Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
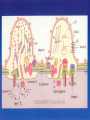
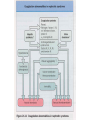
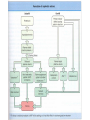
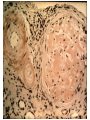
Nephrotic Syndrome Farid Nakhoul M.D. Deputy Director Dept. of Nephrology Rambam Medical Ctr. Faculty of Medicine Haifa Tel: 04-8542590 Fax: 048542946 Email:[email protected] Structure of the Glomerulus(1) • One of the central function of the kidney is to excrete low molecular weight watersoluble plasma waste products into the urine. Whereas macromolecules the size of albumin and larger are retained. The filtration of plasma occurs in specialized filtration units called Glomeruli. Structure of the Glomerulus(2) The glomerular filtration barrier consists of the three layers of the capillary wall: 1. The innermost fenestrated vascular endothelium. 2. The GBM: the GBM is regarded as a primary size and charge-selective molecular sieve of the glomerulus . The GBM contains type IV collagen, laminin, nidogen, and proteoglycans as its main components. 3. Podocyte cell layer facing the urinary space. Proteinuria Is the halmark of glomerular disease !! Clinical Evaluation of Glomerular disease 1. History 2. Physical Examination 3. Laboratory Studies 4. Imaging: Ultrasound 5. Renal Biopsy ( Open, Closed, Laparoscopic). Proteinuria • Asymptomatic Non-nephrotic Proteinuria (<3.5 gr Protein/day). - Glomerular - Non-glomerular • Nephrotic Syndrome: Pathognomonic of glomerular disease Non-Nephrotic Proteinuria • Defined as a urine protein excretion of less than 3.5 gr/d, and is absolutely characteristic of glomerular disease, but may occure in many nonglomerular parenchymal disease. Clinical Classification Isolated Hematuria Proteinuria / Nephrotic Syndrome Acute Nephritic Syndrome Rapidly Progressive Glomerulonephritis Chronic Glomerulonephritis Clinical Features of Glomerular Diseases Proteinuria Hematuria Edema Hypertension Renal Dysfunction Evaluation of Proteinuria – General Practice History, Physical Examination, urinalysis Repeat visit for qualitative proteinuria test Positive Negative Measure U&E, Albumin, Quantify urine protein excretion Transient Proteinuria; Reassure Patient Quantifying protein excretion • Should perform quantitative measure in persistent proteinuria 24 hour urine collection – Readily quantified – Wide understanding – Cumbersome Protein-to-creatinine ratio (PCR) – Simple – Validated Nephrotic Syndrome Definition • Urinary protein level exceeding 3.5 gr per 1.73m2 of body-surface area/day. • Hypoalbuminemia. • Sodium retention (Edema-State). • Hyperlipoproteinemia. • Hypercoagulopathy Prognosis • Depends upon degree of proteinuria • 20 year follow up: – Hypertension in 50% – Renal Insufficiency in 40% Diagnostic Approach 24 urine collection Serum electrolytes, BUN, creatinine, lipid profile, serum albumin Serological work up - depends upon the clinical presentation - common serological tests done are ANA; Anti-ds DNA Complement levels (C3, C4) Hepatitis B and C serologies SPEP; UPEP, ANCA, anti GBM Abs Renal Ultrasound Renal Biopsy - Indications Nephrotic Syndrome Primary Membranous Glomerulopathy Focal Segmental Glomerulosclerosis Minimal Change Disease Membranoproliferative GN Secondary Diabetic glomerulosclerosis Paraproteinemia /Amyloidosis Lupus Nephritis Nephrotic Syndrome Thromboembolic Complications • Major hazard of the nephrotic syndrome • RVT( membranous GN) in 20-30% of adult patients, 10% are symptomatic: flank pain, gross hematuria • Pulmonary emboli(Silent) • DVT(frequent) • Arterial thrombosis is less common • Prophylaxis : Albumin< 2 gr, Uprotein>10 gr/d. decreased level of antithrombin III, Increased fibrinogen, Hypovolemia Nephrotic Syndrome Infections • Decreased levels of immunoglobulin IgG • Decreased the alternative complement factor B • Pneumococcal peritonitis • Recurrent Cellulitis General