Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
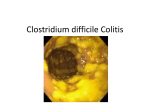
Review Article Indian J Med Res 131, April 2010, pp 487-499 Clinical spectrum & pathogenesis of Clostridium difficile associated diseases Chetana Vaishnavi Department of Gastroenterology, Postgraduate Institute of Medical Education & Research, Chandigarh, India Received April 23, 2009 Clostridium difficile is the major aetiological agent of antibiotic associated diarrhoea and colitis. The majority of hospitalized patients infected by C. difficile are asymptomatic carriers who serve as silent reservoirs for continued C. difficile contamination of the hospital environment. C. difficile associated disease (CDAD) is a serious condition with mortality up to 25 per cent in frail elderly people. C. difficile infection may present itself in several forms with both colonic and extracolonic manifestations. Several factors are involved in determining whether or not a patient develops C. difficile infection. These include factors related to the pathogen as well as the host. Transmission of C. difficile can be endogenous or exogenous. Colonization of the pathogen occurs when the gut flora gets disrupted due to various factors. The main virulence factors for CDAD are the two potent toxins: toxin A and toxin B which share 63 per cent of amino acid sequence homology and act on small guanosine triphosphate binding proteins. The emergence of the global hypervirulent C. difficile strain has been a cause of concern. Diagnosis of CDAD infection can be done by detection of C. difficile toxin in the stool specimen. Vancomycin is the drug of choice for severely ill patient, whereas metronidazole can be used for mild to moderately ill patients. Clinical spectrum, the factors precipitating CDAD, pathogenesis, diagnostic assay and treatment of the disease are reviewed. Key words Clinical spectrum - Clostridium difficile - diagnosis - pathogenesis - predisposing factors - treatment carriers who serve as silent reservoirs for continued C. difficile contamination of the hospital environment6. However C. difficile-associated disease (CDAD) is a serious condition with mortality up to 25 per cent in frail elderly people7. C. difficile is now recognized as the primary cause of hospital acquired colitis in patients who receive antibiotics, chemotherapeutics or other drugs that alter their normal flora. Introduction Clostridium difficile, a Gram-positive spore bearing anaerobic bacteria is the major aetiological agent of diarrhoea and colitis associated with antibiotics. Hall and O’Toole1 originally identified the organism as a component of normal colonic flora of newborn infants. C. difficile is commonly present in the stools of 5 per cent of healthy adults usually in low numbers2 and in about 15-70 per cent of infants3-5. The majority of hospitalized patients infected by C. difficile are asymptomatic When C. difficile was first discovered, infection by the organism was regarded primarily as an outcome of 487 488 INDIAN J MED RES, April 2010 antibiotic intake and not as a life threatening disease. During the recent outbreaks of CDAD in at least 12 hospitals in the entire Estrie region in Quebec, a threefold rise in the incidence of CDAD, and a higher number of cases involving toxic megacolon, colectomy or death have been reported8. The mutant hypervirulent strain was typed as NAP1/BI/027 (North American PFGE type I/restriction endonuclease analysis BI/ribotype 027)9 and was found to produce greater than 16 times toxin A and 23 times toxin B in addition to the binary toxin10. McDonald et al9 found NAP1/BI/027 strain in eight institutions, in six different States in United States and it represented more than 50 per cent of the isolates from five institutions. This global epidemic strain has also been reported to cause outbreaks in parts of continental Europe11, Great Britain, The Netherlands and Belgium12 with increased morbidity and mortality. The risk for CDAD has increased not only by usual factors, as pseudomembranous colitis (PMC), toxic megacolon and perforations in C. difficile was rare before 2002, but their incidence increased dramatically after that, particularly due to emergence of fluroquinolone resistant strains13. C. difficile is also being reported more frequently even from non hospital-based settings, such as from the community14. Domestic as well as wild animals are probably transmitting this as the same ribotypes found in them were found to be associated with human infection. In this review the clinical spectrum, the factors precipitating CDAD, pathogenesis, diagnostic assay and treatment of the disease are discussed. Clinical spectrum C. difficile, like virtually all bacterial enteric pathogens, causes a spectrum of clinical conditions15 with both colonic and extracolonic manifestations. The different manifestations are detailed below: (a) Colonic manifestation (i) Asymptomatic carriage: Colonization with C. difficile is the presence of the organism in a person with no clinical symptoms like diarrhoea. Asymptomatic carriage of C. difficile is quite common in hospitalized patients. Epidemiologic studies have reported that 10-16 per cent of hospital inpatients in high-risk units become carriers after receiving antibiotics16. Symptomatic disease is less often seen in carriers, despite the fact that most of the C. difficile isolates are toxin producing17. Riggs et al18 suggested that asymptomatic carriers of epidemic and non epidemic C. difficile isolates have the potential to contribute significantly to disease transmission in long-term care facilities. Asymptomatic carriage can be predicted by taking into account certain clinical factors such as recent antibiotic exposure or previous occurrence of CDAD. Patients with C. difficile colonization and a serum IgG response to C. difficile enterotoxin usually become asymptomatic carriers. (ii) C. difficile diarrhoea: Usually mild to moderate diarrhoea, sometimes accompanied by lower abdominal cramps is seen with C. difficile infection. Symptoms usually begin during or shortly after antibiotic therapy. Occasionally these may be delayed for several weeks. C. difficile toxins can be usually detected from faecal specimens, even though endoscopic and histologic features may be normal in patients with mild disease. The diarrhoea resolves with the stoppage of antibiotics19. (iii) C. difficile colitis: The most common clinical manifestation of C. difficile infection is colitis without pseudomembrane formation. This is a more serious illness than benign or simple antibiotic-associated diarrhoea and presents as malaise, abdominal pain, nausea, anorexia, and watery diarrhoea. Abdominal pain and cramps in some patients are relieved by passage of stool19. Sometimes dehydration and a low-grade fever with a systemic polymorphonuclear leukocytosis may occur. Levels of lactoferrin released from the secondary granules of intestinal leukocytes, as well as other inflammatory markers rise significantly in patients having advanced CDAD compared to patients with a milder form of the disease20. Faecal lactoferrin assay performed simultaneously with the C. difficile toxin assay can help rule out asymptomatic carriage of C. difficile21. A nonspecific diffuse or patchy erythematous colitis without pseudomembrane may be seen under sigmoidoscopy. (iv) Pseudomembranous colitis (PMC): Finney22 was the first to describe PMC as a post-operative complication of gastroenterostomy. PMC is the classic manifestation of full-blown C. difficile colitis and is accompanied by similar, but often more severe symptoms than those observed in colitis. The classic pseudomembranes, which are raised yellow plaques ranging from 2-10 mm in diameter scattered over the colorectal mucosa are best revealed by sigmoidoscopic examination. White blood cell counts of 20,000 or greater and hypoalbuminaemia of 3.0 g/dl or lower may be observed in severely ill patients23. Most patients with PMC have