Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
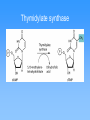
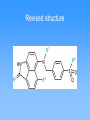
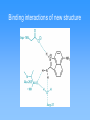
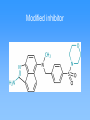
Molecular mechanics • Classical physics, treats atoms as spheres • Calculations are rapid, even for large molecules • Useful for studying conformations • Cannot calculate electronic properties Energy minimization Visualizing molecules Quantum Mechanics • Considers interactions between electrons and neutrons • Can calculate electronic properties • Slower calculations than molecular mechanics • Ab initio vs. semi-empirical Partial charges on histamine Partial charges on protonated histamine Effect of delocalized charge Molecular electrostatic potentials (MEPs) Conformational analysis Molecular Dynamics Structure Comparison (2D) Structure Comparison (3D) Identifying the active conformation of ligand • • • • X-ray crystallography Cambridge Structural Database Protein Data Bank Comparing biological activity of nonrigid ligands with various rigid ligands 3D Pharmacophore Identification • X-ray crystal structure of protein-ligand complex (from PDB) • Comparison of active compounds (when target structure is unknown) • Automated identification of pharmacophores Automated identification of pharmacophores • Generate range of conformers • For each conformer, define set of pharmacophore triangles • Another structure is analyzed • Pharmacophore triangles compared to those for previous structures Pharmacophore plot Use pharmacophore triangles common to all active compounds x,y,z correspond to lengths of three sides of triangles Graphing allows identification of distinct pharmacophores Omit triangles involving non-essential binding groups Docking procedures • X-ray crystal structure of target protein with binding region highlighted • Place ligand within active site with different orientations to identify best orientation • Simplest approach—treat ligand and target as non-flexible DOCK ChemX: Analyzing potential binding centers Compare ligand pharmacophores to those in binding site Bump filter Reject conformations which involve bad steric interactions Constructing protein model • • • • Need primary amino acid sequence Compare to other proteins Need X-ray structure of related protein Arrange new protein to match sequences similar to known protein • Determine structure of connecting sequences by comparison to proteins in databases or with loops Model protein • Side chains added in energetically favorable conformations • Energy minimization • Structure refined with molecular dynamics • Use this model protein to analyze potential ligands Constructing binding site when protein structure is unknown • Range of structurally diverse compounds with varying activities • Align molecules to match up pharmacophores • Potential energy grid with probes to measure interaction energies Potential energy probe to find binding site De novo design • In theory, design drug for target given structure of binding site • In reality, design good lead compound • Used to get drugs unlike natural substrates to minimize side effects Thymidylate synthase Thymidylate synthase cofactor Inhibitors similar to substrate or cofactor CB3717 binding to thymidylate synthase active site • Create empty binding site from X-ray crystal structure of protein plus inhibitor • Found hydrophobic area near where pteridine group is bound De novo design of Thymidylate synthase inhibitor Intended vs. actual interactions Revised structure Binding interactions of new structure Modified inhibitor