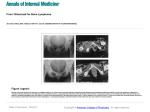
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
ASH NEWS AND REPORTS® Ask the Hematologist COMPENDIUM 2010-2015 This collection contains updated clinical information on malignant and nonmalignant hematology as profiled in The Hematologist. Editor: Jason Gotlib, MD, MS Ask the Hematologist Compendium INTRODUCTION A core feature of the ASH publication The Hematologist: ASH News and Reports is the department known as “Ask the Hematologist” (ATH). The aim of ATH is to provide clinicians with expert opinion about challenging diagnostic and management problems that span the areas of malignant and nonmalignant hematology. In some instances, the question posed in the ATH column is premised on a very specific patient scenario; in others, the question is intended to elicit a broader review of practice-changing diagnostic algorithms or therapeutic advances in hematology. ATH articles commonly touch on disease pathophysiology that helps anchor clinical explanations to the biologic underpinnings of disease. We have found that this translational mix of information has appeal not only to clinicians, but also to physiciancollection updated clinical information on malignant scientistscontains and basic researchers. This and nonmalignant hematology as profiled in The Hematologist. In this compendium, we have compiled ATH articles spanning the years 2010 to 2015. With the exception of some more recent works, we have asked authors to provide state-of-the-art updates to the content where applicable; to that end, authors either have rewritten their ATH articles or have provided an update/commentary that introduces their original piece. We hope that readers find the ATH compendium to be an easily digestible learning companion that serves as a “portable feast” for the practicing hematologist. Jason Gotlib, MD, MS Editor-in-Chief irector of Communications D Jenifer Hamilton, CAE Managing Editor Juana Llorens, MS Editorial Associate Laura Santini Graphic Designer grayHouse design Table of Contents M A L I G N A N T H E M AT O L O GY : 73-Year-Old Male with Stage 0 Chronic Lymphocytic Leukemia with Skin Involvement and No Evidence of Mantle Cell Lymphoma................................ 5 Approach to Patient with a Bladder Tumor with Severe Hematuria and Hemoglobin Level of Eight with Associated Dyspnea............................................. 6 Recommendation of CHOP-Rituximab/More Intensive Regimen Such as Dose-Adjusted EPOCH-Rituximab for Cyclin D1 Expression in DLBCL.......... 7 FDA Approval of the JAK Inhibitor Ruxolitinib and the Management of Patients with Myelofibrosis................................................................................................ 8 Evaluation of Young Adult with Bone Marrow Failure............................................................................................................................................................................... 10 Standard of Care for Treating Older Patients with Chronic Lymphocytic Leukemia........................................................................................................................ 12 Optimal, Upfront Management of Mantle Cell Lymphoma....................................................................................................................................................................... 14 Approach to the Diagnosis and Management of Mastocytosis.............................................................................................................................................................. 16 Assessment and Management of Patients with Chemotherapy-Induced Peripheral Neuropathy................................................................................................ 18 Approach to the Evaluation of Patients with an IgM Monoclonal Protein............................................................................................................................................ 20 Treatment of Elderly Patients with Acute Myeloid Leukemia................................................................................................................................................................... 22 Treatment of Relapsed/Refractory Hairy Cell Leukemia........................................................................................................................................................................... 24 Diagnosis of Immune Pathophysiology in Patients with Low-Risk Myelodysplastic Syndromes................................................................................................... 25 Hematopathologic Overview of Lymphocytosis......................................................................................................................................................................................... 27 N O N M A L I G N A N T H E M AT O L O GY : Specific Thrombophilia Work-Up Approach................................................................................................................................................................................................ 30 Recommendations for Thrombophilia Testing for Women before Oral Contraceptives are Prescribed.................................................................................... 31 Treatment of Pregnant Female with Mesenteric Thrombosis and History of Spontaneous Fetal Losses.................................................................................. 32 Factor Replacement for Surgery in 48-Year-Old Male with Factor XI Deficiency............................................................................................................................. 33 Immune Thrombocytopenia with Pulmonary Embolism and Deep-Vein Thrombosis: Recommendations for Bone Marrow Aspirate and Biopsy......... 34 Heparin-Induced Thrombocytopenia Being Treated with Argatroban with Persistent Thrombocytopenia................................................................................ 35 Approach to the Diagnosis and Management of the Anemia of Chronic Inflammation................................................................................................................... 37 Approach to the Diagnosis and Management of Transfusion-Related Acute Lung Injury............................................................................................................... 39 Criteria for Selecting Patients with Sickle Cell Anemia for Allogeneic Hematopoietic Stem Cell Transplantation.................................................................. 41 Diagnosis and Management of Platelet Alloimmunization........................................................................................................................................................................ 42 Recommendation of Light Transmission Aggregometry as Part of the Evaluation of a Patient with a Suspected Bleeding Disorder............................... 44 Anti-Phospholipid Antibodies and Pregnancy............................................................................................................................................................................................. 46 Treatment Approaches to Castleman Disease with the Advent of Anti-Interleukin-6 Therapy...................................................................................................... 48 Treatment with Direct Oral Anticoagulants.................................................................................................................................................................................................. 50 Ask the Hematologist M A L I G N A N T H E M AT O L O GY This collection contains updated clinical information on malignant and nonmalignant hematology as profiled in The Hematologist. 73-Year-Old Male with Stage 0 Chronic Lymphocytic Leukemia with Skin Involvement and No Evidence of Mantle Cell Lymphoma – March/April 2010 The author has indicated that there has been no update regarding the content of this article since the original publication date in 2010. SUSAN O’BRIEN, MD Associate Director for Clinical Science, Chao Family Comprehensive Cancer Center; Medical Director, Sue & Ralph Stern Center for Clinical Trials and Research; Professor, Division of Hematology/Oncology, School of Medicine, University of California, Orange, CA (Editor’s Note: The original question was submitted to Dr. O’Brien through ASH’s Consult a Colleague, a service for ASH members that helps facilitate the exchange of information between hematologists and their peers. She expanded her answer for print.) The Question I have been following a 73-year-old male with Stage 0 chronic lymphocytic leukemia (CLL) for two years. He has a normal hemoglobin and platelet count with a white blood cell count of around 15,000 and cytogenetics showing 13q deletion and trisomy 12. He recently had two small asymptomatic skin lesions removed from his arm and back. These consisted of a clonal population of B cells that expressed Bcl-2 diffusely and were CD20positive, but CD5- and CD43-negative. This was similar to his original flow cytometry from peripheral blood. This case was interpreted by a referral lab as B-cell lymphoma. The patient is completely asymptomatic with no evidence of liver or spleen enlargement or adenopathy on CT scans. I’m not sure how to put this all together; is this atypical CLL with skin involvement? There is no evidence of mantle cell lymphoma. My Response This case presents two unusual aspects of CLL. The first is that this patient has small skin lesions that appear to be deposits of CLL. The second unusual feature is that this patient’s cells are CD5-negative. Looking at the whole picture, I would agree that this is most likely atypical CLL with skin involvement. I will discuss the phenotype first. CLL is a B-cell malignancy that also expresses CD5. The other well-known lymphoid malignancy that expresses CD5 in addition to B-cell markers is mantle cell lymphoma, which can present in a leukemic phase. Typically, the morphology of mantle cell is significantly different from CLL, although in less classical cases, there may be some blurring of the morphology. CD23 is a useful marker to differentiate them, as this antigen is present on CLL cells and not present on mantle cells. However, in this case we have a patient who appears to have CLL but is CD5-negative. From a practical point of view, the good news is that the approach to MZL is similar to that of CLL — namely watch-and-wait in early-stage asymptomatic patients. One difference, however, is in the approach to treatment. MZL cells have very strong expression of CD20 (as opposed to CLL cells), and these patients have dramatic responses to single-agent rituximab. In patients who need treatment, I generally use 375 mg/m2 rituximab weekly for eight weeks and have had patients in complete remission for years after this single agent, whereas the efficacy of single-agent rituximab in CLL is much weaker. The reason I would diagnose CLL in this case is the presence of a 13q deletion and trisomy 12. These are wellknown abnormalities seen in CLL, and, in fact, the presence of trisomy 12 is commonly associated with atypical features. Skin involvement in CLL is quite rare. Probably the largest series to examine this came from a group of pathologists in Austria, who describe cutaneous infiltrates of CLL in 42 patients. The five-year survival of these patients was 67 percent, suggesting that they had a good, if not a more favorable, prognosis compared to those without skin involvement; there was no attempt to match them for comparable prognostic features to patients without skin involvement.2,3 I’ve been following some patients for years with skin involvement. In particular, I’m thinking of one who has small deposits on both earlobes and has very indolent CLL that’s never required treatment. If these lesions are in a problematic area, low-dose radiation might be an option, but generally this is not an indication for systemic therapy, and it can simply be monitored. 1. Ugo V, Leporrier N, Salaun V, et al. Deciphering leukemic B-cell chronic lymphoproliferative disorders. Leuk Lymphoma. 2006;47:2088-2095. 2. Colburn DE, Welch MA, Giles FJ. Skin infiltration with chronic lymphocytic leukemia is consistent with a good prognosis. Hematology. 2002;7:187-188. 3. Cerroni L, Zenahlik P, Höfler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010. Dr. O’Brien receives research support from Genentech. Other B-cell lymphoproliferative disorders are usually CD5-negative, and the ones that could potentially overlap with CLL are prolymphocytic leukemia (PLL), marginalzone lymphoma/leukemia (MZL), and follicular lymphoma in a leukemic phase. Again, morphologies differ between all these diagnoses, but there can be overlap. The most common overlap is between CLL and MZL. That’s because classically the diagnosis of marginal zone is, as the name implies, based on pathologic examination of lymph nodes. Patients presenting with leukemic disease without significant adenopathy render this diagnosis more difficult to ascertain. A group of investigators in France evaluated 165 leukemic patients with B-cell lymphoproliferative disorders selected on the basis of a score of ≤3 in the Royal Marsden Hospital scoring system, which implies atypical cases that don’t fit clearly into one diagnostic entity.1 Fifty-four cases were CD5-negative. Morphological and phenotypical examinations were able to reveal the diagnosis in 24 of these, including the most common diagnosis of MZL in 11 cases. Thirty CD5-negative cases still could not be fully characterized after morphological and phenotypical examination. Only five of these patients had superficial lymphadenopathy, and in three cases where lymph node biopsy was done, the diagnosis of MZL was established. All in all, 14 were diagnosed as MZL, which was the most common diagnosis, and eight patients remained “unclassified.” These are likely the patients we would call atypical CLL. M A L I G N A N T H E M A T O L O G Y 5 The Hematologist Compendium 2010-2015 Approach to Patient with a Bladder Tumor with Severe Hematuria and a Hemoglobin Level of Eight with Associated Dyspnea – July/August 2010 UPDATE/COMMENTARY I would like to re-state the concept that hemorrhage occurs within Virchow’s triad. To manage bleeding, one must identify and rank the pathophysiological factors affecting vascular integrity, blood flow, and hemostasis. 1) The most important cause of painless gross hematuria in anyone, even when it is self-limited, is vascular injury from bladder cancer. In a patient without known transitional cell carcinoma with painless gross hematuria, one must direct all interventions at getting tissue, and this usually begins with cystoscopy. Between 10 and 20 percent of these patients will be diagnosed with bladder cancer.U1 2) Local hyperfibrinolysis contributes to hematuria in patients with urothelial tumors, but systemic hyperfibrinolysis is unlikely in the absence of disseminated cancer. Recent data from liver transplant recipients indicate that thromboelastometry is two to four times more sensitive than thromboelastography (TEG) for identifying a systemic fibrinolytic state. TEG was 99 percent specific but only 23 percent sensitive, so it cannot be used to rule out systemic hyperfibrinolysis.U2 3) Intractable hematuria from almost any cause – with or without systemic hyperfibrinolysis – should be treated with systemic antifibrinolytic therapy plus local measures, which could include intravesical instillation with formalin, alum, MICHAEL H. KROLL, MD Professor of Medicine, University of Texas MD Anderson Cancer Center, Houston, TX (Editor’s Note: The original question was submitted to Dr. Kroll through Consult a Colleague. He expanded his answer for print.) The Question A 78-year-old male with a bladder tumor had severe hematuria and a hemoglobin level of 8 g/dL with associated dyspnea. Urologists were unable to evaluate the tumor secondary to severe bleeding in the bladder. Coagulation testing revealed a normal prothrombin time (PT) of 10 seconds, but his activated partial thromboplastin time (aPTT) was elevated at 108 seconds, lowering to 56 seconds when mixed with normal plasma. Levels of coagulation factors VIII, IX, X, XI, and XII were normal, although the report noted the possibility of an inhibitor. Von Willebrand factor (VWF) studies were normal, as were assays for anticardiolipin antibodies (ACA) and lupus anticoagulant (LA). The patient continues to bleed and his hemoglobin continues to drop. Should any other work-up be done? Given that he had no benefit from fresh frozen plasma infusions, should I give factor VII complex? My Response Bleeding is impaired hemostasis and therefore can be evaluated from the same perspective that Virchow organized for thrombosis — as dysfunction within the triad of blood (mainly soluble coagulation factors, fibrinolytic apparatus, or platelets), blood vessel (mainly its structural integrity or permeability), and/or blood flow (mainly the effects of ischemia or hyperperfusion). In most cases of bleeding, a simple, single explanation emerges. But in more complicated cases, like the one in question, I try to identify and rank the hemostatic elements that have broken down. When I see bleeding from the bladder, I focus first on tumor-associated hemorrhage. This is because one must immediately direct therapy toward the cancer, if the fundamental pathophysiological element is tumor-erosive hemorrhage. But how does one establish the diagnosis if hematuria is so severe as to prevent visualization and biopsy of the bladder mucosa? I would retreat slightly and look for disturbed and perhaps related hemostatic elements that could be reversed and thereby permit proper evaluation of the root cause of the hematuria. When there is severe hematuria, perhaps related to transitional cell carcinoma, I ask the question: Is there hyperfibrinolysis? The malignant urothelium is rich in urokinase-type plasminogen activator,1 and local and sometimes systemic hyperfibrinolysis develops and contributes to bleeding in patients with bladder cancer. Systemic hyperfibrinolysis is not apparent by examining the PT and aPTT, but always results in elevated fibrin split products and d-dimers. It can be identified by TEG or by measuring the euglobulin lysis time. Neither test is available in every clinical laboratory, but in our institution we perform TEG. When there is hyperfibrinolysis, or anything else that de-stabilizes polymerized fibrin, the TEG tracing over 30 to 60 minutes shows a decline in the clot tensile strength — the plateau begins to decay or decline.2 When hyperfibrinolysis is identified, one can try to control it with an antifibrinolytic drug such as aminocaproic acid or tranexamic acid.3 The main risk of these treatments in someone with hematuria is ureteral obstruction if blood refluxes and clots in a ureter, so one must monitor urine output and serum creatinine concentration closely. M A L I G N A N T H E M A T O L O G Y 6 or prostaglandin PGF2α (best for low flow bleeding); arterial embolization; or hydrostatic pressure or urinary diversion (as a last resort). For patients with bladder cancer, hypofractionated radiotherapy or chemoperfusion with mitoxantrone often controls bleeding.U3 4) Today, more than five and a half years since this case was first reviewed, there are no new data that recombinant activated factor VII (rFVIIa) should be used for massive hematuria from any condition other than acquired hemophilia or inherited hemophilia with an inhibitor. The therapeutic index of rFVIIa as a nonspecific hemostatic agent is unfavorable.U4 U1. Nielsen M, Qaseem A, High Value Care Task Force of the American College of Physicians. Hematuria as a marker of occult urinary cancer: advice for high-value care from the American College of Physicians. Ann Intern Med. 2016;164:488-497. U2. Abuelkasem E, Lu S, Tanaka K, et al. Comparison between thromboelastography and thromboelastometry in hyperfibrinolysis detection during adult liver transplantation. Br J Anaesth. 2016;116:507-512. U3. Abt D, Bywater M, Engeler DS, et al. Therapeutic options for intractable hematuria in advanced bladder cancer. Int J Urol. 2013;20:651-660. U4. Levi M, Levy JH, Andersen HF, et al. Safety of recombinant activated factor VII in randomized clinical trials. N Engl J Med. 2010;363:1791-1800. In this patient, there may be a weak coagulation factor VIII inhibitor. The isolated elevated aPTT must reflect a deficiency of, or an inhibitor to, either factor XII, XI, IX, or VIII and the incomplete aPTT correction with mixing (from 108 to 56 seconds) indicates that there is an inhibitor. The most commonly seen inhibitor is a lupus anticoagulant, which is an antiphospholipid antibody that prolongs an in vitro clotting assay, which, in this case, would be the aPTT. But lupus anticoagulants are not associated with bleeding — except in the rare case where they have antiprothrombin activity4 — and this patient’s anticardiolipin antibody assay and lupus anticoagulant screen were normal. So the most parsimonious explanation for this case of bleeding and abnormal coagulation studies is the presence of the most common acquired inhibitor encountered in a bleeding patient: an acquired factor VIII inhibitor. It can emerge in patients with solid tumors,5 including bladder cancer.6 It almost always causes clinically significant bleeding that can be controlled with a pharmacological intervention such as rFVIIa.7 And the inhibitor itself (an auto-antibody to factor VIII that causes rapid clearance) is often reduced or eradicated with immunosuppressive therapy.8 To evaluate for this possibility, I suggest repeating the factor VIII assay and measuring the inhibitor titer in Bethesda units, which is the inverse of the dilution of the patient’s plasma that decreases factor VIII activity in control plasma by 50 percent. My speculation is that when this patient’s factor VIII level is repeated, it will be borderline low or low and that he will have an inhibitor level < 5 units. If an inhibitor is identified, I would administer rFVIIa, although some would first try human factor VIII in cases of low titer acquired factor VIII inhibitors.8 Should one use rFVIIa under any circumstances? Assuming that the PT and platelet count remain satisfactory, I would first try bladder irrigation. If this or other urological manuevers don’t work and everyone remains mystified by the pathophysiology, I would begin aminocaproic acid at an infusion rate of 1 gm/h. If aminocaproic acid doesn’t work, I would then add rFVIIa, but only at 20 to 30 mcg/kg and beginning at four- or even six-hour intervals.9 This dose is prohemostatic but probably causes fewer thromboses than the conventional dose of 90 mcg/kg every two hours.10 1. Dubeau L, Jones PA, Rideout III WM, et al. Differential regulation of plasminogen activators by epidermal growth factor in normal and neoplastic human urothelium. Cancer Res. 1988;48:5552-5556. 2. Traverso CI, Caprini JA, Arcelus JI. The normal thromboelastogram amd its interpretation. Semin Thromb Hemost. 1995;21:7-13. 3. Mannucci PM. Hemostatic drugs. New Eng J Med. 1998;339:245-253. 4. Galli M, Barbui T. Antiprothrombin antibodies: detection and clinical significance in the antiphospholipid syndrome. Blood. 1999;93:2149-2157. 5. Hauser I, Lechner K. Solid tumors and factor VIII antibodies. Thromb Haemost. 1999;82:1005-1007. 6. Kreuter M, Retzlaff S, Enser-Weis U, et al. Acquired haemophilia in a patient with gram-negative urosepsis and bladder cancer. Haemophilia. 2005;11:181-185. 7. Huth-Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;94:566-575. 8. Franchini M, Lippi G. Acquired factor VIII inhibitors. Blood. 2008;112:250-255. 9. O’Connell NM, Perry DJ, Hodgson AJ, et al. Recombinant FVIIa in the management of uncontrolled hemorrhage. Transfusion. 2003;43:1711-1716. 10. Mayer SA, Brun NC, Begtrup K, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358:2127-2137. Dr. Kroll is part of Eisai’s Speakers’ Bureau. The Hematologist Compendium 2010-2015 Recommendation of CHOP-Rituximab/More Intensive Regimen Such as DoseAdjusted EPOCH-Rituximab for Cyclin D1 Expression in DLBCL – March/April 2011 UPDATE/COMMENTARY Since this article was originally published in 2011, limited additional data have emerged on diffuse large B-cell lymphoma (DLBCL) expressing cyclin D1. Although most of these data have, as before, been anecdotal, an international consortium study of patients with DLBCL treated with the rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP) regimen, identified cyclin D1 expression in 30 patients of a total of 1,435 included in the study (2.1%). None of these patients had rearrangements of the CCND1 gene and gene expression profiling showed them to be roughly equally distributed between the germinal center B-cell (GCB) and activated B-cell (ABC) subtypes. In terms of clinical behavior, patients with cyclin D1 expression were predominantly male, and were significantly younger than those without cyclin D1. No differences were observed in clinical behavior or overall and progression-free survival between those with and without cyclin D1 expression, suggesting that R-CHOP should remain the standard of care in this group.U1 With respect to the broader question of the clinical utility of molecular subtyping of DLBCL, data from the phase I/II study of ibrutinib have demonstrated an encouraging response rate, largely restricted to patients with the ABC subtype, which is known to overexpress Bruton’s tyrosine kinase.U2 This observation is now being tested in a randomized clinical trial in untreated DLBCL, comparing R-CHOP alone with R-CHOP plus ibrutinib. The importance of evaluating these preliminary observations in randomized trials is underlined by recent results from two studies addressing the inclusion of bortezomib in first-line therapy of DLBCL. Despite the early encouraging results cited in the original article, two recent randomized studies have failed to show a benefit to the inclusion of bortezomib in patients with ABC-type diffuse large B-cell lymphoma.U3,U4 We are still waiting on mature results from a third trial from the United Kingdom, which has now closed. U1.Ok CY, Xu-Monette ZY, Tzankov A, et al. Prevalence and clinical implications of cyclin D1 expression in diffuse large B-cell lymphoma (DLBCL) treated with immunochemotherapy: a report from the International DLBCL Rituximab-CHOP Consortium Program. Cancer. 2014;120:1818-1829. U2.Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922-926. U3.Offner F, Samoilova O, Osmanov E, et al. Frontline rituximab, cyclophosphamide, doxorubicin, and prednisone with bortezomib (VR-CAP) or vincristine (R-CHOP) for non-GCB DLBCL. Blood. 2015;126:1893-1901. U4.Leonard JP, Kolibaba K, Reeves JA, et al. Randomized phase 2 open label study of R-CHOP ± bortezomib in patients (pts) with untreated non-germinal center (non-GCB) subtype diffuse large B-cell lymphoma (DLBCL): results from the Pyramid Trial (NCT 00931918). Blood. 2015;126:811. and overall survival rates independent of the IPI. This difference in prognosis is seen for patients treated both with CHOP and with CHOP-rituximab.3 JOHN W. SWEETENHAM, MD, FRCP, FACP Professor of Medicine, Senior Director of Clinical Affairs, and Executive Medical Director, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT The Question A previously healthy 72-year-old male presented with a new diagnosis of clinical stage IVB DLBCL, with maxillary sinus involvement and involvement of multiple bony sites. His ECOG performance status was 0 and his International Prognostic Index (IPI) score was 3 based on his age, clinical stage, and involvement of multiple extranodal sites, all of which are adverse factors in the IPI. A biopsy from the maxillary sinus confirmed DLBCL by morphology and was positive for cyclin D1. Since cyclin D1 expression in DLBCL is reported to have a poor prognosis, should this patient be treated with R-CHOP or should he receive a more intensive regimen such as dose-adjusted EPOCH-rituximab? My Response This clinical case raises several important questions regarding the management of advanced DLBCL, partly related to diagnostic criteria and to the optimal treatment regimen, but also regarding the broader issue of whether different immunophenotypic or molecular subtypes of DLBCL should be treated differently. Cyclin D1 is a nuclear protein with a well-defined role as a regulator of cell cycle progression from G1. Its deregulation is central to the pathogenesis of some B-cell neoplasms, especially mantle cell lymphoma (MCL), where it is over-expressed in about 90 percent of cases, as well as in myeloma and hairy cell leukemia. In the vast majority of MCLs, cyclin D1 over-expression is associated with the t(11;14), whereas this association is much less common in other B-cell neoplasms.1 Reports of the expression of cyclin D1 in DLBCL have been conflicting, partly because different methodologies are used for detection of cyclin D1 and partly because the morphologic distinction between DLBCL and the blastoid variant of MCL can be problematic, particularly since other immunophenotypic characteristics of DLBCL and MCL can sometimes overlap. Despite these challenges, there are several case reports and some larger series that describe the presence of cyclin D1 in cases of DLBCL that are negative for CD5 but have other classical markers of DLBCL, such as bcl-6 and MUM-1.2 As indicated by the questioner, there is a suggestion from the limited literature that patients with cyclin D1-positive DLBCL have a worse prognosis. In the largest published series, nine of 10 patients had died from progressive disease after a median of 29 months from diagnosis.2 However, these data are anecdotal, and if this represents a true clinicopathologic entity, there are insufficient data to indicate whether it has a meaningfully different prognosis from other cases of DLBCL. In contrast, other immunophenotypic and molecular features of DLBCL have been shown to have defined predictive and prognostic value. Examples include the adverse prognostic effect of bcl-2 expression and lack of bcl-6 expression, both of which can apparently be overcome by the addition of rituximab to standard chemotherapy regimens such as CHOP. Gene expression profiling studies have identified two major molecular subtypes of DLBCL; one has a gene expression profile (GEP) consistent with GCB (GCB-like), and one has a profile consistent with ABC (ABC-like). Molecular subtyping by GEP has also been shown to have prognostic value, as patients with ABC-like GEPs have poorer progression-free M A L I G N A N T H E M A T O L O G Y 7 The clinical utility of these findings has been limited by the lack of routine availability of suitable tissue and resources for conducting GEP studies. As a result, several groups have explored the use of immunohistochemical (IHC) surrogates to assign GCB and ABC subtypes.4 Since the correlation between cell of origin identified by GEP and IHC is high, these algorithms are gaining widespread use as a method of risk stratification in clinical trials. Although the prognostic significance of cell of origin (GCB versus ABC) in DLBCL is well established, the question of whether this or any other biologic predictor of outcome should be used to direct therapy for DLBCL is unknown. The dose-adjusted EPOCH-R regimen mentioned by the questioner uses infusional drug scheduling and pharmacodynamic dosing based on hematologic toxicity to exploit tumor proliferation as a target mechanism. Phase II studies of this regimen have shown impressive results and, interestingly, no difference in progression-free or overall survival according to GCB or non-GCB cell of origin.5 As a result, this regimen is now being compared directly with CHOP-rituximab in a randomized, prospective intergroup study led by the CALGB, which includes an analysis of cell of origin by GEP. The results of this study will not be available for several years, so for now, CHOP-rituximab remains the standard regimen for first line therapy of DLBCL, irrespective of cell of origin or expression of other biomarkers. Early results from some recent studies suggest that this is likely to change. Two recent studies have suggested that the addition of bortezomib to standard chemotherapyrituximab combinations for DLBCL produces improvements in response rates and progression-free survival rates, which may be restricted to patients with the ABC subtype.6,7 Additionally, data are emerging to suggest that some recently identified therapeutic targets such as Bruton’s tyrosine kinase (Btk) are differentially expressed in ABC- versus GCB-like DLBCL, providing a rationale for limiting trials of these agents to those patients with specific molecular subtypes. Future progress in the treatment of DLBCL is likely to emerge from further characterization of specific molecular targets. In the meantime, CHOP-rituximab should be regarded as standard, first-line therapy for all patients with DLBCL outside clinical trials. 