involvement of the rectosigmoid area. However, colonoscopy is required because as many as one third of patients Vaishnavi: C. difficile pathogenesis have pseudomembranes limited to the more proximal colon24. In patients with hypoalbuminaemia or acquired immunodeficiency syndrome a neutrocytic ascites with low serum to ascites albumin gradient may occur25, 26 with ascites being the only presenting manifestation of PMC. (v) Fulminant colitis: C. difficile infections may present as fulminant colitis in approximately 3 per cent of patients and account for most of the serious complications including perforation, prolonged ileus, megacolon and death27. Patients with fulminant colitis complain of severe lower quadrant or even diffuse abdominal pain, diarrhoea, and distension and some of them may exhibit high fever, chills and marked leukocytosis. Diarrhoea may occur as a usual symptom, but may be minimal in patients with ileus as a consequence of which secretions accumulate in the dilated, atonic colon. Severe protein-losing enteropathy may result in hypoalbuminaemia. A patient with toxic megacolon has a dilated colon with signs and symptoms of severe toxicity that include fever, chills, dehydration and high white blood count. On plain abdominal radiograph the patient may also have dilated small intestine with air-fluid levels mimicking an intestinal obstruction or ischaemia or pseudoobstruction28. The risk of perforation and precipitation of megacolon28 deters a barium enema examination. However, computed tomographic scan of the abdomen is most valuable in severe cases and those localized to the proximal colon may reveal colonic distension, thickening, pericolonic inflammation, or free air29, 30. Signs and symptoms of bowel perforation may be present in some patients with fulminant C. difficile infection. Further morbidity and mortality can be prevented in patients with fulminant C. difficile colitis by aggressive diagnostic and therapeutic interventions27. Though the risks of perforation are generally uncommon, limited flexible sigmoidoscopy or colonoscopy may be performed at the bedside31. C. difficile infection has also been reported to be involved in the exacerbation of ulcerative colitis32. Hookman & Barkin33 observed that fulminant colitis is reported more frequently during outbreaks of C. difficile in patients with inflammatory bowel disease (IBD) and carries higher mortality than those without underlying IBD. (vi) Recurrent CDAD: Recurrent CDAD is a difficult clinical problem due to repeated recurrences of the manifestation. The pathophysiology is not quite clear and may be due to persistently altered faecal flora. Repeat 489 antibiotics may subsequently be unable to suppress C. difficile overgrowth. Alternatively, impaired immune response may also be responsible. It has been estimated that approximately 15-20 per cent of patients treated for CDAD, relapse following successful therapy34. This condition is manifested by the sudden re-appearance of diarrhoea and other symptoms usually within a week of stopping treatment with vancomycin or metronidazole. Patients who relapse once are at greater risk of further relapses. McFarland et al35 reported a relapse rate of as high as 65 per cent in patients who had suffered two or more previous relapses. Relapse is generally not related to antibiotic resistance because in some patients re-infection can occur with the same or different strain. The small bowel and the appendix may also act as reservoirs of C. difficile spores that enter the colon and result in relapse36. The basis for variability in response to C. difficile infection is not entirely clear, but host factors appear to be more important than bacterial virulence factors37. It was suggested that there might be an association between the C. difficile strains, production of toxins, and clinical manifestation of the infection38. However, a few studies where C. difficile was not an epidemic strain have shown that there was no difference between strains causing symptomatic cases and asymptomatic carriage39,40. (b) Extracolonic features Recent literature mentions that CDAD is no longer limited to the colon. C. difficile may infrequently cause disease in a variety of other organ systems and except for bowel involvement and reactive arthritis most of the cases do not appear to be strongly related to previous antibiotic exposure though they are preceded by specific or nonspecific gastrointestinal (GI) disease. Some of the features of extracolonic diseases can be summed up as follows: (i) Small bowel: Jacobs et al41 reviewed literature on extracolonic manifestation of C. difficile and revealed that small intestinal C. difficile infections seem to be increasing in incidence. Small bowel CDAD with formation of pseudomembranes on ileal mucosa may occur when previous surgery on it has been carried out and is associated with a high mortality rate. Testore et al42 examined jejunal specimens from 100 patients who died without any immediate history of GI symptoms and mucosal cultures in 3 cases treated with antibiotics were positive for C. difficile. Boland and Thomson43 presented a case of C. difficile enteritis in a 42 yr old 490 INDIAN J MED RES, April 2010 patient with ileal pouch-anal anastomosis and flexible endoscopy revealed copious amounts of mucus with adherent pseudomembranes throughout pouch and distal small bowel. Navaneethan & Giannella44 reported that small bowel involvement is more frequently reported in IBD patients who have undergone total colectomy or in patients with ileal-anal anastomosis. The increase in the number of these patients may actually reflect an increase in the rising incidence of C. difficile infection in general or increasing virulence of the infecting organism. (ii) Bacteraemia: Like with other colonic bacteria, C. difficile is also known to cause bacteraemia with about 20 per cent mortality45. Bacteraemia due to C. difficile has previously been described in 14 patients with underlying GI processes46. These authors also reported a unique case of monomicrobial C. difficile bacteraemia in a young woman with an underlying haematologic malignancy but without any GI symptoms. (iii) Reactive arthritis: C. difficile-related polyarticular kind of reactive arthritis may involve joints of the knee and wrist in about a 50 per cent of the cases47. Reactive arthritis begins an average of 11.3 days after the onset of diarrhoea and is a prolonged illness, taking an average of 68 days to resolve41. Ducroix-Roubertou et al48 reported a case of a monoarticular arthritis of the left knee following PMC in a 45 yr old man, 8 days after the onset of a C. difficile enterocolitis. (iv) Miscellaneous entities: Other extracolonic manifestations due to C. difficile include cellulitis, necrotizing fascitis, osteomyelitis, prosthetic device infections, intra-abdominal abscess, empyema, localized skin infections, etc. Factors precipitating CDAD The following factors determine whether or not a patient develops a C. difficile infection: (a) General factors: An indepth review on established and potential risk factors for CDAD has recently been published49. Briefly these include (i) long duration or multiple antibiotic intake; (ii) the nature of the faecal flora; (iii) the size of the C. difficile population; (iv) production of the requisite cytotoxins; (v) the presence of other organisms that affect toxin expression or activity, and (vi) the presence of host risk factors, including advanced age, presence of a nasogastric tube37, receiving anti-ulcer medication50, severe underlying illness, prolonged hospital stay, use of enemas, GI stimulants and stool softeners. Johnson & Gerding51 derived a model of pathogenesis for infection with C. difficile. They hypothesized that a patient admitted to hospital was at negligible risk for infection until an antimicrobial agent was administered. If during or after treatment such a patient gets subsequently exposed to C. difficile, he/she