Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
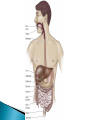
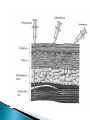
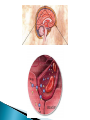
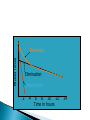
Pharmacokinetics: ◦ How drugs are handled by the body ◦ Overview followed by details!! Lets say you have a headache or a really really really bad reaction to poison ivy and you take some meds – This illustrates the basic processes in the branch of pharmacokinetics 1. the route of administration - how a drug is taken into the body 2. absorption and distribution - factors affecting its absorption and how it gets distributed to the brain 3. metabolism (detoxification or breakdown) how a drug is broken down or made into inactive forms 4. excretion – (elimination) ◦ how the drug is eliminated Knowing about pharmacokinetics tells us critical information about insight into the actions of a drug. Ex. benzodiazepenes ultra short acting, short acting, long acting lorazepam (Ativan) and triazolam (Halcion) – pharmacokinetics lorazepam – persists for at least 24 hr triazolam – 6 – 8 hours midazolam – 1 – 2 hrs Absorption – the process by which a drug enters the bloodstream chemically altered or without being The movement of a drug from its site of application into the blood oral injection ◦ iv, im, sc, intrathecal, intraperitoneal most common, sometimes referred to as po safe, self administered, economical BUT blood levels are often irregular (most complicated route of adm) liquid more readily absorbed than solids soluble and stable in stomach (not destroyed by stomach enzymes more acidic) enter intestine; penetrate lining of intestine, pass into bloodstream and reach site of action absorption favored if the drug is nonionized and more lipophilic ◦ chemicals in stomach must deal with: ◦ stomach acids ◦ digestive enzymes ◦ first pass metabolism through liver ◦ other items in stomach ex. tetracycline ◦ Convenient - can be self- administered, pain free, easy to take ◦ Absorption - takes place along the whole length of the GI tract ◦ Inexpensive - compared to most other parenteral routes disadvantages of oral administration: ◦ ◦ ◦ ◦ ◦ ◦ vomiting/stomach distress variability in dose effect too slow for emergencies unpleasant taste of some drugs unable to use in unconscious patient first pass metabolism First pass metabolism - term used for the hepatic metabolism of a drug when it is absorbed from the gut and delivered to the liver via the portal circulation. The greater the first-pass effect, the less the agent will reach the systemic circulation when the agent is administered orally first pass metabolism disadvantages of oral administration: ◦ ◦ ◦ ◦ vomiting stomach distress variability in dose first pass metabolism ex. buspirone (BuSpar) – antianxiety drug 5% reaches central circulation and is distributed to brain metabolism can be blocked by drinking grapefruit juice (suppresses CYPp450 enzyme) Hours J.Clin. Invest. 99:10, p.2545-53, 1997 Drugs that are destroyed by gastric juice or cause gastric irritation can be administered in a coating that prevents dissolution in acidic gastric contents (however may also preclude dissolution in intestines) Controlled – Release Preps - GI motility- speed of gastric emptying affects rate of absorption ◦ ex. migraine and analgesics vs metoclopramide Malabsorptive States ◦ GI diseases, ex. Crohn’s disease can affect absorption Food ◦ iron, milk alters tetracycline ◦ fats first pass metabolism chemicals delivered with a hypodermic needle; ◦ most commonly - injected into vein, muscle or under the upper layers of skin, in rodents also intraperitoneal cavity requirements for parenteral: must be soluble in solution (so it can be injected) ◦ ◦ ◦ ◦ ◦ ◦ Intravenous Intramuscular Subcutaneous Intracranial Epidural Intraperitoneal absorption more rapid than SC ◦ less chance of irritation; ways to speed up or slow down absorption depot injections - extremely rapid rate of absorption adv: useful when you need rapid response or for irritating substances Disadv: rapid rate of absorption contingent on blood flow SO ◦ IV, intraperitoneal, IM, SC increasing or decreasing blood flow affects drug absorption Drugs leave bloodstream and are exchanged between blood capillaries and body tissues bolus or depot shots related - drugs that accumulate in fat ◦ ex. THC nasal, oral, buccal medications include: nitroglycerine, fentanyl –(1998) , nicotine gum, lozenges, buprenorphine cocaine – snuff, cigars ◦ Advantages: rapid absorption avoid first-pass effect ◦ Disadvantages: inconvenient small doses unpleasant taste of some drugs 1990’s – several medications incorporated into transdermal patches: ◦ estrogen, nicotine, fentanyl, nitroglycerin, scopolamine controlled slow release for extended periods of time usually suppository form for unconscious, vomiting or unable to swallow disadv: not very well regulated dose; irritation (yikes) not really used for psychotropics Route for administration -Time until effect intravenous 30-60 seconds inhalation 2-3 minutes sublingual 3-5 minutes intramuscular 10-20 minutes subcutaneous 15-30 minutes rectal 5-30 