Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
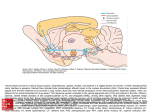
Volume 25, Number 9 October 2011 Drugs & Therapy B U ◆ ◆ L FORMULARY UPDATE The Pharmacy and Therapeutics Committee met September 20, 2011. 7 products were added in the Formulary, and 2 were deleted and designated nonformulary and not available. 3 criteria for uses were approved including 2 restrictions. ◆ADDED Asenapine (Saphris®)* *Restricted: continuation from home & Psychiatry Service Belatacept (Nulojix®)* *Restricted to renal transplant patients Menthol Lozenges (Generic) Prothrombin Complex Concentrate (Profilnine® SD) *Restricted: life-threatening hemorrhage due to warfarin overdose Rilpivirine (Edurant®) Rilpivirine-EmtricitabineTenofovir (Complera®) Rivaroxaban (Xarelto®) ◆ DELETED ◆ L ◆ E ◆ †Nonformulary and not available ◆CRITERIA-FOR-USE CHANGES Brentuximab Vedotin (Adcetris®)* *Added in the Chemotherapy Policy Cytomegalovirus Immune Globulin (Cytogam®)‡ ‡Off-label Use in Pregnant Women Endorsed Vemurafenib (Zelboraf®)* *Added in the Chemotherapy Policy (continued on next page) ◆ I ◆ N PROPOSAL Restrict IV acetylcysteine use for the prevention of radiocontrastinduced nephropathy T he P&T Committee will be considering a proposal to limit the use of intravenous (IV) n-acetylcysteine (NAC) for the prevention of radiocontrast-induced nephropathy (RCIN). Attending physicians who would like to comment on these proposed restrictions should email their comments to [email protected] by October 25, 2011. There will be an effort to let the major users of IV NAC know about this proposed change, but notification in the newsletter is intended to make this proposal known to all current users of this product. Last year, approximately a half a million dollars was spent on this dosage form. ◆ Attending physicians who would like to comment on these proposed restrictions should email their comments by October 25. Cepacol® Lozenges† Telavancin (Vibativ®)† T The current proposal is to limit the use of IV NAC to a single dose unless the patient meets specific criteria regarding the volume of contrast dye used AND the existence of pre-existing renal dysfunction. Data suggest that only very large dye loads (ie, greater than 200 mL) place the patient at risk. For example, patients receiving routine CT scans with dye should NOT receive IV NAC. Patients with pre-existing renal dysfunction are defined by an estimated glomerular filtration rate (eGFR) less than 30 mL per minute or both an eGFR less than 50 mL per minute and either diabetes or congestive heart failure. The Division of Nephrology of the Department of Medicine agrees with this proposal. RCIN is a potentially serious adverse drug reaction that occurs in up to 50% of high-risk patients requiring contrast for coronary angiography and computed tomography. NAC is often given to prevent the development of RCIN despite mixed results from clinical trials. Eleven randomized controlled trials have compared IV NAC to hydration for the prevention of RCIN. There are no studies comparing IV and oral NAC. Trials investigating IV NAC have produced conflicting results. The trials are heterogeneous regarding IV NAC dosing regimens, hydration regimens, volumes of contrast used, and RCIN risk level. For example, the rate of RCIN in control groups ranged from 0% to 33%. Only 3 out of the 11 trials demonstrated a benefit in preventing RCIN. Some trials have a higher incidence of RCIN in the IV NAC arm, further confusing the issue. The trials suggesting possible efficacy for IV NAC help determine which patients are good candidates versus those that are not. IV NAC may be beneficial when administered to patients at high risk for RCIN that receive high volumes of contrast dye. Oral NAC should be given in most cases, if NAC use is deemed necessary. Given the option between therapies with equally poor evidence, it is reasonable to choose the most economical alternative. IV NAC is roughly 10 times more expensive than oral NAC. If the P&T Committee approves the restriction of IV NAC, patients not meeting the dye volume and eGFR requirements would be switched to oral NAC or NAC would be discontinued after the first dose. ◆ INSIDE THIS ISSUE ◆ Breaking news on bisphosphonates 2 Formulary update, from page 1 Asenapine is a sublingual secondgeneration antipsychotic with a labeled indication for acute and maintenance treatment of schizophrenia, as well as monotherapy and adjunctive therapy of manic or mixed episodes for bipolar mania. Asenapine has high affinity and antagonistic action at serotonin, dopamine, alpha-adrenergic, and histamine receptors with no appreciable activity at muscarinic cholinergic receptors. Some hypothesize this activity allows better control of negative symptoms of schizophrenia and decreased anticholinergic adverse