either develops CDAD or becomes colonized without diarrhoea or potentially does not get infected at all. But once established as an asymptomatic carrier, a patient is at decreased risk for CDAD. Patients are at continuous risk of exposure to C. difficile during the period of hospitalization and become vulnerable to infection after they have been exposed to antimicrobials. The two most important components essential for CDAD is exposure to antimicrobials followed by exposure to C. difficile and majority of the patients do not get ill with these till the third additional factor related to host immunity, virulence of infecting C. difficile strain or to type and timing of exposure come into play. (b) Specific factors: Immunosuppressive drugs have also been reported to be associated with the development of CDAD52,53. Faulty immune response to C. difficile toxins has been quoted as one of the major host factors predisposing patients to the development of symptomatic CDAD54,55. Patients receiving immunosuppressive drugs are debilitated and therefore are unable to mount an effective IgG antibody response against C. difficile toxin A thereby increasing the risk for CDAD15. C. difficile colonization is more frequent in intensive care and oncology units, where broad spectrum antibiotics and immunosuppression are wide spread56. Administration of tacrolimus, an immunosuppressive agent resulted in the development of CDAD57. Ulcerative colitis patients unresponsive to corticosteroids58 may require long time immunosuppressive treatment, which may result in multiple infections, inclusive of C. difficile59. Five leukemic patients treated with immunosuppressives, died from secondary complications of PMC60. As the use of immunosuppressives increase, the incidence of CDAD will also rise and may account for as many as 20 per cent of patients of CDAD without prior use of antibiotics61. Gastric acid suppressive use due to raised pH of stomach results in increased risk of CDAD62,63. Proton pump inhibitors (PPI) may thus contribute to the pathogenesis of CDAD, due to increased survival of spores. PPI use was a significant risk Vaishnavi: C. difficile pathogenesis factor for development of CDAD in a retrospective case control study63. However, Pepin et al8 reported that elevated risk of CDAD with PPI occurred only in univariate analysis but not so after adjustment for co-morbidities on multivariate analysis. Kaur et al50 found that BALB/c mice treated with PPI had a higher experimental colonization with C. difficile, enhanced myeloperoxidase activity as well as greater level of epithelial damage, oedema and neutrophilic infiltrates in the colon as compared to control untreated animals. Jayatilaka et al64 in a five year study period found that PPI usage correlated exactly with the overall annual increased CDAD incidence and believed that the widespread prescription of PPI could be responsible. PPI therapy was reported as an independent and the only risk factor associated with reported as an increased length of hospital stay in CDAD patients65. Thus the risk of CDAD in hospitalized patients receiving antibiotics may be compounded by exposure to PPI therapy. Administration of cancer chemotherapeutic agents possessing antibacterial properties may also result in sufficient disturbance of the intestinal microflora to allow colonization with C. difficile. Emoto et al66 reported severe CDAD in 6.1 per cent of patients receiving cisplatin based combination chemotherapy for ovarian malignancies. Resnik & Lefevre67 described development of fulminant C. difficile colitis in a 66 yr old patient with ovarian cancer who received paclitaxel and carboplatin chemotherapy. Kumar et al68 reported that 32.7 per cent patients treated with methotrexate or mesalamine for psoriasis were positive for C. difficile toxins. Exposure to corticosteroids was also significantly associated with an increased risk of CDAD relapse69. Thus the combination of the environmental presence of C. difficile in health care settings and the number of people receiving antibiotics, immunosuppressives, PPI or cancer therapeutics in these settings can result in frequent outbreaks. (c) Factors responsible for recent increase in CDAD in the West: Zerey et al70 demonstrated that the incidence of C. difficile infection was increasing in surgical patients in United States and was most prevalent after emergency operations particularly among patients having intestinal tract resections. The increased incidence of nosocomial CDAD in the West with marked increase in severity of cases requiring colectomy or ending in death was attributed to liberal use of fluroquinolones and cephalosporins71. The NAP1/BI/027 strain poses a great risk as it is also 491 found to be resistant to fluroquinolone. In 2007, severe cases of CDAD with this epidemic strain was detected in Germany for the first time and was strongly associated with receipt of cephalosporins and fluroquinolones in the 3 month before onset of symptoms72. Pathogenetic mechanisms Transmission of C. difficile occurs both endogenously as in the carrier state and exogenously through nosocomial source 73. When an individual is exposed to C. difficile or its spores, an initial disruption of the normal colonic bacterial flora occurs resulting in colonization of the organism74 through surface proteins. Damage to enterocytes due to C. difficile toxins occur as soon as C. difficile colonizes the intestine resulting in cytoskeletal changes and the release of fluids and inflammatory products75. Pathogenic C. difficile produces two high molecular weight potent toxins - A and B - which bind to specific receptors on the luminal aspect of the colonic epithelium76. The role of surface proteins and toxins of C. difficile in the pathogenesis of CDAD are detailed below: (a) Surface proteins of C. difficile: Colonization process is thought to be a necessary preliminary step in the course of C. difficile infection. Calabi et al77 investigated tissue binding of C. difficile surface layer proteins (SLPs) which are the predominant outer surface components encoded by slpA gene. They demonstrated that SLPs play a role both in the initial colonization of the gut by C. difficile and in the subsequent inflammatory reaction. Different adhesins implicated in the colonization process of C. difficile are (i) flagella, composed of the flagellin Fli C and the flagellar cap protein Fli D, involved in cell and mucus attachment (ii) a cell-surface protein with adhesive properties, Cwp 66 (iii) a fibronectin-binding protein, Fbp68 and (iv) s-layer protein. These adhesins are able to induce an immune response, which could play a role in the defense mechanism of the host78. Janoir et al79 observed that cwp84, a surface protein exhibited proteolytic activity which could contribute to the degradation of the host tissue integrity and to dissemination of the infection. (b) Role of C. difficile toxins: The main virulence factors for CDAD are the two potent toxins - toxin A and toxin B that share 63 per cent of amino acid sequence homology. Both the toxins induce mucosal injury and colitis as seen by neutrophil infiltration, which is a prominent feature of CDAD34. 