1. Swerdlow SH, Zukerberg LR, Yang WI, et al. The morphologic spectrum of non-Hodgkin’s lymphomas with BCL1/cyclin D1 gene rearrangements. Am J Surg Pathol. 1996;20:627-640. 2. Ehinger M, Linderoth J, Christensson B, et al. A subset of CD5- diffuse large B-cell lymphomas expresses nuclear cyclin D1 with aberrations at the CCND1 locus. Am J Clin Pathol. 2008;129:630-638. 3. Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large B-cell lymphomas. N Engl J Med. 2008;359:2313-2323. 4. Meyer PN, Fu K, Greiner TC, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol. 2011;29:200-207. 5. Wilson WH, Dunleavy K, Pittaluga S, et al. Phase II study of dose-adjusted EPOCH and rituximab in untreated diffuse large B-cell lymphoma with analysis of germinal center and post-germinal center biomarkers. J Clin Oncol. 2008;26:2717-2724. 6. Dunleavy K, Pittaluga S, Czuczman MS, et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009;113:6069-6076. 7. Ruan J, Martin P, Furman RR, et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol. 2010. 2011;29:690-697. Dr. Sweetenham indicated no relevant conflicts of interest. The Hematologist Compendium 2010-2015 FDA Approval of the JAK Inhibitor Ruxolitinib and the Management of Patients with Myelofibrosis – March/April 2012 UPDATE/COMMENTARY Five-year follow-up data are now available for the COMFORT-I and II trials of ruxolitinib.U1,U2 In COMFORT-II, 53 percent of patients treated with ruxolitinib achieved a ≥ 35 percent reduction in spleen volume from baseline at any time during treatment. U1 Improvement in spleen size was maintained with long-term therapy, with the median duration of maintenance of spleen volume reduction being 3.2 years. The KaplanMeier probability of maintaining this reduction was 51 percent at three years and 48 percent at five years. Median OS was not reached in the ruxolitinib arm and was 4.1 years in the best available therapy (BAT) arm. There was a 33 percent reduction in risk of death with ruxolitinib compared with BAT (HR, 0.67; 95% CI, 0.44-1.02; p = .06). The probability of survival at five years was 56 percent with ruxolitinib and 44 percent with BAT. There were no new or unexpected adverse effects (AEs) with long-term ruxolitinib exposure. The most commonly reported AEs in patients who received ruxolitinib any time were thrombocytopenia (52%), anemia (49%), diarrhea (36%), and peripheral edema (33%); grade 3/4 AEs included anemia (23%), thrombocytopenia (15%), and pneumonia (6%). There were similar rates of evolution to leukemia between the ruxolitinib (5.5%) and BAT (6.8%) arms. The long-term efficacy and safety results of COMFORT-1U2 are very similar to COMFORT-II, and were presented at the ASCO annual meeting in June 2016 at the time of this writing.U2 Ruxolitinib has immunosuppressive properties that are in part related to impairment of dendritic cell, NK-cell, and T-cell function.U3-5 Atypical and/or opportunistic infections have been observed in MF patients treated with ruxolitinib, including reactivation of tuberculosis,U6 viral hepatitis,U7 and herpes zoster. Additionally, there have been case reports of toxoplasmosis and cytomegalovirus retinitis,U8,9 as well as pneumocystis and fungal pneumonias. Although there are no consensus guidelines regarding use of anti-infective prophylaxis (nor is the live zoster vaccine recommended), treating physicians should perform risk assessments for these infections in patients before commencing ruxolitinib, with tailored laboratory screening where appropriate. Phase III clinical development of another JAK inhibitor, fedratinib, was discontinued because of the development of several cases of Wernicke’s encephalopathy. In vitro studies indicate that oral absorption of dietary thiamine update is inhibited by fedratinib, which blocks the human thiamine transporter.U10 Inhibition of the thiamine transporter does not appear to be an issue with the other JAK inhibitors currently being tested. Another JAK inhibitor, pacritinib, has been evaluated in two phase III clinical trials vs. BAT: PERSIST-1 (JAK inhibitor-naive MF patients) and PERSIST-2 (prior JAK2 inhibitors treatment allowed and platelet count < 100 × 109/L required). In the PERSIST-1 trial, pacritinib produced reductions of spleen volume and symptoms and a 26 percent rate of red blood cell transfusion-independence with otherwise minimal myelosuppression.U11 However, in February 2016, the FDA placed a clinical hold on pacritinib due to an interim analysis of the PERSIST-2 trial which demonstrated a detrimental effect on survival in the ruxolitinib compared to BAT arm. Specifically, a relatively higher rate of intracranial hemorrhage, cardiac failure, and cardiac arrest was observed with ruxolitinib therapy.U11 Momelotinib remains the only other JAK inhibitor currently in phase III clinical trial testing, with results expected soon from the SIMPLIFY-1 trial (momelotinib vs. ruxolitinib; 1:1 randomization; no prior JAK inhibitor therapy) and SIMPLIFY-2 trial (momelotinib vs. BAT; 2:1 randomization; a key inclusion criterion is anemia and/or thrombocytopenia with prior ruxolitinib treatment). Although a limited proportion of patients treated with ruxolitinib or the other JAK inhibitors may demonstrate a reduction in bone marrow fibrosis and/or JAK2 V617F allele burden over time,U1 disappearance of fibrosis or complete molecular JASON R. GOTLIB, MD, MS Associate Professor of Medicine (Hematology) and Director, Hematology Fellowship Program, Stanford University School of Medicine and Stanford Cancer Institute, Stanford, CA The Question How does the recent FDA approval of the JAK inhibitor ruxolitinib influence your management of patients with myelofibrosis? My Response Myelofibrosis (primary and post-PV/ET MF) is a Philadelphia chromosome-negative myeloproliferative neoplasm with a natural history characterized by progressive anemia, spleen enlargement due to extramedullary hematopoiesis, and potential for evolution to acute myeloid leukemia (AML). Impairment of quality of life is due to both massive splenomegaly (e.g., early satiety and abdominal discomfort) and inflammatory cytokines, which mediate debilitating symptoms such as night sweats, fevers, muscle/bone pain, and M A L I G N A N T H E M A T O L O G Y 8 remissions is exceedingly rare. More rigorous data are needed to establish the long-term effects of JAK inhibitors on marrow fibrosis improvement or stabilization compared to conventional therapies. Meanwhile, phase I/II trials of ruxolitinib in combination with other therapies with different mechanisms of action are being undertaken to assess whether response rates and/or drug-related anemia can be improved. Such medications include immunomodulatory agents (e.g., lenalidomide; pomalidomide); hypomethylating agents (e.g., azacitidine/decitabine); phosphoinositide-3 kinase inhibitors (e.g., buparlisib; idelalisib); histone deacetylase inhibitors (e.g., panobinostat); hedgehog pathway inhibitors (e.g., sonidegib); and agents to stimulate erythropoiesis (erythropoietin; danazol).U12 Phase II monotherapy trials of antifibrotics (e.g., PRM-151) and the telomerase inhibition (imetelstat) are in progress in intermediate or high risk MF patients who did not respond to, or did not tolerate ruxolitinib. Type II JAK inhibitors, which only inhibit mutant JAK2 (e.g., CHZ868), are in pre-clinical development and may offer the hope of more in-depth responses since currently available JAK inhibitors inhibit both wildtype and mutant JAK2.U13 Lastly, the role of ruxolitinib in the pre-and post-transplant setting is the subject of several ongoing clinical trials. U1. Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term efficacy and safety in COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis: 5-year final study results. Blood. 2015;126:59. U2. Gupta V, Verstovsek S, Mesa RA, et al. Long-term outcomes of ruxolitinib therapy in patients with myelofibrosis: 5-year update from COMFORT-I. ASCO Meeting Abstracts. 2016:7012. U3. Heine A, Held SA, Daecke SN, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122:1192-1202. U4. Schönberg K, Rudolph J, Vonnahme M, et al. JAK Inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res. 2015;75:2187-2199. U5. Parampalli Yajnanarayana S, Stübig T, Cornez I, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. 2015;169:824-833. U6. Hopman RK, Lawrence SJ, Oh ST. Disseminated tuberculosis associated with ruxolitinib. Leukemia. 2014;28:1750-1751. U7. Caocci G, Murgia F, Podda L, et al. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2014;28:225-227 U8. Goldberg RA, Reichel E, Oshry LJ. Bilateral toxoplasmosis retinitis associated with ruxolitinib. New Engl J Med. 2013;369:681-683. U9. von Hofsten J, Johnsson Forsberg M, Zetterberg M. Cytomegalovirus retinitis in a patient who received ruxolitinib. New Engl J Med. 2016;374:296-297. U10. Zhang Q, Zhang Y, Diamond S, et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos. 2014;42:1656-1662. U11. Mesa RA, Egyed M, Szoke A, et al. Results of the PERSIST-1 phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). ASCO. 2015:LBA7006 U12. Mascarenhas J. Looking forward: novel therapeutic approaches in chronic and advanced phases of myelofibrosis. Hematology Am Soc Hematol Educ Program. 2015;2015:329-339. U13. Meyer SC, Keller MD, Chiu S, et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell. 2015;28:15-28. Dr. Gotlib has received (or is pending) research funding from Incyte (manufacturer of ruxolitinib), Sanofi-Aventis (fedratinib), Gilead (momelotinib; idelalisib), Promedior (PRM-151), and imetelstat (Janssen). cachexia. The activating JAK2 V617F mutation is present in 50 to 60 percent of patients, and the MPL mutation (W515L/K), resulting in ligand-independent activation of the thrombopoietin receptor, is identified in an additional 5 to 10 percent of individuals. It has become abundantly clear, however, that MPNs such as MF are more genetically complex. Molecular alterations in additional genes and dysregulation of the epigenetic machinery also contribute to disease pathogenesis. Prognostic scoring systems based on clinical and laboratory factors obtained either at the time of diagnosis (IPSS)1 or during the disease course (dynamic IPSS, or DIPSS)2 have been developed in order to estimate both overall survival and risk of progression to AML. The IPSS uses five adverse prognostic factors: age >65, hemoglobin <10 g/dL, white blood cell count >25,000/mm3, constitutional symptoms, and peripheral blood blasts >1 percent. The DIPSS-Plus refines prognosis assessment by incorporating three additional adverse risk factors: platelet count <100,000/mm3, the need for red blood cell transfusions, and poor-risk cytogenetics. Using the IPSS as an example, patients can be stratified into one of four risk groups: low (score 0), intermediate-1 (score 1), intermediate-2 (score 2), or high (score >3), with a median overall survival among the groups ranging from approximately 11 years to just over two years.1 Age, performance status, and prognostic risk group drive decision making about treatment options. The Table illustrates this point using three patients. Patient 1 is a low-risk patient with excellent performance status, minimally abnormal blood counts and splenomegaly, The Hematologist Compendium 2010-2015 and no constitutional symptoms. Such patients do not warrant immediate treatment and a watch-and-wait approach may be undertaken. At the other end of the spectrum, Patient 3 is a younger, high-risk patient characterized by abnormal blood counts, marked splenomegaly, and poor-risk cytogenetics. The DIPSS-Plus estimate of overall survival is 16 months, and the five- and 10-year leukemia rates are 18 and 31 percent, respectively.2 Given this patient’s younger age and relatively poor prognosis, evaluation for a potentially curative myeloablative hematopoietic stem cell transplant would be encouraged. For patients who require treatment and are not candidates for transplantation, available therapies for MF-related cytopenias, splenomegaly, and symptoms are considered palliative. These options have included chemotherapy such as hydroxyurea; erythropoiesisstimulating agents; immunomodulatory drugs such as thalidomide or lenalidomide, with or without corticosteroids; splenectomy; splenic irradiation; and clinical trials. In November 2011, only four years after commencing clinical trial evaluation, ruxolitinib became the first JAK inhibitor approved by the FDA for MF patients (intermediate- and high-risk). The registration trials for ruxolitinib consisted of two large phase III trials: COMFORT-I was a randomized (1:1), double-blind, multicenter study comparing ruxolitinib 15 or 20 mg twice daily (dose stratified according to baseline platelet count) versus placebo,3 and COMFORT-II was a randomized (2:1), open-label, multicenter trial comparing ruxolitinib 15 or 20 mg bid versus BAT; investigator-selected including no treatment).4 Both trials met the primary endpoint of the percentage of ruxolitinib versus control patients achieving > 35 percent reduction in spleen volume at week 24 (COMFORT-I: 41.9% vs. 0.7%) and week 48 (COMFORT-II: 28.5% vs. 0%). After 24 weeks in the COMFORT-I trial, the proportion of patients with ≥50 percent improvement in total symptom score (using the myelofibrosis symptom assessment form) was 45.9 percent versus 5.3 percent (ruxolitinib vs. placebo, p<0.0001). Anemia and thrombocytopenia were common ruxolitinib-related adverse events but rarely led to drug discontinuation, and the drug was otherwise well tolerated. In an updated analysis of COMFORT-I, there was a significant overall survival benefit with ruxolitinib; at a median follow-up of 51 weeks, there were 13 (8.4%) deaths in the ruxolitinib group and 24 (15.7%) deaths in the placebo arm.5 The implications of these short-term data are unclear since JAK inhibitors exert modest or no impact on fundamental disease-related features such as JAK2 mutant allele burden or marrow fibrosis. Ruxolitinib’s potency as a “spleen shrinker” and “symptom mitigator” is shared by other JAK inhibitors currently in clinical trials (e.g., fedratinib [formerly SAR302503 and TG101308], momelotinib [formerly CYT387], pacritinib [formerly SB1518], etc.). Patients with or without the JAK2 V617F mutation respond similarly. Therefore, first-line treatment with a JAK inhibitor would be an ideal choice for Patient 2 described in the table and could be considered a bridging option for Patient 3 until a transplant is performed. In an ad hoc analysis of the COMFORT-I and COMFORT-II trials, worsening of spleen size as well as symptoms and quality-of-life scores were similar between the placebo and BAT control groups.6 Although it may be premature to abandon conventional treatments such as hydroxyurea, the comparative superiority of ruxolitinib in these trials justifies its frontline use for intermediate- and high-risk MF patients in whom the primary goal is improvement of splenomegaly and constitutional symptoms. For MF patients with anemia or RBC transfusion-dependence as the predominant clinical issue, no standard of care currently exists. In this regard, differences among JAK inhibitors may prove informative in tailoring particular agents to specific patient presentations. For example, the JAK 1/2 inhibitor momelotinib (CYT387) has demonstrated improvements in hemoglobin/transfusiondependence in an ongoing phase II trial.7 Lenalidomide (or pomalidomide, on a trial basis) may also elicit benefits in anemia in ~20 to 30 percent of MF patients. Table. MF Patient Profiles Age/Gender ECOG Performance Status Patient 1 Patient 2 Patient 3 63/F 61/M 48/M 0 1 1 WBC (/mm3) 9,200 18,700 31,000 Hb (g/dL) 11.1 10.5 8.2, Transfusiondependent 155,000 218,000 85,000 0 2 5 Platelet Count (/mm3) PB Blasts (%) Constitutional Symptoms Splenomegaly* Cytogenetics JAK2 V617F Mutation IPSS Score/Risk Group No Yes Yes 4 cm 15 cm 21 cm Normal Normal Monosomy 5 Yes No Yes 0/Low 2/Intermediate-2 4/High PB=peripheral blood *below left costal margin by palpation 1. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895-2901. 2. Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011; 29:392-397. 3. Verstovsek S, Mesa RA, Gotlib JR, et al. Results of COMFORT-I, a randomized double-blind phase III trial of JAK 1/2 inhibitor INCB18424 (424) versus placebo (PB) for patients with myelofibrosis (MF). J Clin Oncol. ASCO Meeting Abstracts. 2011;29:6500. 4. Harrison CN, Kiladjian J, Al-Ali HK, et al. Results of a randomized study of the JAK inhibitor ruxolitinib (INC424) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera-myelofibrosis (PPV-MF) or post-essential thrombocythemia-myelofibrosis (PET-MF). J Clin Oncol. ASCO Meeting Abstracts. 2011;29:LBA6501. 5. Verstovsek S, Mesa RA, Gotlib J, et al. Consistent benefit of ruxolitinib over placebo in spleen volume reduction and symptom improvement across subgroups and overall survival advantage: results from COMFORT-I. Blood. ASH Annual Meeting Abstracts. 2011;118: 278. 6. Mesa RA, Verstovsek S, Cervantes F, et al. Comparison of the efficacy of placebo and best available therapy for the treatment of myelofibrosis in the COMFORT studies. Blood. ASH Annual Meeting Abstracts. 2011;118:1753. 7. Pardanani A, Gotlib J, Gupta V, et al. An expanded multicenter phase I/II study of CYT387, a JAK- 1/2 inhibitor for the treatment of myelofibrosis. Blood. ASH Annual Meeting Abstracts. 2011;118:3849. 8. Bhagwat N, Koppikar P, Kilpivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Blood. ASH Annual Meeting Abstracts. 2011;118:122. Dr. Gotlib receives research funding from Incyte (manufacturer of ruxolitinib), SanofiAventis (manufacturer of SAR302503), YMBiosciences (manufacturer of CYT387), and Celgene (manufacturer of lenalidomide). Given the rapid “on/off” action of JAK inhibitors, caution must be undertaken when stopping these agents because of the potential for return of symptoms in a short period of time. During therapy, disease “persistence” (incomplete regression or return of splenomegaly and symptoms) can occur and may be partly explained biologically by reactivation (phosphorylation) of JAK2 through heterodimerization with JAK family members JAK1 and TYK2.8 Future directions will therefore be focused on combination trials of JAK inhibitors with other targeted targets (e.g., histone deacetylase inhibitors, anti-fibrotics, PI3 kinase/ AKT inhibitors) to improve the quality and duration of responses. New Open-Access Peer-Review Journal, Blood Advances The new bi-weekly journal from ASH, Blood Advances, will make its debut at the 58th ASH Annual Meeting in San Diego in December. Founding editor, Dr. Robert Negrin, describes the journal as “novel approaches for presentation of original content, including approaches that can only be performed using electronic format, including enhanced graphics, video, and discussion.” This journal will complement ASH’s flagship journal by publishing all new content not covered by Blood. For more information, visit www.bloodadvances.org. M A L I G N A N T H E M A T O L O G Y 9 The Hematologist Compendium 2010-2015 Evaluation of Young Adult with Bone Marrow Failure – May/June 2012 UPDATE/COMMENTARY UPDATE BY DARIA V. BABUSHOK, MD, PhD 1 AND TIMOTHY S. OLSON, MD, PhD 2 1. Instructor, Division of Hematology/Oncology, Department of Medicine, Hospital of the University of Pennsylvania; Comprehensive Bone Marrow Failure center, Division of Pediatric Hematology, The Children’s Hospital of Philadelphia, Philadelphia, PA 2. Director, Comprehensive Bone Marrow Failure Center, Division of Pediatric Hematology, The Children’s Hospital of Philadelphia; Assistant Professor of Pediatrics, Perelman School of Medicine, University of Pennsylvania; Blood and Marrow Transplant Section, Division of Pediatric Oncology, The Children’s Hospital of Philadelphia, Philadelphia, PA With the increasing use of next-generation sequencing (NGS), our understanding of the genetics underlying inherited bone marrow failure syndrome (IBMFS) and predisposition to myelodysplastic syndrome (MDS) has grown significantly. The numbers of genes linked to classic IBMF, such as Fanconi Anemia (FA; ≥ 17 genes), Diamond Blackfan Anemia (DBA; ≥ 14 genes), and Dyskeratosis Congenita (DC; ≥ 11), have increased. Additionally, throughout the last five years, five new genes (GATA2, ANKRD26, SRP76, DDX41, and ETV6) have been linked to familial MDS and leukemia (Figure 1). Importantly, it is increasingly recognized that these conditions can initially present in adulthood. With the improved knowledge of the inherited BMF and MDS, the differential diagnosis for a Figure 2. Evaluation young adult presenting with BMF or myelodysplasia should be broadened to include not only the classical IBMFSs such as DC and FA, but also the emerging familial MDS/ AML predisposition syndromes. Although the knowledge of these disorders remains imperfect, we expect that our diagnostic ability will improve as more genes and syndromes are identified. Presently, the comprehensive evaluation of a young adult with a suspected IBMFS or familial MDS/leukemia syndrome (Figure 2) continues to rely on functional testing of affected biological pathways to screen for the classic polygenic IBMFSs DC and FA. With the ready availability of clinically approved comprehensive NGS panels targeting genes known to cause IBMFS and familial MDS/AML, these panels have become a useful and frequently more economical alternative to sequential single-gene testing, particularly in cases in which syndromic features suggestive of a specific diagnosis are not present. of Bone Marrow Failure in Children and Young Adults Figure 1. Timeline of Discovery of the Emerging Inherited Myelodysplastic Syndrome/Acute Myeloid Leukemia Predisposition Syndromes. BEN OSHRINE, MD, 1 MONICA BESSLER, MD, PhD 2 1. Fellow in Hematology-Oncology, Children’s Hospital of Philadelphia, Philadelphia, PA 2. Buck Family Professor in Hematology, Pediatric & Adult Comprehensive Bone Marrow Failure Center, Children’s Hospital of Philadelphia and The Hospital of the University of Pennsylvania, University of Pennsylvania School of Medicine, Philadelphia, PA The Case The patient is a 29-year-old male who presented with a two-year history of progressive pancytopenia (WBC 2.2 x 109/L, ANC 0.83 x 109/L; Hb 9.9 g/dL with 2.9% reticulocytes; platelet count 26 x 109/L). He was of short stature with discrete facial dysmorphisms and mildly impaired cognitively. The family history was negative for BMF, but there was an extensive history of early cancer on the maternal side. Bone marrow examination revealed marked hypocellularity without dysplasia or excess blasts. The patient became transfusion-dependent, so an unrelated bone marrow transplant (BMT) had been recommended as definitive treatment. The Question How do you evaluate a young adult with BMF? What features suggest an underlying IBMFS as the etiology? How do you evaluate for these disorders, and why is it important? Our Response IBMFS is a collection of genetic disorders including FA, DC, DBA, Shwachman-Diamond syndrome (SDS), severe congenital neutropenia, and congenital megakaryocytic thrombocytopenia that are associated with insufficient blood cell production, congenital anomalies, and, in some cases, an increased risk of malignancies including MDS, leukemia, and certain solid tumors.1 IBMFS is not always inherited. Disease may also occur M A L I G N A N T H E M A T O L O G Y 10 sporadically due to a de novo mutation, which may account for more than 50 percent of all cases for some of the IBMFS (e.g., certain genetic forms of DBA or DC). Sporadic appearance of disease may also be due to low penetrance of the causative mutation or genetic anticipation in which clinical phenotype becomes more severe in successive generations as observed in autosomal dominant DC. In this case, family members may carry the mutation but not have clinical symptoms (silent mutation carriers). IBMFS can affect one, two, or all blood cell lineages. Abnormal blood cell counts are frequently not present at birth but develop during childhood, adolescence, or later in life.2 In some cases, BMF never becomes clinically manifest, and the affected individual may seek medical attention because of an unusually early onset of MDS, leukemia, cancer, or other non-hematologic manifestations associated with the condition, such as pulmonary fibrosis in individuals with DC.3 While once thought to be strictly the domain of pediatric hematologists, with improved insights into the spectrum of disease expression and genetic testing, IBMFS is increasingly being diagnosed and cared for in “adult” hematology clinics.4 When? For all patients presenting with BMF, the possibility of an underlying IBMFS should be considered. Such consideration is particularly relevant for patients in whom BMT is being considered as treatment. IBMFS should also be considered in children and young adults presenting with MDS or MDS/leukemia, as these clonal disorders are late complications of IBMFS and may be the first symptom of disease in about 20 percent of cases, typically in the young adult population.3 IBMFS, with disease onset in adults, often lack the classic physical stigmata, such as abnormal thumbs (FA) or dystrophic fingernails (DC), and other extra-hematopoietic manifestations, such as small stature or discrete facial anomalies, may be subtle.4 Therefore, it is difficult to propose an age limit for the exclusion of an IBMFS, as these disorders may be diagnosed even in old age. However, the therapeutic consequences are probably most significant when diagnosed in pediatric and young adult patients. The diagram above suggests the diagnostic genetic workup for the young adult presenting with BMF. The Hematologist Compendium 2010-2015 What? Why? Currently, there are more than 80 different genes that may cause IBMFS.5 With the technology of whole-exome and whole-genome sequencing, the number of genetic abnormalities associated with IBMFS is expected to increase dramatically, making it impractical to perform testing for every gene in every affected individual. Many of the mutated gene products fall into distinct biologic pathways (e.g., the FA DNA repair pathway, the maintenance of telomeres in DC, or ribosome biogenesis in DBA). Some clinically available screening tests investigate the integrity of the affected pathway and, if abnormal, direct a focused and more economical approach to genetic testing. Given the important therapeutic implications associated with diagnosis and the availability of sensitive diagnostic tests, screening for FA and DC is recommended in every patient presenting with BMF or early MDS/leukemia. Due to abnormal DNA repair, chromosomes of dividing cells of patients with FA are hypersensitive to breakage induced by cross-linking agents such as diepoxybutane (DEB) or mitomycin C (MMC), and this characteristic is the basis of the diagnostic test for the disease.6 The DEB/MMC sensitivity test uses patient lymphocytes in the analysis and is available in several major medical centers with a specific interest in FA. Telomeres of nucleated peripheral blood cells are excessively short in DC.7 The natural history, response to treatment, and surveillance requirements differ significantly between patients with acquired BMF and IBMFS. Patients with IBMFS are typically not responsive to immunosuppressive therapies that may be used for acquired aplastic anemia. Furthermore, the presence of IBMFS requires special considerations for BMT, both in terms of family member donor selection and choice of transplant conditioning regimen, as conventional conditioning regimens can be excessively toxic or fatal for some of the IBMFS.8 IBMFS may require surveillance for long-term complications that may be accelerated by the choice of treatment (pulmonary fibrosis, osteopenia, MDS/leukemia, and other non-hematologic malignancies). Finally, the diagnosis of IBMFS requires informed genetic counseling for both the affected individual and the family. Clinical telomere length measurements by PCR, flow cytometry, or Southern blotting are available through several commercial laboratories. Results are usually provided as “percentile” in comparison to normal controls. The interpretation of the clinical significance of the measured telomere lengths can be difficult and often needs to be done in the context of disease manifestations. In general, telomere lengths within the normal distribution exclude DC to be the cause of BMF. In children and young adults with BMF caused by DC, the telomere lengths of total peripheral blood lymphocytes are usually 2 to 3 standard deviations below the telomere lengths distribution of age-matched healthy controls. Short telomere lengths in granulocytes are less specific for DC and can be short in a number of other IBMFS. As telomeres get shorter with age, telomere length becomes less diagnostic for DC in older individuals. If one of these screening tests is abnormal, subsequent genetic testing should be pursued, as only the identification of a disease-causing mutation can confirm the diagnosis of an IBMFS. More specific tests for other IBMFS have to be chosen on an individual basis depending on clinical presentation, family history, and laboratory findings. For example, DBA may be considered in patients who present with macrocytic anemia, low reticulocyte count, and a bone marrow characterized by an isolated erythroid hypoplasia. An elevated erythrocyte adenosine deaminase activity may further support a diagnosis of DBA. A history of symptoms of pancreatic insufficiency in a patient with early-onset MDS may be suggestive for SDS. Genetic testing for mutations in the ribosomal proteins most frequently associated with DBA and for mutations in SBDS (the gene that is mutated in SDS) are available commercially. With wider applicability of novel sequencing technologies, genetic testing for all major IBMFS genes using a single multiplex platform is likely to become available in the not-too-distant future. However, with the increased availability of genetic testing, the interpretation of results and consideration of their clinical significance will become more complicated, requiring management by an experienced team of physicians that includes geneticists, hematopathologists, and hematologists. In the patient presented above, testing of peripheral blood lymphocytes revealed telomere lengths within the normal distribution, excluding DC as the cause of his progressive BMF.9 No increased chromosomal sensitivity to DEB was observed in his peripheral blood lymphocytes and only a mildly increased chromosomal fragility with the formation of few radials using MMC was observed. These findings were not sufficient for a firm diagnosis but raised our suspicion of FA and led to further testing of skin fibroblasts that demonstrated significant chromosomal breakage and numerous radial formations with both DEB and MMC. Together, these results suggested FA as the cause of the patient’s progressive BMF. Genetic reversion (the process by which a lymphohematopoietic cell undergoes spontaneous correction of an FA-causing gene mutation) and blood mosaicism are known to occur in about 15 percent of FA patients.10 This process is the likely cause of the ambiguous FA screening results from the peripheral blood lymphocytes in this patient. 