healthy volunteers. Br J Clin Pharmacol 51, 169-173.
Haginaka, J. (2002). Pharmaceutical and biomedical applications of enantioseparations using liquid
chromatographic techniques. J Pharm Biomed Anal 27, 357-372.
Han, S. M. (1997). Direct enantiomeric separations by high performance liquid chromatography using
cyclodextrins. Biomed Chromatogr 11, 259-271.
Haupt, D. (1996). Determination of citalopram enantiomers in human plasma by liquid chromatographic
separation on a Chiral-AGP column. J Chromatogr B Biomed Appl 685, 299-305.
Heim, M., and Meyer, U. A. (1990). Genotyping of poor metabolisers of debrisoquine by allele-specific PCR
amplification [see comments]. Lancet 336, 529-532.
Hennion, M. C. (1999). Solid-phase extraction: method development, sorbents, and coupling with liquid
chromatography. J Chromatogr A 856, 3-54.
Hermann, M., Christensen, H., and Reubsaet, J. L. (2002). Evaluation of essential parameters in the
chromatographic determination of cyclosporin A and metabolites using a D-optimal design. J Pharm Biomed
Anal 30, 1263-1276.
Herrlin, K. (2001) CYP2C19 catalyzed drug metabolism in different populations, KI, Stockholm.
Hiemke, C., and Hartter, S. (2000). Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther
85, 11-28.
Hinze, W. L., Riehl, T. E., Armstrong, D., DeMond, W., Alak, A., and Ward, T. (1985). Liquid chromatogrpahic
separation of enantiomers using a chiral beta-cyclodextrin-bonded stationary phase and conventional aqueousorganic mobile phases. Anal Chem 57, 237-242.
Hund, E., Vander Heyden, Y., Haustein, M., Massart, D. L., and Smeyers-Verbeke, J. (2000). Robustness testing
of a reversed-phase high-performance liquid chromatographic assay: comparison of fractional and asymmetrical
factorial designs. J Chromatogr A 874, 167-185.
83
Hund, E., Vander Heyden, Y., Massart, D. L., and Smeyers-Verbeke, J. (2002). Derivation of system suitability
test limits from a robustness test on an LC assay with complex antibiotic samples. J Pharm Biomed Anal 30,
1197-1206.
Hyttel, J. (1994). Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin
Psychopharmacol 9 Suppl 1, 19-26.
Hyttel, J., Bogeso, K. P., Perregaard, J., and Sanchez, C. (1992). The pharmacological effect of citalopram
residues in the (S)-(+)- enantiomer. J Neural Transm Gen Sect 88, 157-160.
Ingelman-Sundberg, M., Oscarson, M., and McLellan, R. A. (1999). Polymorphic human cytochrome P450
enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci 20, 342-349.
Isacsson, G., Holmgren, P., Druid, H., and Bergman, U. (1999). Psychotropics and suicide prevention.
Implications from toxicological screening of 5281 suicides in Sweden 1992-1994. Br J Psychiatry 174, 259-265.
Jenkins, A. J., and Gubanich, K. (2002). Disposition of citalopram in biological specimens from postmortem
cases. J Forensic Sci 47, 159-164.
Jeppesen, U., Gram, L. F., Vistisen, K., Loft, S., Poulsen, H. E., and Brosen, K. (1996). Dose-dependent
inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J
Clin Pharmacol 51, 73-78.
Joffe, P., Larsen, F. S., Pedersen, V., Ring-Larsen, H., Aaes-Jorgensen, T., and Sidhu, J. (1998). Single-dose
pharmacokinetics of citalopram in patients with moderate renal insufficiency or hepatic cirrhosis compared with
healthy subjects. Eur J Clin Pharmacol 54, 237-242.
Joubert, A. F., Sanchez, C., and Larsen, F. (2000). Citalopram. Hum Psychopharmacol 15, 439-451.