492 INDIAN J MED RES, April 2010 Toxin A is an enterotoxin that causes haemorrhage and fluid secretion in the intestines of rodents whereas toxin B is a cytotoxin detectable by its cytopathic effects in tissue culture2. However, both toxins affect the cytoskeletal features, even though their activity differs in potency. Toxins A and B have been shown by nucleotide sequencing to be located in close proximity to each other80 within a 19.6 kbp pathogenecity locus (PaLoc) encoded by two separate genes (tcdA and tcdB) on the same chromosome. C. difficile can be divided into 24 toxinotypes based on the changes in both toxin genes81. Some toxinotypes possess a third kind of toxin known as the binary toxin82 described elsewhere in this review. The toxins get transported into the cytoplasm where these act on small guanosine triphosphate binding proteins known as the Rho proteins. The Rho proteins are associated with actin polymerization, maintenance of the cytoskeletal architecture, and the cell movement83, 84 . A severe inflammatory reaction in the lamina propria with the formation of micro-ulcerations of the colonic mucosa that is covered by a pseudomembrane occurs due to the activity of the toxins85. (i) Toxin A - Toxin A is a 308-kDa lethal enterotoxin and minute quantities can stimulate fluid secretion in animal intestinal loops86 similar to cholera toxin though the mechanism of action is quite different87. Toxin A causes extensive damage to the epithelial lining of the intestine and accounts for nearly all of the GI symptoms. The villus tips of the epithelium are initially disrupted followed by damage to the brush border membrane. Katyal et al88 reported a disturbance in the intestinal brush border membrane enzymes in CDAD patients. Denuding of the mucosa eventually is accompanied by extensive neutrophil infiltration resulting in massive inflammation. The fluid response is partly an outcome of the damage to the intestinal epithelium. Toxin A also acts as a cytotoxin resulting in disruption of the tight junctions of the intestinal epithelium and might be an important mechanism of toxin A enterotoxicity89. Toxin A also elicits the production of various cytokines and neurokinins, the biological reactants, which are believed to be playing an important role in pathogenesis90. Purified toxin A has potent effect on human colonic epithelial cells as seen in vitro. Toxin A initially induces cell rounding which results in detachment of the cell from the basement membrane, followed by apoptosis. Toxin A also brings about a rapid loss of resident cells such as macrophages, T cells and eosinophils and induces changes in the shape of adherent polymorphonuclear leukocytes. At least two pathophysiologic pathways are involved in changes in the epithelial cell barrier via glycolysation of the Rho proteins. These are (i) disaggregation of actin microfilaments leading to epithelial cell destruction and opening of tight junctions, and (ii) early release of proinflammatory cytokines from intestinal epithelial cells probably via activation of mitogen-activated protein kinase91. The spherical cells become thin and rope like with rearrangement of F-actin cytoskeleton into aggregates92. Thus the toxins alter the actin cytoskeleton, cause epithelial cell damage and result in increased permeability of the tight junctions. A severe acute necro-inflammatory reaction is produced by toxin A in the intestine resulting in activation of mast cells, vascular endothelium, and immune cells91. (ii) Toxin B - Toxin B is also a very large and potent cytotoxin with 279 kDa molecular weight. It causes a number of non specific in vitro responses in mammalian cells. This includes disorganization of the actin filaments, loss of intracellular potassium, decrease in the level of protein and nucleic acid synthesis93. Under normal physiological conditions toxin B by itself cannot cause damage or a fluid response in intestinal loops94 probably due to its inability to bind to the specific carbohydrate receptors on the intestinal brush border membrane. After toxin A has bound to the receptor initiating the damage, toxin B joins in and gains access to the underlying tissue as supported by animal experimentations94. The cytotoxic activity of toxin B is similar to that of toxin A, but is 1000-fold more potent than the former. There is formation of neurite-like retraction fibers resulting in partial detachment of cells. Next the cell-spanning stress fibers disappear and the remainder of the actin filaments accumulates in the perinuclear space95,96. Both toxins disrupt the function of the Rho family of protein97. Decreased transepithelial resistance and increased flux of paracellular marker such as mannitol and raffinose98 indicate the disruption of the tight junction. (iii) Binary toxin - Another toxin, which is an iotalike toxin, was described from the C. difficile strain CD19699 and has been named binary toxin CDT. Binary toxin contains both toxins A and B and is a product of both toxin genes (cdtB for the binding component and cdtA for the enzymic component). It is located on the chromosome outside the PaLoc and is actin-specific Vaishnavi: C. difficile pathogenesis ADP-ribosyltransferase toxin that could be acting synergistically. It has not been found to be essential for eliciting C. difficile associated colitis. Binary toxin CDT is produced by most of C. difficile isolates with mutations in the tcdA and tcdB genes100 and up to 2 per cent A-B- strains of C. difficile are estimated to produce only binary toxin CDT101. Diagnosis by toxin assay: C. difficile toxins can be detected in the faecal samples by several methods as mentioned below: (i) Tissue culture: Tissue culture can detect as little as 1.0 pg of toxin B making it the most sensitive test available and has therefore been regarded as the gold standard in laboratory diagnosis of C. difficile toxin. Toxin identification can be confirmed with C. difficile antitoxin or antitoxin against C. sordellii, which produces the cross-reacting toxins102. Commercial tests recommend a final dilution of 1:40 to 1:50 of the stool sample103. Cell lines that may be used include Vero, Hep 2, Chinese hamster ovary, HeLa cells and MRC-5 lung fibroblasts. Many disadvantages accompany tissue culture technique because it is the least controlled test. Specimens may at times cause nonspecific cell rounding that is neutralized not only by the specific antitoxin but also by neutral serum. The addition of too much faecal material to the tissue culture well can cause false positive reactions. The maintenance of cell cultures is also very difficult. The procedure is cumbersome, expensive and time consuming and requires a well-developed laboratory. Sometimes a non specific cytopathic effect can also occur in a few of the cases due to a viral agent or another bacterial enterotoxin such as that of C. perfringens, rendering any interpretation difficult. False negatives can occur in stored samples due to several reasons such as (i) toxin degrading enzymes, (ii) delay in transportation of sample, (iii) medication of the patient, and because (iv) some cell lines are less sensitive than others to the cytopathic effect of the toxin. Infact, a negative cytotoxicity assay does not completely rule out C. difficile as the cause of diarrhoea. (ii) Counter immunoelectrophoresis: This method for detection of toxin is expensive, cumbersome, and lacks the required levels of sensitivity and specificity for satisfactory diagnostic test. (iii) Latex agglutination test: Commercially available latex agglutination test (LAT) is rapid, but is 493 unaffordable for routine use because of the high cost per test. Moreover commercially available LAT is known to detect a non toxic marker antigen for C. difficile and therefore frequently results in false positive reactions. Now a days commercial LAT is less frequently available and has been replaced by enzyme-immunoassays. (iv) Enzyme immunoassays: Enzyme-linked immunoassays (ELISA) are commercially available to detect either toxin A alone or both toxins A and B in stool specimens. ELISA has sensitivity and specificity ranges of 50 to 90 per cent and 70 to 95 per cent respectively. About 100 to 1000 pg of toxins must be present for the test to be positive. The advantage of using ELISA is because of the lesser time required for the test. But the high cost per single test may necessitate batching of samples. A high percentage of indeterminate readings may also occur. Most ELISA have a sensitivity of more than 80 per cent compared to that of tissue culture assay. However, ELISA that detect only toxin A may miss out on toxins from A-B+ isolates resulting in wrong interpretation. Thus, ELISA that detect both toxin A and B are