Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
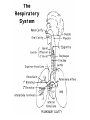
CYSTIC FIBROSIS Presented by Lisa Waseleski, R.N. Learning objectives: • The learner will describe normal structures and functions of the respiratory system. • The learner will describe normal structures and functions of the pancreas. • The learner will describe characteristics of an autosomal recessive disorder in relation to cystic fibrosis. • The learner will describe the pathophysiology and clinical manifestations of cystic fibrosis. • The learner will describe the diagnosis and treatment of cystic fibrosis. Structures of the respiratory system: The respiratory system includes the following structures: nasal cavity pharynx epiglottis larynx carina trachea right and left main stem bronchus segmental bronchi terminal bronchioles acinus thoracic cavity/lungs mediastinum diaphragm Functions of the respiratory system • The air-conducting passages that bring air into the lungs are the nose, pharynx, larynx, trachea, bronchi and bronchioles. • Ciliated mucous membranes line the respiratory tract. Air is filtered, warmed and humidified upon entering the nasal cavity. • Air passes from the pharynx into the larynx. • A triangular space between the vocal cords, the glottis, opens into the trachea and forms the division between the upper and lower respiratory tracts. Continued from the previous slide: • The trachea and bronchi form the tracheobronchial tree. • The trachea branches into the right and left main stem bronchi known as the carina. • The right and left main stem bronchi divide to become the lobar bronchi, then segmental bronchi, then terminal bronchioles. • Beyond the terminal bronchiole is the acinus, the pulmonary functional unit, gas exchange takes place here. • 300 million alveoli or small air sacs are contained within each lung. Continued from the previous slide: • Alveolar lining cells: type 1 and 2 pneumocytes are responsible for the secretion of the lipoprotein surfactant. • The lungs are elastic cone shaped organs that lie within the thoracic cavity. • The central mediastinum separates the lungs. • The diaphragm forms the floor of the thoracic cavity and separates it from the abdominal cavity. • Pulmonary and bronchial blood vessels, bronchi, nerves and lymphatics enter each lung at the hilus to form the root of the lung. • The right lung is divided into three lobes and is larger compared to the left lung which has two lobes. Pulmonary circulation: • The lung has a dual blood supply from the bronchial and pulmonary arteries. • The bronchial circulation provides oxygenated blood from the systemic circulation to meet the metabolic needs of lung tissue. • The bronchial arteries originate from the thoracic aorta and travel along the posterior walls of the bronchi. • The smaller bronchial veins drain into the pulmonary veins. The large bronchial veins empty into the superior vena cava and return blood to the right atrium. • The bronchial circulation does not take part in gas exchange causing a shunt of unoxygenated blood, approximately 2-3% of cardiac output. • The pulmonary artery arising from the right ventricle provides mixed venous blood to the lungs, where gas exchange takes place. Continued from the previous slide: • A network of pulmonary capillaries surround the alveoli providing the necessary exchange of gases between the blood and the alveoli. • Oxygenated blood is returned through the pulmonary veins to the left ventricle where it is distributed to the cells via systemic circulation. Structures and functions of the pancreas: • The pancreas is a compound tubuloalveolar gland, approximately 6-8 inches in length and 1 ½ inches wide. • The pancreas lies retroperitoneal and is divided into three major segments: the head, the body, and tail. • The pancreas resembles a bunch of grapes, the branches of which are the ducts terminating into the main pancreatic duct, or the duct of Wirsung. • Small ducts from each acinus empty into the main ducts. The main duct extends through the length of the gland and joins the common bile duct at the ampulla of Vater before entering the duodenum. Continued from the previous slide: • The pancreas is made up of two types of cells: the exocrine cells and the endocrine cells. • The exocrine cells produce the components of pancreatic juice. • The endocrine cells produce insulin and glucagon. Characteristics of autosomal recessive inheritance and cystic fibrosis: • Gene is on an autosome. • Expressed only in homozygotes (aa); heterozygotes (Aa) are phenotypically normal carriers. • Males and females are equally affected. • Horizontal