Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
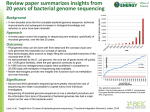
corso di Genomica 2010-2011 lezione 17-18 • laurea magistrale Biotecnologia Industriale Giovedì 9 dicembre 2010 aula 6 orario : Martedì ore 14.00 - 16.00 Giovedì ore 13.00 - 15.00 Lunedì 13 Dicembre seminario sequenziamento c/o Fac.medicina ore 9.30 aula anfiteatro (p -1) D. Frezza In search of human variants The 1000 genomes project Rasmus Nielsen 2010 Nature vol 467 28 Oct. News and views 1050-1051 applicazioni di HapMap Once the information on tag SNPs from the HapMap is available, researchers will be able to use them to locate genes involved in medically important traits. Consider the researcher trying to find genetic variants associated with high blood pressure. Instead of determining the identity of all SNPs in a person's DNA, the researcher would genotype a much smaller number of tag SNPs to determine the collection of haplotypes present in each subject. The researcher could focus on specific candidate genes that may be associated with a disease, or even look across the entire genome to find chromosomal regions that may be associated with a disease. If people with high blood pressure tend to share a particular haplotype, variants contributing to the disease might be somewhere within or near that haplotype. 1000 genomes project Nature. 2010 Oct 28;467(7319):1061-73. A map of human genome variation from population-scale sequencing. The 1000 Genomes Project aims to provide a deep characterization of human genome sequence variation as a foundation for investigating the relationship between genotype and phenotype. Here we present results of the pilot phase of the project, designed to develop and compare different strategies for genome-wide sequencing with high-throughput platforms. We undertook three projects: low-coverage whole-genome sequencing of 179 individuals from four populations; high-coverage sequencing of two mother-father-child trios; and exon-targeted sequencing of 697 individuals from seven populations. We describe the location, allele frequency and local haplotype structure of approximately 15 million single nucleotide polymorphisms, 1 million short insertions and deletions, and 20,000 structural variants, most of which were previously undescribed. We show that, because we have catalogued the vast majority of common variation, over 95% of the currently accessible variants found in any individual are present in this data set. On average, each person is found to carry approximately 250 to 300 loss-of-function variants in annotated genes and 50 to 100 variants previously implicated in inherited disorders. We demonstrate how these results can be used to inform association and functional studies. From the two trios, we directly estimate the rate of de novo germline base substitution mutations to be approximately 10(-8) per base pair per generation. We explore the data with regard to signatures of natural selection, and identify a marked reduction of genetic variation in the neighbourhood of genes, due to selection at linked sites. These methods and public data will support the next phase of human genetic research. utilità ed uso dei 1000 genomes in quale modo si applicherebbe la banca dati del progetto dei 1000 genomi? vedi articolo su Nature vol 467 del 28 Ottobre 2010 la prospettiva nel restante 95% del genoma genoma si ricomincia da capo nell’interattoma va inserito il genoma e si allargano le prospettive nuove tecniche, metodi, strumenti P.Fraser & W.Bickmore Nature vol.447, 24 May 2007; 413-417 Nuclear organization of the genome and the potential for gene regulation lettura articolo news and views “in search of human variants” 1000 genomes project strategies, objectives, goals discussion and work in progress preliminary results Rasmus Nielsen: Nature vol 467, 28 October 2010 pp 1050-1051