Kaliszan, R., Nasal, A., and Turowski, M. (1995). Binding site for basic drugs on alpha 1-acid glycoprotein as
revealed by chemometric analysis of biochromatographic data. Biomed Chromatogr 9, 211-215.
Karlsson, A., and Aspegren, A. (2000). Enantiomeric separation of amino alcohols on protein phases using
statistical experimental design. A comparative study. J Chromatogr A 866, 15-23.
Kemp, W. (1975). Organic Spectroscopy (London, The MacMillan Press Ltd).
Kirchheiner, J., Brosen, K., Dahl, M. L., Gram, L. F., Kasper, S., Roots, I., Sjoqvist, F., Spina, E., and
Brockmoller, J. (2001). CYP2D6 and CYP2C19 genotype-based dose recommendations for antidepressants: a
first step towards subpopulation-specific dosages. Acta Psychiatr Scand 104, 173-192.
Kobayashi, K., Chiba, K., Yagi, T., Shimada, N., Taniguchi, T., Horie, T., Tani, M., Yamamoto, T., Ishizaki, T.,
and Kuroiwa, Y. (1997). Identification of cytochrome P450 isoforms involved in citalopram N- demethylation
by human liver microsomes. J Pharmacol Exp Ther 280, 927-933.
Kosel, M., Amey, M., Aubert, A., and Baumann, P. (2001). In vitro metabolism of citalopram by monoamine
oxidase B in human blood. Eur Neuropsychopharmacol 11, 75-78.
Kosel, M., Eap, C. B., Amey, M., and Baumann, P. (1998). Analysis of the enantiomers of citalopram and its
demethylated metabolites using chiral liquid chromatography. J Chromatogr B Biomed Sci Appl 719, 234-238.
Kragh-Sorensen, P., Overo, K. F., Petersen, O. L., Jensen, K., and Parnas, W. (1981). The kinetics of citalopram:
single and multiple dose studies in man. Acta Pharmacol Toxicol (Copenh) 48, 53-60.
Kroemer, H. K., Fromm, M. F., and Eichelbaum, M. (1996). Stereoselectivity in drug metabolism and action:
effects of enzyme inhibition and induction. Ther Drug Monit 18, 388-392.
Kugelberg, F. C., Apelqvist, G., Carlsson, B., Ahlner, J., and Bengtsson, F. (2001). In vivo steady-state
pharmacokinetic outcome following clinical and toxic doses of racemic citalopram to rats. Br J Pharmacol 132,
1683-1690.
Laine, K., Tybring, G., and Bertilsson, L. (2000). No sex-related differences but significant inhibition by oral
contraceptives of CYP2C19 activity as measured by the probe drugs mephenytoin and omeprazole in healthy
Swedish white subjects. Clin Pharmacol Ther 68, 151-159.
84
Lam, Y. W., Gaedigk, A., Ereshefsky, L., Alfaro, C. L., and Simpson, J. (2002). CYP2D6 inhibition by selective
serotonin reuptake inhibitors: analysis of achievable steady-state plasma concentrations and the effect of
ultrarapid metabolism at CYP2D6. Pharmacotherapy 22, 1001-1006.
Lamba, J. K., Lin, Y. S., Schuetz, E. G., and Thummel, K. E. (2002). Genetic contribution to variable human
CYP3A-mediated metabolism. Adv Drug Deliv Rev 54, 1271-1294.
Landi, M. T., Sinha, R., Lang, N. P., and Kadlubar, F. F. (1999). Human cytochrome P4501A2. IARC Sci Publ,
173-195.
Lane, R., Baldwin, D., and Preskorn, S. H. (1995). The SSRIs: advantage, disadvantage and difference. J
Psychopharmacology 9, 585-6163-6178.
Lapinsh, M., Prusis, P., Gutcaits, A., Lundstedt, T., and Wikberg, J. E. S. (2001). Development of proteochemometrics: a novel technology for the analysis of drug-receptor interactions. Biochimica et Biophysica Acta
(BBA) - General Subjects 1525, 180-190.