recommended to detect these atypical isolates. Such tests will also take care of specimens containing low levels of toxin A and B. (v) Dot immunobinding assay: The test is carried out on the surface of individual membrane cassette employing the principle of enzyme immunoassay (EIA). Stool supernatant is made to pass through a filter onto the membrane and then made to react to mouse monoclonal antibody to C. difficile toxin. Appropriate enzyme conjugate and substrate are added to visualize the blue coloured dot. Sometimes the presence of excessive amount of debris can cause difficulty in interpretation of the results. (vi) Rapid membrane tests: These are lateral flow devices with coloured conjugates or flow through formats that require multistep processing. Such tests utilize peroxidase tagged antibodies and a wash step followed by the addition of a substrate. The sample preparation for these tests requires centrifugation or filtration. These tests have sensitivity in the range of 60 to 89 per cent. These tests are toxin A specific and therefore do not detect A- B+ isolates. (vii) Polymerase chain reaction: The polymerase chain reaction (PCR) technique is used to detect enterotoxin or toxin B gene in isolates or faeces and has sensitivity similar to cytotoxin testing. The advantage of PCR 494 INDIAN J MED RES, April 2010 is the rapidity of the test. But PCR needs appropriate infrastructure and technical expertise, and is timeconsuming. Most assays target only one of the two genes, potentially missing isolates carrying only one of them. Feces may contain PCR inhibitory components, which can cause difficulties in the assay. Moreover, PCR detects even minute number of C. difficile genome copies present even in healthy individuals thereby overemphasizing the aetiology. (viii) Immunochromatography assay: This technique is a single test EIA for detection of toxins A and B in faecal samples. It can be done within 20 min and without any requirement of pre-treatment. (ix) Loop mediated isothermal amplification: Loop mediated isothermal amplification (LAMP) is a rapid and simple method for detecting toxin B gene in stool samples as well as in isolates. Detection of tcdB by LAMP from overnight cultures in cooked meat medium could be an alternative method of diagnostic testing at clinical laboratories without special apparatus. Even though the technique is easier to perform it is not as sensitive as the PCR104. Therapy and management Withdrawal of the antibiotic therapy that precipitated the disease or at least changing antibiotic regimens results in early resolution of the diarrhoeal symptoms even in some cases of established PMC. Fluid replacement and electrolyte balance maintenance is important. About 25 per cent of patients respond within a few days to these simple measures105. In case of non response, they should be treated with specific antimicrobial therapy, which is crucial to prevent the progression of C. difficile pathogenesis. The drug of choice for seriously ill patients is oral vancomycin because it has no side effect and is not absorbed by the intestine. Diarrhoea generally resolves over an average of 5 days even though up to 50 per cent relapse rate may occur after vancomycin treatment106. Vancomycin administered in the form of capsules, help to camouflage its bitter taste. However, in seriously ill patients, oral suspensions help to achieve high concentrations in the colon more quickly. Vancomycin can also be administered by nasogastric tube, a long intestinal tube, by enema, or by direct instillation through a colostomy or ileostomy in patients too ill for oral therapy. Intravenous administration of vancomycin to patients unable to take oral medication can be done with the hope that some of the drug will reach the colonic lumen through the inflamed mucosal surface. Other desperate measures in patients who continue to do poorly are cecostomy or colectomy. Some patients develop a series of relapses, thereby extending the illness. Use of vancomycin in routine is discouraged because it is expensive and there is a risk of development of vancomycin-resistant enterococci. Oral metronidazole can be used in place of vancomycin and is favoured because it is less expensive. Randomized trials show excellent initial responses in approximately 95 per cent of patients treated with metronidazole107. But, the disadvantage of the drug is the near complete absorption such that the levels achieved in the colon are virtually nil. Resistance to metronidazole has been found in some isolates of C. difficile. Metronidazole therefore is used for patients with mild or moderate illness but should not be used for critically ill patients. Antibiotics that are active against C. difficile include ampicillin, bacitracin, fusidic acid and teicoplanin, which have been tried with little success. C. difficile also shows good in vitro susceptibility to various antimicrobials, such as rifaximin, ramoplanin and nitazoxanide and these could be used in future trials. Antimicrobial treatment also kills the normal bacterial flora thereby causing the disease to recur. Once the colon has been injured, it seems to be more susceptible to reinfection. Nelson108 suggested that in order to improve the patient’s clinical condition and prevent the spread of C. difficile infection to other patients if one does decide to treat, one should choose the antibiotic that brings both symptomatic and bacteriologic cure and that teicoplanin appears to be the best choice. Limited success can be achieved by administration of intravenous gamma globulin. Probiotics like Lactobacilli species or Saccharomyces boulardii can also be used to replace the pathogenic C. difficile flora. Even rectal instillation of fresh stool in saline from living related donors and rectal instillation of mixed broth cultures of stool flora have been tried to replenish the normal flora109. Binding agents like cholestyramine, Isabgol husk and tolevamer may also bind to administered drugs and delay elimination of C. difficile toxins2. Parenteral administration of C. difficile toxoid vaccine might protect high-risk individuals against CDAD by development of antibody response. Indian experience with CDAD The prevalence of C. difficile-associated colitis is global and the incidence varies considerably from place Vaishnavi: C. difficile pathogenesis to place. In India, the studies on C. difficile-associated diarrhoea have been limited, probably due to the lack of technology and the difficulty in culturing the pathogen. Available reports from India estimate a prevalence of 15-30 per cent of paediatric and adult patients taking antibiotics110-114. Gupta & Jadav115 reported 25.3 per cent isolation of C.difficile from diarrheal patients of all age groups. Niyogi et al116 reported C. difficile in 8.4 per cent and cytotoxin in 7 per cent of faecal samples in children between 0-14 yr of age. C. difficile was the only pathogen in 7.3 per cent of patients with acute diarrhoea whereas 3.1 per cent control children without diarrhoea harboured the organism. Niyogi et al117 isolated C. difficile in 11 per cent hospitalized patients with diarrhoea and 2.9 per cent non diarrhoeic controls; 87 per cent isolates produced cytotoxin even though the diarrhoeic patients had no history of antibiotic use. Bhattacharya et al118 investigated 233 patients with acute diarrhoea and isolated C. difficile as a sole pathogen from 7.3 per cent, of which, 82.4 per cent produced cytotoxin. In another study Niyogi119 reported that of the 43 C. difficile isolates, 100 per cent were inhibited by low concentrations of metronidazole, penicillin G, tetracycline and ampicillin, but were highly resistant to gentamycin, trimethoprim, sulphamethoxazole, nalidixic acid, cycloserine and cefotaxime. Kochhar et al120 demonstrated that infectious agents like C. difficile were responsible for some of the exacerbations in ulcerative colitis patients without any history of recent exposure to antimicrobial