1. Bessler M, Mason PJ, Link DC, Wilson DB. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT, eds. Nathan’s and Oski’s hematology of infancy and childhood. 7th edition. Philadelphia: W.B. Saunders Co; 2008;307-395. 2. Shimamura A. Clinical approach to marrow failure. Hematology (ASH Education Program Book). 2009:329-337. 3. Seif AE. Pediatric leukemia predisposition syndromes: clues to understanding leukemogenesis. Cancer Genet. 2011;204:227-244. 4. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101-122. 5. Parikh S, Bessler M. Recent insights into inherited bone marrow failure syndromes. Curr Opin Pediatr. 2012;24:23-32. 6. Cervenka J, Arthur D, Yasis C. Mitomycin C test for diagnostic differentiation of idiopathic aplastic anemia and Fanconi anemia. Pediatrics. 1981;67:119-127. 7. Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dykeratosis congenita. Blood. 2007;110:1439-1447. 8. Mehta P, Locatelli F, Stary J, et al. Bone marrow transplantation for inherited bone marrow failure syndromes. Pediatr Clin North Am. 2010;57:147-170. 9. Du HY, Pumbo E, Ivanovich J, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113:309-316. 10. Gregory JJ Jr., Wagner JE, Verlander PC, et al. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA. 2001;98:2532-2537. Drs. Oshrine and Bessler indicated no relevant conflicts of interest. The Hematologist: ASH News and Reports Podcasts The Hematologist offers a series of podcasts that enhance the audience’s reading experience by presenting key results and additional insights from published articles in a convenient audio format. Add this podcast to your favorite RSS feed and follow us on SoundCloud and iTunes. For a full listing of podcasts, visit www.hematology.org/thehematologist/podcasts/. M A L I G N A N T H E M A T O L O G Y 11 The Hematologist Compendium 2010-2015 Standard of Care for Treating Older Patients with Chronic Lymphocytic Leukemia – July/August 2012 UPDATE/COMMENTARY Since our original article was published, the treatment landscape of chronic lymphocytic leukemia (CLL) has evolved rapidly. The recently approved agents for front-line CLL therapy (obinutuzumab and ibrutinib) demonstrate a favorable toxicity profile, making them excellent treatment options for elderly CLL patients. Obinutuzumab, an Fcγ-engineered anti-CD20 monoclonal antibody, combined with chlorambucil, improved progression-free survival (PFS) compared with chlorambucil alone in untreated elderly patients with CLL and comorbid conditions. U1 The regimen was well tolerated. Therefore, obinutuzumab plus chlorambucil was approved for front-line CLL treatment. In elderly untreated patients with CLL without del(17p13.1), ibrutinib, an oral inhibitor of Bruton’s tyrosine kinase, demonstrated significantly improved overall response and overall survival rates compared with chlorambucil-treated patients.U2 In other studies, similar efficacy was noted in ibrutinib-treated CLL patients with and without del(17p13.1).U3 Ibrutinib was well tolerated. Uncommon but serious (≥ grade 3) adverse events in ibrutinibtreated patients were atrial fibrillation (≈ 6%) and major bleeding (1-4%, especially in patients receiving warfarin). Subsequently, ibrutinib was approved for all frontline CLL patients. Based on the availability of these novel therapies, our previous treatment algorithm must be revised. If an elderly untreated CLL patient does not 1) require anticoagulation with warfarin, 2) have uncontrolled atrial fibrillation, or 3) have malabsorption, ibrutinib should be considered. If the patient has one of the listed comorbid conditions and does not express del(17p13.1), obinutuzumab plus chlorambucil or the less-favored bendamustine plus rituximab regimen should be considered. In the setting of comorbid conditions and expression of del(17p13.1), high-dose methylprednisolone plus rituximab therapy should still be considered. In summary, approval of the novel agents obinutuzumab and ibrutinib have established a new standard of care for elderly patients with CLL. U1.Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101-1110. U2.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373:2425-2437. U3.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213-223. Standard frontline therapy for younger patients with cytogenetic abnormalities other than del(17p13.1) or del(11q22.3) is the combination of fludarabine, cyclophosphamide, and rituximab (FCR) or fludarabine and rituximab. However, a subset of CLL patients ≥ age 60 in a large study of frontline therapy with FCR were more likely to require early treatment discontinuation due to myelosuppression and other non-hematologic toxicities,3 which likely limits the clinical benefit of the regimen in this population. In addition, Eichhorst et al. compared firstline CLL therapy consisting of either fludarabine or chlorambucil in a group of 193 patients ≥ 65 years. Although fludarabine improved the overall response rate (ORR), improved the percentage of complete remissions (CR), and increased the time to treatment failure, there was no difference in PFS or overall survival (OS) between the two groups. Notably, the fludarabine group demonstrated a shorter median survival time and higher rate of toxicity, indicating that there is no major clinical benefit of using fludarabine over chlorambucil in the older population.4 These findings were confirmed by Woyach DEBORAH M. STEPHENS, DO, 1 AND JOHN C. BYRD, MD 2 1. Assistant Professor, Division of Hematology and Hematologic Malignancies, University of Utah, Salt Lake City, UT 2. D. Warren Brown Chair of Leukemia Research, Professor, and Director of the Division of Hematology, The Ohio State University, Columbus, OH The Question Is there a standard of care for treating older patients with CLL? Our Response Despite recent progress in the understanding and management of CLL, the disease remains essentially incurable. Incidence of CLL is significantly higher in the elderly (those > 65 years old), as the median age at diagnosis is 72 and an estimated 70 percent of newly diagnosed patients are over 65.1 Despite the predominance of elderly CLL patients, this group has been under-represented in clinical trials because many have major medical co-morbidities or are perceived likely to tolerate therapy poorly. This omission leaves clinicians who treat CLL without definitive data on which therapy to choose for the large population of older adults. Figure Therapeutic Recommendations for Elderly Patients with Chronic Lymphocytic Leukemia who Meet Criteria for Therapy Eligible for clinical trial? At initial presentation, the clinical spectrum of CLL ranges from an asymptomatic patient, identified by routine blood work, to a symptomatic patient who experiences rapid progression to death from complications of CLL. Therefore, the first decision to make is whether or not treatment is required. Indications for consideration of treatment include: clinical symptoms (fevers, night sweats, weight loss, or painful lymphadenopathy or splenomegaly), non-immune-mediated cytopenias (hemoglobin < 11g/dL or platelets < 100 × 1012/L), autoimmune hemolytic anemia, or thrombocytopenia (ITP) that is poorly responsive to standard immunosuppressive therapy and rapidly progressive disease (lymphocyte count rising to > 300 × 109/L or rapidly enlarging lymph nodes, spleen, or liver). Isolated mild thrombocytopenia (platelets 70-100 × 1012/L) often represents chronic ITP and can be followed closely without treatment if symptoms are absent. Treatment decisions can be informed by identifying factors that influence response rates, duration of response, and prognosis, including specific interphase cytogenetic abnormalities such as del(17p13.1) and del(11q22.3), complex cytogenetics (> 3 abnormalities) determined by stimulated metaphase karyotype, unmutated immunoglobulin heavy chain gene status, Rai stage III/ IV, Zap-70 expression in 20 percent or more of peripheral blood lymphocytes, and elevated serum β2–microglobulin concentration. Trials by the German CLL Study Group have factored in co-morbid features assessed by the CIRS-G scale2 for choosing therapy in elderly patients; however, this score is subjective, and, to date, no study has documented the benefit of this scale over simpler methods. Only performance status and the presence of del(17p13.1) or del(11q22.3) influence our treatment choice (Figure). If an elderly CLL patient requires therapy and is eligible for and willing to participate, we recommend enrollment in a clinical trial. Outside of a trial, if the patient’s performance status will allow therapy, he or she should be assigned to one of three groups according to the presence of FISH/cytogenetic abnormalities: del(17p13.1), del(11q22.3), and all others. Yes No Enroll in clinical trial Functional performance status acceptable for treatment? Yes No Cytogenetics other than Del(11q) or Del(17p) expressed Del(11q) expressed Del(17p) expressed Chlorambucil +/rituximab FCR,* PCR,** or bendamustine + rituximab High-dose methylprednisolone + rituximab Consider chlorambucil, rituximab + chlorambucil, or palliative care *FCR=fludarabine, cyclophosphamide, and rituximab; **PCR=pentostatin, cyclophosphamide, and rituximab M A L I G N A N T H E M A T O L O G Y 12 The Hematologist Compendium 2010-2015 and colleagues5 who reviewed the experience of elderly CLL patients across all completed Cancer and Leukemia Group B trials and demonstrated no benefit for fludarabine treatment in patients > 70 years. In contrast, patients of all age groups appeared to benefit from the addition of rituximab. Recently, Hillmen and colleagues published a phase II trial in which rituximab was combined with chlorambucil in 50 patients (median age = 70.5 years). ORR was 84 percent, with infection and neutropenia being the most common adverse effects.6 Therefore, for an elderly CLL patient without either del(17p13.1) or del(11q22.3), we recommend therapy with chlorambucil (10 mg/m2 orally days 1-7 of a 28-day cycle) +/rituximab (375 mg/m2 day 1, cycle 1 and 500 mg/m2 day 1, cycles 2-6). 1. Howlader N, Noone AM, Krapcho M, et al(eds). SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations), SEER Stat Fact Sheet: Chronic Lymphocytic Lymphoma, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/statfacts/html/clyl.html, based on November 2011 SEER data submission, posted to the SEER web site, April 2012. Accessed June 1, 2012. Patients with del(11q22.3) do not have favorable outcomes in the absence of fractionated alkylator-based therapy. For older CLL patients with del(11q22.3), we recommend therapy with FCR, PCR (pentostatin, cyclophosphamide, and rituximab) or bendamustine (70 mg/ m2 days 1 and 2 of 28-day cycle) and rituximab (375 mg/m2 first cycle, 500 mg/m2 subsequent 5 cycles). Using the latter regimen, Fischer et al. reported an ORR of 90.9 percent in 117 patients (median age = 64 years) with minimal major toxicities. A subgroup analysis of patients with del(11q22.3) showed a 90.5 percent ORR.7 5. Woyach JA, Ruppert AS, Peterson B, et al. Impact of age on outcomes following initial therapy with various chemotherapy and chemoimmunotherapy regimens in patients with chronic lymphocytic leukemia: Results of CALGB studies. Blood. ASH Annual Meeting. 2011;118:289. Treatment options are limited for older patients with del(17p13.1). Many propose the use of alemtuzumab, but we don’t recommend this therapy due to significant risk of infectious complications and limited response duration. Instead, we recommend rituximab (375 mg/ m2 weekly for 12 weeks) in combination with high-dose methylprednisolone (1 g/m2 days 1-3 of each 4-week cycle). Using this regimen, James and colleagues reported that in a relatively small study of 28 patients (29% ≥ age 70) the ORR was 96 percent (100% in the > 70-yearold subset) with CR observed in 32 percent (38% in > 70 years). Adverse effects were found to be minimal.8 In general, for patients being treated with this regimen, we recommend hospitalization during the first three days of cycle 1 as tumor lysis syndrome, metabolic aberrations, and fluid retention can occur. 2. Miller MD, Paradis CF, Houck PR, et al. Rating chronic medical illness burden in geropsychiatric practice and research: application of the Cumulative Illness Rating Scale. Psychiatry Res. 1992;41:237-248. 3. Keating MJ, O’Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23:4079-4088. 4. Eichhorst BF, Busch R, Stilgenbauer S, et al. First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood. 2009;114:3382-3391. 6. Hillmen P, Gribben JG, Follows A, et al. An open-label phase II study to investigate the safety and efficacy of rituximab plus chlorambucil in previously untreated patients with CD20-positive B-cell chronic lymphocytic leukemia. Blood. ASH Annual Meeting. 2009;114:3428. 7. Fischer K, Cramer P, Stilgenbauer S, et al. Bendamustine combined with rituximab in first-line therapy of advanced CLL: a multicenter phase II trial of the German CLL Study Group. Blood. ASH Annual Meeting. 2009;114:205. 8. James AF, Castro JE, Sandoval-Sus JD, et al. Rituximab and high-dose methylprednisolone for the intial treatment of chronic lymphocytic leukemia is associated with promising clinical activity and minimal hematologic toxicity. Blood. ASH Annual Meeting Abstracts. 2008;112:47. Drs. Stephens and Byrd indicated no relevant conflicts of interest. For elderly, unfit patients without del(17p13.1), for whom we have concerns that therapy will be poorly tolerated, we typically recommend chlorambucil or rituximab alone or palliative supportive care. Elderly, unfit patients with del(17 p13.1) are unlikely to respond to either of these therapies and should be directed to supportive care. WATCH THE EXPERTS In summary, due to paucity of definitive data on management of the elderly CLL population, we encourage enrollment of these patients in clinical trials. However, when enrollment in a clinical study is not feasible, we use a treatment approach based on FISH/ cytogenetic analysis together with our assessment of the individual patient’s capacity to tolerate therapy (Figure). The CLL treatment landscape is evolving rapidly. Promising new therapies that are currently undergoing clinical trials include lenalidomide, ibrutinib, GS1101, ofatumumab, GA101, ABT-199, and TRU-016. While we await the outcome of these and other studies, a carefully considered approach to the management of elderly patients with CLL is essential. Jennifer Woyach, MD Dr. Jennifer Woyach discusses the latest drug treatments for CLL patients in this video from the ASH Impact Series available on ASH On Demand. To watch this video and more, visit www.ashondemand.org/FreeContent/Videos. M A L I G N A N T H E M A T O L O G Y 13 The Hematologist Compendium 2010-2015 Optimal, Upfront Management of Mantle Cell Lymphoma – November/December 2012 The authors have provided a rewrite of the original article to present all content updates since the publication date in 2012. OREOFE O. ODEJIDE, MD, MPH, 1 AND ANN S. LACASCE, MD, MSc 2 1. Physician, Dana-Farber Cancer Institute, Boston, MA 2. Assistant Professor of Medicine, Harvard Medical School, Boston, MA Question What is the optimal, upfront management of mantle cell lymphoma (MCL)? Our Response MCL is a rare type of lymphoma, representing about 6 percent of all cases of non-Hodgkin lymphoma.1 Median age at diagnosis is 68 years, and the vast majority of patients are diagnosed with advanced-stage disease. Patients with MCL typically present with generalized lymphadenopathy and extranodal involvement, particularly in the bone marrow, spleen, and gastrointestinal tract. Cyclin D1 expression as a result of t(11;14) (q13;q32) translocation is present in more than 95 percent of cases. Notably, MCL has a poor prognosis, though the median overall survival (OS) has improved over time and is now approximately seven to eight years. Given the small number of randomized clinical trials in previously untreated patients with MCL, none of which have demonstrated an improvement in OS, the optimal initial approach to MCL has not been clearly established. Subsequently, patient participation in well-designed clinical trials is highly recommended. This article will focus on treatment strategies that have been shown to offer the best chance of prolonged remission. A Watch-and-Wait Strategy Is Appropriate for a Subset of Patients Given the burden of disease, the majority of patients with MCL require therapy at diagnosis. However, a subset of patients with splenomegaly and circulating lymphoma cells with a low ki-67, typically without significant lymphadenopathy, may have an indolent course. Data from Weill Cornell Medical Center also suggest that, for selected patients with asymptomatic disease, there is no survival disadvantage to expectant management.2 For those requiring therapy, patients may be divided into categories of young and fit (≤ 65 years without significant comorbidity) or older (> 65 years with inability to tolerate high-dose therapy). Young and Fit Adults with MCL Although the addition of rituximab to conventional chemotherapy regimens has significantly improved prognosis in nearly all subtypes of B-cell non-Hodgkin lymphoma, its addition to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) in MCL has not translated into improved outcomes. The German Low Grade Lymphoma Study Group found that, due to the short durability of the response, rituximab with CHOP (R-CHOP) compared with CHOP did not improve progression-free survival (PFS) despite an improvement in overall response rate (94% vs. 75%).3 Given the disappointing outcome with R-CHOP therapy alone in younger patients, emphasis has been placed on intensification of induction therapies and consolidation strategies with high-dose therapies followed by autologous stem cell rescue (ASCR). six cycles of CHOP to R-CHOP/rituximab with DHAP, suggest both a PFS and OS advantage for patients receiving the intensified regimen.7 In terms of post-transplantation strategies, preliminary results from a randomized study from Europe demonstrated an improved threeyear event-free survival of 88 percent versus 73 percent in patients receiving maintenance rituximab versus observation.8 Another approach to increase the durability of remission in MCL patients uses intensified doses of chemotherapy including cytarabine without stem cell transplantation. A phase II trial of R-HyperCVAD (rituximab, fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine) by the MD Anderson Cancer Center group showed a high complete remission rate of 87 percent and three-year failure-free survival of 64 percent. However, this therapy has significant toxicity, with 8 percent treatment-related mortality.9 The regimen has been tested in the cooperative group setting by the Southwest Oncology Group, with demonstrated three-year PFS and OS of 66 percent and 81 percent respectively.10 Of note, 39 percent of patients were unable to complete all therapy due to significant myelosuppression and infection. Therefore, this therapy should be reserved for young and fit patients with close monitoring for toxicity. Finally, a comparative effectiveness study from the National Comprehensive Cancer Network NHL database comparing initial MCL therapy in younger patients demonstrated that R-CHOP alone was inferior to aggressive therapies such as R-HyperCVAD or R-CHOP followed by HDT and ASCR.11 In general, we favor an intensive treatment approach consisting of an induction regimen followed by HDT and ASCR for patients who are 65 years or younger without significant comorbidity. The optimal induction chemotherapy has not been established, and options include standard or intensified R-CHOP with or without the addition of cytarabinecontaining regimens. Post-transplant maintenance rituximab may be considered, but it is associated with neutropenia and risk of sinopulmonary infections. Older Adults with MCL The use of dose-intensified, cytarabine-containing induction regimens followed by HDT and ASCR is not feasible for most elderly patients, due to excessive toxicity. Therefore, treatment strategies focus on regimens that improve PFS while minimizing toxicity. Results of a double randomized trial comparing R-CHOP versus R-FC (rituximab, fludarabine, and cyclophosphamide) in patients older than 60 years who were ineligible for HDT, with second randomization of responders (complete or partial response) to either maintenance rituximab or maintenance interferon-α showed a clear OS benefit as well as less toxicity for R-CHOP compared with R-FC. The remission duration was also doubled in the maintenance rituximab versus the interferon-α arm. Moreover, among patients who had a response to Figure Recommendations for Initial Management of Mantle Cell Lymphoma A randomized trial comparing myeloablative radiochemotherapy and ASCR versus maintenance interferon-α in patients ≤ 65 years achieving a partial or complete response with CHOP-like induction, with or without rituximab, demonstrated an improved PFS in the radiochemotherapy arm (median PFS, 39 months vs. 17 months) though there was no OS difference.4 This study is the basis for adoption of high-dose therapies and ASCR as consolidative strategies in younger patients. The Nordic Lymphoma Group in the MCL2 study adopted a cytarabinecontaining induction regimen consisting of six cycles of dose-intensified CHOP alternating with high-dose cytarabine with rituximab in cycles two through six, followed by highdose therapy (HDT) and ASCR. The four- and 10-year PFS were highly encouraging at 73 percent and 55 percent, respectively.5 A median event-free survival of 84 months was reported by the French group in a phase II trial using an induction regimen consisting of three cycles of CHOP and three cycles of rituximab with DHAP (dexamethasone, highdose cytarabine, and cisplatin) followed by HDT and ASCR.6 Preliminary results of a randomized phase III trial comparing M A L I G N A N T H E M A T O L O G Y 14 KEY HDT: High-dose therapy ASCR: Autologous stem cell rescue R-CHOP: Rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone BR: Bendamustine and rituximab VR-CAP: Bortezomib, rituximab, cyclophosphamide, doxorubicin, prednisone The Hematologist Compendium 2010-2015 R-CHOP induction, maintenance rituximab significantly improved OS when compared with maintenance interferon-α (4-year OS, 87% vs. 63%), while it had no influence in the group that received R-FC induction.12 1. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma: The Non-Hodgkin’s Lymphoma Classification Project. Blood. 1997;89:3909-3918. Bendamustine is also a highly active drug in MCL. When compared to R-CHOP, bendamustine plus rituximab (BR) resulted in an improved PFS of 35 months compared to 22 months in a randomized study from Europe.13 In early results from a small study in the upfront setting, the combination of lenalidomide plus rituximab yielded overall and complete response rates of 92 and 64 percent respectively with a two-year PFS of 85 percent.14 In a randomized study comparing bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) to R-CHOP in transplant-ineligible patients, VR-CAP resulted in a superior median PFS of 24.7 months compared with 14.4 months with R-CHOP.15 However, there were significantly increased rates of toxicity with VR-CAP, and it is not clear whether its superior PFS will be sustained in the setting of rituximab maintenance after R-CHOP. An ongoing intergroup study in the United States is currently comparing BR with or without bortezomib followed by random assignment to maintenance rituximab with or without lenalidomide. Until these data are mature, we favor R-CHOP or BR therapy followed by maintenance rituximab as upfront therapy in older patients with MCL. 3. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23:1984-1992. 2. Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27:1209-1213. 4. Dreyling M, Lenz G, Hoster E, et al. Early consolidation by myeloablative radiochemotherapy followed by autologous stem cell transplantation in first remission significantly prolongs progression-free survival in mantle-cell lymphoma: results of a prospective randomized trial of the European MCL Network. Blood. 2005;105:2677-2684. 5. Geisler CH, Kolstad A, Laurell A, et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood. 2008;112:2687-2693. 6. Delarue R, Haioun C, Ribrag V, et al. CHOP and DHAP plus rituximab followed by autologous stem cell transplantation in mantle cell lymphoma: a phase 2 study from the Groupe d’Etude des Lymphomes de l’Adulte. Blood. 2013;121:48-53. 7. Hermine O, Hoster E, Walewski J, et al. Alternating courses of 3x CHOP and 3x DHAP plus rituximab followed by a high dose ARA-C containing myeloablative regimen and autologous stem cell transplantation (ASCT) increases overall survival when compared to 6 courses of chop plus rituximab followed by myeloablative radiochemotherapy and asct in mantle cell lymphoma: final analysis of the MCL younger trial of the European Mantle Cell Lymphoma Network. Blood. 2012;623:151. 8. Le Gouill S, Deconinck E, Ghesquieres H, et al. Rituximab maintenance versus WW after R-DHAP plus autologous stem cell transplantation in untreated patients with MCL: Interim analysis of the LYMA trial, a LYSA study. Haematologica. 2015:387-387. 9. Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005;23:7013-7023. 10. Bernstein SH, Epner E, Unger JM, et al. A phase II multicenter trial of hyperCVAD MTX/Ara-C and rituximab in patients with previously untreated mantle cell lymphoma; SWOG 0213. Ann Oncol. 2013;24:1587-1593. 11. LaCasce AS, Vandergrift JL, Rodriguez MA, et al. Comparative outcome of initial therapy for younger patients with mantle cell lymphoma: an analysis from the NCCN NHL Database. Blood. 2012;119:2093-2099. 12. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med. 2012;367:520-531. 13. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381:1203-1210. 14. Ruan J, Martin P, Shah B, et al. lenalidomide plus rituximab as initial treatment for mantle-cell lymphoma. N Engl J Med. 2015;373:1835-1844. 15. Robak T, Huang H, Jin J, et al. bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372:944-953. WATCH THE EXPERTS Dr. Odejide and Dr. LaCasce indicated no relevant conflicts of interest. John C. Byrd, MD Dr. John Byrd of The Ohio State University explains how the identification of novel targets in leukemia cells has impacted patients’ lives at the 2015 ASH Meeting on Hematologic Malignancies in Chicago. Watch the video at www.hematology.org/ Malignancies/Speakers. Read an article by Dr. Byrd in this Compendium on page 12. M A L I G N A N T H E M A T O L O G Y 15 The Hematologist Compendium 2010-2015 Approach to the Diagnosis and Management of Mastocytosis – January/February 2013 UPDATE/COMMENTARY Several new developments have emerged in the field of systemic mastocytosis (SM) since the publication of the “Ask The Hematologist” article on this subject in 2013. Classification In the revised 2016 World Health Organization Classification of hematolymphoid neoplasms, “mastocytosis” was removed as one of the subtypes under the major category of “myeloproliferative neoplasms” and is now classified as its own major category.U1 Smoldering systemic mastocytosis is now a full variant of SM, and no longer a provisional subvariant under indolent SM. Additionally, “systemic mastocytosis with an associated hematologic non-mast cell lineage disease” (SM-AHNMD) has been given a “name makeover”; the designation “SM with an associated hematologic neoplasm” (SM-AHN), a simpler designation, can now be used in place of, or interchangeably with, SM-AHNMD.U1 More attention is being focused on how to better capture the clinical heterogeneity within SM subtypes. For example, for patients with aggressive systemic mastocytosis (ASM) and increased numbers of neoplastic mast cells on a bone marrow aspirate (e.g., 5-19%), the term ASM-t (ASM in transformation) has been proposed to reflect the worse prognosis of these patients and higher risk of transformation to mast cell leukemia (MCL) compared to individuals with ASM patients with <5 percent mast cells on a marrow aspirate.U2 Although the life expectancy of MCL is often less than six months, such patients may also exhibit variable clinical outcomes. The terms “acute MCL” (one or more signs of organ damage) and “chronic MCL” (no evidence of organ damage) have been proposed as variants that respectively denote relatively worse and better outcomes within the spectrum of MCL.U3 However, retrospective and prospective data are needed to confirm whether the absence of presence of organ damage adequately discriminates prognosis between these two MCL subgroups. Biology Although the KIT D816V mutation is identified in approximately 90 percent of SM patients, SM-AHNMD is usually characterized by multiple myeloid mutations, the most common being TET2, SRSF2, ASXL1, CBL, and RUNX1.U4 Patients with a mutation in one or more of these genes (and especially SRSF2, ASXL1, or RUNX1) exhibit a worse prognosis compared to patients who only exhibit the KIT D816V mutation. U5 Sequencing of granulocyte-macrophage colony-forming progenitor cells (CFU-GM) indicates that such mutations often precede the acquisition of KIT D816V.U6 This parallels the finding that TET2 mutations often arise before JAK2 V617F in Philadelphia chromosome-negative MPNs.U7 Notably, CFU-GM from ISM or SSM patients only exhibited the KIT D816V mutation, indicating that indolent SM may be primarily driven by this mutation alone.U6 Treatment In the fully accrued global trial of the multikinase/KIT inhibitor midostaurin in 89 evaluable patients with advanced SM (ASM, SM-AHNMD, and MCL), the overall response rate was 60 percent; 45 percent of patients achieved a major response, defined by complete resolution of ≥1 type of organ damage.U8 The median duration of response was 24.1 months. Median overall survival was 28.7 months and progression-free survival was 14.1 months. Response rates were similar regardless of advanced SM subtype, KIT mutation status, or prior therapy. The serum tryptase level and bone marrow mast cell burden decreased by >50 percent in the majority of patients. In addition to reducing splenomegaly, midostaurin elicited significant improvement of patient-reported symptoms and quality of life that may be related to JASON GOTLIB, MD, MS Associate Professor of Hematology, Stanford University School of Medicine, Stanford Cancer Institute, Stanford, CA The Question What is your approach to the diagnosis and management of mastocytosis? Defining Mastocytosis Mastocytosis is characterized by the accumulation and proliferation of neoplastic mast cells in one or more organs (e.g., skin, bone marrow, spleen, lymph nodes, liver, and gastrointestinal tract). Flushing, diarrhea, anaphylaxis, and neuropsychiatric symptoms resulting from mast cell release of bioactive molecules such as histamine, leukotrienes, and various inflammatory cytokines can impose a significant burden on a patient’s quality of life. Such mediator symptoms may be triggered by physical stimuli, exercise, alcohol, NSAIDs, opioids, insect stings, or certain foods. In advanced disease, mast cell infiltration of tissues can lead to organ damage and shortened survival. The World Health Organization (WHO) includes mastocytosis within the category of myeloproliferative neoplasms (MPN) and divides these disorders into cutaneous and systemic forms.1 The diagnosis of systemic mastocytosis (SM) is based on consensus M A L I G N A N T H E M A T O L O G Y 16 the combined inhibitory effects of midostaurin on neoplastic mast cell proliferation and mediator release. In the MCL patients, where median survival of <6 months is typical, the median overall survival was 9.4 months and not reached in the eight of 16 MCL patients who responded. Further studies are warranted to evaluate the role of combining midostaurin with other drugs with activity in advanced SM (e.g. 2-chlorodeoxadenosine; 2-CdA) and in the pre- and post-transplant setting. New KIT D816V-specific inhibitors (e.g. BLU-285) have recently commenced clinical trial evaluation of patients with advanced SM. Recent data from Dr. Celalettin Ustun and colleagues have provided more clarity on the role of hematopoietic stem cell transplantation (HSCT) in advanced SM which heretofore has been primarily based on case reports and small series.U9 In a large, multicenter retrospective analysis published in 2014, Dr. Ustun and colleagues evaluated the outcomes of 57 SM patients (SM-AHNMD, n=38; ASM, n=7; and MCL, n=12) who underwent allogeneic HSCT. Responses were observed in 70 percent of patients, including a 16 percent complete remission (CR) rate. The remaining 30 percent of responses were split between stable disease (21%) and primary refractory disease (9%). All 38 patients with SM-AHNMD achieved CR of the AHNMD component, but 10 subsequently relapsed with AHNMD, and half of these patients died. The median overall survival for all patients at three years was 57 percent, for all patients, consisting of 74 percent for patients with SM-AHNMD, and 43 and 17 percent for ASM and MCL patients, respectively. The strongest risk factor for worse overall survival was a diagnosis of MCL. Additionally, lower survival was observed in patients undergoing reduced intensity vs. myeloablative conditioning. These data suggest that transplantation can provide extended survival in selected patients, particularly for patients with SM-AHNMD. U1.