Leinonen, E., Lepola, U., and Koponen, H. (1996). Substituting carbamazepine with oxcarbazepine increases
citalopram levels. A report on two cases. Pharmacopsychiatry 29, 156-158.
Leinonen, E., Lillsunde, P., Laukkanen, V., and Ylitalo, P. (1991). Effects of carbamazepine on serum
antidepressant concentrations in psychiatric patients. J Clin Psychopharmacol 11, 313-318.
Levine, B., Jenkins, A. J., Queen, M., Jufer, R., and Smialek, J. E. (1996). Distribution of venlafaxine in three
postmortem cases. J Anal Toxicol 20, 502-505.
Levy, R. H. (1995). Cytochrome P450 isozymes and antiepileptic drug interactions. Epilepsia 36, S8-13.
Lin, J. H., and Lu, A. Y. (1998). Inhibition and induction of cytochrome P450 and the clinical implications. Clin
Pharmacokinet 35, 361-390.
Linder, M. W., Valdes, R., Jr., and Prough, R. A. (1999). Pharmacogenetics in the practice of laboratory
medicine
Pharmacogenetics: a laboratory tool for optimizing therapeutic efficiency. Mol Diagn 4, 365-379.
Lindsay, S. (1992). High performance liquid chromatography, second edn (New York, John Wiley&Sons, Inc).
Lipkowitz, K. B. (2001). Atomistic modeling of enantioselection in chromatography. J Chromatogr A 906, 417442.
Lundqvist, E., Johansson, I., and Ingelman-Sundberg, M. (1999). Genetic mechanisms for duplication and
multiduplication of the human CYP2D6 gene and methods for detection of duplicated CYP2D6 genes. Gene
226, 327-338.
Lundstedt, T., Seifert, E., Abramo, L., Thelin, B., Nystrom, A., Pettersen, J., and Bergman, R. (1998).
Experimental design and optimization. Chemometrics and Intelligent Laboratory Systems 42, 3-40.
Maier, N. M., Franco, P., and Lindner, W. (2001). Separation of enantiomers: needs, challenges, perspectives. J
Chromatogr A 906, 3-33.
Masand, P. S., and Gupta, S. (1999). Selective serotonin-reuptake inhibitors: an update. Harv Rev Psychiatry 7,
69-84.
Mason, S. (1986). The origin of chirality in nature. Trends Pharmacol Sci 7, 20-23.
Mason, S. F. (1991). From Pasteur to parity violation: cosmic dissymmetry and the origins of biomolecular
handedness. Ambix 38, 85-99.
Meyer, U. A., Amrein, R., Balant, L. P., Bertilsson, L., Eichelbaum, M., Guentert, T. W., Henauer, S., Jackson,
P., Laux, G., Mikkelsen, H., et al. (1996). Antidepressants and drug-metabolizing enzymes--expert group report.
Acta Psychiatr Scand 93, 71-79.
Molinoff, P. B., and Ruddon, R. W., eds. (1996). Goodman and Gillman´s The pharmacological basis of
therapeutics, ninth edn (New York, McGraw-Hill).
85
Montgomery, S. A., Loft, H., Sanchez, C., Reines, E. H., and Papp, M. (2001). Escitalopram (S-enantiomer of
citalopram): clinical efficacy and onset of action predicted from a rat model. Pharmacol Toxicol 88, 282-286.
Muldoon, C. (1996). The safety and tolerability of citalopram. Int Clin Psychopharmacol 11, 35-40.
Murphy, T. K., Bengtson, M. A., Tan, J. Y., Carbonell, E., and Levin, G. M. (2000). Selective serotonin reuptake
inhibitors in the treatment of paediatric anxiety disorders: a review. Int Clin Psychopharmacol 15 Suppl 2, S4763.
Naranjo, C. A., Sproule, B. A., and Knoke, D. M. (1999). Metabolic interactions of central nervous system
medications and selective serotonin reuptake inhibitors. Int Clin Psychopharmacol 14 Suppl 2, S35-47.