drugs or hospitalization. Vaishnavi et al112 reported 30 per cent positivity for C. difficile toxin in hospitalized patients of all age group receiving single to multiple antibiotics for various ailments, but only in 7 per cent of samples from patients not receiving antibiotics. When only adult population were investigated, the positivity for C. difficile toxin was 19.4 per cent in the antibiotic receiving hospitalized patients121. Kang et al122 reported that C. difficile-associated diarrhoea was more common in the post transplantation period in India than in developed countries. Gogate et al123 observed that C. difficile was an important pathogen for antibiotic associated diarrhea in children of age group 5-12 yr. Vaishnavi et al124 reported the association of C. perfringens with antibiotic associated diarrhoea either by itself or in synergy with C. difficile infection. Balamurugan et al125 reported overgrowth of C. difficile in the stool of Indian patients with ulcerative colitis compared to healthy controls using real time PCR. A decrease in the number of C. 495 difficile positive cases has been reported during a 5 year study period and attributed the reduction to stringent surveillance and an improved antibiotic policy adopted in the hospital126. Conclusions C. difficile associated disease is a growing nosocomial and public health problem. Hospitalized patients receiving antibiotics for their ailments are at great risk of acquiring CDAD. Clinical suspicion is more important than ever before because stool assays for diagnosing CDAD are not widely available. Wherever available it is fraught with inherent problems and therefore diagnosis may be missed or delayed. Several measures significantly reduce the incidence of CDAD. Infection control procedures that should be followed to prevent spread of the disease include environmental hygiene, washing hands with ordinary soap and water or using 0.03 per cent Triclosan and isolating patients with CDAD. Environmental cleaning should be done with phenolic disinfectant. Isolated patients should have equipment dedicated to them to reduce cross-contamination. Infection control plays a key role in controlling CDAD outbreaks. The spores of C. difficile can survive in the hospital environment for months, providing a reservoir for new infections. Active and aggressive surveillance activity is the key to reduce incidence. Monitoring should enable the development and implementation of policies and procedures that minimize the risk of this nosocomial pathogen. Preventing C. difficile infection offers a potentially significant improvement in patient’s outcomes, as well as a reduction in hospital costs and resource expenditures. References 1. Hall IC, O’Toole E. Intestinal flora in newborn infants with description of a new pathogenic anaerobe. Am J Dis Child 1935; 49 : 390-402. 2. Fekety R, Shah AB. Diagnosis and treatment of Clostridium difficile colitis. JAMA 1993; 269 : 71-5. 3. George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinnagawa, N et al. Identification of Clostridium difficile as a cause of pseudomembranous colitis. BMJ 1978; 1 : 695. 4. Larson HE, Price AB, Honour P, Borriello, SP. Clostridium difficile and the etiology of pseudomembranous colitis. Lancet 1978; 1 : 1063-6. 5. Falson E, Kaijder B, Nehls L, Nygreu B, Svedham A. Clostridium difficile in relation to enteric bacterial pathogens. J Clin Microbiol 1980; 12 : 297-300. 6. Johnson S, Gerding DN. Clostridium difficile. In: May Hall GC, editor. Hospital epidemiology and infection control, 3rd ed. Philadelphia: Lippincott Williams &Wilkins; 2004. p. 623-32. 496 INDIAN J MED RES, April 2010 7. Crogan NL, Evans BC. Clostridium difficile: an emerging epidemic in nursing homes. Geriatr Nurs 2007, 28 : 161-4. 8. Pepin J, Saheb N, Coulombe M, Alary M, Corriveau M, Authier S, et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhoea: a cohort study during an epidemic in Quebec. Clin Infect Dis 2005; 41 : 254-60. 9. McDonald LC, Killgore GE, Thompson A, Owens RC, Khazakova Jr, Sambol SV, et al. An epidemic, toxin genevariant strain of Clostridium difficile. N Engl J Med 2005; 353 : 2433-41. 10. Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, et al. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in NorthAmerica and Europe. Lancet 2005; 366 : 1079-84. 11. Kuijper EJ, Coignard B, Tull P. Emergence of Clostridium difficile associated disease in North America and Europe. Clin Microbiol Infect 2006; 12 : 2-18. 12. Cloud J, Kelly CP. Update on Clostridium difficile associated disease. Curr Opin Gastroenterol 2007; 23 : 4-9. 13. Riley TV. Epidemic Clostridium difficile. Med J Australia 2006; 185 : 133-4. 14. Dial S, Kezouh A, Dascal A, Barkun A, Suissa S. Patterns of antibiotic use and risk of hospital admission because of Clostridium difficile infection. Can Med Assoc J 2008; 179 : 767-72. 15. Kyne L, Warny M, Qamar A, Kelly CP. Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 2001; 357 : 189-93. 16. Johnson S, Kent SA, O’Leary KJ, Merrigan MM, Sambol SP, Peterson LR, et al. Fatal pseudomembranous colitis associated with a variant Clostridium difficile strain not detected by toxin A immunoassay. Ann Intern Med 2001; 135 : 434-8. 17. Shim JK, Johnson S, Samore MH, Bliss DZ, Gerding, DN. Primary symptomless colonisation by Clostridium difficile and decreased risk of subsequent diarrhoea. Lancet 1998; 351 : 633-6. 18. Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RL, Donskey CJ. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis 2007; 45 : 992-8. 19. Farrell RJ, LaMont JT. Pathogenesis and clinical manifestations of Clostridium difficile diarrhea and colitis. Curr Top Microbiol Immunol 2000; 250 : 109-25. 20. Steiner TS, Flores CA, Pizarro TT, Guerrant RL. Fecal lactoferrin, interleukin-1beta, and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immunol 1997; 4 : 719-22. 21. Vaishnavi C, Bhasin D, Kochhar R, Singh K. Clostridium difficile toxin and faecal lactoferrin assays in adult patients. Microbes Infect 2000; 2 : 1827-30. 22. Finney JM. Gastroenterostomy for cicatrizing ulcer of the pylorus. Bull John Hopkins Hosp 1893; 4 : 53-5. 23. Gebhard RL, Gerding DN, Olson MM, Peterson LR, McClain CJ, Ansel HJ, et al. Clinical and endoscopic findings in patients early in the course of Clostridium difficile-associated pseudomembranous colitis. Am J Med 1985; 78 : 45-8. 24. Tedesco FJ. Treatment of recurrent antibiotic-associated pseudomembranous colitis. Am J Gastroenterol 1982; 77 : 220-1. 25. Zuckerman E, Kanel G, Ha C, Kahn J, Gottesman BS, Korula J. Low albumin gradient ascites complicating severe pseudomembranous colitis. Gastroenterology 1997; 112 : 991-9. 26. Jafri SF, Marshall JB. Ascites associated with antibioticassociated pseudomembranous colitis. South Med J 1996; 89 : 1014-7. 27. Rubin MS, Bodenstein, LE, Kent KC. Severe Clostridium difficile colitis. Dis Colon Rectum 1995; 38 : 350-4. 28. Tedesco FJ, Barton RW, Alpers DH. Clindamycin associated colitis. A prospective study. Ann Intern Med 1974; 81 : 42933. 29. Drapkin MS, Worthington MG, Chang TW, Razvi SA. Clostridium difficile colitis mimicking acute peritonitis. Arch Surg 1985; 120 : 1321-2. 30. Yankes JR, Baker ME, Cooper C, Garbutt JCT. Appearance of focal pseudomembranous colitis. J Comput Assist Tomogr 1988; 12 : 394-6. 31. Bartlett JG. Clostridium difficile: clinical considerations. Rev Infect Dis 1990; 12 : S243-51. 32. Vaishnavi C, Kochhar R, Bhasin, DK, Thennarasu K, Singh K. Simultaneous assay for Clostridium difficile and fecal lactoferrin in ulcerative colitis. Trop Gastroenterol 2003; 24 : 13-6. 33. Hookman P, Barkin JS. Clostridium difficile associated infection, diarrhea and colitis. World J Gastroenterol 2009; 15 : 1554-80. 34. Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med 1994; 330 : 257-62. 35. McFarland LV, Surawicz CM, Greenberg RN, Fekety R, Elmer GW, Moyer KA. A randomized placebo-controlled trial of Saccharomyces boulardii in combination with standard antibiotics for Clostridium difficile disease. JAMA 1994; 271 : 1913-8. 