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405. U2.Valent P. Diagnosis and management of mastocytosis: an emerging challenge in applied hematology. Hematology Am Soc Hematol Educ Program. 2015;2015:98-105. U3.Valent P, Sotlar K, Sperr WR, et al. Refined diagnostic criteria and classification of mast cell leukemia (MCL) and myelomastocytic leukemia (MML): a consensus proposal. Ann Oncol. 2014;25:1691-1700. U4.Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood. 2013;122:2460-2466. U5.Jawhar M, Schwaab J, Schnittger S, et al. Additional mutations in SRSF2, ASXL1, and/or RUNX1 identify a high-risk group of patients with KIT D816V(+) advanced systemic mastocytosis. Leukemia. 2016;30:136-143. U6.Jawhar M, Schwaab J, Schnittger S, et al. Molecular profiling of myeloid progenitor cells in multimutated advanced systemic mastocytosis identified KIT D816V as a distinct and late event. Leukemia. 2015;29:1115-1122. U7.Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. New Engl J Med. 2015;372:601-612. U8.Gotlib J, Kluin-Nelemans HC, George TI, et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N Engl J Med. 2016;374:2530-2541. U9.Ustun C, Reiter A, Scott BL, et al. Hematopoietic stem-cell transplantation for advanced systemic mastocytosis. J Clin Oncol. 2014;32:3264-3274. Dr. Gotlib is Chairman of the Study Steering Committee for the global trial of midostaurin and receives reimbursement for travel expenses from Novartis; Dr. Gotlib receives research funding for administration of the BLU-285 trial. diagnostic criteria (Table) and requires one major plus one minor criterion or three minor criteria. Clinicopathologic criteria are used to further classify SM into subtypes that generally reflect clinical severity and prognosis. In indolent SM (ISM), where bone marrow mast cell burden is relatively low, survival is similar to age-matched populations, and clinical issues are usually related to mediator symptoms.2 Smoldering SM (SSM) is a subtype of ISM that reflects SM in transition toward more advanced disease and is characterized by two or more so-called “B-findings” (Figure), including organomegaly, and indicators of a relatively higher mast cell burden (e.g., > 30% bone marrow mast cells and serum tryptase level > 200 ng/ml). Advanced SM collectively refers to SM subtypes in which organ damage (referred to as “C” findings) is present. This includes the WHO-defined subtypes ASM, MCL, and SM-AHNMD. Approximately 90 percent of AHNMDs represent a myeloid neoplasm such as myelodysplastic syndrome (MDS), MPN, MDS/MPN (e.g., chronic myelomonocytic leukemia), eosinophilic disorders, or acute myeloid leukemia (AML). Rarely, a lymphoid neoplasm may be concurrently diagnosed with SM. Mastocytosis is a challenging disease for many reasons. In the normal state, mast cells are not identified on the peripheral blood differential and represent a small fraction of the myeloid lineage on visual inspection of the marrow aspirate. The idiom “out of sight, out of mind” is apt in the case of SM and, together with its rare incidence, tests the physician to think about this neoplasm in the first place. If present, prototypic mediator symptoms such as flushing, diarrhea, and anaphylaxis may make this task easier. In advanced disease, organ damage such as cytopenias, liver dysfunction, ascites, lytic bone lesions, and hypoalbuminemia with weight loss due to mast cell infiltration of the gastrointestinal tract are sufficiently protean to conceal a unifying diagnosis. Because SM often partners with a myeloid disease, immunohistochemical (IHC) stains (e.g., tryptase, CD117 [KIT], and CD25) are necessary to identify and quantify neoplastic mast cells in trephine biopsies and can The Hematologist Compendium 2010-2015 additionally help unmask SM when it lives in the shadow of other neoplasms. Clinical and biologic heterogeneity, even within specific SM disease subtypes, can make it difficult to gauge disease trajectory and prognosis. In patients with an AHNMD, prognosis is usually related to the associated myeloid neoplasm, highlighting the need to fully characterize and stage the concomitant disease. Diagnosis Table World Health Organization Diagnostic Criteria for Systemic Mastocytosis* Major criterion Multifocal dense infiltrates of mast cells (>15 mast cells in aggregates) detected in sections of bone marrow and/ or other extracutaneous organ(s) Minor criteria a. In biopsy sections of bone marrow or other extracutaneous organs, >25% of the mast cells in the infiltrate are spindle shaped or have atypical morphology or, of all mast cells in bone marrow aspirate smears, >25% are immature or atypical. b. Detection of an activating point mutation at codon 816 in KIT in bone marrow, blood, or another extracutaneous organ In addition to a high index of suspicion, obtaining the c. Mast cells in bone marrow, blood, or other extracutaneous organs express CD2 and/or CD25 in addition to normal expertise of a cadré of subspecialists (hematologists, mast cell markers dermatologists, and allergists/immunologists) as well as d. Serum total tryptase persistently exceeds 20 ng/ml (unless there is an associated clonal myeloid disorder, in which hematopathologists versed with the nuances of mast cell case this parameter is not valid) disease is important in establishing the correct diagnosis. A combination of clinical, morphologic, immunophenotypic, *Requires 1 major + 1 minor criterion or 3 minor criteria and molecular studies is required to establish the diagnosis of SM and its subtype. Bone marrow mast cell burden is best symptoms who do not fulfill criteria for SM (e.g., only one or two minor criteria and/or exhibit quantified by morphologic analysis, and use of the aforementioned IHC stains on the core a low burden of atypical mast cells without aggregates on biopsy) have been given the biopsy is essential. The major criterion for SM requires demonstration of multifocal mast provisional diagnosis of mast cell activation syndrome (MCAS).2 The diagnostic criteria for cell aggregates in the bone marrow (or other extracutaneous organ), of which > 25 percent this mast cell disorder are still evolving and its natural history is not well-defined. are atypical, often spindle-shaped mast cells. In MCL, mast cells account for > 20 percent of nucleated cells on the BM aspirate and form a diffuse infiltrate on the core biopsy with or without circulating mast cells. Multi-parameter flow cytometry of the BM aspirate can also Treatment be used to quantify mast cells in the marrow and complements morphologic evaluation. The overwhelming majority of SM patients (> 80%) carry the activating KIT D816V mutation, which Individuals with symptomatic skin-only disease or SM with mediator symptoms are should be obtained from the bone marrow aspirate or from a core biopsy that is preserved in educated to avoid known triggers and are encouraged to carry an Epipen®, particularly formalin to avoid degradation of DNA for PCR analysis. Determination of KIT D816V mutation those with a history of anaphylaxis or anaphylactoid symptoms. Antihistamines (H1- and status is critical to the diagnostic evaluation of SM, but it also guides treatment decisions. H2-blockers) serve as the foundation for symptom palliation; leukotriene antagonists (e.g., Although KIT D816V is an imatinib-resistant mutation, this tyrosine kinase inhibitor has monteleukast) and mast cell stabilizers (e.g., cromolyn sodium) are typically used for been reflexively misapplied and overused in the treatment of SM. However, a small minority refractory mediator symptoms. Osteoporosis is treated with conventional approaches, and of patients (< 5%) may exhibit juxtamembrane KIT mutations, which exhibit sensitivity to radiotherapy and/or IV bisphosphonates may be used to treat osteolyses or pathologic imatinib in vitro as well as in clinical practice. Such cases illustrate that sequencing the fractures. Patients with advanced SM exhibit shortened survival, and cytoreductive therapy remainder of the KIT gene in SM patients who are negative for codon 816 mutations may prove is used in an attempt to reverse organ damage. Interferon-α with or without corticosteroids fruitful. Total serum tryptase levels generally reflect the increased burden of mast cells in and 2-chloro-deoxyadenosine demonstrate overall response rates in the range of 30 to patients with SM. A serum tryptase level > 20 ng/ml is an additional minor criterion for the 60 percent, including major responses denoting normalization of organ dysfunction.3 diagnosis of SM. Although not always concordant, it is the most useful blood marker to assess Intensive chemotherapy has been used in MCL with modest benefit, and there is a paucity changes in mast cell burden in response to cytoreductive therapy. In advanced disease, of published data regarding the utility of stem cell transplantation for advanced SM. The staging studies include CT of the abdomen to assess hepato/splenomegaly, lymphadenopathy, generally short-lived responses and tolerability of these approaches have reinforced the and ascites; metastatic skeletal survey to evaluate osteolyses and/or pathologic fractures; need to enroll patients in clinical trials evaluating novel agents. The tyrosine kinase inhibitors and DEXA scans to follow osteoporosis, which is common in SM. Some patients with mediator dasatinib and midostaurin exhibit in vitro activity against D816V-mutated KIT, with the latter demonstrating encouraging activity in an ongoing international, multi-center clinical trial.4 In patients with SM-AHNMD, the clinical approach for such patients has been to treat the SM Figure component as if the myeloid neoplasm were not present and to treat the myeloid neoplasm as if SM were not present. However, in clinical practice, priority should be given to treating the disease component that is contributing to the most urgent clinical concerns. Because KIT D816V or other pathogenetic abnormalities may reside in both the abnormal mast cell and associated myeloid clonal cell populations, the future availability of agents that inhibit shared therapeutic targets may make the distinction between the two disease compartments less relevant. Next-generation sequencing approaches of sorted cell populations should inform this approach. The recent identification of CD30 expression on neoplastic mast cells5 also provides an opportunity for testing anti-CD30 antibody approaches (e.g., brentuximab vedotin) in advanced SM. 1. Horny HP, Akin C, Metcalfe DD, et al. Mastocytosis. In: Swerdlow S, Campo E, Lee Harris N, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press. 2008:53-63. 2. Akin C, P, Metcalfe DD. Mast cell activation syndrome: Proposed diagnostic criteria. J Allergy Clin Immunol. 2010;126:1099-1104. 3. Lim KH, Pardanani A, Butterfield JH, et al. Cytoreductive therapy in 108 adults with systemic mastocytosis: Outcome analysis and response prediction during treatment with interferon-alpha, hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol. 2009;84:790-794. 4. Gotlib J, Kluin-Nelemans, George TI, et al. KIT inhibitor midostaurin in patients with advanced systemic mastocytosis: Results of a planned interim analysis of the global CPKC412D2201 trial. Blood. ASH Annual Meeting Abstracts. 2012;120:799. 5. Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol. 2011;24:585-595. Dr. Gotlib is the chairman of the Study Steering Committee for the Novartis-sponsored global trial of midostaurin in advanced systemic mastocytosis. He also receives funding for administration of the clinical trial and payment for travel expenses from Novartis. Dr. Gotlib also serves as the principal investigator on, and receives funding for a trial of brentuximab vedotin in systemic mastocytosis. M A L I G N A N T H E M A T O L O G Y 17 The Hematologist Compendium 2010-2015 Assessment and Management of Patients with ChemotherapyInduced Peripheral Neuropathy – May/June 2014 UPDATE/COMMENTARY The causes, presentation, and National Cancer Institute–Common Terminology Criteria for Adverse Events (NCI-CTCAE) classification of disability from chemotherapy-induced neuropathy have not changed since the original publication of this article in 2014. Unfortunately, no new proven therapies have been identified.U1 Herbal medicines are also ineffective.U2 A pilot study of an electrical method (the “Scrambler”) was positive, and phase III trials are being planned.U3 JANET L. ABRAHM, MD Professor of Medicine at Harvard Medical School; Institute Physician at Dana-Farber Cancer Institute, Boston, MA U1.Schloss J, Colosimo M, Vitetta L. Herbal medicines and chemotherapy induced peripheral neuropathy: a critical literature review. Crit Rev Food Sci Nutr. 2015. [Epub ahead of print]. U2.Majithia N, Temkin SM, Ruddy KJ, et al. National Cancer Institute-supported chemotherapyinduced peripheral neuropathy trials: outcomes and lessons. Support Care Cancer. 2016;24:1439-1447. U3.Pachman DR, Weisbrod BL, Seisler DK, et al. Pilot evaluation of Scrambler therapy for the treatment of chemotherapy-induced peripheral neuropathy. Support Care Cancer. 2015;23:943-951. Diagnosis of CIPN is clinical, and nerve conduction studies, EMGs, and skin biopsies are not needed for diagnosis or management. Chemotherapy doses are generally adjusted for patients with grade 3 or greater neuropathy based on NCI-CTCAE. NCI-CTC for Neuropathy Peripheral Sensory Neuropathy The Case Grade 1: Asymptomatic or loss of deep tendon reflexes or paresthesia The patient is a 54-year-old woman with IgA lambda multiple myeloma, receiving dexamethasone, lenalidomide, and bortezomib therapy. She was healthy before her diagnosis but had mild hypercalcemia and mild renal insufficiency at diagnosis, both of which have resolved with treatment. Grade 3: Severe symptoms limiting self-care ADL She is about to begin her third treatment cycle, but reports painful (pain score of 7/10) numbness and paresthesias in her fingers and feet. The former problem causes clumsiness when using her touch-screen tablet and phone because she cannot accurately feel her fingers touch the screens, and she needs the light on at night because she cannot reliably feel when her feet touch the floor. Her Zubrod (ECOG) performance status is 1. She had similar, but less severe, symptoms at the beginning of her last chemotherapy cycle, but she did not mention them to anyone for fear that treatment would be delayed or discontinued. Grade 2: Moderate symptoms limiting instrumental activities of daily living (ADL) Grade 4: Life-threatening consequences; urgent intervention indicated Grade 5: Death Peripheral Motor Neuropathy Grade 1: Asymptomatic clinical or diagnostic observations only; intervention not indicated Grade 2: Moderate symptoms limiting instrumental ADL Grade 3: Severe symptoms limiting self-care ADL; assistive device indicated Grade 4: Life-threatening consequences; urgent intervention indicated Grade 5: Death During her exam, she was anxious, alert, and oriented. She denied both spine pain and bowel or bladder incontinence. Her exam was unremarkable except for the neurologic examination. She had impaired proprioception, decreased vibration and light touch in her hands to the wrists and feet to the ankles, 4/5 hand grip and finger strength bilaterally, and 4/5 ankle strength. Her ankle deep tendon reflexes (DTRs) were absent (flaccid). The patient under discussion had grade 2 sensory neuropathy by these criteria, which underestimated her disability and functional losses. The NCI-CTC are not as sensitive to patient-reported symptoms or functional loss as are some other scoring systems, but no consensus has yet been reached on a standard for grading neurotoxicity.4,5 Quality-of-life scales may be more relevant (e.g., the CIPN 20 or FACT/GOG-Ntx).6 Laboratory studies showed normal potassium, BUN, creatinine, glucose, calcium, and magnesium. B12 concentration and thyroid function studies were normal. Her paraprotein concentration showed a continuing decrease in response to treatment. Treatment The Question What is your approach to assessment and management of patients with chemotherapyinduced peripheral neuropathy? My Response Presentation/Physical Findings The patient’s presentation is typical of chemotherapy-induced peripheral neuropathy (CIPN) that is characterized by tingling (71%), numbness (58%), and paresthesias or dysesthesias (45%), or spontaneous burning, cramping, or aching pain (40%) that is usually symmetric, begins peripherally and progresses proximally.1 Patients often report allodynia (i.e., pain from something that is usually not painful, like cold temperatures or the touch of clothing), or hyperalgesia (i.e., experiencing more pain than usual from a mildly painful stimulus). The mechanisms of axonal nerve injury vary by agent and include damage to mitochondria (bortezomib, platinum compounds, vincristine, paclitaxel), glial cells and macrophages (bortezomib, oxaliplatin, vincristine, paclitaxel), vasa vasorum (thalidomide, paclitaxel), endoplasmic reticulum (bortezomib), or microtubules (bortezomib, taxanes, and vinca alkaloids).2,3 CIPN induced by vinca alkaloids causes numbness, not pain, and loss of DTRs, proprioception, and motor impairment, such as foot drop.2 Patients receiving thalidomide, bortezomib, or platinum have a mainly sensory neuropathy, which may include loss of vibration, proprioception, and DTRs.2 Taxanes cause both a sensory neuropathy and proximal weakness.2 Only CIPN induced by thalidomide is irreversible. Evaluation In a patient whose CIPN appears only after chemotherapy, the evaluation should include laboratory testing for B12 deficiency, hypothyroidism, hypocalcemia, hypomagnesemia, and hyperglycemia (Figure 1). Because patients with leptomeningeal carcinomatosis or spinal cord compression can present with these symptoms, an MRI of the spine with contrast is recommended for patients with back pain. If the test is negative, an MRI of the brachial or lumbosacral plexus, depending on the location of the symptoms and clinical suspicion, may be warranted. M A L I G N A N T H E M A T O L O G Y 18 Dose reduction or drug discontinuation is the only specific therapy for most CIPNs; patients may tolerate higher doses of bortezomib if the drug is given by a subcutaneous injection rather than by IV infusion. Symptomatic therapies for CIPN are limited (Figure 1). Of the adjuvant agents effective for patients with neuropathic pain, only duloxetine (30-60 mg/day) has clearly shown efficacy for the CIPN induced by taxanes and platinum.7 Despite the lack of proven efficacy of other agents in CIPN, insurance companies usually require that patients first fail a trial of gabapentin or pregabalin before authorizing duloxetine. Occupational therapy or physical therapy is recommended for patients with ≥ grade 2 sensory or motor toxicity to improve function and quality of life. Patients should also be assessed for depression, which often accompanies uncontrolled chronic pain.8 While vitamin E, vitamin B6, magnesium, calcium, acetyl-L-carnitine, glutamine, glutathione, n-acetyl cysteine, omega-3 fatty acids, alpha lipoic acid, and cannabinoids each have shown efficacy in non-randomized, non-placebo-controlled trials, none can be recommended yet as standard therapy.1, 2, 9 Amitriptyline does not prevent or ameliorate chemotherapy-induced neuropathy from vinca alkaloids, platins, or taxanes.1 Venlafaxine, gabapentin, the N-methyl-d-aspartate (NMDA) receptor antagonist dextromethorphan, the oral anesthetics memantine and mexiletine, and low-dose continuous capsaicin treatment are also ineffective.2 Opioids are recommended for CIPN patients with severe pain.10 Patients will usually require a long-acting agent to provide basal pain relief (e.g., sustained-release morphine or a fentanyl patch) and short-acting opioids for breakthroughs. Methadone (Figure 2) is particularly helpful for basal pain relief. Methadone includes d- and l-isomers that act as opioid-receptor agonists and NMDA receptor antagonists.11 Because NMDA antagonizes the activity of the opiate receptors, blocking NMDA receptors enhances the analgesic effect of externally administered opioids. Methadone also inhibits the reuptake of serotonin and norepinephrine; therefore, it functions as a serotonin-norepinephrine reuptake inhibitor (SNRI). Methadone is usually given orally bid to tid. Dose adjustment is needed for hepatic failure but not for renal failure. The steady state is not reached until 72 hours after the initial dose or after a dose increase. Therefore, short-acting opioids should be used to control symptoms in the interim. If pain level falls to ≤ 3 within the first 24 to 48 hours of initiating methadone, reduce the dose by half immediately to avoid excessive sedation and respiratory depression that may otherwise occur in the next 24 hours. This delay in reaching steady-state plasma levels, along with potential cardiac toxicity (prolongation of the rate corrected QT [QTc] interval) and its interactions with inducers and inhibitors of the cytochrome P450 system, CYP1A2, CYP3A4, and CYP2D6 make using methadone safely somewhat complex. Palliative care specialists can be helpful in managing initiation The Hematologist Compendium 2010-2015 Evidence-Based Algorithm for Evaluation and Symptomatic Treatment of Patients with CIPN Figure 1 First, exclude spinal or leptomeningeal metastases and plexopathies and correct vitamin or hormone abnormalities, if present. For patients who have NCI-CTC grade ≥ 3 CIPN, follow standard protocol guidelines for dose adjustments of anti-neoplastic agents. Use duloxetine for patients with unacceptable pain levels who are receiving either platinum or taxanes. For refractory pain, add an opioid. If standard opioids are not effective, or if they cause excessive sedation, myoclonus, or other side effects, consider using methadone as the “basal” opioid, and use the standard opioids for breakthrough pain. Figure 2 To safely prescribe methadone, monitor the QTc. If the QTc is prolonged, correct contributing metabolic abnormalities or discontinue agents that can prolong the QTc. If the patient requires concomitant therapy with a CYP3A4 competitor (such as voriconazole), use lower doses of methadone. See text for other agents that require adjustments of methadone doses. Increase methadone doses no more than every 72 hours. *Despite the lack of proven efficacy, insurance companies may require that patients first fail a trial of gabapentin or prebabalin before authorizing duloxetine. 1. Finnerup NB, Sindrup SH, Jensen TS. Management of painful neuropathies. Handb Clin Neurol. 2013;115:279-290. or adjustment of methadone doses or for consulting on patients with seemingly refractory pain syndromes. Patients who, at baseline, have a prolonged QTc interval, or who need high doses of methadone, have developed cardiac arrhythmias (torsades de pointes; polymorphic ventricular tachycardia). Fatalities have been reported but were associated with doses of > 600 mg per day. Common drugs used in hematology patients that also prolong the QTc include the quinolone antibiotics (especially levofloxacin), typical and atypical antipsychotics (such as haloperidol and olanzapine), selective serotonin reuptake inhibitors (SSRIs) (but not SNRIs), and metoclopramide. Methadone’s drug interactions are listed on websites including www.drugs.com or www. qtdrug.org. Fluconazole, voriconazole, fluoxetine, and fluvoxamine raise methadone levels. SSRIs may raise methadone levels in CYP2D6 rapid metabolizers but SNRIs (e.g., venlafaxine) do not. Additionally, grapefruit juice and acute alcohol ingestion can increase methadone levels. Drug levels of desipramine (or other tricyclic antidepressants) and zidovudine increase when methadone is added. Antiretrovirals, phenobarbital, carbamazepine, phenytoin, rifampin, somatostatin, spironolactone, and risperidone lower the levels of methadone and have precipitated withdrawal symptoms. Conclusion The patient continued therapy, but bortezomib delivery was switched from IV infusion to SQ injection. Her pain symptoms fell to tolerable levels on 2 mg tid of methadone such that she was able to work, and her functional level was further enhanced by participation in an occupational therapy program. 2. Ferrier J, Pereira V, Busserolles J, et al. Emerging trends in understanding chemotherapy-induced peripheral neuropathy. Curr Pain Headache Rep. 2013;17:364-372. 3. Diezi M, Buclin T, Kuntzer T. Toxic and drug-induced peripheral neuropathies: updates on causes, mechanisms and management. Curr Opin Neurol 2013;26:481-488. 4. Alberti P, Rossi E, Cornblath DR, et al. Physician-assessed and patient-reported outcome measures in chemotherapy-induced sensory peripheral neurotoxicity: two sides of the same coin. Ann Oncol. 2014;25:257-264. 5. Driessen CML, de Kleine-Bolt KME, Vingerhoets AJJM, et al. Assessing the impact of chemotherapyinduced peripheral neurotoxicity on the quality of life of cancer patients. Support Care Cancer. 2012;20:877-881. 6. Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro Oncol. 2012;14:iv45-iv54. 7. Smith EML, Pang H, Cirrincione C, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309:1359-1367. 8. Glover J, Dibble SL, Dodd MJ, et al. Mood states of oncology outpatients: does pain make a difference? J Pain Symptom Manage. 1995;10:120-128. 9. Schloss JM, Colosimo M, Airey C, et al. Nutraceuticals and chemotherapy induced peripheral neuropathy (CIPN): a systematic review. Clin Nutr. 2013;32:888-893. 10. Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacologic management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85:S3-S14. 11. Rhondali W, Tremellat F, Ledoux M, et al. Methadone rotation for cancer patients with refractory pain in a palliative care unit: an observational study. J Pall Med. 2013;16:1382-1387. Dr. Abrahm receives royalties from her book, A Physician’s Guide to Pain and Symptom Management in Cancer Patients, 2nd edition, 2005, from Johns Hopkins University Press. She is the paid section editor for UpToDate on the topic of pain in palliative care patients. She also receives expense reimbursement and honoraria from Knowledge to Practice for her lectures on palliative care. CIPN can be a disabling complication of chemotherapy. Patient education that encourages reporting relevant symptoms, chemotherapy dose adjustment, occupational therapy, physical therapy, and symptomatic therapy using duloxetine and opioids, including methadone, can often help patients maintain or regain function and minimize pain and disability. M A L I G N A N T H E M A T O L O G Y 19 The Hematologist Compendium 2010-2015 Approach to the Evaluation of Patients with an IgM Monoclonal Protein – September/October 2014 UPDATE/COMMENTARY Since this article was published there has indeed been a very important development in multiple myeloma (MM) that has implications for IgM monoclonal proteins. Historically, MM was defined by the presence of “CRAB” criteria (elevated calcium, renal insufficiency, anemia and bony disease). However, three additional criteria have now been added to this definition as endorsed by the International Myeloma Working Group, published in late 2014.U1 pending organ damage, and if untreated, that damage may be permanent. It has therefore shifted a small subset of smoldering myeloma patients to active myeloma warranting treatment. I remember these three criteria with the acronym “SLiM” (60 percent plasma cells; more than 100 light chains involved/uninvolved; MRI evidence of one or more focal lesions) now creating the overall acronym SLiM CRAB for multiple myeloma. These three criteria include clonal bone marrow plasma cell percentage of ≥ 60 percent; involved/uninvolved serum-free light chain (FLC) ratio ≥ 100 (involved FLC level must be ≥ 100 mg/L); and more than one focal lesion on magnetic resonance imaging (MRI) studies (at least 5 mm in size). This new definition of MM, therefore, affects my approach to IgM monoclonal proteins; in the algorithm, I ask the question, “does the patient have end-organ damage attributable to the IgM monoclonal protein?” Instead of only considering CRAB criteria, one should now consider SLiM CRAB criteria, as that would invoke the IgM myeloma diagnosis if present, as opposed to IgM monoclonal gammopathy of undetermined significance (MGUS). Each of these criteria was validated by at least two large databases. They do reflect a change in the concept that one must have “established end organ damage” to be treated for myeloma; this is important, as patients with these three criteria have U1.