effects; however, whether these differences translate to clinically important effects compared with other agents is unproven. Efficacy in schizophrenia is inconsistent. Attrition among asenapine patients has been more than with comparators due to adverse events and lack of efficacy. No dose-response effect has been shown with flexible dosing regimens. Asenapine has not shown better negative symptom control in the acute and maintenance treatment of schizophrenia. In the treatment of bipolar mania, asenapine was not consistently better than placebo in response and remission rates. No currently available study has been designed to compare asenapine to another active treatment. Exclusion of patients with common comorbid conditions (eg, substance abuse, rapid cycling) limits external validity for general psychiatry patients. Food and liquid intake restrictions, as well as twice-daily dosing, may impose additional barriers to compliance in an already noncompliant population. Low bioavailability when swallowed is an obstacle to therapeutic efficacy. Based on current evidence, asenapine does not appear to provide an advantage over other antipsychotics. A similar agent listed in the Formulary, olanzapine orally disintegrating tablets (Zyprexa® Zydis®), consistently outperformed asenapine in indirect comparisons for schizophrenia maintenance and acute bipolar treatment. Asenapine was added in the Formulary but restricted to patients who fail treatment with other atypical antipsychotics. The Psychiatric Service is required for initiation of therapy. Patients admitted already on asenapine may continue therapy. Asenapine cannot be used for agitation in general medicine patients or critically ill patients. Belatacept is a potent antagonist of CD80 and CD86 ligands present on antigen-presenting cells, which are responsible for CD28 activation. Produced by CD28, the co-stimulation pathway of T-cell activation and proliferation is essential to the immune system’s ability to identify a transplanted organ as foreign. After phase II trials revealed that belatacept may provide adequate immunosuppression compared to calcineurin inhibitors (eg, cyclosporine or tacrolimus) while avoiding some of the chronic toxicities associated with calcineurin inhibitors, larger phase III studies were performed. The Belatacept Evaluation of Nephroprotection and Efficacy as First-Line Immunosuppression Trial (BENEFIT) and Belatacept Evaluation of Nephroprotection and Efficacy as First-Line Immunosuppression Trial-EXTended criteria donors (BENEFIT-EXT) were some of the largest studies ever performed in kidney transplant recipients. In both standard criteria and extended criteria kidney donors, belatacept was associated with similar patient/graft survival when compared to the calcineurin inhibitor cyclosporine. Belatacept-treated patients had better renal function and more favorable cardiovascular profiles compared to cyclosporine. However, belatacept was associated with an increased incidence of acute rejection and cases of post-transplant lymphoproliferative disorder (PTLD), a rare and serious adverse event occurring after transplantation. PTLD occurred most often in patients who were EpsteinBarr Virus seronegative. Because the cost of each vial of belatacept is high (approximately $700) and each transplanted patient will receive approximately 6 vials during their hospital stay, the use of belatacept will add considerably to pharmaceutical expenditures. Whether improvements in renal function and cardiovascular markers will result in improved long-term outcomes and the safety data on this new drug class remains largely unknown. There are concerns about rare but serious adverse effects (PTLD and progressive multifocal leukoencephalopathy). Therefore, belatacept was added in the Formulary and restricted to use in renal transplant recipients. After a year, the impact of this agent on reimbursements for renal transplantation and utilization data will be reviewed by the P&T Committee. Menthol is a common ingredient in oral throat lozenges. Until recently, Cepacol® Lozenges were listed in the Formulary. Cepacol® Lozenges were deleted from the Formulary and designated nonformulary and not available after they were reformulated to contain benzocaine. A plain menthol-containing lozenge will now be listed in the Formulary. In March 2010, benzocaine-containing lozenges (Chloraseptic® Lozenges) and spray (Hurricaine® Spray) were deleted from the Formulary and designated nonformulary and not available. These decisions were made to decrease the risk of methemoglobinemia that has been associated with the mucosal use of benzocaine. A menthol-containing lozenge, like Cepacol®, that did not contain benzocaine was offered as an alternative to Chloraseptic® Lozenges. Chloraseptic® Spray, which uses phenol as an active