36. Mahajan LA, Hupertz V, Mahajan, S, Lisa F, John D. The appendix: A possible reservoir for Clostridium difficile. Am J Gastroenterol 2006; 101 : S392. 37. Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect 1998; 40 : 1-15. 38. Pantosti A, Cerquetti M, Gianfrilli PM. Electrophoretic characterization of Clostridium difficile strains isolated from antibiotic-associated colitis and other conditions. J Clin Micobiol 1988; 26 : 540-3. 39. McFarland LV, Elmer GW, Stamm WE, Mulligan ME. Correlation of immunoblot type, enterotoxin production, and cytotoxin production with clinical manifestations of Clostridium difficile infection in a cohort of hospitalized patients. Infect Immun 1991; 59 : 2456-62. 40. Cheng SH, Lu JJ, Young TG, Perng CL, Chi WM. Clostridium difficile-associated disease: comparison of symptomatic infection versus carriage on the basis of risk factors, toxin production and genotyping results. Clin Infect Dis 1997; 25 : 157-8. Vaishnavi: C. difficile pathogenesis 41. Jacobs A, Barnard K, Fishel R, Gradon JD. Extracolonic manifestations of Clostridium difficile infections. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2001; 80 : 88-101. 42. Testore GP, Nardi F, Babudieri S, Giuliano M, Di Rosa R, Panichi G. Isolation of Clostridium difficile from jejunum: identification of a reservoir for disease? J Clin Pathol 1986; 39 : 861-2. 43. Boland E, Thompson JS. Fulminant Clostridium difficile enteritis after proctocolectomy and ileal pouch-anal anastomosis. Gastroenterol Res Pract 2009; 2008 : 1-5. 44. Navaneethan U, Giannella RA. Thinking beyond the colonsmall bowel Involvement in Clostridium difficile infection. Gut Pathog 2009; 19 : 7. 45. Daruwala C, Mercogliano G, Newman G, Ingerman MJ. Bacteremia due to Clostridium difficile: Case report and review of the literature. Clin Med Case Reports 2009; 2 : 5-9. 46. Libby DB, Bearman G. Bacteremia due to Clostridium difficile - review of the literature. Int J Infect Dis 2009; 13 : e305-9. 47. Birnbaum J, Bartlett JG, Gelber AC. Clostridium difficile: an under recognized cause of reactive arthritis? Clin Rheumatol 2008; 27 : 253-5. 48. Ducroix-Roubertou S, Genet C, Rogez JP, Weinbreck P, Denes E. Reactive arthritis due to Clostridium difficile. Med Mal Infect 2005; 35 : 419-21. 49. Vaishnavi C. Established and potential risk factors for Clostridium difficile infection. Indian J Med Microbiol 2009; 27 : 291-302. 50. Kaur S, Vaishnavi C, Prasad KK, Ray P, Kochhar R. Comparative role of antibiotic and proton pump inhibitor in experimental Clostridium difficile infection in mice. Microbiol Immunol 2007; 51 : 1209-14. 51. Johnson S, Gerding DN. Clostridium difficile associated diarrhoea. Clin Infect Dis 1998; 26 : 1027-36. 52. West, M., Pirenne, J., Chavers, B Gillingham, K, Sutherland, DE, Dunn, DL, et al. Clostridium difficile colitis after kidney and kidney-pancreas transplantation. Clin Transplant 1999; 13 : 318-23. 53. Keven K, Basu A, Re L. Clostridium difficile colitis in patients after kidney and pancreas-kidney transplantation. Transpl Infect Dis 2004; 6 : 10-14. 54. Bartlett JG. Clinical practice: Antibiotic associated diarrhoea. N Engl J Med 2002; 346 : 334-9. 55. Dallal RM, Harbrecht BG, Boujoukas AJ, Sirio CA, Farkas LM, Lee KK, et al. Fulminant Clostridium difficile: an under appreciated and increasing cause of death and complications. Ann Surg 2002; 235 : 363-72. 56. Manian FA, Meyer L. CDAD rates. Infect Control Hosp Epidemiol 1995; 16 : 63-5. 57. Sharma AK, Holder FE. Clostridium difficile diarrhea after use of tacrolimus following renal transplantation. Clin Infect Dis 1998; 27 : 1540-1. 497 virus and Clostridium difficile. Zeitschriff fur Gastroenterologie 2008; 46 : 780-3. 60. Milligan DW, Kelley JK. Pseudomembranous colitis in a leukemia unit: a report of five fatal cases. J Clinical Pathol 1979; 32 : 1237-43. 61. Chen FC, Woods R. Pseudomembranous panenteritis and septicaemia in a patient with ulcerative colitis. Aust N Z J Surg 1996; 66 : 565-7. 62. Williams C. Occurrence and significance of gastric colonization during acid-inhibitory therapy. Best Pract Res Clin Gastroenterol 2001; 15 : 511-21. 63. Cunningham R, Dale B, Undy B, Gaunt N. Proton pump inhibitors as a risk factor for Clostridium difficile diarrhoea. J Hosp Infect 2003; 54 : 243-5. 64. Jayatilaka S, Shakov R, Eddi R, Bakaj G, Baddoura WJ, DeBari VA. Clostridium difficile infection in an urban medical center: five-year analysis of infection rates among adult admissions and association with the use of proton pump inhibitors. Ann Clin Lab Sci 2007; 37 : 241-7. 65. Nachnani JS, Bulchandani D, Allen MJ. Proton pump inhibitors are an independent risk factor for an increased length of hospital stay in patients with Clostridium difficile infection. Indian J Gastroenterol 2008; 27 : 171-2. 66. Emoto M, Kawarabayashi T, Hachisuga T, Eguchi F, Shirakawa K. Clostridium difficile colitis associated with cisplatin-based chemotherapy in ovarian cancer patients. Gynecol Oncol 1996; 61 : 369-72. 67. Resnik E, Lefevre CA. Fulminant Clostridium difficile colitis associated with paclitaxel and carboplatin chemotherapy. Internat J Gynecol Cancer 2001; 9 : 512-4. 68. Kumar B, Vaishnavi C, Sandhu K, Kaur I. Clostridium difficile toxin assay in psoriatic patients. Trop Gastroenterol 2004; 25 : 164-7. 69. Gellad ZF, Alexander BD, Liu JK, Griffith BC, Meyer AM, Johnson JL, et al. Severity of Clostridium difficile-associated diarrhea in solid organ transplant patients. Transpl Infect Dis 2007; 17 : 1-5. 70. Zerey M, Paton BL, Lincourt AE, Gersin KS, Kercher KW, Heniford BT. The burden of Clostridium difficile in surgical patients in the United States. Surg Infect (Larchmt) 2007; 8 : 557-66. 71. Loo VG, Poirier L, Miller MA, Oughton M, Libman M, Michaud S, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 2005; 353 : 2442-9. 72. Weiss B, Kleinkauf N, Neumann M, Eckmanns T, Michels H, Jansen A. Risk factors for Clostridium difficile ribotype 027 infection in Germany: preliminary results of a retrospective case-control study. 18th European Congress of Clin. Microbial & Infectious Diseases, Barcelona, Spain Abst 2008. p. 1480. 73. Tabaqchali S, Jumma P. Diagnosis and management of Clostridium difficile infection. BMJ 1995; 310 : 1375-80. 58. Sohi S, Cohen RD. Management of refractory ulcerative colitis. Curr Treat Options Gastroenterol 2006; 9 : 234-45. 74. Borriello SP, Barclay FE. Protection of hamsters against Clostridium difficile ileocaecitis by prior colonisation with non-pathogenic strains. J Med Microbiol 1984; 19 : 339-50. 59. Arnold C, von Sanden S, Theilacker C, Blum HE. Ulcerous colitis and infection with cytomegalovirus, herpes simplex 75. Rupnik M, Kato N, Grabnar M, Kato H. New types of toxin A-negative, toxin B-positive strains among Clostridium 498 INDIAN J MED RES, April 2010 difficile isolates from Asia. J Clin Microbiol 2003; 41 : 111825. 76. Krivan HC, Clark GF, Smith DF, Wilkins TD. Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3 Gal beta 1-4GlcNAc. Infect Immun 1986; 53 : 573-81. 77. Calabi E, Calabi F, Phillips AD. Fairweather NF. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun 2002; 70 : 5770-8. 78. Pechine S, Gleizes A, Janoir C, Gorges-Kergot R, Bare M-C, Delmee M, et al. Immunological properties of surface proteins of Clostridium difficile. J Med Microbiol 2005; 54 : 193-6. 79. Janoir C, Pechine S, Grosdidier C, Collignon A. Cwp 84, a surface associated protein of Clostridium difficile is a cysteine protease with degrading activity on extracellular matrix proteins. J Bacteriol 2007; 159 : 7174-80. 80. Pothoulakis C. Pathogenesis of Clostridium difficile-associated diarrhea. Eur J Gastroenterol Hepatol 1996; 8 : 1041-7. 81. Rupnik M, Brazier JS, Duerden BI, Grabnar M, Stubbs, SL. Comparison of toxiotyping of Clostridium difficile strains and description of novel toxinotypes. Microbiol 2001; 147 : 439-47. 