Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
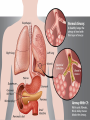
CYSTIC FIBROSIS BY: TERESA KRASZEWSKI BACKGROUND AND HISTORY • Late 16th century babies who had “salty skin” when kissed were likely to die • 1938 Dr. Dorothy Andersen produced first description of cystic fibrosis in medical literature • 6 month life expectancy • Only thought as mucus disorder • 1959 sweat test • Larger than just mucus disorder • 1989 discovery of CFTR gene • Made treatment easier • Life expectancy grew CAUSE AND INHERITANCE • Autosomal recessive • Most common in whites • Defect in cysitc fibrosis transmembrane regulator (CFTR) gene on chromosome 11 • Makes protein that controls movement of salt and water in cells • Most common mutation F508 • 2/3 • Bodily secretions thick/sticky • Blocks tubes, ducts, passageways • Life threatening • 20-30 yrs • Small % 40-50 yrs SYMPTOMS • • • • Salty skin Clubbed fingers/toes Diabetes Respiratory: • • • • • Persistent cough (phlegm) Wheezing Breathlessness Repeated lung infections Sinus infections/nasal polyps • • • • • Foul-smelling stool Poor weight gain Intestinal blockage Severe constipation Rectal prolapse (end of the large intestine protrudes outside anus) • Digestive: DIAGNOSIS • 70% diagnosed by age 2 • Screening newborns required in all 50 states • Blood sample from heel • Checks for high levels of immunoreactive trypsinogen (released by the pancreas) • Sweat chloride test • Sweat-producing chemical applied skin • Collected sweat tested to see if higher chloride concentration than normal • DNA samples from blood/saliva • Checks for defects on gene DIAGNOSIS (CONTINUED) • People who weren’t screened: • • • • • X-rays, CT scans and MRI (show damage to lungs/intestines) Lung function tests Sputum culture (analyzed for bacteria) Blood tests (health of your pancreas/liver) Tested for diabetes regularly (ages 10+) TREATMENT • No cure • Chest physical therapy • Drugs: • • • • Antibiotics (prevent lung infections) Mucus-thinning drugs Bronchodilators (relax muscles around bronchial tubes) Oral pancreatic enzymes (help digestive tract absorb nutrients) • • • • • • Nasal polyp removal Oxygen therapy (prevent high blood pressure in lungs) Endoscopy (suction mucus out of airways) Lung transplant (controversial) Feeding tube Bowel surgery • Surgery: SUMMARY Autosomal recessive inherited disease Defect in CFTR gene Life threatening Now diagnosed by screening newborns Symptoms mostly affect respiratory and digestive systems • Treatments available for symptoms but no cure • • • • • WORKS CITED "CYSTIC FIBROSIS." Cystic Fibrosis. N.p., n.d. Web. 1 Dec. 2013. <http://learn.genetics.utah.edu/content/disorders/whataregd/cf/>. "Cystic Fibrosis Since 1938." (thoracic). N.p., n.d. Web. 1 Dec. 2013. <http://www.atsjournals.org/doi/full/10.1164/rccm.200505-840OE#.UqUaeSe5pmg>. "Cystic Fibrosis Symptoms, Treatment, Life Expectancy, Genes, Testing - MedicineNet." MedicineNet. N.p., n.d. Web. 1 Dec. 2013. <http://www.medicinenet.com/cystic_fibrosis/article.htm>. "Cystic Fibrosis: Overview." Cystic Fibrosis. N.p., n.d. Web. 1 Dec. 2013. <http://www.nationaljewish.org/healthinfo/conditions/cysticfibrosis/>. "January/February 2014." Discover Magazine. N.p., n.d. Web. 1 Dec. 2013. <http://discovermagazine.com/2013/september/14-doorway-to-a-cure>. staff, Mayo. "Definition." Mayo Clinic. Mayo Foundation for Medical Education and Research, 13 June 2012. Web. 1 Dec. 2013. <http://www.mayoclinic.com/health/cystic-fibrosis/DS00287>.