ingredient, is another alternative to benzocaine-containing lozenges. Hall’s Sugar-Free Triple-SoothingAction lozenges were selected because they contained a similar amount of menthol (5 mg) per lozenge and are sugar-free [but do not contain sorbitol, which can cause diarrhea]. Profilnine® SD is a 3-factor prothrombin complex concentrate (PCC) containing primarily factors II, IX, and X. PCC is also known as Factor IX Complex, but it will be listed in EPIC as prothrombin complex concentrate in an attempt to prevent confusion with other factors (eg, recombinant Factor IX). Despite its labeled indication for the prevention and control of bleeding in patients with Factor IX deficiency due to hemophilia B, it is used primarily for the management of serious bleeding associated with excessive anticoagulation with warfarin. When a 3-factor PCC is used to treat excessive anticoagulation, fresh frozen plasma (FFP) is usually given at the same time to supplement the amount of Factor VII (FVII). For life-threatening bleeding, guidelines from the American College of Chest Physicians (ACCP) recommend holding warfarin therapy and administering FFP, PCC, or recombinant FVIIa (NovoSeven® RT) along with 10 mg IV vitamin K, repeating if needed, depending on the INR. The available evidence for the use of PCC for the treatment of excessive warfarin anticoagulation is limited. The ideal evidence for the use of Profilnine® SD for the treatment of warfarin toxicity would be randomized controlled trials assessing the safety and efficacy of products (ie, PCC vs FVIIa). Unfortunately, no such evidence exists for the available PCC products or FVIIa. One recently completed, but unpublished, non-randomized trial used a parallel design to compare FVIIa, PCC, and FFP in intracranial hemorrhage with the primary outcome of INR normalization as defined by an INR less than 1.4. If this study is published, it may provide additional evidence. The evidence for products currently on the market mostly comes from observational studies and case series. Recent evidence suggests that FVIIa can correct elevated INR, but (continued on next page) Formulary update, from page 2 may not decrease bleeding time or blood loss. Viral (hepatitis A, B, C, HIV, HTLV1, and parvovirus B19) transmission is possible with PCCs as they are derived from human plasma. Since FFP is used with 3-factor PCC, the risks associated with transfusion-related acute lung injury must be considered. It is characterized by acute onset of noncardiogenic pulmonary edema and occurs in about 1 of every 5000 transfusions; it is the leading cause of transfusion-related deaths in the US (~50%). The use of PCC plus FFP is similar in cost to FVIIa. The doses of FVIIa and Profilnine SD® can greatly vary between patients. While treating a patient with PCC and FFP appears to be less expensive than FVII alone, if a patient needs a higher PCC dose or more units of FFP, it can be more costly. In addition, miscellaneous costs (extra personnel, etc.) in handling the additional administration of FFP may be an issue. Profilnine® SD was added in the Formulary for use in life-threatening hemorrhages due to warfarin overdose. Each use will be reviewed immediately after it is administered to assure appropriate use. Rilpivirine is a novel human immunodeficiency virus type-1 (HIV-1) non-nucleotide reverse transcriptase inhibitor (NNRTI). Rilpivirine has a labeled indication for the treatment of HIV-1 in treatment-naïve patients. Rilpivirine non-competitively binds to and inhibits HIV-1 reverse transcriptase. It is best absorbed when taken with meals and is metabolized by CYP3A. Rilpivirine does not inhibit or induce CYP enzymes. Only 1% of a rilpivirine dose is eliminated unchanged in the urine. The approval of rilpivirine was based upon a dose-ranging study and 3 clinical trials. The dose-ranging study showed that 25 mg of rilpivirine daily was shown to be equivalent to 75 mg and 150 mg daily regimens, and have efficacy comparable to efavirenz. The efficacy of 25 mg rilpivirine daily was compared to 600 mg efavirenz daily. Rilpivirine was found to be non-inferior to efavirenz in suppressing viral load, based upon a time-to-loss-of-virological-response algorithm. However, an increased risk of virological failure with rilpivirine was observed. The observed virological failure rate in rilpivirinetreated subjects conferred a higher rate of overall treatment resistance and cross-resistance to the NNRTI class compared to efavirenz. Rilpivirine failures result in etravirine and efavirenz resistance in most patients (89%). This is a major disadvantage for rilpivirine. The most common adverse effects observed in clinical trials were nausea and dizziness. The incidences of rash and central nervous system adverse effects with rilpivirine appear to be less than with