Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
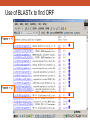
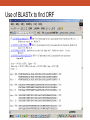
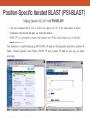
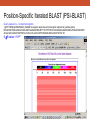
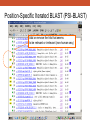
Database Similarity Searching BLAST Global alignment of a pair of seqs., in which all residues from both seqs. are included. BLAST – local alignment Interpreting BLAST output -Smith and Waterman algorithm guaranteed to find the best local alignment of two seqs. -Too slow in practice !! -BLAST heuristic search method that is not guaranteed to find the best local alignment, but has been especially effective in practice -e.g. S45649 (from a fossilized insect) >gi|256517|gb|S45649.1| 16S rRNA [Mastotermes electrodominicus=termites, amber-preserved fossil, Mitochondrial, 94 nt] AATAAAATTTTAATAAATATAAAGATTTATAGGGTCTTCTCGGCCTTTAAAAATA TTTTAGCCTTTTGAC AAAAAAAAAAAAATCTACAAAAAA BLAST http://www.ncbi.nlm.nih.gov/BLAST/ E-value, with the most significant hits listed first E-value is the number of hits with the same level of similarity that you would expect by chance E = 0.01 occur once every 100 searches even when there is no true match in the database E-value is similar in spirit to the p-value of statistical hypothesis tests. P-value is the probability of finding a seq. similarity as similar as the observed match if there were really no true matches in the database. E-value ≠ p-value E-value ~ p-value when it is small (say < 0.1) Since we are interested in unusual hits, it is safe to interchange E-value with p-value. E-value – the lower the better the alignment, matches above 0.001 are often close to the twilight zone (not significant) Score (bits) – the higher the better the alignment, score below 50 are unreliable BLAST The BLAST output may not be the same every time due to the upgrade of several components : Database, the BLAST program, the default parameters of the server E-value, similarity and homology Protein : >25 %, > 100 a.a., < 10-4 DNA : >70%, > 100 bp, < 10-4 Gap penalties - constant penalty independent of the length of gap, A - proportional penalty, penalty is proportional to the length L of the gap, BL - Affine (『數』遠交的,『化學』親和的) gap penalty, gap-opening penalty + gap-extension penalty = A+BL Remark • Prediction using similarity is a powerful idea in bioinformatics • homologue seqs. evolved by divergence from a common ancestor, therefore to say two seqs. share 50% homology is nonsense; to say two seqs. share 50% similarity and that they indicate possible homology is the correct usage of the terms • Similarity NOT necessary implied homology BLAST (choosing the parameters) BLAST - Most highly cited paper >12000 times alternative methods seeds + dynamics programming speed up, faster not guaranteed to find the best alignment less accurate BLAST (Sequence filters) http://www.ncbi.nlm.nih.gov/BLAST/ BLAST What is a coiled-coil? Coiled-coil domains are characterized by a heptad (成七的一組) repeat pattern in which residues in the first and fourth position are hydrophobic, and residues in the fifth and seventh position are predominantly charged or polar. This pattern can be used by computational methods, such as MultiCoil (MIT) or SOCKET (University of Sussex)to predict coiled-coil domains in amino acid sequences. BLAST programs BLASTing DNA sequences Use of BLASTx to find ORF AE008569 Use of BLASTx to find ORF Frame = +1 Frame = -2 Use of BLASTx to find ORF Use of BLASTx to find ORF Use of BLASTx to find ORF BLAST procedures BLAST ls • The E-value of the BLAST is given by E kmne • where k (depend on the scoring matrix and gap penalty combination) and l are constants, m and n denote the seqs. length, s is the scaling factor for the scoring matrix used • Gumbel extreme value distribution for alignment scores kmne lx • P 1 e http://www.itl.nist.gov/div898/handbook/eda/section3/eda366g.htm Position-Specific Iterated BLAST (PSI-BLAST) Position-Specific Iterated BLAST (PSI-BLAST) Query sequence – human hemoglobin >gi|57013850|sp|P69905|HBA_HUMAN Hemoglobin alpha subunit (Hemoglobin alpha chain) (Alpha-globin) MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPN ALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSK YR 0 ≦E-value < 10-40 Position-Specific Iterated BLAST (PSI-BLAST) Query sequence – human hemoglobin >gi|57013850|sp|P69905|HBA_HUMAN Hemoglobin alpha subunit (Hemoglobin alpha chain) (Alpha-globin) MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHA HKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSK YR Gene or Structure information Position-Specific Iterated BLAST (PSI-BLAST) More seqs. are identified than Iteration 1 Position-Specific Iterated BLAST (PSI-BLAST) Add or remove the hits that seems to be relevant or irrelevant (non-human seq.) Position-Specific Iterated BLAST (PSI-BLAST) B~C