Management Edema - salt restriction, diuretics Hyperlipidemia - lipid lowering agents ACE/ARB – decrease Pgc and decrease proteinuria – Renal protective Hypercoagulable - ASA Specific treatment based on biopsy Minimal Change Disease MCD Introduction and Definition Minimal Change Disease Pathogenesis - unknown, most likely autoimmune Treatment – 85% respond to steroids – If resistant to steroid may require cyclophosphamide Minimal Change Nephropathy (MCD) • Minimal change disease (MCD) :is defined by the absence of histologic glomerular abnormality, other than evidence of epithelial cell foot process fusion (EM). Minimal Change Disease MCD Pathology Minimal Change Disease Nephrotic Syndrome Minimal Change Disease • Is the major cause of the nephrotic syndrome in children(85-90%) and 10% in adults • Malignancies account for 10% of cases of idiopathic nephrotic syndrome in adults • LM: Normal glomeruli • EM: Fusion of the epithelial cell foot Processes ( KNB). Minimal Change Disease MCD Etiology and Pathogenesis Minimal Change Disease • Idiopathic • Secondary: Malignancy Drugs Infections Minimal Change Disease MCD Clinical manifestations Minimal Change Nephropathy MCD • Patients with Minimal change disease present with edema that develops over a short period of time, with fluid retention exceeding 3% of the body weight. • Up to two-thirds of presentation and relapses follow an infection, most commonly upper respiratory tract infection. Minimal Change Nephropathy • Clinical Feature: Heavy proteinuria, low serum albumin, edema formation and elevated serum cholesterol. • Normal urinary sediment • Normal C3, C4 • Negative ANA and Cryoglobulins • MCD patients are at risk of venous thromboembolism. • Hypovolemia and ARF, especially patients with diuretic therapy. Minimal Change Disease Natural History Minimal Change Disease • There is a tendency for patients with MCD to run a relapsing-remitting course and this is more frequent in children. • Long-term remission can be expected in 75% of initial responders who do not relapse within 6 months. • MCD does not progress to Renal Failure Membranous GN Definition Membranous Nephropathy(MN) • Membranous Nephropathy is a glomerular disease in which immune deposits of IgG and complement components develop predominantly or exclusively on the subepithelial surface of the glomerular capillary wall. • Deposition is associated with a marked increase in glomerular permeability to protein, which manifest clinically as nephrotic syndrome. • The pathogenesis of human MN is not known. Membranous Nephropathy ETIOLOGY Membranous Nephropathy Most common cause of idiopathic nephrotic syndrome in Caucasian adults. Heavy proteinuria is common. Hypertension and azotemia develops as disease progresses. Increased incidence of renal vein thrombosis. Membranous nephropathy • 2nd most common cause of nephrotic syndrome in adults (~30%) • Usually idiopathic • Associated with – Autoimmune diseases – Hepatitis B – Carcinoma – Drugs (eg penicillamine, captopril, NSAID) • Outcome very variable – 1/3 spontaneous remission – 1/3 partial remission or very slow progression – 1/3 progressive renal impairment • Higher incidence of thromboembolism • Therapy very difficult Membranous Nephropathy • Idiopathic • Secondary GN: • Immunological: -SLE, MCTD -Sjogren Syndrome • Rheumatoid Arthritis • Neoplasms: -Ca of lung -Ca of breast, Kidney • Non-Hodgin Lymphoma Membranous Nephropathy • Infectious: -Hepatitis B -Malaria -Schistosomiasis • Medications: -NSAIDs, Gold, Penicillamine -Capoten Pathology • The pathologic feature of MN evolve from the initial formation of subepithelial immune complexes of IgG and complement. • Initial stage on EM ranging from minimal change with only small deposits( stage 1) through the evolution to thickened basement membrane with resolution of deposits (stage IV). Membranous Nephropathy Membranous Nephropathy