Nemeroff, C. B., DeVane, C. L., and Pollock, B. G. (1996). Newer antidepressants and the cytochrome P450
system [see comments]. Am J Psychiatry 153, 311-320.
Neuvonen, P. J., Pohjola-Sintonen, S., Tacke, U., and Vuori, E. (1993). Five fatal cases of serotonin syndrome
after moclobemide-citalopram or moclobemide-clomipramine overdoses. Lancet 342, 1419.
Noble, S., and Benfield, P. (1997). Citalopram. A review of its pharmacolcogy, clinical efficacy and tolerability
in treatment of depression. CNS Drugs 8, 410-431.
Ohman, D., Carlsson, B., and Norlander, B. (2001). On-line extraction using an alkyl-diol silica precolumn for
racemic citalopram and its metabolites in plasma. Results compared with solid- phase extraction methodology. J
Chromatogr B Biomed Sci Appl 753, 365-373.
Okamoto, M. (2002). Reversal of elution order during the chiral separation in high performance liquid
chromatography. J Pharm Biomed Anal 27, 401-407.
Olesen, O. V., and Linnet, K. (1999). Studies on the stereoselective metabolism of citalopram by human liver
microsomes and cDNA-expressed cytochrome P450 enzymes. Pharmacology 59, 298-309.
Ostrom, M., Eriksson, A., Thorson, J., and Spigset, O. (1996). Fatal overdose with citalopram. Lancet 348, 339340.
Otani, K., and Aoshima, T. (2000). Pharmacogenetics of classical and new antipsychotic drugs. Ther Drug Monit
22, 118-121.
Otto, M. (1988). Fuzzy theory explained. Chemometrics and Intelligent Laboratory Systems 4, 101-120.
Otto, M. (1999). Chemometrics. Statistics and computer application in analytical chemistry (Weinheim, WileyVCH).
Owens, M. J., Knight, D. L., and Nemeroff, C. B. (2001). Second-generation SSRIs: human monoamine
transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry 50, 345-350.
Owens, P. K., Fell, A. F., Coleman, M. W., and Berridge, J. C. (1996). Method development in liquid
chromatography with a charged cyclodextrin additive for chiral resolution of rac-amlodipine utilising a central
composite design. Chirality 8, 466-476.
Pacher, P., Ungvari, Z., Nanasi, P. P., Furst, S., and Kecskemeti, V. (1999). Speculations on difference between
tricyclic and selective serotonin reuptake inhibitor antidepressants on their cardiac effects. Is there any? Curr
Med Chem 6, 469-480.
Personne, M., Persson, H., and Sjoberg, E. (1997). Citalopram toxicity. Lancet 350, 518-519.
Poolsup, N., Li Wan Po, A., and Knight, T. L. (2000). Pharmacogenetics and psychopharmacotherapy. J Clin
Pharm Ther 25, 197-220.
Popa, R. (1997). A Sequential Scenario for the Origin of Biological Chirality. J Mol Evol 44, 121-127.
Preskorn, S. H. (1998). Debate resolved: there are differential effects of serotonin selective reuptake inhibitors
on cytochrome P450 enzymes. J Psychopharmacol 12, S89-97.
86
Raimundo, S., Fischer, J., Eichelbaum, M., Griese, E. U., Schwab, M., and Zanger, U. M. (2000). Elucidation of
the genetic basis of the common 'intermediate metabolizer' phenotype for drug oxidation by CYP2D6.
Pharmacogenetics 10, 577-581.
Rang, H. P., Dale, M. M., and Ritter, J. M., eds. (1999). Pharmacology, fourth edn (Edinburgh, Churchill
Livingstone).
Rasmussen, S. L., Overo, K. F., and Tanghoj, P. (1999). Cardiac safety of citalopram: prospective trials and
retrospective analyses. J Clin Psychopharmacol 19, 407-415.