efavirenz. Rilpivirine is classified as pregnancy category B. Rilpivirine use is contraindicated when taking inducers of CYP3A or proton pump inhibitors. Concomitant administration of these drugs may decrease rilpivirine exposure and increase the chance for virological failure or development of resistance. In general, any substrates, inhibitors, or inducers of CYP3A have the potential to interact with rilpivirine. Rilpivirine was added to the Formulary for continuity of care and to prevent the development of resistance, which would be particularly problematic for this agent. It is “non-inferior” to efavirenz, but is more expensive and can cause cross-resistance with other NNRTIs. Rilpivirine could be considered when efavirenz is not tolerated or when pregnancy is a concern. Complera® is a combination product containing emtricitabine [a nucleoside reverse transcriptase inhibitor or NRTI], rilpivirine [a non-nucleoside reverse transcriptase inhibitor or NNRTI], and tenofovir [a NRTI] with a labeled indication for the treatment of human immunodeficiency virus (HIV) infections in treatment-naïve patients. This rilpivirine-containing combination product was also added in the Formulary. Rivaroxaban is one of several new agents receiving attention as an alternative to warfarin as an oral anticoagulant. Many of these agents offer “advantages” over older anticoagulants. However, evidence-based literature regarding the safety and efficacy of these new agents needs further development and careful review before they establish a more prominent role in therapy. Rivaroxaban is the first and only oral direct factor Xa inhibitor available in the US. Currently, rivaroxaban has a labeled indication only for the prevention of deep vein thrombosis (DVT) in patients following knee or hip replacement surgery. Randomized controlled data from 4 large ‘RECORD’ trials (Regulation of Coagulation in Orthopedic Surgery to Prevent DVT and PE) indicate superiority of rivaroxaban over enoxaparin in the composite outcome of any DVT, nonfatal PE, or all-cause mortality for the labeled indication. The trials observed a similar major bleeding risk in the rivaroxaban treatment arms compared to enoxaparin; there was a nonsignificant trend toward increased major bleeding in the rivaroxaban treatment arms. Being a new agent, rivaroxaban does not have an extensively documented safety profile. Rivaroxaban is available only as a 10-mg tablet. The labeled dosage is 10 mg once daily with or without food starting 6-8 hours after hip or knee replacement surgery. It is almost completely bioavailable and exhibits a linear pharmacokinetic profile. Rivaroxaban, unlike some older anticoagulants, does not require frequent monitoring or dosage adjustments. Ongoing and recently published trials are aiming to expand rivaroxaban’s labeled indications to include stroke prophylaxis in atrial fibrillation, treatment of symptomatic DVTs, and extended prophylaxis following a symptomatic DVT event. Rivaroxaban should be avoided in patients with renal impairment (CrCl less than 30 mL/min) and patients with moderate to severe hepatic impairment. Bleeding is the most common adverse effect. Increases in hepatic enzymes have been reported. Rivaroxaban has a boxed warning for epidural or spinal hematomas in patients receiving epidurals. Rivaroxaban is competitively priced for DVT prophylaxis. Similar agents at Shands include fondaparinux 2.5 mg SC daily (which is 4 times more expensive), enoxaparin 40 mg SC daily (which is approximately 3 times more expensive), enoxaparin 30 mg SC daily (which is approximately 4 times more expensive), and heparin 5000 units SC every 8 hours (which is about 25% less expensive). Recently published data showed efficacy for the use of rivaroxaban for the off-labeled use for the prevention of stroke in patients with atrial fibrillation. An FDA advisory committee recommended that this indication be added in the labeling; however, final FDA action is still pending. Telavancin is a lipoglycopeptide derivative of vancomycin that received FDA approval in September 2009 for the treatment of adult patients with complicated skin and skin structure infections (cSSSI) caused by susceptible gram-positive bacteria. In November 2010, Shands at UF added telavancin in the Formulary. Telavancin’s spectrum of activity is similar to vancomycin, with activity against a wide range of aerobic and anaerobic gram-positive bacteria, including some multidrug-resistant strains. Telavancin has a dual mechanism of action: inhibition of peptidoglycan synthesis and disruption of membrane potential. This dual mechanism accounts for its enhanced activity as well as rapid bactericidal properties. (continued on next page) 3 ALGORITHM FOR DIAGNOSIS AND MANAGEMENT OF CONGENITAL CMV INFECTION Diagnosis suspected on basis of clinical illness 1.Obtain CMV IgG and IgM serology 2. Assess IgG avidity (low vs high) 3. Assess for virus in maternal blood and urine via quantitative CMV PRC Diagnosis of primary maternal infection confirmed 1.Perform amniocentesis at ≥ 15 weeks to assess for virus in amniotic fluid by quantitative PCR or culture. 