Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Specialty drugs in the United States wikipedia , lookup
Compounding wikipedia , lookup
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Plateau principle wikipedia , lookup
Drug design wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Psychopharmacology wikipedia , lookup
Drug discovery wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription costs wikipedia , lookup
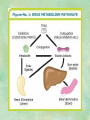
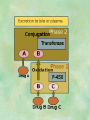
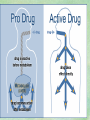
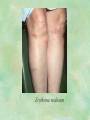
Clinical Pharmacokinetics. Clinical Pharmacodynamics. Drugs’ Interaction. Adverse Effects of Drugs DRUG NAMES The generic name often indicates the drug group (eg, drugs with generic names ending in “cillin” are penicillins). The trade name is designated and patented by the manufacturer. For example, amoxicillin is manufactured by several pharmaceutical companies, some of which assign a specific trade name (eg, Amoxil, Trimox) and several of which use only the generic name. Amoxicillin Metronidazole Ranitidin . ENTRY AND MOVEMENT OF DRUG MOLECULES THROUGH THE BODY TO SITES OF ACTION, METABOLISM, AND EXCRETION ABSORPTION is the process that occurs from the time a drug enters the body to the time it enters the bloodstream to be circulated. Onset of drug action is largely determined by the rate of absorption; intensity is determined by the extent of absorption. Dosage form is a major determinant of a drug’s bioavailability (the portion of a dose that reaches the systemic circulation and is available to act on body cells). An intravenous drug is virtually 100% bioavailable; an oral drug is virtually always less than 100% bioavailable because some is not absorbed from the GI tract and some goes to the liver and is partially metabolized before reaching the systemic circulation. Most oral drugs must be swallowed, dissolved in gastric fluid, and delivered to the small intestine (which has a large surface area for absorption of nutrients and drugs) before they are absorbed. Liquid medications are absorbed faster than tablets or capsules because they need not be dissolved. Sustained blood levels The size and frequency of dosing is determined by the pharmacodynamic and pharmacokinetic properties of the drug. The slower the rate of absorption, the less the blood concentrations fluctuate within a dosing interval. This enables higher doses to be given less frequently. For drugs with relatively short half-lives, the use of extendedrelease products may maintain therapeutic concentrations over prolonged periods Drug transport pathways. Drug molecules cross cell membranes to move into and out of body cells by directly penetrating the lipid layer, diffusing through open or gated channels, or attaching to carrier proteins. Plasma proteins, mainly albumin (A), act as carriers for drug molecules (D). Bound drug (A–D) stays in bloodstream and is pharmacologically inactive. Free drug (D) can leave the bloodstream and act on body cells. Half-life of a drug Elimination of a hypothetical drug with a half-life of 5 hours. The drug concentration decreases by 50% every 5 hours (i.e., T1/2 5 hours). The slope of the line is the elimination rate (Ke). THE LIVER IS THE PRINCIPAL ORGAN OF DRUG METABOLISM. Other tissues that display considerable activity include the gastrointestinal tract, the lungs, the skin, and the kidneys. Following oral administration, many drugs (eg, isoproterenol, meperidine, pentazocine, morphine) are absorbed intact from the small intestine and transported first via the portal system to the liver, where they undergo extensive metabolism. This process has been called a first-pass effect. Pharmacodynamic Variables Clearance is the single most important factor determining drug concentrations. Clearance is readily estimated from the dosing rate and mean steady-state concentration. Blood samples should be appropriately timed to estimate steady-state concentration. Вioavailability is defined as the fraction of a given drug dose that reaches the circulation in unchanged form and becomes available for systemic distribution. The larger the presystemic elimination, the smaller is the bioavailability of an orally administered drug. Сеll membrane contains receptors for physiologic substances such as hormones (H) and neurotransmitters (NT). These substances stimulate or inhibit cellular function. Drug molecules (Da and Db) also interact with receptors to stimulate or inhibit cellular function Presystemic elimination Factors that influence on drug metabolism Factor Reaction type Age (newborns, Decreasing of metabolism speed children, elderly) Pregnancy Increasing of metabolism speed Genetic factor Various reactions Liver pathology Decreasing of excreation speed of drugs, depending on their kinetics, type and stage of liver disease, increasing of bioavailability and decreasing of excretion speed of orally administered drugs with high hepatic clearence GI pathology Changes in metabolism in GI epithelium Nutrition character Increasing of metabolism speed of certain drugs in case of diet with dominance of proteins and carbohydrates Decreasing of metabolism speed in case of heavy digestive disorders linked with starvation (total or protein) Environment Alcohol — one time consumption — chronic consumption Smoking Way of excretion Increasing of metabolism speed if in contact with chlorine insecticides Depressing of enzymes that metabolise drugs Induction of enzyme system Increasing of metabolism of certain drugs (i.e. theophyllin) Metabolism in liver before entering system circulation (first going-through effect) after peroral administration of drugs Circade changes in drugs metabolism Time of introduction of drugs Interaction of Stimulation drugs reaction and depression of enzyme Client-Related Variables Age Body Weight Genetic and Ethnic Characteristics Gender (еxcept during pregnancy and lactation, gender has been considered a minor influence on drug action). Pathologic Conditions Psychological Considerations TOLERANCE AND CROSS-TOLERANCE Drug tolerance occurs when the body becomes accustomed to a particular drug over time so that larger doses must be given to produce the same effects. Tolerance may be acquired to the pharmacologic action of many drugs, especially opioid analgesics, alcohol, and other CNS depressants. Tolerance to pharmacologically related drugs is called cross-tolerance. For example, a person who regularly drinks large amounts of alcohol becomes able to ingest even larger amounts before becoming intoxicated—this is tolerance to alcohol. If the person is then given sedative-type drugs or a general anesthetic, larger-than-usual doses are required to produce a pharmacologic effect—this is cross-tolerance. Tolerance and cross-tolerance are usually attributed to activation of drug-metabolizing enzymes in the liver, which accelerates drug metabolism and excretion. They also are attributed to decreased sensitivity or numbers of receptor sites. 1. Drug-drug interaction 2. Drug-food interaction 1. Drug-others(herbs, supplements and DNA) Fixed drug eruptions Erythema nodosum THANK YOU