Reist, M., Carrupt, P. A., Francotte, E., and Testa, B. (1998). Chiral inversion and hydrolysis of thalidomide:
mechanisms and catalysis by bases and serum albumin, and chiral stability of teratogenic metabolites. Chem Res
Toxicol 11, 1521-1528.
Rochat, B., Amey, M., and Baumann, P. (1995a). Analysis of enantiomers of citalopram and its demethylated
metabolites in plasma of depressive patients using chiral reverse-phase liquid chromatography. Ther Drug Monit
17, 273-279.
Rochat, B., Amey, M., Gillet, M., Meyer, U. A., and Baumann, P. (1997). Identification of three cytochrome
P450 isozymes involved in N- demethylation of citalopram enantiomers in human liver microsomes.
Pharmacogenetics 7, 1-10.
Rochat, B., Amey, M., Van Gelderen, H., Testa, B., and Baumann, P. (1995b). Determination of the enantiomers
of citalopram, its demethylated and propionic acid metabolites in human plasma by chiral HPLC. Chirality 7,
389-395.
Rochat, B., Baumann, P., and Audus, K. L. (1999). Transport mechanisms for the antidepressant citalopram in
brain microvessel endothelium. Brain Res 831, 229-236.
Rochat, B., Kosel, M., Boss, G., Testa, B., Gillet, M., and Baumann, P. (1998). Stereoselective biotransformation
of the selective serotonin reuptake inhibitor citalopram and its demethylated metabolites by monoamine oxidases
in human liver. Biochem Pharmacol 56, 15-23.
Rogde, S., Hilberg, T., and Teige, B. (1999). Fatal combined intoxication with new antidepressants. Human
cases and an experimental study of postmortem moclobemide redistribution. Forensic Sci Int 100, 109-116.
Rouan, M. C., Buffet, C., Masson, L., Marfil, F., Humbert, H., and Maurer, G. (2001). Practice of solid-phase
extraction and protein precipitation in the 96-well format combined with high-performance liquid
chromatography-ultraviolet detection for the analysis of drugs in plasma and brain. J Chromatogr B Biomed Sci
Appl 754, 45-55.
Sanchez, C., and Hyttel, J. (1999). Comparison of the effects of antidepressants and their metabolites on
reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol 19, 467-489.
Schneiderman, E., and Stalcup, A. M. (2000). Cyclodextrins: a versatile tool in separation science. J Chromatogr
B Biomed Sci Appl 745, 83-102.
Schwab, M., Eichelbaum, M., and Fromm, M. F. (2002). Genetic Polymorphisms of the Human MDR1 Drug
Transporter. Annu Rev Pharmacol Toxicol 17, 17.
Scordo, M. G. (2003) Cytochrome P450 2C9, 2C19 and 2D6 genetic polymorphisms. Evaluation of genotyping
as a tool for individualised treatment, KI, Stockholm.
Scordo, M. G., and Spina, E. (2002). Cytochrome P450 polymorphisms and response to antipsychotic therapy.
Pharmacogenomics 3, 201-218.
Scott, A. K. (1993). Stereoisomers and drug toxicity. The value of single stereoisomer therapy. Drug Saf 8, 149159.
Scriba, G. K. (2002). Selected fundamental aspects of chiral electromigration techniques and their application to
pharmaceutical and biomedical analysis. J Pharm Biomed Anal 27, 373-399.
Sidhu, J., Priskorn, M., Poulsen, M., Segonzac, A., Grollier, G., and Larsen, F. (1997). Steady-state
pharmacokinetics of the enantiomers of citalopram and its metabolites in humans. Chirality 9, 686-692.
87
Simonyi, M., Fitos, I., and Visy, J. (1986). Chirality of bioactive agents in protein binding storage and transport
processes. Trends Pharmacol Sci 7, 112-116.
Sindrup, S. H., Brosen, K., Hansen, M. G., Aaes-Jorgensen, T., Overo, K. F., and Gram, L. F. (1993).
Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther
Drug Monit 15, 11-17.