2. Perform comprehensive ultrasound examination No virus detected/ normal ultrasound Virus detected with normal ultrasound Virus detected with abnormal ultrasound Repeat ultrasound in 2–3 weeks; consider repeat amniocentesis Prophylactic Cytogam at 200 mg/kg Termination versus Cytogam at 200 mg/kg Serial ultrasounds Reassess findings Retreat as indicated No treatment 4 Formulary update, from page 3 Telavancin has been compared to vancomycin for the treatment of cSSSI and hospital-acquired pneumonia (HAP) in several non-inferiority studies. For the treatment of cSSSI, telavancin was compared to vancomycin in 2 parallel, randomized, double blind, controlled, phase III trials (known as the ATLAS trials). Patients received either telavancin or vancomycin for 7-14 days. Telavancin was shown to be non-inferior to vancomycin based on clinical cure rates. In a subanalysis of the ATLAS trials, clinical cure rates in the telavancin treatment group were lower in patients with impaired renal function defined as a CrCl less than 50 mL/min, compared to patients with normal renal function. The ATTAIN studies were randomized, double blind, phase III trials in patients with HAP secondary to suspected or documented gram-positive pathogens. Once again, telavancin proved to be non-inferior to vancomycin for the treatment of HAP. Telavancin has also been used off-label for the treatment of bacteremia and endocarditis. Rapid infusions can lead to Red-Man Syndrome; thus, telavancin should be administered over 60 minutes to reduce this risk. The most common adverse events reported are taste disturbance, nausea, vomiting, and foamy urine. The drug is classified as pregnancy category C and carries a black-box warning about potential fetal risks. Precaution should also be taken when prescribing telavancin to patients with a prolonged baseline QTc interval or taking drugs known to prolong the QT-interval. Telavancin was added in the Formulary and restricted to approval by the Infectious Diseases or the Antimicrobial Management Program for the management of gram-positive infections in patients who fail or who are intolerant to other therapies (ie, vancomycin). Since its inclusion in the Formulary, telavancin has been used in only 1 patient. In addition, product has been discarded due to expiration. Therefore, telavancin was deleted from the Formulary and designated nonformulary and not available due to low use and high risk of waste. (continued on other side) 4 Formulary update, from page 4 Brentuximab vedotin is a CD30directed antibody-drug conjugate with labeled indications for the treatment of patients with Hodgkin's lymphoma after failure of autologous stem cell transplant or after failure of at least 2 prior multi-agent chemotherapy regimens in patients who are not stem cell transplant candidates and for the treatment of patients with systemic anaplastic large cell lymphoma after failure of at least 1 prior multi-agent chemotherapy regimen. Brentuximab vedotin was added in the Chemotherapy Policy. Cytomegalovirus immune globulin (CMV IVIG) is an intravenous immune globulin (IgG) with a standardized amount of antibody to cytomegalovirus. It has a labeled indication for the prophylaxis of CMV disease associated with solid-organ transplantations. Treatment of CMV disease is an off-label use. It is estimated that 5000 to 8000 children per year develop disabilities (hearing or vision loss, cognitive impairment) secondary to congenital CMV infection. During pregnancy, a small number of pregnancies have seroepidemiologic evidence of CMV primary infection and approximately 1% of newborns are infected. Approximately 40% of woman with a primary infection during pregnancy will transmit CMV to the fetus. The highest transmission rates are typically seen in the third trimester; however, the greatest risk of fetal injury occurs in the first trimester or early second trimester. Few treatment options exist for preventing congenital CMV infection or disease. Antiviral therapy and CMV specific immunoglobulin can be considered to mitigate complications associated with primary CMV infection. CMV IVIG has been shown to suppress CMV replication within the placenta and may prevent placental dysfunction through reductions in direct placental damage and improvements in uteroplacental perfusion. Nigro and colleagues evaluated the effects of primary CMV infection in 157 pregnant women. A subgroup of 45 women who underwent amniocentesis and were CMV-positive were offered CMV IVIG, no immunotherapy, or abortion. Of