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538-e548. JOSEPH MIKHAEL, MD, MEd, FRCPC Professor of Medicine, Mayo College of Medicine, Mayo Clinic, Phoenix, AZ The Question What is your approach to the evaluation of patients with an IgM monoclonal protein? My Response Benign monoclonal proteins were first described by Dr. Jan Waldenström in 1960 after he detected abnormal narrow hypergammaglobulinemia bands in serum protein electrophoresis (SPEP) samples from healthy individuals.1 The term MGUS was coined by Dr. Robert Kyle in 1978 to describe an asymptomatic plasma cell dyscrasia characterized by a monoclonal protein of <3 gm/dL, bone marrow plasma cells <10 percent, and the absence of end-organ damage commonly associated with MM.2 MGUS is a relatively common condition with a prevalence of three to five percent in the adult population over the age of 50 years.3 SPEP is a frequently performed laboratory test ordered by primary-care physicians evaluating patients with anemia, by nephrologists evaluating patients with renal insufficiency (possibly with associated proteinuria), and by neurologists evaluating patients with peripheral neuropathy. When the SPEP reveals a monoclonal protein, referral to a hematologist usually follows. Therefore, it is part of routine practice for hematologists to see patients with monoclonal proteins that are initially identified as the result of screening assessments. The hematologist must then decide which additional diagnostic studies are warranted, and based upon those results, develop management and follow-up plans. Although it accounts for only 15 to 20 percent of MGUS cases (that also include IgGMGUS, IgA-MGUS, and light chain-MGUS), IgM-MGUS poses a unique diagnostic challenge because of the association of monoclonal IgM proteins with B-cell lymphoproliferative disorders (particularly Waldenström macroglobulinemia [WM], amyloidosis, and peripheral neuropathy).4 This review is intended to provide the practicing hematologist with a focused diagnostic approach to patients with a monoclonal IgM protein that takes into account its associations with other disease processes. MGUS Figure 1 Three distinct classes of MGUS Natural History of MGUS are recognized: Non-IgM-MGUS (essentially IgG-MGUS or IgA-MGUS MGUS as both IgD-MGUS and IgE-MGUS are fleetingly rare), light chainMGUS, and IgM-MGUS5, 6 (Figure 1). Non IgM IgM Light Chain Distinguishing among these subgroups is important, as doing so directs both the diagnostic plan and MM +/WM MM +/the follow-up recommendations, Amyloid Amyloid Amyloid IgM Myeloma facilitates identification of diseases associated with MGUS, and impacts on management recommendations and prognosis. Although the majority of patients with IgM-MGUS will have a benign course, it is critical for the clinician to rule out a concurrent associated disease and to monitor for progression or transformation into a distinct entity that requires specific therapy. The Risk of MGUS Progression Unstratified patients with non-IgM MGUS have approximately a one percent per-year risk of their disease transforming into MM; however, the risk of transformation is double in patients M A L I G N A N T H E M A T O L O G Y 20 with IgM MGUS. Risk for transformation can be more precisely stratified based on three parameters: IgG subtype versus non-IgG subtype, monoclonal protein concentration <1.5 gm/ dL versus ≥1.5 gm/dL, and normal versus abnormal serum free light chain ratio.7 Stratification of risk can help guide the diagnostic evaluation and follow-up recommendations. IgM MGUS not only has a higher risk of transformation than non-IgM MGUS, but also the spectrum of diseases associated with IgM MGUS transformation is broader than that of nonIgM MGUS. Whereas non-IgM MGUS can progress into smoldering and active MM and AL amyloidosis, IgM MGUS can transform into WM, AL amyloidosis, and less commonly, IgM smoldering myeloma or IgM MM.8 For this reason, patients with IgM MGUS require closer follow-up than patients with non-IgM MGUS, and from a conceptual perspective, IgM-MGUS can be thought of as a “lymphoproliferative” MGUS while non-IgM MGUS behaves as a “plasma cell proliferative” MGUS. Special Concerns for IgM MGUS a. Association with neuropathy – Neurologists routinely screen patients with peripheral neuropathy for the presence of a monoclonal protein, and approximately five to 10 percent of such patients will be found to have a monoclonal protein by SPEP. A causal relationship between MGUS and peripheral neuropathy is supported by association of peripheral neuropathy with other plasma cell dyscrasias including WM, MM, AL amyloid, and POEMS (polyneuropathy, organomegaly, endoocrinopathy, monoclonal gammopathy, skin changes). The peripheral neuropathy of MGUS is classically bilateral, peripheral, and sensory with electrophysiologic studies showing a demyelinating pattern; and in patients with progressive, debilitating disease, biopsy may reveal axonal loss. Although peripheral neuropathy can occur with all forms of MGUS, it is most commonly associated with IgM-MGUS. Demonstration of IgM anti-myelin-associated glycoprotein (MAG) antibodies supports a causal relationship between IgM-MGUS and polyneuropathy but is not essential for the diagnosis. Because peripheral neuropathy can be caused by other processes that may co-exist with IgM-MGUS, the clinician is often faced with the dilemma of whether to assign the neuropathy to MGUS, and if so, what to do about it. The decision can be guided by the observations that the neuropathy associated with IgM-MGUS is characteristically a relatively benign, slowly progressive sensory process (although some cases can be severe and debilitating). Management is challenging as effective therapy is lacking. A minority of patients respond to rituximab, and other immunomodulatory treatments are generally ineffective.9 As is the case with other plasma cell dyscrasiaassociated neuropathies, the monoclonal protein concentration in IgM-MGUS-associated peripheral neuropathy does not correlate with disease severity, arguing against the use of myeloma-directed therapy to reduce the plasma cell burden as a treatment strategy for the neuropathy of IgM-MGUS. b. Association with AL amyloid – This complex disease can be associated with any form of myeloma, although usually one of the two processes dominates the clinical picture. That is to say that patients typically have either a myeloma-phenotype manifested by some combination of hypercalcemia, renal insufficiency, anemia, and skeletal involvement or an amyloid phenotype characterized by organ (liver, heart, kidney) infiltration by the pathologic immunoglobulin light chain. AL amyloid is more commonly associated with IgM-MGUS than other forms of MGUS. Thus, it is imperative that AL amyloid be carefully considered when evaluating patients with an IgM monoclonal protein. c. Association with WM – The hallmark of this disease is an IgM monoclonal protein, lymphoadenopathy, hepatosplenomegally, and bone marrow involvement by plasmacytoid lymphocytes. Frank disease may be preceded by a relatively asymptomatic smoldering phase in which IgM MGUS is the only apparent clinical manifestation. d. Association with other B-cell lymphoproliferative disorders – Although IgM-MGUS is more commonly associated with WM, it can be observed in association with another B-cell lymphoproliferative disease such as CLL or non-Hodgkin lymphoma. As indicated in the algorithm (Figure 2), determining if organ damage is attributable to the monoclonal protein is critical, as in most cases the monoclonal protein simply “co-exists” with the lymphoproliferative disorder. Although the monoclonal protein may be discovered first as part of a laboratory evaluation, it is not usually the presenting clinical manifestation of lymphoproliferative diseases other than WM. The Hematologist Compendium 2010-2015 e. Association with IgM MM – This is a rare form of myeloma that is genuinely distinct from WM. IgM MM is distinguished from WM primarily by skeletal involvement (lytic bone lesions) in the former, but genetic studies may also be informative (e.g., t[4:14] in IgM MM and somatic mutation of MYD88 in WM).10, 11 Figure 2 Algorithm for Investigation of IgM Monoclonal Protein IgM Monoclonal Protein Detected Suggested Approach to the Evaluation of Patients with an IgM Monoclonal Protein The strategy that I use in the evaluation of patients with an IgM monoclonal protein is described below and illustrated in Figure 2. 1. History – This remains a critical aspect of the evaluation, as symptoms elicited from a careful history focus attention on specific issues that require further investigation. Indeed, even subtle symptoms become important when the differential diagnosis includes such a wide spectrum of disorders as amyloidosis, POEMS, MM, and lymphoma. Key symptoms to address include the following: a.Constitutional (weight loss, extreme fatigue) – Such symptoms suggest AL amyloid or lymphoma. History/Physical + Labs* + Skeletal Imaging** + Bone Marrow Evaluation◆ Does the patient have end organ damage attributable to the IgM Monoclonal Protein? YES Are there osteolytic lesions or t(11;14)? NO Are there ≥ 10% Plasma Cells or is the M Spike ≥ 3gm/dL? Are there findings consistent with AL Amyloidosis? YES IgM Myeloma Obtain echo and fat aspirate +/organ biopsy b.Gastrointestinal – Upper GI bleeding, early satiety, and chronic diarrhea raise the possibility of GI amyloid. NO IgM MGUS If positive c.Cardiac – Progressive shortness of breath, presyncope/syncope, and chest pain are consistent with cardiac amyloid. Smoldering Waldenström Macroglobulinemia✝ d.Neurologic – Bilateral, sensory neuropathy is consistent with the neuropathy associated with plasma cell dyscrasias; and vision changes, headache, vertigo, or dizziness raise the possibility of hyperviscosity associated with WM. AL Amyloid e.Skeletal pain – This symptom suggests IgM myeloma. f. Skin – Urticarial rash raises the possibility of Schnitzler syndrome.12 Waldenström Macroglobulinemia✝ 2. Physical Exam – The physical exam can be informative as patients with an IgM monoclonal protein and WM or other non-Hodgkin lymphoma may present with lymphadenopathy and heptosplenomegaly. Patients with concurrent AL amyloid may have hepatosplenomegally or macroglossia, while patients with IgM myeloma may have discrete sites of skeletal pain. A careful neurologic exam is essential for identifying and characterizing a concurrent neuropathy. 3. Labs a. The following lab studies should be obtained: i. CBC, serum calcium, creatinine, β-2-microglobulin, LDH, liver enzymes ii. Serum protein electrophoresis with immunofixation electrophoresis iii. Serum free light-chain assay iv. Quantitative immunoglobulins v.Spot urine sample for protein quantitation, protein electrophoresis, and immunofixation electrophoresis. For patients with significant proteinuria and patients with a monoclonal light chain (Bence Jones protein) detected in the spot urine, a 24-hour urine sample should be collected to quantitate total protein and light-chain excretion. b.Other laboratory tests that should be considered based on the clinical circumstances: i. Cardiac function biomarkers (NT-proBNP and troponin) for suspected amyloid ii. Anti-MAG antibodies in patients with a sensory neuropathy iii. Peripheral blood flow cytometry for suspected CLL 4. Radiologic and Other Diagnostic Testing a.Skeletal survey (radiographic or MRI myeloma protocol) is recommended for patients with bone pain and for patients in whom IgM MM is a consideration. b.Other diagnostic studies that may be considered based on clinical circumstances: i. Abdominal US to assess for hepatic amyloid ii. Echocardiogram and ECG to assess for cardiac amyloid iii. Abdominal/pelvic CT to assess for non-Hodgkin lymphoma including WM iv. PET scan to assess for lymphoma 5. Bone marrow evaluation – Although many patients with MGUS do not require this procedure, in higher-risk cases (monoclonal-protein concentration >1.5 gm/dL and IgM-MGUS), bone marrow aspirate and biopsy is recommended and may also include cytogenetic, myeloma FISH, flow cytometric analyses, and targeted gene sequencing.13 I do not, however, recommended bone marrow analysis for all patients with a monoclonal protein. Specifically, I may defer bone marrow analysis in the very elderly, in those in whom the monoclonal-protein concentration is low (< 0.5 gm/dL), and in patients with an inflammatory condition, as low-risk MGUS is relatively common in these settings. a.Myeloma FISH and targeted gene sequencing – Recommended if marrow findings are consistent with MM or lymphoma. Finding t(11;14) by cytogenetics or FISH suggests IgM MM, and mutant MYD88 supports a diagnosis of WM. b.Flow cytometry – This analysis is informative in patients suspected of having a B-cell lymphoproliferative disorder including CLL, non-Hodgkin lymphoma, or WM. 6. Fat pad aspirate – I do not routinely subject patients to this procedure; however, if there is clinical suspicion of AL amyloid, fat pad aspirate with congo red staining is an excellent screening tool. I have a slightly higher index of suspicion for AL amyloid when the IgM monoclonal protein is lambda-restricted. If the fat pad aspirate is negative for AL amyloid but suspicion remains high, targeted organ biopsy with congo red staining is warranted. M A L I G N A N T H E M A T O L O G Y 21 * Laboratory studies include CBC, serum calcium, creatinine, b-2microglobulin, LDH, liver enzymes, serum protein electrophoresis with immunofixation, serum free light chain assay, quantitative immunoglobulins, 24-hour urine testing for protein, and urine electrophoresis. ** Skeletal survey is the preferred first-line imaging choice. ◆ Marrow evaluation includes immunohistochemical studies, flow cytometry, conventional cytogenetics, and myeloma FISH. ✝ Confirm by MYD88 sequence analysis. Follow-Up If after the initial evaluation a patient is diagnosed with IgM MGUS, regular surveillance is imperative as risk of progression/transformation is a continuous variable. I recommend that patients be seen twice annually with a clinical assessment, along with the following laboratory studies: CBC, comprehensive metabolic panel, SPEP, serum-free light-chain assay, and quantitative immunoglobulins.14 I do not recommend repeat imaging or bone marrow analysis unless there is suspicion of progression. Conclusion Monoclonal IgM is associated with a diverse set of diseases that range from a generally benign process requiring no specific therapy (IgM-MGUS) to overt disease requiring a specific management plan. Approaching the initial evaluation systematically and comprehensively will enable the clinician to accurately characterize the disease process. 1. Waldenstrom J. Studies on conditions associated with disturbed gamma globulin formation (gammopathies). Harvey Lect. 1960;56:211-231. 2. Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med. 1978;64:814-826. 3. Kyle RA, Therneau TM, Rajkumar V, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354:1362-1369. 4. Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003;102:3759-3764. 5. Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc. 2010;85:945-948. 6. van de Donk NW, Palumbo A, Johnsen HE, et al. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. 2014;99:984-996. 7. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106:812-817. 8. Baldini L, Goldaniga M, Guffanti A, et al. Immunoglobulin M monoclonal gammopathies of undetermined significance and indolent Waldenstrom’s macroglobulinemia recognize the same determinants of evolution into symptomatic lymphoid disorders: proposal for a common prognostic scoring system. J Clin Oncol. 2005;23:4662-4668. 9. Latov N. Diagnosis and treatment of chronic acquired demyelinating polyneuropathies. Nat Rev Neurol. 2014;10:435-446. 10. Schuster SR, Rajkumar, Dispenzieri A, et al. IgM multiple myeloma: disease definition, prognosis, and differentiation from Waldenstrom’s macroglobulinemia. Am J Hematol. 2010;85:853-855. 11. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367:826-833. 12. Simon A, Asil B, Braun-Falco M, et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562-568. 13. Kyle RA, Durie BG, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121-1127. 14. Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood. 2011;117:5573-5581. Dr. Mikhael indicated no relevant conflicts of interest. The Hematologist Compendium 2010-2015 Treatment of Elderly Patients with Acute Myeloid Leukemia – January/February 2015 Due to the recent publication date of this article, an update has not been requested. These data suggest that age alone is a poor predictor of treatment intent and outcomes, and is likely a surrogate for other covariates associated with inferior prognosis. BRUNO MEDEIROS, MD Associate Professor of Medicine (Hematology) at the Stanford Cancer Institute, Stanford, CA The Question What is your approach to the treatment of elderly patients with acute myeloid leukemia? My Response Acute myeloid leukemia (AML) is not a significant public health hazard, accounting for less than 2 percent of all cancers diagnosed yearly in the United States. The American Cancer Society estimates that 18,860 new cases of AML were diagnosed in 2014.1 Nonetheless, AML is second only to chronic lymphocytic leukemia as the most common subtype of leukemia in adults. The median age at diagnosis is 67 years, and more than 60 percent of newly diagnosed patients are older than 60 years.2 The management of elderly patients with AML poses unique therapeutic challenges. These individuals disproportionally account for greater than 75 percent of AML deaths yearly. In addition, recent data from the Surveillance, Epidemiology, and End Results (SEER) Program demonstrate that 50 to 60 percent of newly diagnosed AML patients older than 65 years do not receive any form of antileukemia therapy.3 Therapeutic Options for Elderly Patients with AML In routine clinical practice, it remains challenging to know how to best utilize these data to inform patients and families on the optimal therapeutic approach for an individual with AML. Although these predictive models provide some guidance on the potential risks and benefits associated with conventional induction chemotherapy, treatment decisions regarding therapeutic intensity remain a highly individualized exercise. In my practice, I direct patients with low treatment-related mortality scores or favorable AML risk prediction scores to intensive standard or investigational therapy; conversely, I consider patients with high treatmentrelated mortality scores or unfavorable AML risk profiles for lower-intensity investigational regimens (Figure). Results from population studies and randomized clinical trials add to the challenge of deciding optimal therapeutic intensity. Recent SEER-Medicare linked data showed that, after using a propensity score-matched survival analysis to account for all confounders, similar reductions in the risk of death were observed for elderly patients receiving either intensive chemotherapy or hypomethylating agents.3 Similar results have been reported in randomized clinical trials with similar overall survival between single agent hypomethylating agents (decitabine or azacitidine) and conventional care regimens.9,10 The Future is Now: The Rise of Targeted Therapies “It was the best of times, it was the worst of times, it was the age of wisdom, it was the age of foolishness...” This opening sentence from A Tale of Two Cities describes the sentiment of hematologists regarding the current state of management of AML in older patients. While our understanding of the molecular alterations responsible for leukemogenic transformation has flourished in recent years, none of these discoveries has yet translated into approved treatment strategies for patients beyond "7+3" conventional induction chemotherapy. However, characterization of the mutational landscape of AML has accelerated development of targeted therapeutics that may soon be within the reach of patients. Activating mutations in signaling pathways, including FLT3, KIT, RAS, and others, have been described in roughly 60 percent of AML patients.11 Clinical trials testing the activity of FLT3, KIT and MEK inhibitors (as single agents or in combinations) are available in most academic cen- Definition of “Elderly” in Patients with AML Older patients can be classified into three categories of chronological age: 1) young-old patients are 65 to 75 years of age; 2) old patients are 76 to 85 years of age; and 3) oldestold patients are older than 85 years.4 In AML, clinical outcomes worsen with advancing age.2 Recognizing this relationship between older age and outcomes, contemporary AML treatment protocols have typically used an arbitrary cutoff of 55 to 65 years to distinguish between younger and older subjects. However, chronologic age by itself is not reliable in estimating life expectancy, functional reserve, or the risk of treatment complications. Comprehensive geriatric assessments (CGA) are multidisciplinary, in-depth evaluations designed to assess life expectancy and attendant morbidity and mortality risks in older patients. CGAs include tools to predict the functional age based on functional status, comorbidities, Figure polypharmacy, nutritional status, and geriatric syndromes.5 Treatment Algorithm For this reason, pre-treatment CGAs or more focused geriatric assessment tools may replace chronologic age as an New diagnosis of AML identifier of “functionally older” AML patients ineligible for aggressive treatment approaches. AML and Non–AML-Related Factors Influencing Treatment Decisions Recent studies suggest that age may be a suboptimal sole criterion for allocation to intensive AML treatment protocols, and that additional variables may improve the ability to predict toxicity and outcomes. Population-based data demonstrate that older age was only one of the several covariates (including history of antecedent hematologic disorder [sAML], higher comorbidity score, poor performance indicators, marital status, and lower household incomes) associated with lack of antileukemia therapy in newly diagnosed older patients with AML.3 A retrospective analysis showed that a pre-treatment geriatric assessment, focused on cognitive and physical function, improved the prediction of survival among older adults with AML treated with conventional induction chemotherapy.6 Two large retrospective studies have identified several AML and non-AML covariates that predict early outcomes, such as induction mortality and likelihood of complete remission, following intensive chemotherapy. In the first, investigators developed a simplified early death score with moderate discriminatory power that incorporates performance status at diagnosis, age, platelet count, albumin, sAML, white blood cell count, percentage of peripheral blood blasts, and serum creatinine (cstaging. fhcrc-research.org/TRM/Default.aspx).7 Interestingly, removal of age as a covariate had minimal impact on the predictive power of this model. Similarly, investigators from the Study Alliance Leukemia demonstrated that the risk of induction mortality and the chance of complete remission could be predicted in older patients with AML using a combination of pre-treatment covariates, including age, hemoglobin, platelet count, fibrinogen, type of AML, karyotype and limited molecular abnormalities at diagnosis (www.aml-score.org).8 for Older Patients with AML • Age Patient-related characteristics • Performance status • Renal and hepatic function • Coagulopathy High treatment-related mortality score or unfavorable AML score • Karyotype AML-related characteristics • sAML (t-AML or AML-MRC) • Blood counts at diagnosis (WBC, Hb, platelets) Acceptable treatment-related mortality score or favorable AML score Non-intensive treatment Vulnerable Focused geriatric assessment or comorbidity index • Investigational therapy • Hypomethylating agents • Low-dose cytarabine Fit Intensive treatment • Investigational therapy • Conventional induction therapy Abbreviations: AML, acute myeloid leukemia; sAML, antecedent hematologic disorder; t-AML, therapy-related AML; AML-MRC, AML with myelodysplastic syndrome--related changes. M A L I G N A N T H E M A T O L O G Y 22 The Hematologist Compendium 2010-2015 ters and may reshape treatment strategies for these patients. Mutations in the DNMT3A and TET2 genes have been linked to increased sensitivity to hypomethylating agents.12,13 Examples of novel agents in clinical development include small molecule inhibitors of mutant IDH1 and IDH2,14,15 DOT1-L inhibitors for patients with MLL rearranged acute leukemia, novel epigenetic modifiers such as the bromodomain (BRD4) inhibitors, and development of second-generation anti-CD33 conjugated monoclonal antibodies. Also, addition of multikinase (sorafenib) or aurora kinase inhibitors to conventional induction chemotherapy is associated with promising antileukemic activity and improved survival. Finally, improved patient selection (genomic profile, AML-MRD, or adverse-risk karyotype) or combination with novel agents (pracinostat or pevonedistat) may optimize the clinical benefit of DNA methyltransferase inhibitors such as azacitidine. At last, physicians caring for older patients with AML may say farewell to the age of foolishness when patients and physicians were painstakingly faced with choosing between nonspecific and toxic regimens or palliative care. Instead, they can usher in the age of wisdom wherein genomic-driven targeted and risk-adapted treatment strategies are incorporated into the management of AML. Current enthusiasm in the field is married to the promise that the best of times will soon be upon us. 1. American Cancer Society. Cancer Facts & Figures 2014. www.cancer.org/research/cancerfactsstatistics/ cancerfactsfigures2014. Accessed December 2014. 2. National Cancer Institute. SEER Stat Fact Sheets. seer.cancer.gov/statfacts/html/amyl.html. Accessed November 2013. 3. Medeiros BC, Satram-Hoang S, Hurst D, et al. Big data analysis of treatment patterns and outcomes among elderly acute myeloid leukemia patients in the United States. 2014. Manuscript in preparation. 4. Hurria A, Browner IS, Cohen HJ, et al. Senior adult oncology. J Natl Compr Cancer Netw. 2012;10:162-209. 5. Extermann M, Hurria A. Comprehensive geriatric assessment for older patients with cancer. J Clin Oncol. 2007;25:1824-1831. 6. Klepin HD, Geiger AM, Tooze JA, et al. Geriatric assessment predicts survival for older adults receiving induction chemotherapy for acute myelogenous leukemia. Blood. 2013;121:4287-4294. 7. Walter RB, Othus M, Borthakur G, et al. Prediction of early death after induction therapy for newly diagnosed acute myeloid leukemia with pretreatment risk scores: a novel paradigm for treatment assignment. J Clin Oncol. 2011;29:4417-4423. 8. Krug U, Röllig C, Koschmieder A, et al. Complete remission and early death after intensive chemotherapy in patients aged 60 years or older with acute myeloid leukaemia: a web-based application for prediction of outcomes. Lancet. 2010;376:2000-2008. 9. Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30:2670-2677. 10. Dombret H, Seymour JF, Butrym A, et al. Results of a phase 3, multicenter, randomized, open-label study of azacitidine (AZA) VS conventional care regimens (CCR) in older patients with newly diagnosed acute myeloid leukemia (AML). Paper presented at 19th Congress of the European Hematology Association. June 12-15, 2014. Milan, Italy. 11. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059-2074. 12. Metzeler KH, Walker A, Geyer S, et al. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106-1107. 13. Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705-2712. 14. Study of orally administered AG-120 in subjects with advanced hematologic malignancies with an IDH1 mutation. ClinicalTrials.gov Identifier: NCT02074839. 15. Phase 1 study of AG-221 in subjects with advanced hematologic malignancies with an IDH2 mutation. ClinicalTrials.gov Identifier: NCT01915498. Dr. Medeiros indicated no relevant conflicts of interest. Check Out the ASH Image Bank The ASH Image Bank is a public web-based image library that offers a comprehensive collection of high-quality, peer-reviewed images relating to a wide range of hematologic topics. The Image Bank was re-launched March 2016 and contains more than 2,100 images. Recently added features include the Atlas, which provides a quick way to view images of blood cells in a normal and abnormal state, and reference cases describing common blood disorders through image sets and descriptive text. ASH encourages new submissions, subject to review by the Image Bank Editor, and new images are continuously being published to the collection. To browse the Image Bank, submit, or download images, members need to log in with their standard ASH username and password and nonmembers need to create an account, currently free of charge. To submit comments, questions, and requests for the inclusion of specific images, send an email to imagebank@ hematology.org. To view the ASH Image Bank, go to imagebank.hematology.org. M A L I G N A N T H E M A T O L O G Y 23 The Hematologist Compendium 2010-2015 Treatment of Relapsed/Refractory Hairy Cell Leukemia – July/August 2015 Due to the recent publication date of this article, an update has not been requested. ROBERT J. KREITMAN, MD Chief, Clinical Immunotherapy Section, Laboratory of Molecular Biology, National Cancer Institute, National Institutes of Health, Bethesda, MD The Question What is your management approach to patients with relapsed/refractory hairy cell leukemia? My Response Hairy cell leukemia (HCL) is an indolent B-cell malignancy, originally with a median survival of approximately four years.1 Single-agent purine analog therapy with pentostatin or cladribine achieves complete remission (CR) rates of 70 to 90 percent and median relapse-free survivals in excess of 15 years.2-4 However, minimal residual disease (MRD) often remains, and may be associated with relapse, Figure particularly in young patients with longer follow-up.5 Thus, in the past 25 to 30 years, many patients have neither died nor been cured, leading to the increasingly common question of how to treat relapsed and refractory patients. The answer requires accurate diagnosis, understanding the history of response and toxicity to prior agents, and a strategy for prioritizing options including exciting new targeted therapies. Diagnosis of HCL and Recognition of Variant Disease both regimens and allows crossover to the other regimen if needed. Most can achieve MRDnegative CR, but with more toxicity than patients receiving non-chemotherapy approaches such as moxetumomab pasudotox. Targeted Approaches in HCL In the BRAF pathway of normal cells, BRAF phosphorylates MEK, phospho-MEK phosphorylates ERK, and phospho-ERK leads to cell proliferation. BRAF containing the V600E mutation, present in nearly half of malignant melanomas, in lower percentages of other tumors, but in nearly all cases of classic HCL, exhibits uncontrolled phosphorylation leading to the malignant phenotype. Inhibition of this mutant BRAF with vemurafenib, or with dabrafenib combined with the MEK inhibitor trametinib, is approved for malignant melanoma and is currently being tested in HCL. Oral BRAF inhibitors in HCL can rapidly reverse severe cytopenias but have not yet been reported to clear MRD and prevent relapse. The oral agent ibrutinib, which targets the Bruton’s tyrosine kinase pathway, is also being tested in patients with HCL and HCLv. Though most patients achieve stable disease, this can have palliative benefit, particularly for patients with HCLv. Summary and Algorithm for Treatment Approach to Patients with Relapsed/ Refractory HCL The Figure shows our algorithm for prioritization of clinical trials for relapsed HCL; additional options can be used off-protocol. Patients with once-relapsed HCL after purine analog can be retreated with the same or different purine analog as a single-agent, particularly if the first remission duration was long, but we prefer combining cladribine with rituximab on study to eliminate MRD. For patients with multiply relapsed HCL, moxetumomab pasudotox is our preferred option due to its unique ability to achieve MRDnegative CR without chemotherapy toxicities. For patients who responded to anti-CD22 recombinant immunotoxins (BL22 or moxetumomab pasudotox) but need additional therapy, we target CD25 with recombinant immunotoxin LMB-2. For patients ineligible for moxetumomab pasudotox or LMB-2, we use BRAF or BRAF/MEK inhibition, provided patients are able to handle toxicities and understand that remission durations may be limited if MRD persists. For patients with HCL/HCLv ineligible for these approaches, we prefer either the ibrutinib protocol or our protocol using rituximab combined with either bendamustine or pentostatin. Ibrutinib offers oral therapy without chemotherapy toxicity, while the rituximabchemotherapy combination offers a likely path to high MRD-negative CR rates and the potential (as in patients after moxetumomab pasudotox) of not needing therapy for many years. Further follow-up and testing will be needed to determine the long-term benefit of these approaches and whether MRD-negative CR can translate into cure. HCL diagnosis today is most accurately made by flow cytometry showing strong expression of B-cell antigens CD20 and CD22; strong CD11c; and positive CD25, CD103, and CD123.6 Bone marrow immunohistochemistry is positive for CD20, tartrate-resistant acid phosphatase (TRAP), annexin 1A (Anxa1), and for the V600E mutated form of BRAF.7 Variant HCL (HCLv) has a similar Newly diagnosed HCL patients typically receive a purine analog, most immunophenotype but lacks CD25, Anxa1, CD123, commonly cladribine in five daily doses, less commonly pentostatin in six 6 and TRAP. Additionally, BRAF is wild type. HCLv is to 12 biweekly doses. Newly diagnosed or once-relapsed patients may more aggressive, with worse splenomegaly, malignant receive repeat courses of single-agent purine analog, but at MD Anderson and the NIH, are also eligible for protocol treatment with rituximab and lymphocytosis rather than cytopenias, and exhibits a cladribine (CDAR). At the NIH, multiply relapsed HCL/HCLv patients poor response to single-agent purine analog; it also may eligible for moxetumomab pasudotox receive that option (also available require adding rituximab. Although classified in 2008 by in other centers), while ineligible patients may receive LMB-2, BRAF the World Health Organization as a separate disorder, inhibition, ibrutinib, and BRPR (bendamustine-rituximab vs pentostatinHCLv is also discussed here. Another aggressive variant is rituximab) in that order of priority. Common off-protocol options for characterized by an immunophenotype of HCLv or classic multiply-relapsed HCL include repeat courses of single-agent purine analog, single-agent rituximab, and pentostatin-rituximab. Splenectomy HCL, but with the IGHV4-34 type of immunoglobulin and even splenic radiation can have palliative benefit at least for a limited rearrangement that is more than 98 percent homologous period of time. (unmutated) to its germline sequence.8 Even with the classic immunophenotype, these patients resemble those with HCLv with malignant lymphocytosis, purine analog resistance, and wild-type BRAF.8,9 By shipping blood samples and unstained bone marrow 1. Golomb HM, Catovsky D, Golde DW. Hairy cell leukemia: a clinical review based on 71 cases. Ann Intern slides to an expert center, accurate diagnosis is also possible without travel. Treatment Med. 1978;89:677-683. should only be initiated after accurate diagnosis is established. 2. Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol. 1995;13:974-982. Treatment of Patients in First Relapse With some exceptions, remission rates are lower after second-line versus after first-line purine analog therapy3,4; however, if the length of the first remission is at least two to four years, many will recommend repeating the initial purine analog. The shorter the treatment-free interval, the greater the risk of overlapping toxicity. Patients relapsing after shorter intervals often receive the anti-CD20 MAb rituximab alone in four to eight weekly doses. While single-agent rituximab can achieve response without overlapping chemotherapy toxicity, response rates are relatively low in patients with prior purine analog therapy who have cytopenias requiring treatment. A more effective strategy is to combine a purine analog with rituximab. Elimination of MRD, detectable by immunohistochemistry of the bone marrow biopsy, as well as by flow cytometry of the blood or bone marrow aspirate, is possible with rituximab one month after cladribine.10 We treat once-relapsed HCL/HCLv with rituximab, starting immediately with cladribine to effect synergy,11 or delayed at least six months so MRD status can be determined. Therapy of Multiply Relapsed HCL Several options are being tested for multiply relapsed HCL. In order to avoid chemotherapy toxicities, but to retain cytotoxic power, we use recombinant immunotoxins containing an Fv fragment of a Mab and a truncated form of Pseudomonas exotoxin. High CR rates in multiply relapsed HCL were reported in 2001 using the anti-CD22 recombinant immunotoxin BL22,12 including patients with HCLv. Mutations were made in the VH domain of BL22 that improve its binding to CD22, and the resulting affinity-matured recombinant immunotoxin moxetumomab pasudotox achieved CRs in approximately 50 percent of patients with multiply relapsed HCL, many without MRD.13 Another strategy to achieve MRD-negative CR is to use six cycles of either pentostatin plus rituximab,4 or bendamustine plus rituximab.14 Our protocol at the National Institutes of Health (NIH) prospectively evaluates and randomizes patients between M A L I G N A N T H E M A T O L O G Y 24 3. Saven A, Burian C, Koziol JA, et al. Long-term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood. 1998;92:1918-1926. 4. Else M, Dearden CE, Matutes E, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145:733-740. 5. Rosenberg JD, Burian C, Waalen J, et al. Clinical characteristics and long-term outcome of young hairy cell leukemia patients treated with cladribine: a single-institution series. Blood. 2014;123:177-183. 6. Matutes E, Wotherspoon A, Catovsky D. The variant form of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16:41-56. 7. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305-2315. 8. Arons E, Suntum T, Stetler-Stevenson M, et al. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114:4687-4695. 9. Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119:3330-3332. 10. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood. 2011;118:3818-3823. 11. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res. 2013;19:6873-6881. 12. Kreitman RJ, Wilson WH, Bergeron K, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241-247. 13. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30:1822-1828. 14. Burotto M, Stetler-Stevenson M, Arons E, et al. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res. 2013;19:6313-6321. Dr. Kreitman is the co-inventor on the NIH patent for Moxetumomab pasudotox. The Hematologist Compendium 2010-2015 Diagnosis of Immune Pathophysiology in Patients with Low-Risk Myelodysplastic Syndromes – September/October 2015 Due to the recent publication date of this article, an update has not been requested. patients owing to age-related fatty changes in the BM. Therefore, hematologists should keep in mind that BM hypercellularity does not necessarily preclude a diagnosis of immune-mediated BM failure. SHINJI NAKAO, MD, PHD Professor of Cellular Transplantation Biology (Hematology/Respirology), Kanazawa University Graduate School of Medical Science, Kanazawa, Japan Laboratory Markers Representing Immune Pathophysiology of BM Failure The Question The presence of predominant thrombocytopenia is the most fundamental feature of immunemediated BM failure that should prompt further examinations of other laboratory markers. An increased percentage of paroxysmal nocturnal hemoglobinuria (PNH) –type cells, which can be detected using high-sensitivity flow cytometry (FCM), represents a reliable marker of immune pathophysiology.7,8 The usefulness of detecting increased PNH-type cells for predicting the response to immunosuppressive therapy (IST) in AA patients was confirmed in recent prospective studies utilizing high-sensitivity FCM-based methods that can precisely measure 0.01 percent or more PNH-type cells.9,10 Figure 1 shows the results of FCM in the current case, which revealed 0.126 percent of granulocytes and 0.001 percent of erythrocytes to be PNH-type cells. Such results can be obtained on the same day as the patient’s visit, and if those results are positive, physicians have the option of commencing IST shortly thereafter. The presence of HLA-DRB1*15:01 has been reported to predict a good response to IST in patients with MDS.11 However, in our previous study using a multivariate analysis of several factors, including the presence or absence of DRB1*15:01, only the detection of PNH-type cells was associated with a good response to IST in patients with AA.12 Therefore, DRB1 typing to clarify the immune pathophysiology of BM failure is unnecessary if high-sensitivity FCM is available and detects PNH-type cells. What is your approach to the diagnosis and treatment of patients with low-risk myelodysplastic syndrome (MDS) related to immune pathophysiology? Case A 53-year-old man was found to have pancytopenia during follow-up for hyperuricemia. The laboratory findings were as follows: WBC, 2.44×109/L with 26 percent neutrophils, 1 percent eosinophils, 8 percent monocytes, and 65 percent lymphocytes; RBC, 1.49×1012/L; hemoglobin, 5.7 g/dL; mean corpuscular volume (MCV), 112.8 fL; platelets, 22×109/L; reticulocytes, 45×109/L; LDH=195 IU/L. The patient’s bone marrow (BM) was normocellular with 2 percent blasts and showed signs of dysplasia in erythroid precursors and granulocytes without increased ring sideroblasts. The megakaryocyte count was decreased. The karyotype of the BM cells was 46,XY in 20 dividing cells. The patient was therefore diagnosed as having refractory cytopenia with multilineage dysplasia (RCMD) with an International Prognostic Scoring System (IPSS) risk group of intermediate-1 (IPSSrevised risk category: intermediate). His physician suggested that he receive red blood cell transfusions and possibly hematopoietic stem cell transplantation or azacitidine therapy if the pancytopenia progressed further. Figure 1 Detection of PNH-type cells using high sensitivity flow cytometry Q2 Q2 10 4 10 0.001% 3 P5 10 10 3 P6 10 CD11bPE-A Q1 5 0.126% 4 10 5 Q1 10 10 2 Diagnosing MDS is often challenging in patients with pancytopenia or bicytopenia who do not show definitive poor prognostic markers, such as karyotypic abnormalities or increased blasts or ring sideroblasts in the BM, as in the present case. The differential diagnosis of such cases of gray zone BM failure includes refractory cytopenia with unilineage dysplasia, RCMD, idiopathic cytopenia(s) of undetermined significance, and moderate aplastic anemia (AA).1 It is generally difficult to make an exact diagnosis based on the established diagnostic criteria because these disease entities overlap, and each diagnosis relies on a subjective judgement of the cell morphology by physicians and/or pathologists.2-4 Giving a disease name to such an equivocal condition has virtually little value. Instead, it is clinically relevant to distinguish benign subsets of BM failure that are likely to respond to immunosuppressive therapy (IST) from cases of non–immunemediated BM failure, including those associated with pre-leukemic features and inherited BM failure syndromes. Glytophorin APE-A Distinguishing Cases of ImmuneMediated BM Failure 2 My Response Another solid marker of immune pathophysiology in patients with BM failure is the presence of HLA-A allele–lacking leukocytes (HLA-LLs) derived from HSCs that develop copy number–neutral loss of heterozygosity of the HLA haplotype owing to uniparental Q3 1 0 10 Q4 2 10 3 4 10 10 5 10 Q3 M -1,639 Q4 0 3 10 4 10 FLAER FITC-A CD55+59 FITC-A Granulocytes Erythrocytes 5 10 Detection of paroxysmal nocturnal hemoglobinuria (PNH) -type cells using high-sensitivity flow cytometry. The patient possessed increased PNH-type cells only in granulocytes. Difficulties in Evaluating BM Cellularity in Patients with BM Failure BM aspiration and trephine biopsies are essential for obtaining the differential diagnosis of BM failure syndromes, and the detection of a hypocellular BM is a prerequisite for diagnosing AA.5 However, assessing BM cellularity in patients with BM failure is often difficult, particularly when the cytopenias are not severe. Even when the BM of one bone site is grossly replaced with fat tissue as a result of the immune-mediated destruction of hematopoietic stem cells (HSCs), some hematopoietic nests remain in other bone sites and may show hypercellularity due to increased BM activity that compensates for the decreased hematopoiesis.6 BM aspiration or biopsies of these hot spots can sometimes produce erroneous results. When pathological reports of BM examinations show hyperor normal cellularity, the physician may not generally consider the differential diagnosis of AA. Hematologists should be careful with respect to cellularity when interpreting the results of BM examinations, considering that BM cellularity cannot be accurately determined based on BM biopsies of limited sites in the iliac bone. Magnetic resonance imaging (MRI) of the thoracolumbar spine can be used to supplement an assessment of cellularity with a BM biopsy. However, MRI often produces equivocal results in elderly M A L I G N A N T H E M A T O L O G Y 25 disomy of the short arm of chromosome 6 (6pUPD).13 Detecting 6pUPD(+) leukocytes requires a single nucleotide polymorphism array analysis, which takes several weeks. On the other hand, HLA-LLs can be easily detected using monoclonal antibodies specific to HLA-A alleles with FCM if the patient is heterozygous for the HLA-A allele. Another useful examination that does not require advanced techniques is measurement of the plasma thrombopoietin (TPO) level. Patients with MDS who have a TPO level of 320 pg/mL or greater (TPOhigh patients) exhibit a high progression-free survival rate and good response to IST, similar to cases of AA.14 Virtually all patients having increased PNHtype cells or HLA-LLs fall into the TPOhigh group. Accordingly, if data for the TPO level are available, physicians may not need to examine the peripheral blood in patients with BM failure to determine the presence of other markers. The recent National Comprehensive Cancer Network guidelines recommend using hypomethylating agents for the treatment of MDS associated with thrombocytopenia; however, this option may be hazardous to TPOhigh patients, as BM failure in these cases is not based on the presence of abnormal stem cells with pre-leukemic features.15 The Hematologist Compendium 2010-2015 Figure 2 illustrates the mutual relationship of the markers that are potentially useful for diagnosing immune pathophysiology in patients who do not show any karyotypic abnormalities, increased blasts or ring sideroblasts in the BM. The precise role of these markers in the management of gray zone BM failure must be evaluated in a large prospective study because the predictive values of HLA-LLs and plasma TPO levels have only been assessed in Japanese patients with BM failure. When deciding on treatment, physicians also need to take patient age and disease duration into account, both of which are known to affect the response to IST.11 Patient Follow-up Based on the positive results for PNH-type granulocytes, we treated the patient with cyclosporine at a dose of 4 mg/kg/day. The pancytopenia gradually improved in response to this therapy, and the blood cell count seven months after the initiation of treatment was as follows: WBC, 3.0×109/L with 44 percent neutrophils; RBC, 2.79×1012/L; hemoglobin, 9.4 g/dL; MCV, 101.4 fL; platelets, 57×109/L; reticulocytes, 75×109/L. He continues to receive cyclosporine without any signs of significant toxicities. 1. Valent P, Horny HP, Bennett JM, et al. Definitions and standards in the diagnosis and treatment of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res. 2007;31:727-736. Figure 2 2. Matsuda A, Jinnai I, Iwanaga M, et al. Correlation between dysplastic lineage and type of cytopenia in myelodysplastic syndromes patients with refractory anemia according to the FAB classification. Am J Clin Pathol. 2013;140:253-257. 3. Yamazaki H, Nakao S. Border between aplastic anemia and myelodysplastic syndrome. Int J Hematol. 2013;97:558-563. 4. Parmentier S, Schetelig J, Lorenz K, et al. Assessment of dysplastic hematopoiesis: lessons from healthy bone marrow donors. Haematologica. 2012;97:723-730. TPOhigh 5. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509-2519. HLA-DRBI*15:01(+) 6. Nishimura R, Mase S, Araki R, et al. Massive hyper-reactive hematopoietic nests in bilateral iliac bones in a patient with mild aplastic anemia. Pediatr Blood Cancer. 2014;61:1903-1904. 7. Wang H, Chuhjo T, Yasue S, et al. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome. Blood. 2002;100:3897-3902. PNH-type cells(+) HLA-A allelelacking cells (+) TPOlow 8. Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. 2006;107:1308-1314. 9. Kulagin A, Lisukov I, Ivanova M, et al. Prognostic value of paroxysmal nocturnal haemoglobinuria clone presence in aplastic anaemia patients treated with combined immunosuppression: results of two-centre prospective study. Br J Haematol. 2014;164:546-554. 10. Tutelman PR, Aubert G, Milner RA, et al. Paroxysmal nocturnal haemoglobinuria phenotype cells and leucocyte subset telomere length in childhood acquired aplastic anaemia. Br J Haematol. 2014;164:717-721. 11. Sloand EM, Wu CO, Greenberg P, et al. Factors affecting response and survival in patients with myelodysplasia treated with immunosuppressive therapy. J Clin Oncol. 2008;26:2505-2511. 12. Sugimori C, Yamazaki H, Feng X, et al. Roles of DRB1 *1501 and DRB1 *1502 in the pathogenesis of aplastic anemia. Exp Hematol. 2007;35:13-20. 13. Katagiri T, Sato-Otsubo A, Kashiwase K, et al. Frequent loss of HLA alleles associated with copy numberneutral 6pLOH in acquired aplastic anemia. Blood. 2011;118:6601-6609. 14. Seiki Y, Sasaki Y, Hosokawa K, et al. Increased plasma thrombopoietin levels in patients with myelodysplastic syndrome: a reliable marker for a benign subset of bone marrow failure. Haematologica. 2013;98:901-907. WATCH THE EXPERTS Mutual relationship of the markers that are potentially useful for diagnosing immune pathophysiology in myelodysplastic syndrome patients who do not show any karyotypic abnormalities, increased blasts, or ring sideroblasts in the bone marrow. Each colored circle represents a subset of patients with immune pathophysiology, such that the darker the area is, the more likely immune pathophysiology is involved. TPOhigh, patients with a plasma thrombopoietin (TPO) level ≥320 pg/mL; TPOlow, patients with a plasma TPO level <320 pg/mL; PNH-type cells (+), patients possessing an increased percentage of paroxysmal nocturnal hemoglobinuria (PNH) –type cells; HLA-A allele-lacking cells (+), patients possessing HLA-A allele lacking leukocytes as a result of uniparental disomy of the short arm of chromosome 6. 15. Greenberg PL, Attar E, Bennett JM, et al. Myelodysplastic syndromes: clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2013;11:838-874. Dr. Nakao receives research funds and honoraria from Alexion. Joseph Mikhael, MD, Med, FRCPC Dr. Joseph Mikhael recaps the “Social Media for Hematologist” session at the 2015 ASH Annual Meeting. Attendees discussed the professional benefits of social media and how it can be used by professional hematology research and clinical communities to promote conversations that could have a positive impact on patient outcomes. Watch the video on the ASH Youtube page. Learn about ASH’s social media presence at www.hematology. org/Newsroom/465.aspx. Read an article by Dr. Mikhael in this Compendium on page 20. M A L I G N A N T H E M A T O L O G Y 26 The Hematologist Compendium 2010-2015 Hematopathologic Overview of Lymphocytosis – November/December 2015 Due to the recent publication date of this article, an update has not been requested. Figure 2 TRACY I. GEORGE, MD Associate Professor of Pathology; Director of the Hematopathology Fellowship Program, University of New Mexico School of Medicine, Albuquerque, NM Small, round nuclei Folded or cleaved nuclei The Question DIFFERENTIAL DIAGNOSIS CLL MBL MCL T-PLL Flow cytometry CLL FISH panel FISH CCND1/IGH FL MCL Atypical CLL T-cell lymphomas Pertussis* Flow cytometry FISH CCND1/IGH, BCL2 Tissue biopsy What is your approach to lymphocytosis? Convoluted nuclei Case A 71-year-old man with a history of atypical chronic lymphocytic leukemia (CLL) last treated in 2007 with a rituximab and chlorambucil-based regimen presents with an increasing M protein of 4.1 g/dl (IgGκ). The laboratory findings were as follows: WBC, 7.4 × 109/L with 29 percent neutrophils, 66 percent lymphocytes, and 5 percent monocytes; RBC, 3.94 × 1012/L; hemoglobin, 11.0 g/dL; mean corpuscular volume, 83 fL; platelets, 91 × 109/L. The patient’s bone marrow was hypercellular (90%) with a marked lymphoid infiltrate present in nodular (paratrabecular and interstitial) and focal diffuse patterns involving 75 percent of bone marrow cellularity. Lymphocytes were small and round with condensed chromatin and occasional plasmacytoid lymphocytes were also observed. The karyotype of the bone marrow was 46,XY,add(9)(p24),der(11)del(11)(p13)del(11)(q23) in four cells with a sideline containing all of these abnormalities and +13 in two cells, and an unrelated clone showing 45,X,–Y in six cells, with 46,XY in seven cells. Fluorescence in situ hybridization (FISH) found a deletion of the 13q14.3 region and was negative for deletions of TP53, ATM, and LAMP1, and aneuploidy for chromosome 12. Examining the Blood Smear A slide review is appropriate in all patients with an unexplained lymphocytosis in order to confirm the automated cell counts or to perform a manual differential for leukocyte classification. In manually prepared blood smears, larger white blood cells tend to collect at the edges of the smear and in the feathered edge. Good practice for slide review requires assessments of all cell types (leukocytes, red blood cells [RBCs], and platelets) in both quantity and quality. It is not uncommon for fragile leukocytes such as in CLL, infectious mononucleosis, or acute leukemia to smudge on blood smears. In these situations, a few drops of albumin can be added to peripheral blood before preparing the blood smear. These “albumin smears” allow for proper identification of leukocytes and reduce the number of “smudge” or “basket” cells. However, examination of RBCs and platelets should still be performed on the original blood smear because the albumin can affect platelet and erythrocyte morphology. Reactive Lymphocytosis Separating a monomorphic lymphocytosis from a pleomorphic lymphocytosis can help distinguish a lymphoproliferative disorder from a reactive lymphocytosis, respectively.1 Most reactive lymphocytoses show a wide range of sizes and shapes in lymphocytes. The classic example of a pleomorphic lymphocytosis is infectious mononucleosis, where the lymphocytes range in size from small and round, to intermediate with abundant cytoplasm (reactive lymphocytes), to frank immunoblasts. Figure 1 It is this spectrum of morphology that points to a greater likelihood that a patient has a reactive lymphocytosis; younger age is also a helpful clue. The causes of a reactive lymphocytosis are extensive and include infections (viral, bacterial, and parasitic), autoimmune disease, vaccination, drug hypersensitivity, endocrine disorders, stress (trauma, Bordetella pertussis infection in a child showing three reactive cardiac, extreme lymphocytes that are small with deeply cleaved nuclei and scant exercise), smoking, and cytoplasm, in contrast with the monocyte in the lower right-hand malignancy. corner. Reprinted with permission from I Pereira, TI George, While most of these reactive lymphocytoses are pleomorphic, a few M A L I G N A N T H E M A T O L O G Y 27 Sézary syndrome Adult T-cell leukemia Flow cytometry T-cell clonality HCL SMZL HCLV Flow cytometry BRAF T-PLL LPL Plasmacytoid LPL Plasma cell myeloma Plasma cell leukemia Flow cytometry SPEP/UPEP MYD88 L265P Myeloma FISH panel Granules T-LGL NK cell leukemia Flow cytometry T-cell clonality KIR profile T-PLL B-PLL HCLV MCL Flow cytometry Cytogenetics Burkitt Leukemia DLBCL MCL ALCL Flow cytometry FISH MYC CCND1/IGH ALK Prominent nucleoli My Response DA Arber. Atlas of Peripheral Blood. The Primary Diagnostic Tool. Wolters Kluwer, Lippincott Williams and Wilkins, Inc., Philadelphia, PA (2012), page 130, Figure 13.5. Villous cytoplasm ANCILLARY TESTS Large cells The differential diagnosis for a neoplastic lymphocytosis based on morphology in the peripheral blood smear. The bolded diagnosis represents the image shown. Further recommended ancillary testing is listed in the third column. CLL, chronic lymphocytic leukemia; MBL, monoclonal B-cell lymphocytosis; MCL, mantle cell lymphoma; T-PLL, T-prolymphocytic leukemia; FL, follicular lymphoma; HCL, hairy-cell leukemia; SMZL, splenic marginal-zone lymphoma; HCLV, hairycell leukemia variant; LPL, lymphoplasmacytic lymphoma, T-LGL, T large granular lymphocytic leukemia; B-PLL, B-prolymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; ALCL, anaplastic large-cell lymphoma. Adapted with permission from Chabot-Richards D, George TI. Leukocytosis. Int J Lab Hematol 2014;36:279-88, page 282, Figure 2. *Pertussis infection is a reactive cause of monomorphic lymphocytosis. important exceptions are worth mentioning. The first is Bordetella pertussis, the causative agent of whooping cough. The lymphocytes of B. pertussis are small and deeply clefted with mature chromatin as shown in Figure 1. As this is commonly seen in the pediatric and pregnant populations, clinical correlation will readily separate this from lymphomas, which can show similar morphologic features (e.g., follicular lymphoma or Sézary syndrome). The second exception is polyclonal B-lymphocytosis, which typically shows lymphocytes with distinct nuclear clefts but will demonstrate a spectrum of morphologic changes including nuclear lobation and binucleate forms. This uncommon disorder is found in young to middle-aged female smokers with a high association with human leukocyte antigen DR7, and several genetic abnormalities have also been documented.2 The final exception is a large granular lymphocytosis. Increased numbers of large granular lymphocytes (reactive lymphocytes with scattered azurophilic granules) are commonly seen with viral infections, malignancy, after bone marrow transplantation, and following chemotherapy. These populations of large granular lymphocytes will wax and wane. However, persistence of a large granular lymphocytosis with accompanying neutropenia and variable anemia should raise suspicion for large granular lymphocytic leukemia.3 This is typically T cell in origin, though a chronic lymphoproliferative disorder of natural killer cells is also well described. Flow cytometry is recommended in these cases, followed by either T-cell clonality or KIR analysis, if involving T cells or natural killer cells, respectively. Neoplastic Lymphocytosis Lymphoma cells tend to be monomorphic in appearance. While a blood smear may contain a subset of lymphoma cells, these cells will resemble one another and stand out against a background of normal bland lymphocytes. While CLL is the most common leukemia in adults in the western world and is frequently seen in peripheral blood (or its monoclonal B-cell lymphocytosis counterpart), peripheral blood involvement by bone marrow lymphoma is found in up to 30 percent of subjects in some studies.4 Lymphoma cells will show a wide variety of morphologic appearance, and this appearance raises a differential diagnosis as shown in Figure 2.5 Further identification of the type of lymphoproliferative disorder typically proceeds with flow cytometry. Although each laboratory has its own cocktail of antibodies used for flow cytometry, consensus guidelines have been published.6 While the results from flow cytometry narrow down one’s differential diagnosis to a short The Hematologist Compendium 2010-2015 list, additional genetic or other ancillary studies are typically needed for confirmation, such as FISH for CCND1/IGH to evaluate for mantle cell lymphoma. Additionally, bone marrow biopsy or biopsy of another involved site is often necessary for a final diagnosis. A common question is when should flow cytometry be performed. Some studies have looked at this question in adults. In one study, the authors retrospectively reviewed flow cytometry results of 71 patients 50 years of age and older with an absolute lymphocyte count of 4 × 109/L or greater that had been called suspicious for a lymphoproliferative disorder after smear review by a pathologist.7 Using receiver operating characteristic (ROC) analysis, they found that an absolute lymphocyte count greater than 6.7 × 109/L for patients 50 to 67 years of age, and 4 × 109/L or greater for patients older than 67 years had a 95 percent sensitivity and 76 percent specificity for predicting an abnormal flow cytometry phenotype. A more recent retrospective single-center study examined 71 adults with newly detected lymphocytosis greater than 5 × 109/L in a consecutive three-month period and found that 6.8 × 109/L was the best cut-off value for predicting a lymphoproliferative disorder with ROC analysis (sensitivity 90%, specificity 59%).8 In my own practice, other triggers for flow cytometry include a persistent unexplained lymphocytosis or a morphology that does not correlate with the diagnosis, as discussed below. This case was presented at a multidisciplinary tumor board conference. While the immunophenotype of the lymphocytes switched from CD5 to CD10 expression, the IGH data supported that the same neoplastic clone was present in both the 2007 and current bone marrow. Thus, a low-grade B-cell lymphoma with plasmacytic differentiation was found, raising a differential diagnosis of lymphoplasmacytic lymphoma, versus a marginal zone lymphoma with plasmacytic differentiation. While the lack of a MYD88 L265P mutation argues against lymphoplasmacytic lymphoma, my colleagues have found this to be 96 percent sensitive for a diagnosis of lymphoplasmacytic lymphoma in a study of 317 cases of low-grade B-cell lymphomas.10 A diagnosis of atypical CLL seemed less likely, as the lymphocytes lacked LEF1 expression – a marker identified based on gene expression profiling data that has been described as nearly 100 percent sensitive and specific for CLL.11 The lymphocytes also lacked expression of CD200 by flow cytometry, another marker overexpressed in CLL as well as lymphoplasmacytic lymphoma, though few cases of the latter were tested.12 While the patient lacked splenomegaly, a diagnosis of marginal zone lymphoma with plasmacytic differentiation was considered. The patient is scheduled to receive ibrutinib. 1. George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program. 2012;2012:475-484. 2. Mossafa H, Tapia S, Flandrin G, et al. Chromosomal instability and ATR amplification gene in patients with persistent and polyclonal B-cell lymphocytosis (PPBL). Leuk Lymphoma. 2004;45:1401-1406. Patient Follow-up Review of the patient’s original diagnostic material confirmed that the atypical CLL diagnosis was given based on immunophenotypic expression of FMC7, in addition to the usual phenotype for CLL (CD20+, CD5+, CD23+). Morphology in the current blood and bone marrow showed lymphocytes and plasmacytoid cells and not the usual small round lymphocytes with coarsely clumped chromatin (“soccer balls”) of CLL; the plasmacytoid cells were not readily identified on the earlier bone marrow smears. I performed flow cytometry on the patient’s bone marrow and identified a κ light chain–restricted B-cell population that expressed CD19, CD20, CD10, and CD23, and lacked expression of CD5 and CD200. Additionally, a κ light chain–restricted plasma cell population was identified. Immunohistochemistry was performed on the bone marrow clot section. Cyclin D1 and SOX-11 were negative in the B-cells, excluding the diagnosis of mantle cell lymphoma; SOX11 is a newer marker for mantle cell lymphoma that has been found to be expressed even in mantle cell lymphomas that lack overexpression of cyclin D1.9 LEF1 was negative, providing no support for a diagnosis of CLL. Markers of follicle center cell origin, BCL6 and LM02, were also negative, providing no support for a lymphoma of follicle center cell origin. The cytogenetic karyotype, while abnormal, was not specific for any particular B-cell lymphoma; the lack of t(11;14) and t(14;18) argued against both mantle cell lymphoma and follicular lymphoma. Molecular testing for MYD88 L265P mutation was performed and was negative. DNA polymerase chain reaction analysis for immunoglobulin heavy chain gene (IGH) was performed on the 2007 bone marrow and on the current 2015 bone marrow. A clone was detected in both samples that was identical in amplicon size. 3. Dhodapkar MV, Li CY, Lust JA, et al. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994;84:1620-1627. 4. Arber DA, George TI. Bone marrow biopsy involvement by non-Hodgkin’s lymphoma: frequency of lymphoma types, patterns, blood involvement and discordance with other sites in 450 specimens. Am J Surg Pathol. 2005;29:1549-1557. 5. Chabot-Richards DS, George TI. Leukocytosis. Int J Lab Hematol. 2014;36:279-288. 6. Béné MC, Nebe T, Bettelheim P, et al. Immunophenotyping of acute leukemia and lymphoproliferative disorders: a consensus proposal of the European LeukemiaNet Work Package 10. Leukemia. 2011;25:567-574. 7. Andrews JM, Cruser DL, Myers JB, et al. Using peripheral blood smear review, age and absolute lymphocyte count as predictors of abnormal peripheral blood lymphocytoses diagnosed by flow cytometry. Leuk Lymphoma. 2008;49:1731-1737. 8. Sun P, Kowalski EM, Cheng CK, et al. Predictive significance of absolute lymphocyte count and morphology in adults with a new onset peripheral blood lymphocytosis. J Clin Pathol. 2014;67:1062-1066. 9. Soldini D, Valera A, Solé C, et al. Assessment of SOX11 expression in routine lymphoma tissue sections: characterization of new monoclonal antibodies for diagnosis of mantle cell lymphoma. Am J Surg Pathol. 2014;38:86-93. 10. Insuasti-Beltran G, Gale JM, Wilson CS, et al. Significance of MYD88 L265P mutation status in the subclassification of low-grade B-cell lymphoma/leukemia. Arch Pathol Lab Med. 2015;139:1035-1041. 11. Tandon B, Peterson L, Gao J, et al. Nuclear overexpression of lymphoid-enhancer-binding factor 1 identifies chronic lymphocytic leukemia/small lymphocytic lymphoma in small B-cell lymphomas. Mod Pathol. 2011;24:1433-1443. 12. Challagundla P, Medeiros LJ, Kanagal-Shamanna R, et al. Differential expression of CD200 in B-cell neoplasms by flow cytometry can assist in diagnosis, subclassification, and bone marrow staging. Am J Clin Pathol. 2014;142:837-844. WATCH THE EXPERTS Dr. George indicated no relevant conflicts of interest. Eytan Stein, MD Dr. Eytan Stein discusses molecularly targeted therapies for acute myeloid leukemia (AML) in this video from the ASH Impact Series available on ASH On Demand. To watch this video and more, visit www.ashondemand.org/FreeContent/Videos. M A L I G N A N T H E M A T O L O G Y 28 The Hematologist Compendium 2010-2015 Ask the Hematologist N O N M A L I G N A N T H E M AT O L O GY This collection contains updated clinical information on malignant and nonmalignant hematology as profiled in The Hematologist. Specific Thrombophilia Work-Up Approach – March/April 2010 UPDATE/COMMENTARY Breast cancer is associated with an increased risk for venous thromboembolism (VTE). Among more than 13,000 women with breast cancer in four U.K. databases, the rate of VTE was 3.5 times that of age-matched controls.U1 Similarly, among more than 100,000 patients with breast cancer in two California databases, the one- and two-year incidences of VTE were 0.9 and 1.2 percent, with the incidence greatest in the first six months after diagnosis.U2 VTE incidence also increased while patients were receiving chemotherapy, in the month after therapy was discontinued, and during the initial three months of treatment with tamoxifen.U1 Nonetheless, compared with cancers of the pancreas, stomach, ovary, lung, and kidney, as well as gliomas and lymphomas, this risk is relatively low.U3 There remains a paucity of data regarding thromboprophylaxis in ambulatory patients with cancer in general and with breast cancer in particular. One relevant study, TOPIC-1, was halted prematurely because of no difference in VTE incidence between ambulatory breast cancer patients treated with low-molecular-weight heparin or placebo.U4 It is noteworthy that neither the American Society of Clinical Oncology,U5 the American College of Chest Physicians,U6 nor the European Society of Medical Oncology3 currently recommend thromboprophylaxis in ambulatory patients with any cancer, unless the individual patient is considered to be at high risk. JOEL S. BENNETT, MD Professor of Medicine, Hematology/Oncology Division, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA (Editor’s Note: The original question was submitted to Dr. Bennett through Consult a Colleague. He expanded his answer for print.) Clinical Problem I have been treating a 54-year-old woman with a resected breast cancer with adjuvant docetaxel, cyclophosphamide, and trastuzumab. She developed a central line thrombosis during treatment and was treated with warfarin for three months, after which the central line was removed. I plan to discontinue the warfarin in the next several weeks, assuming it is safe to do so while she is receiving trastuzumab. Should the patient undergo a work-up for thrombophilia despite a negative family history, and should warfarin be restarted if she needs another central line to complete a one-year course of trastuzumab? In 2010, I discussed the treatment of an ambulatory patient with breast cancer with adjuvant chemotherapy who developed a central-line thrombosis. Based on the information available in 2016, I would not change my recommendations. U1. Walker AJ, West J, Card TR, et al. When are breast cancer patients at highest risk of venous thromboembolism? A cohort study using English health care data. Blood. 2016;127:849-857. U2. Chew HK, Wun T, Harvey DJ, et al. Incidence of venous thromboembolism and the impact on survival in breast cancer patients. J Clin Oncol. 2007;25:70-76. U3. Mandala M, Falanga A, Roila F, et al. Management of venous thromboembolism (VTE) in cancer patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2011;22 Suppl 6:vi85-92. U4. Haas SK, Freund M, Heigener D, et al. Low-molecular-weight heparin versus placebo for the prevention of venous thromboembolism in metastatic breast cancer or stage III/IV lung cancer. Clin Appl Thromb Hemost. 2012;18:159-165. U5. Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013;31:2189-2204. U6. Kahn SR, Lim W, Dunn AS, et al. Prevention of VTE in nonsurgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidencebased clinical practice guidelines. Chest. 2012;141:e195S-226S. As I mentioned earlier, I think the central venous catheter itself, as a foreign body in the circulation, was the proximate cause of the thrombosis in your patient. So, it would seem logical to ask whether prophylactic anticoagulation could prevent this from happening again. There have been a number of studies addressing this question. However, a recently reported meta-analysis of eight randomized controlled trials of thromboprophylaxis in patients with cancer and central venous catheters concluded that thromboprophylaxis had no significant effect on the risk of catheter-related thrombosis.6 Similarly, in the WARP study of warfarin thromboprophylaxis in cancer patients with central venous catheters,7 it was found that neither fixed-dose warfarin at 1 mg per day nor dose-adjusted warfarin to maintain an INR of 1.5 to 2.0 reduced the incidence of catheter-related thromboses. Thus, the available evidence does not support the use of prophylactic anticoagulation should the central line be re-inserted. Although your patient has several recognized predispositions to thrombosis, the currently available evidence does not support the use of thromboprophylaxis for her clinical situation. Further, on the basis of this conclusion, as well her age and history, I don’t see the utility of a thrombophilia work-up. My Response 1. Blom JW, Doggen CJ, Osanto S, et al. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293:715-722. This is a common but complex situation. This patient has several predispositions to venous thrombosis, including breast cancer, chemotherapy, and an indwelling venous catheter. The thrombosis she experienced could have resulted from the cumulative effect of these factors, but I suspect the presence of the indwelling catheter was the major precipitating cause. 3. Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:381S-453S. As you know, cancers – in particular malignancies of the brain, adenocarcinomas of the lung and gastrointestinal tract, and hematologic malignancies – are associated with a sixfold to sevenfold increase in the risk for venous thromboembolism (VTE).1,2 Although this risk is greatest in the months following diagnosis and declines with time, the risk remains higher than in individuals without cancer even two years after diagnosis. Moreover, the risk for patients with metastatic disease is increased further compared to patients without. Although the risk for VTE in the first six months after a diagnosis of breast cancer is twofold to fourfold less than for patients with the malignancies noted above, the cumulative incidence of VTE for patients with breast cancer approaches that of the others, because patients with breast cancer live substantially longer. This raises the question whether prophylactic anticoagulation in a patient with breast cancer would be beneficial. This issue was addressed in the most recent American College of Chest Physicians Evidence-Based Clinical Practice Guidelines.3 After reviewing the available data, the authors of the guidelines concluded that the evidence does not support routine thromboprophylaxis for the primary prevention of VTE in ambulatory patients such as this one. 2. Blom JW, Vanderschoot JP, Oostindiёr MJ, et al. Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost. 2006;4:529-535. 4. Levine M, Hirsh J, Arnold A, et al. Double-blind randomised trial of a very-low-dose warfarin for prevention of thromboembolism in stage IV breast cancer. Lancet. 1994;343:886-889. 5. Khorana AA. Cancer and thrombosis: implications of published guidelines for clinical practice. Ann Oncol. 2009;20:1619-1630. 6. Chaukiyal P, Nautiyal A, Radhakrishnan S, et al. Thromboprophylaxis in cancer patients with central venous catheters. A systematic review and meta-analysis. Thromb Haemost. 2008;99:38-43. 7. Young AM, Billingham LJ, Begum G, et al. Warfarin thromboprophylaxis in cancer patients with central venous catheters (WARP): an open-label randomised trial. Lancet. 2009;373:567-574. Dr. Bennett indicated no relevant conflicts of interest. Chemotherapy administration and hormonal manipulation are also associated with an increased risk of VTE. Therefore, it is surprising that there is a paucity of studies addressing the efficacy of thromboprophylaxis in ambulatory cancer patients who are receiving chemotherapy or hormonal therapy. One widely cited study from 1994 reported that giving low-dose warfarin (INRs of 1.3-1.9) in patients with stage IV breast cancer receiving chemotherapy resulted in a significant reduction in VTE.4 However, these results were not replicated in subsequent studies,5 and thromboprophylaxis is not currently recommended for patients with cancer receiving chemotherapy or hormonal therapy.3 With specific regard to trastuzumab, after reviewing the prescribing information and searching the literature, I could not find evidence that VTE is a common adverse event related to the use of this drug. N O N M A L I G N A N T H E M A T O L O G Y 30 The Hematologist Compendium 2010-2015 Recommendations for Thrombophilia Testing for Women before Oral Contraceptives are Prescribed – November/December 2010 The authors have provided a rewrite of the original article to present all content updates since the publication date in 2010. DONNA DIMICHELE, MD, 1 AND ANDRA H. JAMES, MD, MPH 2 1. Deputy Director, Division of Blood Diseases and Resources, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 2. Consulting Professor of Obstetrics & Gynecology, Duke University, Durham, NC The Question A healthy 14-year-old girl presented with a hemoglobin of 2.9 g/lL, MCV 61 lL, and reticulocyte count of 0.9 percent. Pregnancy test was negative. Menarche began at age 11; periods were somewhat irregular and lasted five to seven days with clots. A diagnosis of severe iron deficiency due to menorrhagia was made and appropriately managed with transfusion and iron. Should an evaluation be done for an underlying bleeding disorder? What tests? What about such an evaluation if the anemia due to menorrhagia recurs three years later? The patient is now interested in contraception. A paternal aunt and maternal grandmother have a vague history of thrombosis requiring treatment. What thrombophilia testing should be undertaken in this patient before contraception is prescribed? Indeed, should thrombophilia testing be recommended for all women before oral contraceptives (OCs) are prescribed? What form of contraception and medication for regularization of periods should be advised? Our Response This case represents a clinical situation encountered frequently by pediatric hematologists. Unfortunately, clinical management decisions for adolescents must still be largely extrapolated from published data and recommendations on and for women.1 However, in a 2015 Committee Opinion endorsed by the American Academy of Pediatrics, the American College of Obstetricians and Gynecologists revised and updated their guidance on menstruation in girls and adolescents: menstrual cycle duration is typically 21 to 45 days; menstrual flow is seven days or less; and menstrual product use is three to six pads or tampons per day. 2 Menorrhagia or heavy menstrual bleeding is defined as greater than 80 mL of blood per cycle and can be predicted on the basis of clots ≥ 1 inch diameter or greater, low ferritin, and “flooding,” defined as a change of pad or tampon more frequently than hourly.3 In the case of the 14-year-old girl, heavy menstrual bleeding could be inferred on the basis of clots, cycles of up to seven days and severe iron-deficiency anemia in the absence of any other source of bleeding. The causes of heavy menstrual bleeding include acquired abnormalities of the uterine cavity (polyps, adenomyosis; leiomyoma; malignancy and hyperplasia; other endometrial abnormality) ovulatory dysfunction; iatrogenic causes (usually hormonal); causes not yet classified; and coagulopathy.4 However, in an adolescent, the cause is less likely to be an abnormality of the uterus and more likely to be ovulatory dysfunction, a hormonal cause, or possibly, an underlying bleeding disorder. A bleeding disorder may be the sole cause of heavy menstrual bleeding, or it may be a contributing cause. Consequently, an adolescent presenting with heavy menstrual bleeding requires a thorough evaluation that includes a thorough history and physical examination, an assessment of the uterus, an endocrine evaluation if there is anovulation or hypo-ovulation, and possibly, an evaluation for an underlying bleeding disorder.4 If anemia due to menorrhagia recurs three years later, a re-evaluation would be indicated. Not only is heavy menstrual bleeding more prevalent among women and girls with bleeding disorders, but bleeding disorders are more prevalent among those with heavy menstrual bleeding. In adolescents who present with heavy menstrual bleeding, the prevalence of von Willebrand disease (vWD) has been reported to be 5 to 36 percent; the prevalence of platelet dysfunction (depending on how it was defined) was 2 to 44 percent; the prevalence of factor VIII deficiency was 8 percent; and the prevalence of thrombocytopenia 13 to 20 percent. Dr. Claire S. Philipp and colleagues5 reported on the importance of various signs and symptoms as predictors of a bleeding disorder in women with heavy menstrual bleeding. After administering a 12-page questionnaire of bleeding symptoms, the investigators were able to develop a screening tool that was considered to be positive if one of the following four criteria was met: • Duration of menses greater than or equal to seven days and flooding or impairment of daily activities with most periods The laboratory evaluation for an underlying bleeding disorder would include a platelet count, mean platelet volume (MPV), and review of the blood smear to rule out thrombocytopenia and platelet morphologic disorders; prothrombin (PT), partial thromboplastin (aPTT), and thrombin times along with fibrinogen activity to screen for clotting factor deficiency; and a von Willebrand factor profile and platelet function studies. Additionally, specific factor activity levels would be performed if dictated by a prolonged PT or aPTT or otherwise suggested by high ethnic prevalence or an X-linked family history of a hemorrhagic diathesis. Any history or physical signs of connective tissue laxity should also prompt some consideration of collagen evaluation. If the bleeding is severe and the initial evaluation is negative, evaluation of the fibrinolytic system should be performed. Hormonal therapy is a frequent and usually effective approach to treating heavy menstrual bleeding in women and adolescents.1 That this adolescent is asking for contraception makes this an ideal first-line therapy. The choice of hormonal therapy is made more complicated by the family history of thrombosis and raises the question of thrombophilia screening prior to prescribing contraceptives. The substantially increased risk of venous thromboembolism (VTE) with either thrombophilic risk factor(s) or OCs is synergistically amplified when underlying thrombophilia and OCs are combined.6 Nevertheless, with a low-baseline VTE risk in young women of < 0.8 per 10,000 women years, even a 30-fold risk increase in women with factor V Leiden on OCs would translate into a very low overall event rate, limiting the cost effectiveness of routine screening prior to initiating OCs.6 Furthermore, the psychosocial consequences in adolescents of positive screening for thrombophilia or denial of access to OCs remain unexplored, but are probably significant. In the absence of an effective predictive screening strategy, alternative approaches include counseling for risk-factor avoidance (especially smoking), intermittent thromboprophylaxis during periods of high thrombosis risk (immobilization, lower extremity injury, surgery), and minimization of risk through the prescription of less thrombogenic OCs. These conservative measures would be warranted for this patient. OC preparations containing low-dose estrogen and a second-generation progestin (levonorgestrel) would be preferred. Although there are limited data on their use in adolescents and women with bleeding disorders, combined hormonal contraception in the form of a ring or patch would likely be equally effective in managing heavy menstrual bleeding, but both the ring and the patch are associated with an increased risk of VTE compared to OCs.7 An alternative given this patient’s family thrombosis history would be a levonorgestrel-containing intrauterine device (IUD), which has been demonstrated to decrease menstrual blood loss in women with bleeding disorders,8 but does not increase the risk of VTE.7 Although this patient is quite young, there are reports of the levonorgestrel IUD’s successful use in controlling menstrual blood flow in adolescents.1,9 Hemostasis prophylaxis prior to IUD insertion should be considered in adolescents with severe bleeding disorders. Finally, for those adolescents with a specific bleeding diagnosis who fail gynecologic/hormonal management of heavy menstrual bleeding, hemostatic therapy at the time of menses would be the next step. Oral tranexamic acid is FDA-approved for this indicaton in adults, but has been used in adolescents as well.10 1. James AH. Bleeding disorders in adolescents. Obstet Gynecol Clin North Am. 2009;36:153-162. 2. ACOG Committee Opinion No. 651: menstruation in girls and adolescents: using the menstrual cycle as a vital sign. Obstet Gynecol. 2015;126:e143-e146. 3. Warner PE, Critchley HO, Lumsden MA, et al. Menorrhagia I: measured blood loss, clinical features, and outcome in women with heavy periods: a survey with follow-up data. Am J Obstet Gynecol. 2004;190:1216-1223. 4. Munro MG, Critchley HO, Broder MS, et al. FIGO classification system (PALM-COEIN) for causes of abnormal uterine bleeding in nongravid women of reproductive age. Int J Gynaecol Obstet. 2011;113:3-13. 5. Philipp CS, Faiz A, Dowling NF, et al. Development of a screening tool for identifying women with menorrhagia for hemostatic evaluation. Am J Obstet Gynecol. 2008;198:163.e1-163.e8. 6. Wu O, Greer IA. Is screening for thrombophilia cost-effective? Curr Opin Hematol. 2007;14:500-503. 7. Lidegaard O, Nielsen LH, Skovlund CW, et al. Venous thrombosis in users of non-oral hormonal contraception: follow-up study, Denmark 2001-10. BMJ. 2012;344:e2990. 8. Kingman CE, Kadir RA, Lee CA, et al. The use of levonorgestrel-releasing intrauterine system for treatment of menorrhagia in women with inherited bleeding disorders. BJOG. 2004;111:1425-1428. 9. Silva CD, Geraldes F, Silva IS. Levonorgestrel intrauterine system as a treatment option for severe menorrhagia in adolescent with type III von Willebrand disease. BMJ Case Rep. 2013;2013. pii:bcr2013008833. 10. Srivaths LV, Dietrich JE, Yee DL, et al. Oral tranexamic acid versus combined oral contraceptives for adolescent heavy menstrual bleeding: a pilot study. J Pediatr Adolesc Gynecol. 2015;28:254-257. Dr. DiMichele and Dr. James indicated no relevant conflicts of interest. • History of treatment of anemia • Family history of a diagnosed bleeding disorder • History of excessive bleeding with tooth extraction, delivery or miscarriage, or surgery The screening tool alone had a sensitivity of 82 percent for bleeding disorders. Although the results would not be available at an initial visit, adding a pictorial blood assessment chart score > 100 increased the sensitivity of the screening tool to 95 percent. The patient in this case was positive for two of the criteria (7-day periods and anemia). Therefore, an evaluation for an underlying bleeding disorder would be indicated. N O N M A L I G N A N T H E M A T O L O G Y 31 The Hematologist Compendium 2010-2015 Treatment of Pregnant Female with Mesenteric Vein Thrombosis and History of Spontaneous Fetal Losses – January/February 2011 UPDATE/COMMENTARY In 2011, the question was posed concerning treatment of heparin-induced thrombocytopenia (HIT) during pregnancy. While there are no new treatment options for this patient, a couple of points are worth making. In terms of treatment, options are actually more limited for the acute setting, as the intravenous direct thrombin inhibitor lepirudin is no longer available. More data on the use of fondaparinux in treatment of HIT have been reported, and this remains a good option for treatment of outpatients and stable inpatients with HIT.U1 The major caveats remain its renal clearance and long half-life without a reversal agent. The patient present had a history of both pregnancy loss and mesenteric vein thrombosis (MVT) during her prior pregnancy. The indication for anticoagulation in this pregnancy was the history of MVT. Since 2011, more data have been published showing that anticoagulation does not prevent pregnancy loss.U2,U3 The evidence is strong in women without documented thrombophilia. In women with thrombophilia, we await the results of an ongoing clinical trial; however, based on current data, if there is an effect on pregnancy loss, it is small. While we have more oral anticoagulant options than in 2011, they remain without indication in pregnancy and, depending on the drug, animal data have demonstrated pregnancy loss and fetal harm.U4 Dabigatran crosses the placenta, and given that the other drugs are small molecules, they also would be expected to cross the placenta. Excretion in breast milk is unknown. There are accumulating data reported in women with some exposure during pregnancy,U4 but they are not sufficient to assess safety, and thus these drugs (dabigatran, rivaroxaban, apixaban, edoxaban) should not be initiated during pregnancy. U1. Kang M, Alahmadi M, Sawh S, et al. Fondaparinux for the treatment of suspected heparininduced thrombocytopenia: a propensity score-matched study. Blood. 2015;125:924-929. U2. Pasquier E, de Saint Martin L, Bohec C, et al. Enoxaparin for prevention of unexplained recurrent miscarriage: a multicenter randomized double-blind placebo-controlled trial. Blood. 2015;125:2200-2205. U3. Rodger MA. Recurrent pregnancy loss: drop the heparin needles… Blood. 2015;125:2179-2180. U4. Burnett AE, Mahan CE, Vazquez SR, et al. Guidance for the practical management of the direct oral anticoagulants (DOACs) in VTE treatment. J Thromb Thrombolysis. 2016;41:206-232. Dr. Konkle’s spouse has equity ownership in GlaxoSmithKline and Johnson and Johnson. bed thrombosis. It is unknown whether the JAK2 V617F mutation is associated with thrombosis in the setting of pregnancy. BARBARA A. KONKLE, MD Director, Clinical and Translational Research, Bloodworks Northwest; Professor of Medicine/ Hematology, University of Washington, Seattle, WA The Question A 34-year-old woman had mesenteric vein thrombosis during her last pregnancy in 2007 and two prior second trimester spontaneous fetal losses. She now presents at 13 weeks of pregnancy after being started on low-molecular-weight heparin (LMWH) by another hematologist 14 days prior. Her baseline platelet count was 160,000, but today it is 80,000. A PF4 antibody assay sent to rule out HIT was positive. Is it reasonable to stop the LMWH and put her on warfarin, since she has completed the first trimester? Danaparoid is not available, and fondaparanux is not on the American College of Chest Physicians’ list for HIT treatment. My Response HIT is uncommon during pregnancy. In a retrospective cohort study of 488 heparin-treated women (244 pregnant), there were no cases of HIT in the 10 pregnant women who became thrombocytopenic, but 10 cases in the 26 thrombocytopenic non-pregnant women.1 A study of 31 pregnant women receiving LMWH found that none were positive for anti-heparin/ PF4 antibodies or developed antibodies when followed prospectively throughout their pregnancies.2 However, case reports and clinical experience document the occurrence of HIT with and without thrombosis during pregnancy. Although HIT may be less common in patients treated with LMWH versus unfractionated heparin (UFH), it can occur and must be considered in pregnant women receiving LMWH who have a > 50 percent decrease in the platelet count. In choosing an anticoagulant for a pregnant woman with HIT, one has to consider the effects on both the woman and the fetus. Concerns with anticoagulants that cross the placenta include teratogenicity and bleeding. Data are limited and mostly reflect predictions based on drug characteristics and/or animal studies. Danaparoid, the drug with which there is the most experience in pregnancy, is unavailable in the United States. Fondaparinux, argatroban, and lepirudin have been reported in treatment of pregnant women with HIT.3,4 All are FDA Class B, indicating that animal studies have not shown harm in pregnancy, but there are no data from human studies. Outside the setting of pregnancy, fondaparinux, unlike argatroban and lepirudin, is not FDA-approved for HIT, but there are reports of its use in this setting.5 Patients who receive this drug may develop anti-heparin/PF4 antibodies, but it is not clear that these are associated with the development of HIT. Fondaparinux is a pentasaccharide that, with antithrombin, inhibits factor Xa. An advantage of this drug is its long half-life (~17 hrs.) allowing once-daily dosing. Disadvantages include managing a drug with a long half-life around delivery. Also, the drug has been detected in cord blood but at concentrations unlikely to cause bleeding,6 and no adverse outcomes have been reported in infants. The short-acting direct thrombin inhibitors (DTI) argatroban, lepirudin, and bivalirudin require intravenous administration. Successful short-term use of argatroban and lepirudin has been reported in pregnant women with HIT.4 Based on their pharmacokinetic properties, little or no drug would be expected to cross the placenta, although one study showed minor transplacental passage of lepirudin.6 The patient presented above has a history of MVT in her prior pregnancy. Pregnancy is a known risk factor for splanchnic bed thrombosis, and, in women with such a history, it is appropriate to provide at least prophylactic anticoagulation during subsequent pregnancies. The decision to test for additional conditions should be based on clinical and laboratory indicators of underlying disease. We would check a platelet count remote from pregnancy, because essential thrombocythemia is associated with both pregnancy loss and splanchnic N O N M A L I G N A N T H E M A T O L O G Y 32 When a pregnant woman develops HIT, the heparin should be discontinued; she should be evaluated for thrombosis by vascular ultrasound studies, and, unless contraindicated, alternative anticoagulation should be initiated. This patient needs continued anticoagulation because of the history of mesenteric vein thrombosis and her increased risk of thrombosis in the setting of HIT. As in non-pregnant patients, warfarin should not be started in the setting of acute HIT. The patient could be transitioned to warfarin later after the acute HIT resolves. Teratogenic effects have not been demonstrated when warfarin is ingested after 12 weeks gestation; however, it does cross the placenta and complications related to fetal hemorrhage have been reported. Two new oral anticoagulants, dabigatran and rivaroxiban, will not be useful alternatives, as both demonstrated reproductive toxicity and excretion in breast milk in animals.7 Given the need for continued outpatient anticoagulation, it is reasonable to switch her to fondaparinux, recognizing the need to discontinue the drug in anticipation of labor given its long half-life. In a patient on prophylactic therapy without a recent thrombotic event, it may be possible to stop the drug three days prior to scheduled induction or cesarean section. Epidural or spinal anesthesia could be given if an anti-Xa measurement demonstrates drug clearance. A woman at higher risk for thrombosis could be transitioned to an intravenous short-acting DTI, but since there will be anticoagulant effects in the mother and possibly the fetus, it should be discontinued in anticipation of a scheduled delivery to prevent bleeding complications. The best approach to delivery should be carefully considered in collaboration with maternal fetal medicine specialists. After delivery, anticoagulation should be re-initiated with either fondaparinux or a DTI with transition to warfarin. Fondaparinux is excreted in breast milk in animals, while argatroban, bivalirudin, and lepirudin are not predicted to be excreted in breast milk. Data from one lactating woman on therapeutic doses of lepirudin demonstrated no measureable drug in her breast milk. Thus, if the patient plans to breast feed, lepirudin with transition to warfarin, which also is not excreted in breast milk, may be the best post-partum option. While HIT in pregnancy is rare, it does occur and management requires knowledge of disease and drug effects on the mother and developing fetus, although data to inform these decisions are limited. Potential risks and benefits of different approaches should be discussed with the patient and her other health-care providers. 