Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Hepatitis C wikipedia , lookup
Neonatal infection wikipedia , lookup
Hospital-acquired infection wikipedia , lookup
Marburg virus disease wikipedia , lookup
Meningococcal disease wikipedia , lookup
Human cytomegalovirus wikipedia , lookup
Oesophagostomum wikipedia , lookup
Onchocerciasis wikipedia , lookup
Eradication of infectious diseases wikipedia , lookup
Chagas disease wikipedia , lookup
Leptospirosis wikipedia , lookup
Leishmaniasis wikipedia , lookup
Visceral leishmaniasis wikipedia , lookup
Schistosomiasis wikipedia , lookup
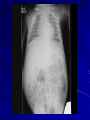
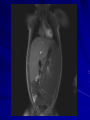
Cases from Virginia: A Neonate and an Infant with Splenomegaly Lauren Youell MD Chief Resident UVA Pediatrics Patient 1: C.M. Patient C.M. Term male G3, now P3 mother No pregnancy complications Fetal decels during labor, thin meconium ROM 12 hours Apgars of 7 and 8 Patient admitted to the newborn nursery Maternal Labs Blood type O+, Ab negative RPR NR Hepatitis B negative GC negative Chlamydia treated during pregnancy with test of cure during first trimester HIV negative GBS negative Rubella immune First exam BW 2674g (10th %) Length 50cm (50th %) HC 33 cm (10th %) Exam normal – palpable liver edge 2 fingers below costal margin NBN Course Initial hypoglycemia that resolved with feeds Persistent jitteriness Mild hypocalcemia Hyperbilirubinemia requiring phototherapy DOL 5 Liver noted to be 2.5cm below costal margin Spleen enlargement also noted on exam Patient otherwise doing well Further Labs and Studies WBC 7, HCT 50, PLT 69 BMP normal except potassium of 5.7 ALP 392, ALT 14, AST 55 Abdominal U/S- Liver not enlarged, caudate lobe prominent. Spleen prominent (6.5cm in length). Small ascites, GB contracted. Differential of Hepatosplenomegaly in the Neonate Congenital Infections Elevated portal pressures (CHF, hepatic vein thrombosis) Obstruction (tumors, stones, cysts) Storage disease (glycogen, lipids, metals, abnormal proteins) Infiltration (neoplasms, metastases, extramedullary hematopoiesis) Algorithm for the Work-up of Neonates with Hepatomegaly Wolf, A. D. et al. Pediatrics in Review 2000;21:303-310 Copyright ©2000 American Academy of Pediatrics Differential of Thrombocytopenia in the Neonate Non-immune platelet destruction Immune platelet destruction Decreased platelet production Non-Immune Platelet Destruction Disseminated intravascular coagulation Bacterial sepsis Intrauterine viral infection Pregnancy induced hypertension Necrotizing enterocolitis Drugs/toxins Immune Thrombocytopenia Maternal Immune Thrombocytopenia Neonatal Alloimmune Thrombocytopenia (NAIT) Maternal Systemic Lupus Erythematosus Decreased Platelet Production Thrombocytopenia absent radii (TAR) syndrome Neonatal leukemia Aplastic anemia Merging the Differentials Congenital Infection Metabolic disease with sequestration Congenital Infection- CMV Most common Risk of transmission greatest with primary maternal infection 10% of congenitally infected infants will have clinical evidence of disease – IUGR – Hepatosplenomegaly – Hematologic abnormalities(thrombocytopenia) – CNS changes CNS Changes of CMV Microcephaly Ventriculomegaly Cerebral atrophy Chorioretinitis Sensorineural hearing loss Intracerebral calcifications (periventricular) Toxoplasmosis Infection during 1st trimester – Chorioretinitis – Encephalitis with intracerebral calcifications – Microcephalus – Hydrocephalus – Hepatosplenomegaly – Maculopapular rash – Thrombocytopenic purpura Our Patient Urine CMV negative Toxoplasmosis IgG positive, IgM negative Head U/S- no calcifications Eye exam- no evidence of chorioretinitis (or cherry red macule) Hematology Consult BM biopsy to rule out leukemia – Trilineage hematopoiesis with normal cellular maturation – No evidence of lipid storage disease or neoplasm Prolonged bleeding time after the BM biopsy A Definitive Diagnosis Liver biopsy necessary for tissue diagnosis Not without risks – Age – Platelet dysfunction? – Liver disease Pathology calls… Foam Cells Niemann-Pick Disease Heterogeneous group of lysosomal lipid storage diseases All have autosomal recessive inheritance Niemann-Pick Disease Niemann-Pick Type A and B – Acid Sphingomyelinase deficiencyabnormal storage of sphingomyelin – Type A=neuronopathic, earlier onset – Type B=non-neuronopathic, milder course Niemann Pick Disease Niemann-Pick Type C – Impaired cholesterol esterification – Sequestration of unesterified cholesterol in lysosomes – Acid Sphinogmyelinase is secondarily reduced – Can present in infants, children