these 45 women, 31 chose to receive therapy, 14 declined therapy (control group), and 10 chose abortion. CMV IVIG was administered for one 200-mg/kg (maternal weight) dose with subsequent doses administered pending ultrasound evidence of persistent infection. Following receipt of CMV IVIG, 1/31 infants had CMV disease versus 7/14 in the control group. These data suggest that CMV IVIG may have a role in decreasing the incidence of congenital CMV in babies born to pregnant women with primary CMV infection during pregnancy. In separate analyses, LaTorre and Nigro identified that CMV IVIG was associated with significant reduction in placental thickness, placental inflammation, or placental viral load in pregnant women with primary infection. The P&T Committee endorsed the use of the treatment of CMV IVIG for this use based on the algorithm on page 4. Prior to infusion, the patient would be premedicated with acetaminophen, diphenhydramine, and methylprednisolone. Epinephrine will be available in case of anaphylaxis. The protocol recommends that Informed Consent be obtained prior to the use of CMV IVIG administration, which will highlight the risks and potential benefits of the product. Vemurafenib is an oral kinase inhibitor with a labeled indication for the treatment of patients with unresectable or metastatic melanoma with BFAFV600E mutation as detected by an FDA-approved test. It is not recommended for use in patients with the wild-type BRAF melanoma. Vemurafenib was added in the Chemotherapy Policy. ADVERSE DRUG REACTIONS Bisphosphonates and atypical femur fractures: A bone of contention R 5 5 ecently, the FDA released a warning regarding a possible increased risk of atypical thigh bone fracture in patients who take bisphosphonates (BPs).1 The Warnings and Precautions section of the labeling will be revised and medication guides will now be given to inform patients about this possible risk. Oral and intravenous BPs approved for osteoporosis will be affected; changes will not apply to BPs approved for Paget’s disease, cancer, or hypercalcemia. Atypical fractures are so named because they occur in locations not typically fractured (subtrochanteric and/or proximal diaphyseal femur), and they occur secondary to minimal or no trauma. Atypical fractures comprise roughly 8% of osteoporotic fractures. Femoral fractures, typically grouped within “other” fracture types in incident analyses, comprised 33% of all osteoporotic fractures in 2005, 25% of which were atypical.2,3 The current concern seems contradictory given the drugs’ indication but consistent with the mechanism of bone strengthening. BPs inhibit bone turnover to strengthen bones. However, normal turnover repairs microcracks; thus, inhibiting this process may lead to increased bone density without increased resilience to trauma, leaving a “more brittle” bone.4 The overarching question is the strength of association between BP use and atypical fracture risk. Data from several observational and randomized trials suggest a significant increase, although conclusions are difficult because of small sample sizes and methodological limitations. Before widespread BP use, Salminem and colleagues characterized an incidence of 1 low-energy fracture per 10,000 patient-years.2 Observational safety studies later showed a significantly increased incidence of atypical fracture with BP use at roughly 2.5/10,000 patient-years.5 Estimates of the association between the use of BPs and atypical fractures vary widely and are based on retrospective data. For example, one case-control study matched atypical fracture cases to controls with lowenergy fractures in other areas. BP use was documented in 15/41 (37%) cases versus 9/82 (11%) controls, producing an odds ratio (OR) of 4.4, (95% CI 1.8-11.4).6 Another study found 19/25 (76%) patients with atypical fracture were receiving alendronate versus 1/45 (2.2%) without atypical pattern, an OR of 139 (19-939).7 Though these observational findings merit consideration, cause and effect conclusions cannot be made because of study limitations, which may result in misclassification bias from unavailability of confirmatory radiographs and confounding from uncharacterized comorbidites and medications. Black and colleagues performed a secondary analysis of femoral fractures in 3 large RCTs that assessed efficacy of BPs.8 The FIT, FLEX, and HORIZON trials, performed over a maximum 5 years, showed 12 atypical fractures in 51,000 patient-years. Contrary to observational findings, relative risk of atypical fracture with BP treatment (continued on page 6) Drugs & Therapy B ◆ U ◆ L ◆ L ◆ E ◆ T ◆ I ◆ N Volume 25, No. 9 October 2011 This publication is produced by the Drug Information and Pharmacy Resource Center under the direction of the Department of Pharmacy Services and the Pharmacy and