Clinical manifestation Membranous Nephropathy • Is the most common cause of the idiopathic nephrotic syndrome in adults(30-50%) • Nephrotic Syndrome is the common presentation • Hypertension 30% • In children: 50% spontaneous remission, 10% to ESRD • Stage 1 >>>good prognosis • Stage 4>>>>bad Prognosis Membranous Nephropathy Natural History and Prognosis Membranous GN • The prognosis of untreated MN is somewhat worse in adults: 20% will have progressed to ESRD at 5-10 years followup. . Bad outcome: male, age>50, HTN, Pcr high at presentation. • Recurrence after Renal Transplantation Membranous Nephropathy Treatment Treatment • Steroides • Cyclosporine • Imuran • Cyclophosphamide • Chlorambucil Treatment of Membranous Nephropathy Rule of 1/3 1/3 – Spontaneous remission 1/3 – Partial remission / slow deterioration 1/3 – Progress to ESRD Steroids alone are not very effective Methylprednisolone alternating with chlorambucil, Methylprednisolone alternating with cyclophosphamide. Cyclosporin Focal Segmental Glomerulosclerosis Introduction and Definition Focal Segmental Glomerulosclerosis Etiology and Pathogenesis Focal Segmental Glomerulosclerosis (FSGS) • Primary (Idiopathic) • Secondary: -Sickle cell disease -Heroin Nephropathy -Morbid obesity -HIV Nephropathy -Aging Kidney -Vesico-Ureteral Reflux - Pamidronate FSGS • Most common idiopathic nephrotic syndrome in adults (33%) • Increasing incidence • More common in blacks • Treatment very difficult Focal Segmental Glomerulosclerosis Most common primary renal disease in African-Americans Etiology: Idiopathic Drugs – Intravenous heroin Infections – HIV Others – reflux nephropathy, obesity Patient usually hypertensive. Usually progresses to ESRD over 5-20 years. FSGS Pathology Focal Segmental Glomerulosclerosis (FSGS) • FSGS is defined on histologic criteria by segmental capillary obliteration with increased mesangial matrix deposition, intracapillary hyaline deposits, and focal adhesions of the capillary tuft to Bowman’s capsule. • Primary FSGS occur in patient with nephrotic syndrome in the absence of any of the known causes of secondary FSGS. FSGS Mild Normal Collapsing Moderate Associations Idiopathic Morbid obesity Heroin abuse HIV infection NSAID (Minimal change disease) FSGS • In FSGS, evidence for a circulating factor is more substantial, mainly based on the high incidence of recurrence following transplantation, with heavy proteinuria developing sometimes within hours. • Plasma exchange can lead to remission in tranplant recurrence of FSGS. FSGS Clinical Manifestations FSGS • Typically patients present with edema that develops over a short period of time. • Very rapid onset of nephrotic syndrome • Microscopic hematuria 14-30% • Arterial Hypertension especially with renal insufficiency. • Increased incidence pf venous thromboembolism. Focal Segmental Glomerulosclerosis • Microscopic Hematuria • Proteinuria >>20-30 gram/daily • Both children and adults usually develops ESRD 5-20 yrs from presentation. • Idiopathic FSGS may recur in the transplanted kidney with severe proteinuria and Nephrotic Syndrome and rapid course to ESRD. Collapsing FSGS • Collapsing FSGS is a histologic variant of FSGS characterized by extensive focal or global glomerular capillary tuft collapse, podocyte hypertrophy and hyperplasia. • Varying degree of tubulointerstitial injury. • African Americans • Severe nephrotic syndrome at presentation, steroid resistance, and rapid developing ESRD (median time of 13 month). FSGS-Treatment • 1mg/kg Prednisone daily for 6 month • PO Cyclosporine • Cytotoxic Treatment (Cytoxan, Imuran) • Symptomatic treatment with ACEI • CellCept (Mycophenolate Mofetil). Nephrotic Syndrome Secondary To Systemic Disease • Diabetes Mellitus—Type I, Type II • Amyloidosis Diabetes Mellitus INTRODUCTION Diabetic Nephropathy Diabetic Nephropathy is a common problem that is most likely to occur in