82. Stubbs SL, Rupnik M, Gilbert M, Brazier JS, Duerden BI, Popoff MR. Production of actin-specific ADPribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett 2000; 186 : 307-12. 83. Just I, Selzr J, Wilm M, von Eichel-Streiber C, Mann M, Akatories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995; 375 : 500-3. 84. Just I, Wilm M, Selzr J, Rex G, von Eichel-Streiber C, Mann M, et al. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 1995; 270 : 13932-6. 85. Linevsky JK, Pothoulakis C, Keates S, Warny M, Keates, AC, LaMont JT, et al. IL-8 release and neutrophil activation by Clostridium difficile toxin-exposed human monocytes. Am J Physiol Gastrointest Liver Physiol 1997; 273 : G1333-40. 86. Lima AA, Lyerly DM, Wilkins TD, Innes DJ, Guerrant RL. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect Immun 1988; 56 : 582-8. integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol 2001; 280 : G178-83. 92. Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis 2002; 186 : 1438-47. 93. Altaie SS, Meyer P, Dryja D. Comparison of two commercially available enzyme immunoassays for detection of Clostridium difficile in stool specimens. J Clin Microbiol 1994; 32 : 51-3. 94. Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. Effect of Clostridium difficile toxins given intragastrically to animals. Infect Immun 1985; 35 : 1147-50. 95. Fiorentini C, Thelestam M. Clostridium difficile toxin A and its effects on cells. Toxicon 1991; 29 : 543-67. 96. Siffert JC, Baldacini O, Kuhry JG, Wachsmann D, Benabdelmoumene S, Faradji A, et al. Effects of Clostridium difficile toxin B on human monocytes and macrophages: possible relationship with cytoskeletal rearrangement. Infect Immun 1993; 61 : 1082-90. 97. Just I, Fritz G, Giry M, Popoff MR, Boquet P, Hegenbarth S, et al. Clostridium difficile toxin B acts on the GTP-binding protein Rho. J Biol Chem 1994; 269 : 10706-12. 98. Hecht G, Koutsouris A, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin B disrupts barrier function of T84 monolayers. Gastroenterology 1992; 102 : 416-23. 99. Popoff MR, Rubin EJ, Gill DM, Boquet P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun 1988; 56 : 2299-306. 100. Rupnik M. How to detect Clostridium difficile variant strains in a routine laboratory. Clin Microb Infect Dis 2001; 7 : 417-20. 101.Geric B, Johnson S, Gerding DN, Grabnar M, Rupnik M. Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J Clin Microbiol 2003; 41 : 5227-32. 102.Bartlett JG, Cang TW, Gurwith M, Gorbach SL, Onderdonk AB. Antibiotic associated pseudomembranous colitis due to toxin producing clostridia. N Engl J Med 1978; 298 : 531-4. 103.Wilkins TD, Lyerly DM. Clostridium difficile testing: after 20 years, still challenging. J Clin Microbiol 2003; 41 : 531-4. 87. Triadafilopoulos G, Pothoulakis C, Weiss R, Giampaolo C, LaMont JT. Comparative study of Clostridium difficile toxin A and cholera toxin in rabbit ileum. Role of prostaglandins and leukotrienes. Gastroenterology 1987; 92 : 1174-80. 104.Kato H, Yokoyama T, Kato H, Arakawa Y. Rapid and simple method for detecting the toxin B gene of Clostridum difficile in stool specimens by loop mediated isothermal amplification. J Clin Microbiol 2005; 43 : 6108-12. 88. Katyal R, Vaishnavi C, Singh K. Faecal excretion of brush border membrane enzymes in patients with Clostridium difficile diarrhea. Indian J Med Microbiol 2002; 20 : 178-82. 105.Teasley PG, Gerding DN, Olson MM, Peterson LR, Gebhard RL, Schwartz MJ, et al. Prospective randomized trial of metronidazole versus vancomycin for Clostridium difficileassociated diarrhea and colitis. Lancet 1983; 2 : 1043-6. 89. Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 1988; 82 : 1516-24. 90. Castagliuolo I, Keats AC, Wang CC, Pasha A, Valenick L, Pothoulakis C, et al. Substance P receptor expression in intestinal epithelium in Clostridium difficile toxin A enteritis in rats. Am J Physiol 1998; 275 : G68-75. 91. Pothoulakis C, LaMont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The 106.Delmee M, Buts J-P. Clostridium difficile-associated diarrhoea in children. In: Buts JP, Sokal EM, editors. Management of digestive and liver disorders in infants and children. Elsevier Science Publishers; 1993. p. 371-9. 107.Barbut F, Petit J-C. Epidemiology of Clostridium difficile associated infections. Clin Microbiol Infect 2001; 7 : 405-10. 108.Nelson R. Antibiotic treatment for Clostridium difficileassociated diarrhea in adults. Cochrane Database Syst Rev 2005; CD004610. Vaishnari: C. difficile pathogenesis 109.Aas J, Gessert CE, Bakken JS. Recurrent Clostridium difficile colitis; case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin Infect Dis 2003; 36 : 580-5. 110. Dutta P, Niyogi SK, Mitra U, Rasardy R, Bhattacharya MK, Chakraborty S. Clostridium difficile in antibiotic associated pediatric diarrhea. Indian Pediatr 1994; 31 : 121-6. 111. Dhawan B, Chaudhry R, Sharma N. Incidence of Clostridium difficile infection: a prospective study in an Indian hospital. J Hosp Infect 1999; 43 : 275-80. 112. Vaishnavi C, Kochhar R, Bhasin DK, Thapa BR, Singh K. Detection of Clostridium difficile toxin by an indigenously developed latex agglutination assay. Trop Gastroenterol 1999; 20 : 33-5. 113. Vaishnavi C, Bhasin DK, Singh K. Faecal lactoferrin assay as a cost effective tool for intestinal inflammation. Am J Gastroenterol 2000; 95 : 3002. 114. Vaishnavi C, Thapa BR, Thennarasu K, Singh K. Faecal lactoferrin assay as an adjunct to Clostridium difficile diarrhea. Indian J Pathol Microbiol 2002; 45 : 69-74. 115. Gupta U, Jadav RN. Clostridium difficile in hospital patients. Indian J Med Res 1985; 82 : 398-401. 116. Niyogi SK, Dutta P, Dutta D, Mitra U, Sikdar S. Clostridium difficile and its cytotoxin in hospitalized children with acute diarrhea. Indian Paediatrics 1991; 28 : 1129-32. 117. Niyogi SK, Bhattacharya SK, Dutta P, Naik TN, De SP, Sen D, et al. Prevalence of Clostridium difficile in hospitalised patients with acute diarrhoea in Calcutta. J Diarrhoeal Dis Res 1991; 9 : 16-9. 118. Bhattacharya MK, Niyogi SK, Rasaily R, Bhattacharya SK, Dutta P, Nag A, et al. Clinical manifestation of Clostridium 499 difficile enteritis in Calcutta. J Assoc Physicians India 1991; 39 : 683-4. 119. Niyogi SK. Antimicrobial susceptibility of Clostridium difficile strains isolated from hospitalised patients with acute diarrhoea. J Diarrhoeal Dis Res 1992; 10 : 156-8. 120.Kochhar R, Ayyagari A, Goenka MK, Dhali GK, Aggarwal R, Mehta SK. Role of infectious agents in exacerbation of ulcerative colitis in India: A study of Clostridium difficile. J Clin Gastroenterol 1993; 16 : 26-30. 121.Vaishnavi C, Bhasin DK, Kochhar R, Singh K. Clostridium difficile toxin and faecal lactoferrin assays in adult patients. Microbes Infect 2000; 2 : 1827-30. 122.Kang G, Srivastava A, Pulimood AB, Dennison D, Chandy M. Etiology of diarrhea in patients undergoing allogeneic bone marrow transplantation in South India. Transplantation 2002; 73 : 1247-51. 123.Gogate A, De A, Nanivadekar R, Mathur M, Saraswathi K, Jog A, et al. Diagnostic role of stool culture and toxin detection in antibiotic associated diarrhoea due to Clostridium difficile in children. Indian J Med Res 2005; 122 : 518-24. 124.Vaishnavi C, Kaur S, Singh K. Clostridium perfringens type A and antibiotic associated diarrhoea. Indian J Med Res 2005; 122 : 52-6. 125.Balamurugan R, Balaji V, Ramakrishna BS. Estimation of faecal carriage of Clostridium difficile in patients with ulcerative colitis using real time polymerase chain reaction. Indian J Med Res 2008; 127 : 472-7. 126.Chaudhry R, Joshy L, Kumar L, Dhawan B. Changing pattern of Clostridium difficile associated diarrhoea in a tertiary care hospital: a 5 year retrospective study. Indian J Med Res 2008; 127 : 377-82. Reprint requests:Dr C. Vaishnavi, Additional Professor (GE Microbiology), Department of Gastroenterology, Postgraduate Institute of Medical Education & Research, Chandigarh 160 012, India e-mail: [email protected], [email protected]