Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Polysubstance dependence wikipedia , lookup
Orphan drug wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Compounding wikipedia , lookup
Neuropharmacology wikipedia , lookup
Pharmacognosy wikipedia , lookup
Theralizumab wikipedia , lookup
Pharmaceutical industry wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Prescription costs wikipedia , lookup
Prescription drug prices in the United States wikipedia , lookup
Drug design wikipedia , lookup
Drug discovery wikipedia , lookup
Drug interaction wikipedia , lookup
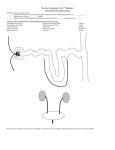
Since drugs can bind with proteins their distribution volume in the body can be affected Drug (Cu) + Protein (Cp) ↔ Drug-Protein (Cb) Total drug concentration C = Cu + Cb At equilibrium then K = Cb / (Cu Cp), but Cp is basically constant So we let K’ = K Cp = Cb/Cu The fraction unbound is defined as fu = Cu / C = Cu / (Cu + Cb) and we can Use K’ to eliminate Cb and hence we obtain that fu = 1 / (1+ K’) and then Cb = Cu (1 – fu) / fu and C = Cu / fu important to remember that only unbound drug is available to move around in the body Acidic drugs Basic drugs Neutral drugs testosterone The volume into which a drug distributes within the body is called the (apparent) distribution volume given by V or Vapp Since usually it is the plasma concentration that we know, ie. C, then the volume of distribution is really just a “volume” when multiplied by C that gives the total amount of drug in the body at a particular time Volume of Distributi on Amount of drug in body Plasma drug concentration Apparent distribution volume linked to fluid volumes as well as the effect of drug binding to proteins, so a drug that binds strongly with plasma proteins will approach a distribution volume of 3 liters, on the other hand a drug that binds tightly with extravascular proteins will result in a very low plasma concentration and to account for the mass of drug that means the value of V may greatly exceed the physical volumes of fluid actually in the body, in the extreme could be 7000 L ! V = apparent distribution volume Vp = plasma volume, 3 L/70kg VE = extracellular fluid volume less plasma volume, 12 L/70kg VR = remainder of the fluid volume in the body fU = fraction unbound in plasma, Cu/C fUT = fraction unbound in R RE/I = ratio of total drug binding sites in the fluids outside of plasma to those within the plasma, ~ 1.4 Oie – Tozer Equation VE VR f U V VP 1 R E / I f U VP R E / I f UT VP Using typical values for Vp and VE and R/EI we get this equation fU V 7 8 f U VR f UT Some special cases: 1. If drug only goes to extracellular spaces and cannot enter the cells, then VR = 0 and V = 7 + 8 fu and V has its smallest value and depends only on the fraction unbound in the plasma, so if drug is totally unbound (fu = 1) then V = 15 L, the extracellular fluid volume; if it is completely bound (fu = 0) then the distribution volume cannot be less than 7 L regardless of how tightly bound to albumin 2. Intracellular water (VR) is about 25-27 L so if the drug enters the cells but is not significantly bound (fUT =1 and fU = 1), then the volume of distribution is that of the total body water, ie. V = 40-42L, example would be alcohol 3. Note that as fUT 0, then V gets really big ADMET – absorption, distribution, metabolism, elimination, toxicity Common routes for drug metabolism include oxidation, reduction, hydrolysis, and conjugation. Can have many simultaneous pathways Primary site for drug metabolism is the liver and sometimes this is the only place metabolism occurs; other sites include the kidneys, lungs, blood, and GI wall Metabolism is good since it limits the time of drug action, may produce the active form of the drug, metabolites may also be active drugs as well, can even be a bioactivation making them more active or toxic than the parent compound (aka prodrug) Metabolism is the result of enzymatic reactions that occur within the cells and the rate of metabolism can be described by Michaelis-Menten kinetics Vmax C rmetabolic k metabolic C Km C Usually C << Km so the kinetics are just 1st order in drug concentration Basic functional unit of the kidney is the Nephron About 1 million per kidney Blood flow to kidneys is about 1100ml/min, and the glomerulus produces (GFR) about 120ml/min of plasma filtrate, fortunately most of the water is reabsorbed producing a urine flow of about 1-2ml/min Unbound drug will be filtered from the blood flowing thru the glomerulus GFR is the rate of filtration of plasma across the fenestrated capillaries of the glomerulus due to hydraulic and osmotic pressure differences, so at the glomerulus