Small, T. S., Fell, A. F., Coleman, M. W., and Berridge, J. C. (1995). Central composite design for rapid
optimisation of ruggedness and chiral separation of amlodipine in capillary electrophoresis. Chirality 7, 226-234.
Spina, E., and Scordo, M. G. (2002). Clinically significant drug interactions with antidepressants in the elderly.
Drugs Aging 19, 299-320.
Sproule, B. A., Naranjo, C. A., Brenmer, K. E., and Hassan, P. C. (1997). Selective serotonin reuptake inhibitors
and CNS drug interactions. A critical review of the evidence. Clin Pharmacokinet 33, 454-471.
Stahl, S. M. (2000). Essential psychopharmacology of depression and bipolar disorders. In (Cambridge,
Cambridge university press).
Steinacher, L., Vandel, P., Zullino, D. F., Eap, C. B., Brawand-Amey, M., and Baumann, P. (2002).
Carbamazepine augmentation in depressive patients non-responding to citalopram: a pharmacokinetic and
clinical pilot study. Eur Neuropsychopharmacol 12, 255-260.
Stinson, S. C. (1999). Chiral drug interactions. Chem Eng News 77, 101-120.
Strong, M. (1999). FDA policy and regulation of stereoisomers: paradigm shift and the future of safer, more
effective drugs. Food Drug Law J 54, 463-487.
Subramanian, G., ed. (1994). A practical approach to chiral separations by liquid chromatography, first edn
(Weinheim, VCH Verlagsgesellschaft).
Takeuchi, T., and Haginaka, J. (1999). Separation and sensing based on molecular recognition using molecularly
imprinted polymers. J Chromatogr B Biomed Sci Appl 728, 1-20.
Tamminga, W. J., Wemer, J., Oosterhuis, B., Weiling, J., Wilffert, B., de Leij, L. F., de Zeeuw, R. A., and
Jonkman, J. H. (1999). CYP2D6 and CYP2C19 activity in a large population of Dutch healthy volunteers:
indications for oral contraceptive-related gender differences. Eur J Clin Pharmacol 55, 177-184.
Tanaka, E. (1999a). Clinically significant pharmacokinetic drug interactions between antiepileptic drugs. J Clin
Pharm Ther 24, 87-92.
Tanaka, E. (1999b). Gender-related differences in pharmacokinetics and their clinical significance. J Clin Pharm
Ther 24, 339-346.
Tanaka, E., and Hisawa, S. (1999). Clinically significant pharmacokinetic drug interactions with psychoactive
drugs: antidepressants and antipsychotics and the cytochrome P450 system. J Clin Pharm Ther 24, 7-16.
Tanigawara, Y. (2000). Role of P-glycoprotein in drug disposition [In Process Citation]. Ther Drug Monit 22,
137-140.
Testa, B. (1986). Chiral aspects of drug metabolism. Trends Pharmacol Sci 7, 60-64.
Tournel, G., Houdret, N., Hedouin, V., Deveau, M., Gosset, D., and Lhermitte, M. (2001). High-performance
liquid chromatographic method to screen and quantitate seven selective serotonin reuptake inhibitors in human
serum. J Chromatogr B Biomed Sci Appl 761, 147-158.
Tsang, P. K., Larew, J. S., Larew, L. A., Miyakawa, T. W., and Hofer, J. D. (1998). Statistical approaches to
determine analytical variability and specifications: application of experimental design and variance component
analysis. J Pharm Biomed Anal 16, 1125-1141.
Tucker, G. T. (2000). Chiral switches. Lancet 355, 1085-1087.
Wainer, I. W., ed. (1993). Drug stereochemistry, second edn (New York, Marcel Deker, Inc).
88
Walters, F. (1999). Sequential simplex optimization an update. Analytical Letters 32, 193-212.
van der Weide, J., and Steijns, L. S. (1999). Cytochrome P450 enzyme system: genetic polymorphisms and
impact on clinical pharmacology. Ann Clin Biochem 36, 722-729.
