Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
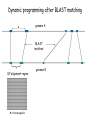
Whole genome alignments Genome 559: Introduction to Statistical and Computational Genomics Prof. James H. Thomas Review • What a score matrix is and how to calculate and use one. • Why an affine gap penalty is desirable. • How to align sequences using dynamic programming. • How to calculate and interpret p-values and E-values for pair alignments and database searches. Whole genome alignments Why? individual genome alignments, darker = higher scoring known gap in assembly sequence present but unalignable UCSC Browser track averaged conservation for 17 genomes alignment discontinuity (e.g. translocation break point) questionable alignment segment GQSQVGQGPPCPHHRCTTCCPDGCHFEPQVCMCDWESCCEEG GQSEVRQGPQCPYHKCIKCQPDGCHYEPTVCICREKPCDEKG How are genome-wide alignments made? • mouse and human genomes are each about 3x109 nucleotides. • how many calculations would a dynamic programming alignment have to make? • at a minimum - 3 integer additions and 3 inequality tests for each DP matrix position (by the way, there are other problems too, including assuming colinearity) Making large searches faster • Most common method is the BLAST search (Basic Local Alignment Search Tool). Only the initial step is substantially different from dynamic alignment. • Search sequence is broken into small words (usually 3 residues long for proteins). 20 * 20 * 20 = 8,000 words. These act as seeds for searches. • The target dataset is pre-indexed to indicate the positions in the database sequences that match each search word above some score threshold (using a global score matrix such as BLOSUM62). BLAST searches (cont.) • For example, the search sequence word “WVH” might score above threshold with these indexed sequences: Indexed word WVH WIH WVY WIY Score 23 22 17 16 • Target sequences around each indexed word hit are retrieved and the initial match is extended in both directions: ...VFEWVHLLP... WIY your sequence database (many sites) Schematic of indexed matches Result – instead of aligning these 3 amino acids to everything, they are aligned only with the tiny fraction of sequence regions that are good candidates for a valid alignment. (note- blast actually looks for two such matches close to each other) Extension and scoring ...QSVFEWVHLLPGA... ..WIY.. ...QSVFEWVHLLPGA... ..WIYQ.. ...QSVFEWVHLLPGA... ..WIYQK.. ...QSVFEWVHLLPGA... ..WIYQKA.. Match Score: Total Score: 16 16 -3 13 -2 11 -1 10 [mention gap variant] Extension termination • Extension is continued until the cumulative score drops below some threshold (usually 0). • This permits the match to cross a region of marginal similarity or frank mismatching (e.g. a small intron in tblastn) if it flanks a region of high similarity. • Extensions whose maximal cumulative score is above some threshold are kept for reporting to user. • For web interfaces, various formatting, links, and overviews are added and reported according to user settings (it is also fairly easy to download and run your own blast). Key to speed: word matching and prior indexing • Though gapped blast local alignment is slow (like dynamic programming), only a very small part of total search space is analyzed. • Because the positions of all database word matches are indexed and stored prior to the blast search, the relevant parts of search space are reached quickly. • Tradeoff is in accuracy and certainty – occasionally matches will be missed (when they are distant enough and dispersed enough that no local word pairs match well enough). Dynamic programming after BLAST matching genome A BLAST matches DP alignment region M x N manageable genome B Defining what a “tree” means rooted tree (all real trees are rooted): unrooted tree (used when the root isn’t known): sequences (leaves or tips) branch points root ancestral sequence branches time vaguely radiates out from somewhere near the center time …divergence time is the sum of (horizontal) branch lengths A tree has topology and distances Are these different trees? The number of tree topologies grows extremely fast 3 leaves 3 branches 1 internal node 1 topology (3 insertions) In general, an unrooted tree with N leaves has: 2N – 3 branches N – 2 internal nodes ~ O(N!) topologies 3 5 7 ... 2 N 5 4 leaves 5 branches 2 internal nodes 3 topologies (x3) (5 insertions) 5 leaves 7 branches 3 internal nodes 15 topologies (x5) (7 insertions) There are many rooted trees for each unrooted tree For each unrooted tree, there are 2N - 3 times as many rooted trees, where N is the number of leaves (# internal branches = 2N – 3). 20 leaves - 564,480,989,588,730,591,336,960,000,000 topologies