or adults Age at Time of Diagnosis Vanier and Millat, Clinical Genetics, 2003; 64:269-281 Niemann-Pick Type C Classic presentation – 60-70% of cases – Mid-late childhood – Insidious onset – Death typically occurs in the second or third decade Classic Niemann-Pick Type C First manifestation- often ataxia Neurologic onset- typically decreased school performance, fine motor skills Organomegaly- up to 90% Cataplexy- 20% Seizures- about 50% Dysphagia is also common Exam Clues Massive HSM (not required) Vertical supranuclear gaze palsy Cherry red spot on macula Niemann-Pick Type C Perinatal onset – Liver disease is major sign – May have history of fetal ascites or hydrops – May also have prolonged cholestatic jaundice – Most children with “rapidly fatal neonatal cholestatic form” die before age of 6 months Niemann-Pick Type C Infantile onset – Can present with ascites, severe liver disease or respiratory failure – Infants without liver or lung involvement may have just hypotonia and developmental delay Niemann-Pick Type C Adult onset – More likely to present with progressive dementia or psychiatric symptoms – Movement disorders – Organomegaly less common Diagnosis Biochemical testing Most have mutations in the NPC1 gene Molecular gene testing of NPC1 and NPC2 will detect mutations in approximately 94% of individuals with NPC Our Patient Homozygous gene mutation in the NPC1 gene Particular mutation not previously reported in the literature or disease specific databases Management Largely symptomatic – Treat seizures, dystonia – Physical therapy – Gastrostomy tube – Aggressively treat intercurrent infections Managing our Patient Required NG feeds at discharge Family counseled and decided against Gtube placement Actigall for cholestasis Palliative care involved at discharge Update on C.M. Now 5 months of age Followed by genetics and palliative care Not requiring NG feeds, growing well Continues to be very hypotonic on exam with massive hepatosplenomegaly Remains on actigall for cholestasis Patient 2: A.B. Patient A.B. 6 mo female Full term PMH: bronchiolitis at 1 month of age, feeding difficulties with emesis x 1 week Developmental: +social smile, not rolling over yet, sits with assistance per mom Splenomegaly noted by PCP Admit Exam Weight: 6.64 kg (25%) Length: 64 cm (25-50%) HC: 41.5 cm (25%) Exam normal except for: – Thrush – Spleen palpable 4 cm below costal margin Admit Labs and Studies WBC 6, Hgb 10, PLT 98 Smear: microcytosis Upper GI series: positive for reflux, no structural abnormalities Abdominal U/S: confirms splenomegaly, otherwise normal Infectious Work-up EBV CMV Parvovirus All serologies negative Hematology consulted Family history of hereditary spherocytosis on paternal side Osmotic fragility testing-normal Bone marrow biopsy performed – Trilineage hematopoiesis – Lipid laden macrophages Gaucher Cells Gaucher Disease Glucocerebrosidase deficiency Represents a continuum of disease Autosomal recessive inheritance Gaucher Disease- Type 1 “Adult-type”-may present in childhood Bone disease Hepatosplenomegaly Anemia Thrombocytopenia Lung disease **No CNS disease Gaucher Disease-Type 2 Neuropathic Onset before age of 2 years Limited development Rapidly progressive course Death by age 2-4 is common Gaucher Disease-Type 3 Neuropathic Onset may also be before 2 years Slowly progressive May live into the third or fourth decade Subtype Type 1 Primary CNS Involvement No Type 2 (acute or infantile) Bulbar signs Pyramidal signs Cognitive impairment Type 3 (subacute; juvenile) Oculomotor apraxia Seizures Progressive myoclonic epilepsy Bone Disease Other Yes Splenomegaly Hepatomegaly Cytopenia Pulmonary disease No Hepatomegaly Splenomegaly Cytopenia Pulmonary disease Dermatologic changes Yes Hepatomegaly Splenomegaly Cytopenia Pulmonary disease Diagnosis Characteristic bone marrow changes Demonstration of deficient enzyme activity in peripheral blood leukocytes Identification of 2 disease causing alleles in GBA (only gene linked to Gaucher disease) Our Patient Presented with concerns for neurologic involvement in the first year of life Gene testing positive for mutation in GBA Type 2 disease diagnosed based on clinical presentation Management Enzyme replacement therapy – Decreases spleen size – Does not affect neurologic deterioration Partial or total splenectomy Analgesics, oral bisphosphonates and calcium for bone changes in Type 1 Update on A.B. Now 2 ½ years old Being treated with enzyme therapy Tracheostomy and G-tube dependent Frequent pulmonary infections Seizure disorder Global developmental delay Final Thoughts