Verweij, A. M., Hordijk, M. L., and Lipman, P. J. (1996). Liquid chromatographic-thermospray tandem mass
spectrometric quantitative analysis of some drugs with hypnotic, sedative and tranquillising properties in whole
blood. J Chromatogr B Biomed Appl 686, 27-34.
West, W. L., Knight, E. M., Pradhan, S., and Hinds, T. S. (1997). Interpatient variability: genetic predisposition
and other genetic factors. J Clin Pharmacol 37, 635-648.
Wieling, J., Hendriks, G., Tamminga, W. J., Hempenius, J., Mensink, C. K., Oosterhuis, B., and Jonkman, J. H.
(1996). Rational experimental design for bioanalytical methods validation. Illustration using an assay method for
total captopril in plasma. J Chromatogr A 730, 381-394.
Wikell, C., Apelqvist, G., Carlsson, B., Hjorth, S., Bergqvist, P. B., Kugelberg, F. C., Ahlner, J., and Bengtsson,
F. (1999). Pharmacokinetic and pharmacodynamic responses to chronic administration of the selective serotonin
reuptake inhibitor citalopram in rats. Clin Neuropharmacol 22, 327-336.
Williams, M. L., and Wainer, I. W. (2002). Role of chiral chromatography in therapeutic drug monitoring and in
clinical and forensic toxicology. Ther Drug Monit 24, 290-296.
Voirol, P., Jonzier-Perey, M., Porchet, F., Reymond, M. J., Janzer, R. C., Bouras, C., Strobel, H. W., Kosel, M.,
Eap, C. B., and Baumann, P. (2000). Cytochrome P-450 activities in human and rat brain microsomes. Brain Res
855, 235-243.
Wold, S. (1991). Chemometrics, why, what and where to next? J Pharm Biomed Anal 9, 589-596.
von Moltke, L. L., Greenblatt, D. J., Giancarlo, G. M., Granda, B. W., Harmatz, J. S., and Shader, R. I. (2001).
Escitalopram (S-citalopram) and its metabolites in vitro: cytochromes mediating biotransformation, inhibitory
effects, and comparison to R-citalopram. Drug Metab Dispos 29, 1102-1109.
Worm, K., Dragsholt, C., Simonsen, K., and Kringsholm, B. (1998). Citalopram concentrations in samples from
autopsies and living persons. Int J Legal Med 111, 188-190.
Yasui-Furukori, N., Hidestrand, M., Spina, E., Facciola, G., Scordo, M. G., and Tybring, G. (2001). Different
enantioselective 9-hydroxylation of risperidone by the two human CYP2D6 and CYP3A4 enzymes. Drug Metab
Dispos 29, 1263-1268.
Zadeh, L. (1965). Fuzzy sets. Information and control 8, 338-353.
Zheng, Z., Jamour, M., and Klotz, U. (2000). Stereoselective HPLC-assay for citalopram and its metabolites.
Ther Drug Monit 22, 219-224.
89
Appendix
tR2
depth
tR1
height
peak 1
height
peak 2
t0
Inject
w1
w2
t0 Designates retention time of an unretained analyte that has travelled through the
column at the speed of the mobile phase
tR1 and tR2 Designate retention time of peaks 1 and 2
w1 and w2 Designate peak width of peaks 1 and 2
k´=( tR- t0)/ t0
The capacity factor, describes the retention of a compound in a chromatogram
Į= k´2/ k´1=(( tR2- t0)/t0)/(tR1- t0)/ t0))
The separation factor or selectivity, describes the separation of compounds relative to
each other (Į1)
Rs=( tR2-tR1)/(0.5(w1+w2)
The resolution, describes the separation of one peak from another (base line separation
obtained if Rs 1.5)
Kaiser peak separation index=depth/((height peak 1+height peak 2)/2)
Defined as the average valley depth expressed as a ratio to the average peak height of
the two adjacent peaks
N=16(tR-w)2
The plate number, used to determine column efficiency
H=L/N
The plate height, measures efficiency for unit column length.
91