minutes ingestion 30-90 minutes transdermal (topical) variable (minutes to hours) The rate at which a drug reaches it site of action depends on: ◦ Absorption - involves the passage of the drug from its site of administration into the blood ◦ Distribution - involves the delivery of the drug to the tissues Factors which influence the rate of absorption ◦ ◦ ◦ ◦ ◦ ◦ routes of administration dosage forms the physicochemical properties of the drug protein binding circulation at the site of absorption concentration of the drug drugs are distributed throughout body by blood very little at site of action at any one time role of passive diffusion, concentration gradient Mostly a passive process ◦ from higher conc to lower (in blood) Concentration Gradient Drug goes from higher concentration to lower concentration [DRUG] receptors ≈ [DRUG] circulation Mostly a passive process ◦ from higher conc to lower (in blood) Binding to plasma proteins ◦ results in a store of bound drug in plasma examples 95-99% - chlorpromazine, diazepam, imipramine 90 - 95% - valproate, propanolol, phenytoin Renal insufficiency last trimester of pregnancy drug interactions (other drugs that bind to proteins) diseases Blood brain barrier◦ layer of thickly packed epithelial cells and astrocytes that restrict access of many toxins/drugs to the brain Lipid solubility – how soluble the drug is in fats ◦ cell membranes are lipid bilayers ◦ similar characteristics allow drugs to cross brain as to cross into cells Lipid solubility Size of molecule Ionization – whether the degree has a charge (+ or -) pKa – the pH at which ½ of the molecules are ionized most drugs are either weakly basic or weakly acidic Basic drugs are highly ionized in acidic environment Acidic drugs are highly ionized in basic environment pKa – the pH at which ½ of the molecules are ionized the closer the pKa of the drug is to the local tissue pH, the more unionized the drug is. ex. morphine – pKa of 8 stomach ~ pH ~ 3 caffeine – pH .5 ◦ Distribution half-life: the amount of time it takes for half of the drug to be distributed throughout the body ◦ Therapeutic level: the minimum amount of the distributed drug necessary for the main effect. Until this time, drug movement has been mostly passive from regions of higher concentration to lower concentration. Elimination of drugs usually requires more of an active process (except gaseous drugs). 1. Biotransformation (metabolism) chemical transformation of a drug into a different compound in the body (metabolite) Most biotransformation takes place in the liver 2. Excretion - removal of drug to outside world ***Drug elimination may be by both or either of these mechanisms role of liver ◦ most significant organ in biotransformation role of liver ◦ most significant organ in biotransformation ◦ largest organ in body ◦ serves many functions transforms molecules via enzymes 1. deactivating the molecule 2. ionize the molecule 3. make it less lipid soluble ** product of biotransformation is called a metabolite Cytochrome p450 enzyme family located primarily in hepatocytes important for metabolism of alcohol, tranquilizers, barbiturates, antianxiety drugs, estrogens, androgens, PCBs and other agents oxidative metabolism – makes drugs more water soluble (so more easily excreted) CYP enzymes ◦ enzyme induction liver produces extra enzyme to break down drug with continued exposure CYP enzymes ◦ enzyme induction liver produces extra enzyme to break down drug with continued exposure Genetics CYP enzymes - ◦ enzyme induction liver produces extra enzyme to break down drug with continued exposure Genetics Liver disease cirrhotic liver In some cases, biotransformation can be to another psychoactive compound ex. benzodiazepenes diazepam nordiazepam oxazepam all drugs not in gaseous state need to use fluid routes of excretion ◦ fluid routes include -sweat, tears, saliva, mucous, urine, bile, human milk ◦ amount of drug excreted in each of these fluids is in direct proportion to amount of fluid excreted SO……. numerous functions – ◦ filters out metabolic products numerous functions – main function – maintain correct balance between water and salt in body fluids ◦ filters out metabolic products ◦ blood continuously flowing through kidneys factors that influence a substance not being resorbed not lipid soluble ionized dialysis – absorption distribution and excretion do not occur independently first pass metabolism blood brain 1. Body weight - smaller size • concentration of drug based on body fluid 2. Sex differences 3. Age 4. Interspecies differences rabbits – belladonna (deadly nightshade) 5. Intraspieces differences 6. Disease states 7. Nutrition 8. Biorhythm Blood level Resultant Elimination Distribution 2 4 6 8 10 12 Time in hours 14 half-life - time takes for the blood concentration to fall to half its initial value after a single dose ½ life tells us critical information about how long the action of a drug will last How long would it take for a drug to reach 12.5% remaining in blood if its ½ life is 2 hours? How long would it take for a drug to reach 12.5% remaining in blood if its ½ life is 100 hours?