patients who have worse glycemic control, Hypertension, glomerular hyperfiltration, Blacks, Mexican, or Pima Indian. Diabetic Nephropathy • Diabetic nephropathy is a leading cause of end-stage renal disease (ESRD) in Western societies • Diabetic nephropathy is a clinical syndrome characterized by persistent albuminuria(>300 mg/24hr or >200ug/min) on at least two occasions separated by 3-6 months. • Patients invariably develop hypertension, progressive increase in proteinuria, and decline in GFR. Diabetic Nephropathy Major Risk Factors For DN 1. Genetic Susceptibility 2. Arterial Hypertension 3. Increased glomerular filtration rate 4. Worse Glycemic Control 5. Blacks, Pima Indian Microalbuminuria . Increased protein excretion is the earlies clinical finding of diabetic nephropathy. • The normal rate of albumin excretion is less than 20 mg/day; persistent values between 30 and 300 mg/day( 20 to 200 ug/min) in a patient with diabetes is called Microalbuminuria . Microalbuminuria and correlation with DN • Great attention to control of both hyperglycemia and hypertension (with ACEI) may also contribute to the apparent improvement in the course of the disease. • Patient who progress are more likely to have higher HbA1c values and higher blood pressure than non progresses. • The incidence of overt hypertension is approximately 15 to 25 percent in all patients with microalbuminuria and much higher as the patient progress to overt nephropathy. Epidemiology • Type 1 diabetic nephropathy, occur in 30-40% of patients with type 1 diabetes after 25-40 years with diabetes • In type 2 diabetes, the cumulative incidence of nephropathy is similar to that seen in type 1 , 25% at 20 years after diagnosis. • Since type 2 diabetes is 10-15 times more common than type 1, the prevalence of type 2 diabetic nephropathy is substantialy higher. Renal Pathology • The kidneys of patients with diabetes are, on average larger than those of nondiabetic control subjects(15%). • Nodular glomerular intercapillary lesions, described by Kimmelsteil and Wilson. • Diffuse glomerular lesion is more frequent than the nodular lesion, with an incidence of over 90% for patients with type 1 DM of over 10 years duration and an incidence of 25-50% in patients with type 2 DM. Diabetic Nephropathy Diabetic Nephropathy Most common cause of nephrotic syndrome in adults. Leading cause of ESRD in USA 30% of patients with Type I and 20% of patients with Type II DM develop diabetic nephropathy. Initially microalbuminuria followed by heavy proteinuria and decline in renal function. Diagnosis usually made on clinical grounds and biopsy not needed. Diabetic Nephropathy Clinical Manifestations and Natural History Treatment of Diabetic Nephropathy Blood glucose control Control of hypertension Use of ACE inhibitors/ARBs Control of hyperlipidemia Smoking cessation Protein Restriction Nephrotic Syndrome Secondary To Systemic Disease: Amyloidosis -Primary Amyloidosis -Secondary Amyloidosis Amyloidosis • The Amyloidosis are a group of disorders in which soluble proteins aggregate and deposit extracellulary in tissues as insoluble fibrils, causing progressive organ dysfunction. The kidney is one of the most frequent sites of amyloid deposition in AA, AL, and several of the hereditary amyloidoses. Amyloidosis • Amyloidosis is defined by the ability of a variety of proteins to form B-pleated sheets. These fibrils can be identified on biopsy specimens both by their characteristic appearance on electron microscopy and by their ability to bind Congo Red(leading to green birefringence under polarized light) and thioflavine-T (producing an intense yellow-green fluorescence). Amyloidosis • Amyloid A in secondary amyloidosis • Immunoglobulin light chains in primary amyloidosis • B2-microglobulin in dialysis-patients associated arthropathy • Amyloid beta protein in Alzheimer’s disease Amyloidosis • Renal involvement