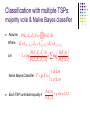Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Classifying Gene Expression
Profiles from Pairwise
mRNA Comparisons
Yan Qi
Biomedical Engineering Department
10/3/2006
Outline
The problem of Molecular
classification
The TSP classifier
Results on three Cancer datasets
Extensions of the TSP classifier
Molecular Classification
Objective: predict class labels, e.g. cancer
subtypes, disease states using gene
expression profiles
Data: G x n matrix, G is number of genes, n
is number of samples, each column is a gene
expression profile
Mathematical formulation
Gene expression profile: X = ( X1, X2, …, XG)
Binary Class label: Y = 1 or Y = 2
Classifier: A mapping f from X to Y
Training dataset:
A = G x n matrix, n = n1+n2
Y: G x 1 vector where n1 entries are 1, n2 entries
are 2
Learning: find a mapping from A to f that
minimizes generalization error e( f ) P( f ( X ) Y )
Challenges to standard
learning methods
Statistical dilemma: n << G
Examples
Consequence: Over-fitting hence poor
generalizability
Practical issue: complex f and DB
Example: ANN, SVM, random forests
Consequence: results are hard to interpret
biologically, inefficient in diagnostic settings
The TSP classifier:
Motivation and strategy
Rank-based scoring exploits the expression
levels of genes relative to each other and
obtains invariance to normalization.
Reduce model complexity by making the
classifier parameter free
Select informative gene pairs by proper
LOOCV and construct intuitive and
biologically interpretable classification rule by
voting.
TSP classifier:
rank-based score
Idea: replace expression value by genes’
ranks within profiles.
Feature: Zij 1{ X X } ,1 i j G,
Score: ij P( X i X j | class 1) P( X i X j | class 2)
i
where
N ij1
n1
j
N ij2
n2
N ijk # of samples in class k that satisfy X i <X j
Gene pair selection
(i* , j * ) arg max ( i , j ) ij
TSP:
The number of TSP and sample size
A few when sample size is not too small ( >102 ).
Many when sample size is small ( < 102 ).
Example: myocardial tissue gene expression profiles
G = 22283; n1=12, n2=10;
2460 statistically significant TSPs
Classification with one gene pair
Let (i* , j* ) be a unique TSP
Suppose
Ni1* j*
n1
Ni2* j*
n2
1 if X i* X j*
TSP classifier: Yˆ fTSP ( X )
2 if X i* X j*
Error on training set: e P( X i2 X 2j ) P( X i1 X 1j )
P( X i2 X 2j ) 1 P( X i1 X 1j )
1 ij
An Example
Classification with multiple
gene pairs – majority vote
Seek a mapping from outputs of multiple single TSP
classifiers to a final prediction.
Let Z1 1{ X X }; Z2 1{ X X }; Z3 1{ X X }
Output from f ( Zi ) represent a vote from TSP i.
Final prediction = class that receives the majority vote
from { f ( Z1 ), f ( Z 2 ), f ( Z3 )}
i1
j1
i2
j2
i3
j3
Assume the features P( Zi 1| k ) are conditionally
independent given the class and equal, a Naïve Bayes
classifier is equivalent to using majority vote
Classification with multiple TSPs:
majority vote & Naïve Bayes classifier
3
Assume
Where
Let
P( Z1 , Z 2 , Z3 | k ) P( Z i | k )
i 1
Z1 1{ X i1 X j1} ; Z2 1{ X i 2 X j 2 } ; Z3 1{ X i 3 X j 3 }
P( Zi |1)
P( Z1 , Z 2 , Z3 |1)
L log
log
P( Z1 , Z 2 , Z3 | 2) i
P
(
Z
|
2)
i
Naïve Bayes Classifier: Yˆ g ( X )
1 if L>0
2 if L<0
Each TSP contribute equally if
P( Z i |1)
p for i=1,2,3
P( Z i | 2)
Loop of cross-validation
Leave-one-out CV
Estimated accuracy: 1 - e/n where e is total
number of errors in cross-validation
Only the TSPs are determined by CV,
unbiased error estimate.
More complicated models, e.g. ANN and
Decision tree need to include both model
topology and parameters in CV loop, more
likely to be biased.
Three bench-mark cancer
datasets
Determine lymphnode status in breast tumor
samples ( West et al. 2001, G=7129, n = 49 )
Classify leukemia subtypes ( Golub et al
1999, G= 7129, n=72)
Distinguish prostate tumors from normal
samples ( Singh et al. 2002, G = 12600,
n=102)
Statistical significance of the score is
evaluated by permutation test
• Repeat e.g. 1000 times
• Keep feature matrix A
• Randomize class labels by keeping
n1 and n2 unchanged
• Get top score Zmax
• Get histogram of Zmax
p P( Z max Z real )
The top scoring gene pairs for
the three cancer studies
What does TSP represent?
ALL
AML
|ALL-AML|
X1
1349
1063
286
X2
1396
1057
339
X3
382
3258
2876
X4
868
162
706
X5
174
861
687
X2-X3
1014
-2201
3215
X1-X3
967
-2195
3162
X4-X5
684
-699
1383
Change weak predictors into strong predictors?
Change reference e.g. gene 3 as reference for gene 1 and gene 2
Combine two markers e.g. x4 and X5 might be individual markers, one for each
class, x4|-|X5 ?
Performance of TSP classifier
compared with previous studies
• Breast cancer: k-nearest neighbors (8-26); DLDA (8-19); DQDA (11-26)
logitboost (9-21); random forests (6-20); SVM (7-29)
• Leukemia: correlation analysis + weighted vote (85% on test set and 95%
on CV set.
• Prostate cancer: k-nearest neighbour to genes chosen by t-statistic
Discussion and extensions
Multiple class classification
Unique TSP: when there are >>1 TSPs, which is the
most informative?
kTSP: there might be many pairs of genes with
informative ordering, combine this information for
more accurate classification?
Normalization invariance: a suitable method to
integrate heterogeneous microarray datasets where
experimental conditions and normalization schemes
differ.