Therapeutics Committee. NON-PROFIT ORG. U.S. POSTAGE PAID GAINESVILLE, FL PERMIT NO. 94 SHANDS Shands at the University of Florida DRUG INFORMATION SERVICE PO Box 100316 Gainesville, FL 32610-0316 EDITOR, DRUGS & THERAPY BULLETIN Randy C. Hatton, PharmD DIRECTOR, PHARMACY SERVICES Alan Knudsen, MS, RPh CHAIRMAN, PHARMACY & THERAPEUTICS COMMITTEE I. David Weiner, MD Professor of Medicine and Physiology and Functional Genomics University of Florida, College of Medicine EDITING, DESIGN, & PRODUCTION Shands HealthCare’s Publication Svcs. © Copyright 2011. All rights reserved. No portion of the Drugs & Therapy Bulletin may be reproduced without the written consent of its editor. FOR MORE INFORMATION, VISIT US ONLINE http://shands.org/professionals/ druginfo/bulletin.asp Adverse drug reactions, from page 5 versus controls was not statistically significant. Grouping these data, however, poses a problem. Although the number of patient years is increased, it does not accurately represent long-term safety. In other words, although the number of patient years may be numerically equivalent, combining data from more patients from shorter studies is not equivalent to that of fewer patients from longer studies. Another important question is the effect of therapy duration on risk for atypical fracture. The study by Lenart and colleagues was remarkable as it showed statistically greater BP therapy duration in subtrochanteric than in intertrochanteric and femoral neck fractures.6 Although the absolute numbers of each fracture type were small (fewer than 10), data over a sixyear period show a trend of increasing risk for subtrochanteric fracture while risk decreases for intertrochanteric and femoral neck fracture. Additionally, Neviaser showed that among cases assessable for therapy duration, BP use was longer at 6.9 years in users with atypical fractures versus 2.5 years in those without.7 6 Product-specific effects may be impossible to determine, if not irrelevant in practice. Although a majority of data reflects alendronate use, data are heterogeneous and FDA’s recommendation is a class-wide warning for all BPs indicated for osteoporosis. FDA may recommend consideration of a drug holiday or a 5-year limit to BP therapy. A sustained, but not increased, benefit has been identified beyond 3-4 years of therapy. Pooled analyses of RCT data for decreasing fracture incidence show similar 5-year efficacy with continued treatment compared to 3 years active treatment followed by placebo.1 Beyond these 5 years, fracture rates are similar, although strong conclusions are muddied as cohort sizes were nearly halved at this time point due to different trial designs. Few data on drug holidays are available from 14 patients in a study that stopped risedronate during year 8 then resumed therapy for 2 subsequent years.1 Fracture rates post-resumption were similar to pre-holiday rates, though the sample size is extremely small and no data are available to guide decisions of appropriate population, duration, and timing of holidays. FDA’s final labeling revision will likely be conservative. For now, FDA recommends health care providers be aware of the possible risk and consider periodic reevaluation of continued therapy beyond 5 years. Patients should report any thigh or groin pain, which should be evaluated for possible femur fracture. Adverse events can be reported to FDA’s MedWatch Reporting program at www.fda.gov/MedWatch. By Ryan Rodriguez, PharmD REFERENCES 1. Center for Drug Evaluation and Research. Background Document for Meeting of Advisory Committee for Reproductive Health Drugs and Drug Safety and Risk Management Advisory Committee. September 9, 2011. Available at http://www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM270958.pdf. 2. Salminen ST, Pihlajamaki HK, Avikainen VJ, et al. Population based epidemiologic and morphologic study of femoral shaft fractures. Clin Orthop Relat Res 2000;2419. 3. Burge R, Dawson-Hughes B, Solomon DH, et al. Incidence and Economic Burden of Osteoporosis-Related Fractures in the United States, 2005–2025. J Bone Miner Res 2007;22:465–475. 4. Ott SM. Editorial: Long-Term Safety of Bisphosphonates. J Clin Endocr Metab 90(3):1897–1899 5. Nieves JW, Bilezikian JP, Lane JM, et al. Fragility fractures of the hip and femur: incidence and patient characteristics. Osteoporos Int 2010;21:399408. 6. Lenart BA, Neviaser AS, Lyman S, et al. Association of lowenergy femoral fractures with prolonged bisphosphonate use: a case control study. Osteoporos Int 2009;20:135362. 7. Neviaser AS, Lane JM, Lenart BA, et al. Lowenergy femoral shaft fractures associated with alendronate use. J Orthop Trauma 2008;22:34650. 8. Black DM, Kelly MP, Genant HK, ET AL. Bisphosphonates and fractures of the subtrochanteric or diaphyseal femur. N Engl J Med. 2010 May 13;362(19):1761-7