occurs in over 90% of patients with either primary or secondary amyloidosis. • Primary forms(> 90%) is a plasma cell dyscrasia and may be associated with MM. • Light Chain Deposition Disease (LCDD) is a disorder of similar pathogenesis. The abnormal protein do not form the B-pleated amyloid fibrils. • Secondary amyloidosis is a consequence of chronic inflammatory diseases such as : Rheumatoid arthritis, FMF, Osteomyelitis, Hypernephroma, PID. Amyloidosis-Secondary • Clinically evident renal involvement occurs only in primary or secondary amyloidosis • Secondary amyloidosis is associated with increased hepatocyte production of the acute phase reactant serum amyloidosis A (SAA); this process may be stimulated by the release of cytokines ( Interleukin-1) from activated macrophages. Cleavage in circulating monocyte/macrophages results in the generation of smaller fragments, called AA protein, that can then deposit in the tissue. Primary Amyloidosis (AL) • The fibrils in AL consist of segments of the variable portions of monoclonal light chains • AL Amyloidosis is a plasma cell dyscrasia in which the malignant characteristic of M.M are usually absent and circulating light chains are frequently demonstrated . • Only a minority of patients who produce an excessive amounts of light-chains (as M.M) develop amyloidosis. • Lambda light-chains are much more likely to produce amyloidosis than Kappa light chains. Dysproteinemia Myeloma Cast Nephropathy – Path shows multiple intraluminal proteinaceous casts – Pathogenesis Disorder of plasma cells with overproduction the antibody light chain – Treatment Hydration, plasmapheresis to remove the abnormal protein and chemotherapy to suppress plasma cells Myeloma Cast Nephropathy Light Chain Deposition Disease Amyloid protein produced in plasma cell dyscrasias is preferentially lamda light chains Kappa chain can occur and termed light chain deposition disease Kappa chain deposits tend to deposit in tubules and glomeruli, spare vessels Kappa more aggressive than lambda Diagnosis • Documentation of tissue deposition of amyloid fibrils. • Kidney biopsy> 90% of patients • Abdominal fat pad> 80-90% • Rectal> 50-80% • Gingival>60% • Once the DX of primary amyloidosis is made, the patient should be evaluated for M.M ! ! ! Pathology • On light microscopy> >>Diffuse deposition of amorphous hyaline material in the mesangium and then capillary loops. • Amyloid deposition in the glomeruli can result in the formation of nodules. • Congo Red induces green birefringence when viewed under polarized light. • Immunofluorescence for immunoglobulin and complement is negative. Key things to Remember Amyloid is hyaline, eosinophils, Congo Red + Green Birefringence when polarized Small non-branching fibrils on EM Amyloidosis • More common in elderly • Two main types of renal amyloid – AL amyloid – AA amyloid Normal Clinical Presentation • Non-specific symptoms, Fatigue, Weight loss, Anemia • Proteinuria, edema>>>Nephrotic Syndrome • Carpal tunnel syndrome • Peripheral neuropathy • Hepato-Splenomegaly, Macroglossia • Renal insufficiency • Tubular involvement : RTA, NDI, Hyperkalemia Course and Treatment • Prognosis depend on type of amyloidosis • MM= bad Prognosis • In primary Amyloidosis usually progress slowly to CRF • Prednisone an Melphalan is the major mode of therapy for primary amyloidosis • Colchicine in AL, AA amyloidosis • In AA the progression depend upon the ability to control the underlying disease. Asymptomatic Non-Nephrotic Proteinuria • Overflow Proteinuria: Multiple Myeloma -Light Chain • Tubular Proteinuria: Tubulointerstitial dis. < 2 gr/day • Glomerular proteinuria Treatment • Steroid Treatment induces remission in almost all patients with MCD. • Adults usually biopsied before treatment (KNB). • Controll first volume overload (Diuretics). Treatment • Steroid responsive • Steroid dependent • Frequent relapsers