unbound drug is removed at the rate given by: Drug removal Rate = GFR x Cu = fu x GFR x C Renal Clearance (CLrenal) = Drug removal rate C Since clearance by definition is that flowrate of the fluid that is totally cleared of the solute CLrenal = fu x GFR So if the drug is only filtered out at the glomerulus, and all of the filtered drug is excreted into the urine (not secreted or reabsorbed in the tubules), then the rate of drug excretion (refers to urine) is the same as the drug removal rate at the glomerulus, or: CLrenal = fu x GFR, and if the drug is unbound (fu = 1), then the renal clearance for the drug is the same as the GFR; for example, inulin is a sugar-like substance with a molecular weight of about 6000 that is used to determine GFR; in addition creatinine is also not secreted or reabsorbed by the tubules and is also unbound and is a product of endogenous protein degradation, its production rate (M) in the body is about 120 mg/min, so at steady state what is produced in the body has to be removed by the kidneys, so we can write that M = GFR x C = U x V or that GFR = U V / C, where U is the concentration in the urine and V is the urine flowrate, also we have that GFR = M/C, so everything else being equal, ie. M constant, then plasma creatinine is a direct measure of kidney function via the GFR as shown on the graph on the next slide Normal GFR is about 125 ml/min Failure need dialysis 5 – 11 mg/100ml severe Most drugs are secreted and reabsorbed so things are a bit more complicated in terms of the renal clearance Secretion may be inferred when the rate of excretion (CLrenal x C) exceeds the rate of drug filtration (fu x GFR x C), or stated in a different manner CLrenal > fu x GFR Tubular reabsorption would be apparent whenever the rate of drug excretion (CLrenal x C) is less than the rate of drug filtration (fu x GFR x C), or CLrenal < fu x GFR Vapparent dC CL renal C dt Which can be integrated from the IC of t=0, C=C0 to give: C( t ) C0 e ( CLrenal / Vapparent ) t C0 e k renal t C krenal is the rate constant CLrenal/Vapparent The drug removed by the kidneys then shows up the urine and we can write that at any time: CL renal C Q urine C urine or CL renal Q urine C urine C Plasma clearance (CLplasma) relates to all possible drug elimination pathways and would include the following: Metabolism CL i Kidneys Elimination rate constants ki Sweating Vapparent Bile i = renal, metabolic, sweat, bile, respiration, feces Respiration Feces k te k i i l Vapparent CL i i CL plasma Vapparent The total elimination rate constant C( t ) C 0 e k te t Biological half life is the time needed for the plasma drug concentration to decrease by ½. For first order processes, this time is related to the first order elimination rate constant by simply letting C/C0 equal 0.5. When solved for t1/2, the following result is obtained. t 1/ 2 0.693 0.693Vapparent k te CL plasma Intravenous Injection of a Drug C0 D C( t ) C 0 e k te t Vapparent AUC 0 0 C( t ) dt AUC 0 dM urine k renal Vapparent C dt M urine k renal C 0 Vapparent k te Measure of drug exposure C0 D D k te Vapparent k te CL plasma k renal C 0 Vapparent k t M urine ( t ) 1 e te k te k renal D AUC 0 CL renal k te Vapparent dC I 0 CL plasma C I 0 Vapparent k te C dt Io k t C( t ) 1 e te k te Vapparent C ss I0 I0 k te Vapparent CL plasma CSS = 873 cpm/ml Example 7.1, inulin infusion study in a laboratory rat i 0 8 number of data points time C time, minutes i inulin concentration, cpm/ml i 30 40 60 70 75 80 90 100 110 849 845 903 888 873 882 565 412 271 CSS 1 6 calculate the steady state inulin concentration 5 C i i0 CSS 873.333 cpm/ml perform linear regression on the data after the infusion stopped at 80 minutes assuming the starting concentration was equal to that of Css C CSS sets the 80 minute value to the average steady state value j 0 3 only have 4 data points after infusion stops including the Cs s value 5 t time j ( 5 j ) 80 y ln C j ( 5 j ) the time since the infusion stopped in minutes transform the concentration data krenal slope( t y ) krenal 0.038 1/min, the renal elimination rate constant C0 exp ( intercept ( t y ) ) r corr( t y ) C0 860.085 r 0.998 cpm/ml, the value of the starting inulin concentration after the infusion stops, compares quite well with the value of Css determined above from the data indicates that we have an excellent fit of the single compartment model that describes the elimination of inulin time 0 30 running time, minutes Cpred( time) C0 exp krenal time Inulin Elimination inulin concentration, cpm 3 110 exp y j Cpred( time) 800 600 400 200 0 10 20 t j time time, minutes 30 the predicted inulin levels after the infusion is stopped