inheritance pattern is present • When both parents are carriers, each offspring has a 50% chance of being a carrier, a 25% chance of having the disease and a 25% chance of being normal. • Early age onset of disease is common. Continued from the previous slide: • Cystic Fibrosis is one of the most common autosomal recessive diseases in Caucasian populations affecting approximately 1 in 2500 individuals. • The gene responsible for CF (cystic fibrosis) is located on the long arm of chromosome 7. The CF gene protein product (CFTR) encodes for a protein that functions as a chloride channel. Pathophysiology of cystic fibrosis: • • • • • CFTR mutations result in abnormalities of chloride transport across epithelial cells on mucosal surfaces. The failure of epithelial cells to conduct chloride and water transport result in dry and viscid secretions in the respiratory tract, pancreas, GI tract, sweat glands and other exocrine tissues. Changes in the chemical properties of mucous may lead to obstructions in the respiratory and digestive systems. In the respiratory system, the viscous mucous obstructs airways and creates conditions that lead to repeated infections in the lungs. CF interferes with the normal functions of the liver, pancreas and other digestive organs, causing problems with digestion and absorption of nutrients. Diagnosis and treatment of cystic fibrosis: • Diagnosis is usually made within the first few years of life. A sweat electrolyte test, sodium >90 chloride>60 is diagnostic for cystic fibrosis. Infants and children may present with a delay in passing meconium, may have foul smelling pale and greasy stools, and have frequent respiratory infections. Nasal polyps and sinusitis are common findings in these patients. • Treatment is aimed at the management of pulmonary dysfunction. Aerosol treatments with bronchodilators and mucolytic agents in conjunction with chest physiotherapy at least twice per day to prevent respiratory infection are crucial. An aerosol recombinant human deoxyribonuclease (DNase) is effective in clearing thick mucous. Continued from the previous slide: • • • • • • • • Prompt treatment of respiratory infection with aerosol or IV antibiotics is necessary. In 1993, researchers developed a mucus-thinning drug called pulmozyme specifically for CF patients. This drug decreases lung infections and increases lung function. (Smith MD, 2001). Regular exercise is helpful to move lung secretions. Pancreatic enzymes given prior to every meal help to enhance digestion. A diet rich in proteins and calories is essential to meet growth needs. Vitamins and other dietary supplements are important to add more nutrients to the diet. Patients with severe digestive problems may require supplemental tube feeding or IV nutrition. Psychosocial support is important to assist the patient cope with this chronic illness. Continued from the previous slide: • Ibuprofen, when given at a dose sufficient to achieve a peak plasma concentration between 50 and 100 ug/ml over several years, has been shown to slow the rate of decline in pulmonary function, especially in children age 5-13. (Merck Manual, Sec. 19, Ch 267). • Heart-lung and bilateral lung transplants have been performed successfully in patients with advanced cardiopulmonary disease. • Mutation analysis can be used for prenatal diagnosis and carrier testing in families with a previously affected child. References/Internet Sources • • • • Guyton, A.C.& Hall, J. E. (2000). Textbook of medical physiology (ioth ed.) Philadelphia: W.B. Saunders Pagana, K.D. & Pagana T.J. (2002). Mosby’s manual of diagnostic and laboratory tests (2nd ed.) St Louis: Mosby Price, S.A. & Wilson, L.M. (2003). Pathophysiology, clinical concepts of disease processes (6th ed.) St Louis: Mosby The Merck Manual of Diagnostics and Therapy, (copyright 1995-2003) section 19. Pediatrics, chapter267. Cystic Fibrosis Topics. Retrieved March 30, 2003 from http://www.merck.com/pubs/mmanual/section19/chapter267/267 a.htm • The Merck Manual of Diagnostics and Therapy, (copyright 19952003) section 12. Immunology: Allergic Disorders, chapter 149. Transplantation Topics. Retrieved March 30, 2003 from http://www.merck.com/pubs/mmanual/section12/chapter149/149 f.htm References/Internet Sources: • Pace, B. (2000). Cystic fibrosis. Journal of American Medical Association, 284:1884. Retrieved March 26, 2003 from http:www.medem.com/search/article_display.cfm?path=n: &mstr=/ZZZIX1MDUEC.html… • Understanding Cystic Fibrosis. ( 2001). WebMD Medical Reference, Retrieved March 29, 2003 from http://aolsvc.health.webmd.aol.com/content/article/10/168 0_54803