1. Fausett MB, Vogtlander M, Lee RM, et al. Heparin-induced thrombocytopenia is rare in pregnancy. Am J Obstet Gynecol. 2001;185:148-152. 2. Gerdsen F, Luxembourg B, Langer F, et al. A prospective analysis of heparin-platelet factor 4 antibodies in pregnant women treated with the low-molecular-weight heparin, dalteparin. Blood Coagul Fibrinolysis. 2008;19:477-481. 3. Mazzolai L, Hohlfeld P, Spertini F, et al. Fondaparinux is a safe alternative in case of heparin intolerance during pregnancy. Blood. 2006;108:1569-1570. 4. Young SK, Al-Mondhiry HA, Vaida SJ, et al. Successful use of argatroban during the third trimester of pregnancy: case report and review of the literature. Pharmacotherapy. 2008;28:1531-1536. 5. Hook KM, Abrams CS. Treatment options in heparin-induced thrombocytopenia. Curr Opin Hematol. 2010;17:424-431. 6. Dempfle CE. Minor transplacental passage of fondaparinux in vivo. N Engl J Med. 2004;350:1914-1915. 7. U.S. Food and Drug and European Medicines Agencies websites: www.pradaxapro.com; www.medicines. org.uk/emc/medicine/21265/SPC/Xarelto+10+mg+film-coated+tablets/#PREGNANCY Dr. Konkle consults and receives research funding from Bayer Corporation, and she has equity ownership in GlaxoSmithKline and Johnson & Johnson. The Hematologist Compendium 2010-2015 Factor Replacement for Surgery in 48-Year-Old Male with Factor XI Deficiency – September/October 2011 No update has been provided U PD ATE/C OM MEN T A R Y for this article. THOMAS L. ORTEL, MD, PhD Professor of Medicine, Chief of the Division of Hematology in the Department of Medicine, and Professor of Pathology, Duke University School of Medicine, Durham, NC (Editor’s Note: The original question was submitted to Dr. Ortel through Consult a Colleague. He expanded his answer for print.) The Question A 48-year-old man with factor XI deficiency (2% activity) was first diagnosed at the age of 17 during evaluation for a prolonged aPTT. He underwent extraction of wisdom teeth without plasma or topical support uneventfully. Periodontal surgery led to oozing for which he was successfully treated with oral aminocaproic acid. He now needs arthroscopic surgery on his left shoulder for repair of a torn rotator cuff. Does he need factor replacement prior to surgery and afterward? Relatively limited information is available describing therapeutic arthroscopy of the shoulder in patients with clotting disorders. A single case series describing the management and outcomes of five patients undergoing a total of six shoulder arthroscopic procedures included one patient with a mild factor XI deficiency (baseline level of 60 U/dL) who underwent arthroscopic subacromial decompression.8 This patient was treated with a combination of intravenous and oral tranexamic acid without any bleeding complications. Although shoulder arthroscopy is performed in a tissue not considered to have high fibrinolytic activity, the impact of even a minimal increase in bleeding during a surgical procedure in an enclosed space can be significant. The severely decreased factor XI activity level in this patient, and the fact that he had previously required antifibrinolytic therapy for periodontal surgery, are concerning for an increased bleeding risk. Consequently, it was recommended that the patient receive fresh frozen plasma to supplement the factor XI level prior to the procedure. Post-operatively, depending on the factor XI activity levels and clinical outcome of the procedure, additional fresh frozen plasma could be administered or antifibrinolytic therapy could be substituted instead. My Response 1. Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Proc Soc Exp Biol Med. 1953;82:171-174. Inherited deficiency of factor XI was first described in 1953 as a mild to moderate bleeding disorder.1 Factor XI deficiency exhibits an autosomal inheritance pattern with a variable clinical penetrance, as described below. It has been described in a wide variety of population groups but is most common in Ashkenazi Jews. In this particular group, it is estimated that one in eight individuals are heterozygous and one in 190 are homozygous for mutations in the factor XI gene.2,3 3. Gomez K, Bolton-Maggs P. Factor XI deficiency. Haemophilia, 2008;14:1183-1189. Spontaneous bleeding, except for menorrhagia, is uncommon in patients with factor XI deficiency. Most bleeding episodes occur following surgical procedures or trauma. In contrast to patients with hemophilia A or B, the correlation between clinical bleeding and the baseline factor XI clotting activity is poor. For example, some patients with severe factor XI deficiency (factor XI level < 10-20 IU/dL) exhibit no increase in hemorrhage, whereas other individuals with levels that are only moderately below the lower limit of the normal range develop bleeding complications after surgery.3 7. Levy O, Haddo O, Sforza G, et al. “Put your ‘extended’ finger on the bleeder”: the use of direct pressure from the shaver blade to achieve hemostasis. J Arthroscopic Related Surgery. 2011;27:867-869. 2. Seligsohn U. Factor XI deficiency in humans. J Thromb Haemost. 2009;7: Suppl 1:84-87. 4. Salomon O, Steinberg DM, Seligsohn U. Variable bleeding manifestations characterize different types of surgery in patients with severe factor XI deficiency enabling parsimonious use of replacement therapy. Haemophilia. 2006;12:490-493. 5. Bolton-Maggs P. Factor XI deficiency – resolving the enigma? Hematology 2009. 2009;97-105. 6. Mannucci PM, Bauer KA, Santagostino E, et al. Activation of the coagulation cascade after infusion of a factor XI concentrate in congenitally deficient patients. Blood. 1994;84:1314-1319. 8. Thomas JMC, Tuddenham EG, Ahrens PM. Therapeutic shoulder arthroscopy in patients with clotting disorders. Haemophilia. 2008;14:859-861. Dr. Ortel indicated no relevant conflicts of interest. Rates of hemorrhagic complication have also been shown to vary depending on the type of surgery being performed.4 In particular, procedures involving sites with increased local fibrinolytic activity, such as the oral mucosa, nose, and genitourinary tract, are more frequently associated with excessive bleeding compared to procedures involving tissues not expressing fibrinolytic activity, such as bones or muscles. This study included six patients who underwent trauma surgery and one total knee replacement, but none of the patients underwent an arthroscopic procedure. The implication from these observations is that conservative use of replacement therapy in patients with even severe factor XI deficiency undergoing certain types of surgery is possible.4 Management of bleeding episodes and prevention of bleeding in relation to surgery is not straightforward and needs to be tailored to the individual patient.5 In many patients with factor XI deficiency, antifibrinolytic therapy alone may be sufficient to prevent bleeding in most surgical settings. Factor XI levels can be raised with fresh frozen plasma, but relatively large volumes may be required. The hemostatic level of factor XI activity to target is debated, but a level of 30 to 45 IU/dL is probably sufficient in patients with severe deficiency.5 Factor XI concentrates have been associated with an increased thrombotic risk in certain patients and are not available in the United States.6 Good hemostasis helps to maintain arthroscopic visual clarity during surgery and is essential for the successful completion of the procedure. A variety of strategies have been used to control arthroscopic bleeding, including using electrocautery or a radiofrequency thermal probe, adding epinephrine to the inflow irrigation solution or increasing the irrigation fluid inflow pressure, and applying direct pressure via the shaver blade.7 N O N M A L I G N A N T H E M A T O L O G Y 33 The Hematologist Compendium 2010-2015 Immune Thrombocytopenia with Pulmonary Embolism and Deep-Vein Thrombosis: Recommendations for Bone Marrow Aspirate and Biopsy – November/December 2011 UPDATE/COMMENTARY In an article written for The Hematologist in 2011, I reviewed the results of several small series and population-based analyses that assessed the risk for thromboembolism in patients with immune thrombocytopenia (ITP). I concluded that “these studies provide evidence that the incidence of thromboembolism is increased in patients with ITP, even in the presence of very low platelet counts.” Since that time, there has been no evidence presented to suggest that that conclusion is not accurate. Indeed, several additional analyses involving large numbers of patients with ITP support the conclusion that ITP is indeed associated with a small, but significant risk of thromboembolism,U1 a risk that many hematologists are now aware of. In most patients with ITP, the increased risk of thrombosis is small (relative risk < 2) and not sufficient to influence management.U2 However, more recent studies suggest that certain subpopulations of patients with ITP may be at a significantly higher risk of thrombotic complications. These include patients who have undergone splenectomy; KEITH MCCRAE, MD Director, Benign Hematology, Department of Hematology and Solid Tumor Oncology; Professor, Molecular Medicine, Department of Cellular and Molecular Medicine, Cleveland Clinic, Cleveland, OH (Editor’s Note: This question was submitted through the Consult-a-Colleague Program. Dr. McCrae was asked to respond.) The Question How often do you see ITP with pulmonary embolism (PE) and deep-vein thrombosis (DVT)? The patient is a 60-year-old woman who had pneumonia about a month ago and now is presenting with a platelet count of 30,000/µl with DVT and PE. A hypercoagulable diagnostic panel, including tests for heparin-induced thrombocytopenia (HIT) and antiphospholipid antibodies, are pending. Would you perform a bone marrow aspirate and biopsy? A pulmonologist inserted an inferior vena cava filter; was that appropriate? My Response ITP is a fascinating disorder with a complex pathogenesis that has only recently begun to be understood in detail.1,2 While a number of immune abnormalities underlie the development of ITP, the disease is largely mediated by antibodies reactive with glycoproteins expressed on platelets and megakaryocytes. These antibodies cause accelerated platelet destruction in the spleen as well as deficient platelet production secondary to impaired maturation of megakaryocytes. several studies, including a large study that identified patients with ITP undergoing splenectomy through administrative codes,U3 suggest that thrombotic risk is increased more than 5-fold in the 90 days after surgery, and 2.7-fold thereafter. Patients older than 60 years are also at increased risk of thromboembolism. Finally, though no definitive data confirm an increased thrombotic risk associated with thrombopoietin receptor agonists, some studies do suggest increased risk with these agents, and this should be considered when treating patients with ITP who have other significant thrombotic risk factors. U1.Ruggeri M, Tosetto A, Palandri F, et al. Thrombotic risk in patients with primary immune thrombocytopenia is only mildly increased and explained by personal and treatment-related risk factors. J Thromb Haemost. 2014;12:1266-1273. U2.Rodeghiero F. Is ITP a thombophilic disorder? Am J Hematol. 2016;91:39-45. U3.Boyle S, White RH, Brunson A, et al. Splenectomy and the incidence of venous thromboembolism and sepsis in patients with immune thrombocytopenia. Blood. 2013;121:4782-4790. antibodies (APLA) appears to be increased in patients with ITP, and several studies suggest that ITP patients with APLA develop thrombosis more frequently. In a prospective study, Diz-Küçükkaya assessed 82 newly diagnosed patients with ITP, finding 31 (37.8%) positive for APLA.8 After five years, the cumulative thrombosis-free survival of APLA-positive and APLA-negative patients was 39 percent and 97.7 percent, respectively (p=0.004). Though current guidelines do not recommend routine screening of ITP patients for APLA,9,10 this should be considered in those who develop thrombi. Additional factors contributing to the development of thrombosis in patients with ITP may include elevated levels of platelet-derived microparticles11 and complement activation on antibody-coated platelets.12 The management of thrombosis in thrombocytopenic patients with ITP is challenging and not addressed by current guidelines. Likewise, there are no evidence-based data on which to draw from for recommendations. Many experts suggest that the risk of anticoagulation is acceptable at platelet counts above 40,000 to 50,000/µl, although the severity of the thrombotic event and clinical characteristics of the patient should be considered. In the case presented here, the clot burden is extensive and the platelet count close to where anticoagulation is reasonable. Risk factors for bleeding in ITP include a prior history of bleeding and age ≥ 60 years, the latter of which is applicable to this patient. I would recommend aggressive treatment of this patient’s ITP, initially using corticosteroids, since agents such as IVIg may potentially predispose to further thrombosis, and I would consider institution of anticoagulation with intravenous heparin or another short-lived anticoagulant once the platelet count increases to ≥ 45,000/µl. The use of an IVC filter is a matter of preference; it may be more strongly justified if there is evidence of progressive thrombosis, or if the thrombocytopenia does not respond quickly. However, given the association of ITP with thrombosis, I would avoid intravascular devices if possible, and if an IVC filter is used, I would recommend a removable model. The most common clinical presentation of ITP is bleeding, the severity of which correlates with the degree of thrombocytopenia.3 However, many patients with ITP have few bleeding complications even with very low platelet counts, an observation that may be related to the presence of young, highly functional platelets, increased levels of plasma microparticles, or other factors. Surprisingly, recent studies have also demonstrated that patients with ITP have an increased risk of thrombosis. The data concerning ITP and thrombosis are derived from several sources. A retrospective study from Aledort et al. suggested an increased risk of thrombosis in patients with ITP, with 18 thromboembolic events occurring in 186 adults (the majority after the diagnosis of chronic ITP).4 In another retrospective study reported in an abstract by Bennett et al., a 6.9 percent cumulative incidence of thromboembolic events in patients with ITP was seen over a 15-month period. Sarpatwari et al. prospectively analyzed the incidence of thromboembolic events in patients with ITP using the UK General Practice Research Database, identifying 1,070 adults >18 years of age with primary ITP that were matched with 4,280 ITP-free patients.5 Over a median of 47.6 months, the adjusted hazard ratio for venous, arterial, or combined thromboembolic events in patients with ITP was 1.58 (95% CI, 1.01-2.48), 1.37 (95% CI, 0.94-2.00), and 1.41 (95% CI, 1.04-1.91), respectively. Subgroup analysis suggested a strikingly increased risk of myocardial infarction in the ITP cohort. Also suggested was a direct relationship between the severity of thrombocytopenia and the risk of thrombosis. Another recent study utilizing a matched ITP cohort obtained from the Danish National Patient Registry observed an incidence rate ratio for venous thromboembolism in patients with ITP of 2.04 (95% CI, 1.45-2.87).6 Two of the ITP patients with VTE had severe thrombocytopenia, although one was ultimately diagnosed with lung cancer. The decision to perform a bone marrow aspirate and biopsy should not be influenced by a thrombotic event. If the peripheral blood smear shows isolated thrombocytopenia and the history and physical examination do not suggest other disorders, bone marrow aspirate would not be required. Taken together, these studies provide evidence that the incidence of thromboembolism is increased in patients with ITP, even in the presence of very low platelet counts. While acute portal vein thrombosis has been reported in up to 15 percent of ITP patients undergoing splenectomy, and an increased risk of subsequent thrombosis has been associated with splenectomy for thalassemia,7 the review from Sarpatwari et al. demonstrates a similar incidence of thrombosis in splenectomized and non-splenectomized ITP patients. 9. Neunert C, Lim W, Crowther M et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011;117:4190-4207. The mechanisms underlying the paradoxical development of thrombosis in patients with ITP have not been defined, though several have been postulated. The incidence of antiphospholipid N O N M A L I G N A N T H E M A T O L O G Y 34 1. Semple JW, Provan D, Garvey MB, et al. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol. 2010;17:590-595. 2. Cines DB, Liebman HA. The immune thrombocytopenia syndrome: a disorder of diverse pathogenesis and clinical presentation. Hematol Oncol Clin North Am. 2009;23:1155-1161. 3. Fogarty PF. Chronic immune thrombocytopenia in adults: epidemiology and clinical presentation. Hematol Oncol Clin North Am. 2009;23:1213-1221. 4. Aledort LM, Hayward CP, Chen MG, et al. Prospective screening of 205 patients with ITP, including diagnosis, serological markers, and the relationship between platelet counts, endogenous thrombopoietin, and circulating antithrombopoietin antibodies. Am J Hematol. 2004;76:205-213. 5. Sarpatwari A, Bennett D, Logie JW et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom General Practice Research Database. Haematologica. 2010;95:1167-1175. 6. Severinsen MT, Engebjerg MC, Farkas DK et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol. 2011;152:360-362. 7. Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood. 2009;114:2861-2868. 8. Diz-Küçükkaya R, Hacehanefioglu A, Yenerel M et al. Antiphospholipid antibodies and antiphospholipid syndrome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood. 2001;98:1760-1764. 10. Provan D, Stasi R, Newland AC et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168-186. 11. Jy W, Horstman LL, Arce M, Ahn YS. Clinical significance of platelet microparticles in autoimmune thrombocytopenias. J Lab Clin Med. 1992;119:334-345. 12. Peerschke EI, Yin W, Ghebrehiwet B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol. 2010;47:2170-2175. Dr. McCrae indicated no relevant conflicts of interest. The Hematologist Compendium 2010-2015 Heparin-Induced Thrombocytopenia being Treated with Argatroban with Persistent Thrombocytopenia – January/February 2012 UPDATE/COMMENTARY There have been a number of reports in the literature where a direct oral anticoagulant has been used in the treatment of heparin-induced thrombocytopenia (HIT). While dabigatran and apixaban have been employed, the most experience is with rivaroxaban.U1-3 Dr. Lori-Ann Linkins and colleaguesU3 conducted a multicenter, single arm, cohort study that included 22 consecutive patients with suspected or confirmed HIT. Participants with a positive local assay were administered rivaroxaban 15 mg bid until platelet recovery or 21 days if there was acute thrombosis at study entry; this was followed by 20 mg daily. Among the 12 patients who proved to be HIT-positive, six had HIT with thrombosis (HITT) at study entry. Platelet recovery was seen in nine of 10 with thrombocytopenia; new thrombosis occurred in one, and one required limb amputation despite platelet recovery. This and other case reports suggest that direct oral anticoagulants such as rivaroxaban are effective in treating HIT. KENNETH A. BAUER, MD Professor of Medicine, Harvard Medical School, Beth Israel Deaconess Medical Center, Boston, MA (Editor’s Note: This question was submitted through the Consult-a-Colleague program. Dr. Bauer was asked to respond.) The Question How do you manage a patient with HIT being treated with argatroban who has persistent thrombocytopenia? The patient is a 45-year-old man with laboratory evidence of HIT (positive serotonin release assay) without thrombosis who has been treated with argatroban for 10 days. The platelet count was 52,000 per microliter at the time of diagnosis and increased to 95,000 per microliter during the first five days of therapy. However, over the past five days, no significant increase in the platelet count has been observed. •Is there a platelet threshold at which you recommend starting warfarin? If so, what evidence supports that determination? Would your recommendation change if the patient had HITT? •If the recommended platelet threshold is not reached, is there a point at which you feel it is safe to start treatment with warfarin? For example, in this patient who has been on argatroban for 10 days, is it acceptable to start warfarin even though his platelet count is less than 100,000 per microliter? Would your recommendation change if the patient had HITT? •Does fondaparinux play a role in management of HIT in this setting? •Is there a role for the new oral anticoagulants (dabigatran and rivaroxaban) in this setting? My Response HIT is a clinical-pathological syndrome that is typically characterized by the onset of thrombocytopenia (≥50% fall from baseline) within five to 10 days of drug initiation. The mean platelet count in patients with this disorder is 60,000 per microliter. Thrombosis occurs in approximately 50 percent of patients and clusters in the first few days following onset of thrombocytopenia (termed HITT). It is mediated by antibodies against platelet factor 4 (PF4)-heparin complexes that activate platelets leading to excessive thrombin generation. Prompt recognition and appropriate treatment of HIT is required to reduce the risk of serious thrombotic events and complications including limb loss. Paradoxically, a major problem with HIT today is its over-diagnosis. Enzyme immunoassays (EIAs) for antibodies against PF4-heparin complexes are currently widely applied for making the diagnosis of HIT; while highly sensitive (>99%), only some antibodies have strong platelet-activating properties in the serotonin-release assay, one of the “gold standard” platelet-activation assays used for diagnostic confirmation. As results of serotonin release assays are rarely available for clinical decision making early in the course of HIT, it is best not to order ELISA tests in patients with low pretest probability scores for HIT (4T score: Thrombocytopenia, Timing of platelet count fall, Thrombosis, oTher causes of thrombocytopenia).1 Also, many labs do not report the optical densities (OD) of positive EIAs; in such instances, it is useful to request this information, as it can be useful diagnostically since the strength of a positive EIA (OD) predicts for a positive SRA.2 After stopping heparin administration in patients with HIT, the median time to achieving a platelet count of >150,000 per microliter is about four days. However, in patients with more severe HIT, the platelet count can take longer (up to two weeks or more) to recover. It is important to keep in mind, however, that patients with HIT often have concurrent medical problems or are on other medications contributing to thrombocytopenia. The recommendation in the 8th Edition of the American College of Chest Physicians EvidenceBased Clinical Practice Guidelines is as follows3: “For patients with strongly suspected or confirmed HIT, we recommend against the use of vitamin K-antagonist (VKA) therapy until N O N M A L I G N A N T H E M A T O L O G Y 35 U1. Ng HJ, Than H, Teo EC. First experiences with the use of rivaroxaban in the treatment of heparin-induced thrombocytopenia. Thromb Res. 2015;135:205-207. U2. Dhakal P, Pathak R, Giri S, et al. New Oral Anticoagulants for the Management of Heparin Induced Thrombocytopenia: A Focused Literature Review. Cardiovasc Hematol Agents Med Chem. 2015;13:87-91. U3. Linkins LA, Warkentin TE, Pai M, et al. Rivaroxaban for Treatment of Suspected or Confirmed Heparin-Induced Thrombocytopenia Study. J Thromb Haemost. 2016;14:1206-1210. Dr. Bauer has served as a consultant to Janssen, BMS, Daiichi Sankyo, and Instrumentation Laboratory. after the platelet count has substantially recovered (i.e., usually to at least 150 × 109/L) over starting VKA therapy at a lower platelet count (Grade 1B); that VKA therapy be started only with low, maintenance doses (maximum, 5 mg of warfarin or 6 mg of phenprocoumon) rather than with higher initial doses (Grade 1B); and that the non-heparin anticoagulant (e.g., lepirudin, argatroban, danaparoid) be continued until the platelet count has reached a stable plateau, the INR has reached the intended target, and after a minimum overlap of at least five days between non-heparin anticoagulation and VKA therapy rather than a shorter overlap (Grade 1B).” Despite a platelet count that has only risen to 95,000 per microliter, I would transition this patient with HIT without thrombosis (termed isolated HIT) off a parenteral direct thrombin inhibitor (i.e., argatroban) after 10 days of therapy. Management options for this patient include: 1) initiation of warfarin carefully under the cover of continued IV argatroban; 2) initiation of fondaparinux alone at therapeutic doses (provided the patient’s creatinine clearance is greater than 30 mL/min) in lieu of argatroban; or 3) initiation of an oral direct thrombin or factor Xa inhibitor (either dabigatran etexilate or rivaroxaban) at therapeutic doses. The initiation of warfarin causes a rapid decline in protein C levels and a slower decline in the levels of longer-lived vitamin K-procoagulant factors (especially prothrombin) augmenting the preexistent hypercoagulable state due to HIT. This mechanism is contributory to the development of venous limb gangrene, which is estimated to occur in 12 percent of patients with HITT4; its frequency is likely less in patients with isolated HIT. Fondaparinux, a synthetic pentasaccharide that selectively inhibits factor Xa after binding to antithrombin, has an attractive profile for the treatment of HIT based on in vitro studies; it is administered subcutaneously once daily. Small case series indicate generally favorable outcomes with initial use of this agent in HIT; in a recent retrospective series of 16 patients with the diagnosis confirmed by serotonin-release assay, nine of whom had thrombosis, none developed new or recurrent thrombosis.5 However, fondaparinux is not approved for this indication; it is approved in the United States for the prophylaxis of VTE following major orthopedic and general surgery and the initial treatment of VTE. If the continuation of argatroban is the only reason for continued hospitalization, I would employ fondaparinux alone at this juncture provided the patient could self-inject the medication and drug acquisition was not a problem (i.e., inability to gain insurance approval or prohibitive out-of-pocket cost). The total duration of anticoagulant treatment has not been defined by prospective studies, but I would treat isolated HIT for a minimum of six weeks given the high risk of thrombosis within the first 30 days after diagnosis. The use of fondaparinux to complete treatment would obviate the use of warfarin entirely and the burden of INR monitoring with attendant dose adjustments. Alternatively, warfarin could be initiated after the platelet count had risen to more than 150,000 per microliter under the cover of fondaparinux overlap for a minimum of five days until the INR reached the target range of 2-3. This would also circumvent complexities relating to INR monitoring during argatroban therapy if this option were chosen.3 For patients with HITT, I would treat with anticoagulation for three to six months. It should be pointed out that the efficacy of non-heparin anticoagulants for HIT and their approval by the U.S. FDA was not based on prospective, randomized controlled clinical trials. Vitamin K antagonists have been used for the treatment of HIT for many years and adopted by evidence-based guidelines after initial treatment with a non-heparin anticoagulant, as they have up until recently been the only class of oral anticoagulants available. Based on our mechanistic understanding of venous-induced limb gangrene in HIT, a strong case can be made that we should be moving away from using warfarin in the initial phase of HITT (and HIT) treatment (up until 30 days after diagnosis) given that alternative oral anticoagulants that do not lower protein C levels are available.6 These oral agents selectively target thrombin or factor Xa, have a rapid onset of action, and do not require coagulation monitoring; however, there is not yet any reported experience with these agents in this patient population, and they have no specific antidote. Dabigatran and rivaroxaban have gained FDA approval for stroke prevention in atrial fibrillation7,8 and rivaroxaban is The Hematologist Compendium 2010-2015 approved for the prophylaxis of VTE following total hip or knee replacement9; they have shown promising result for the treatment of symptomatic VTE but have not yet been approved for this indication in the United States. Thus, caution should be exercised if either dabigatran or rivaroxaban is used in a patient such as this; if chosen, they should be used at therapeutic doses and limited to adult patients with satisfactory renal function (creatinine clearance > 30 mL/min). Furthermore, given that HIT can result in serious complications including limb loss and subsequent litigation against health-care providers, hematologists choosing to use any of the new anticoagulants (dabigatran, rivaroxaban, or fondaparinux) should carefully document in the medical record their rationale for choosing the new agent. Both the infrequent occurrence of HIT confirmed by validated platelet-activation assays and the clinical heterogeneity of affected patients make it difficult to perform trials of new agents in HIT. It is therefore unlikely that the new oral anticoagulants will be studied in controlled trials so as to gain FDA approval for management of this disorder in the near future. Hopefully, the reporting of well-characterized cohorts of patients with HIT (or HITT) treated with these agents will lead to favorable outcomes that will improve, as well as simplify, management of this disorder. 1. Lo GK, Juhl D, Warkentin TE , et al. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4:759-765. 2. Warkentin TE, Sheppard JI, Moore JC, et al. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost. 2008;6:1304-1312. 3. Warkentin TE, Greinacher A, Koster A, et al. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:340S-380S. 4. Warkentin TE, Elavathil LJ, Hayward CP, et al. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann Int Med. 1997;127:804-812. 5. Warkentin TE, Pai M, Sheppard JI, et al. Fondaparinux treatment of acute heparin-induced thrombocytopenia confirmed by the serotonin-release assay: a 30-month, 16-patient case series. J Thromb Haemost. 2011;9:2389-2396. 6. Krauel K, Hackbarth C, Fürll B, et al. Heparin-induced thrombocytopenia: in vitro studies on the interaction of dabigatran, rivaroxaban, and low-sulfated heparin, with platelet factor 4 and anti-PF4/ heparin antibodies. Blood. 2012;119:1248-1255. 7. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Eng J Med. 2009;361:1139-1151. 8. Patel MR, Mahaffey KW, Garg J, et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med. 2011;365:883-891. 9. Turpie AG, Lassen MR, Eriksson BI, et al. Rivaroxaban for the prevention of venous thromboembolism after hip or knee arthroplasty. Pooled analysis of four studies. Thromb Haemost. 2011;105:444-453. Dr. Bauer has served as a consultant to GSK, Bayer Healthcare, and Johnson & Johnson. The Hematologist Video Series A new video series for The Hematologist titled “Conversations with Innovators” is available on YouTube; it features three videos by Dr. Omar Abdel-Wahab from Memorial Sloan Kettering Cancer Center, Dr. Neal Young from the National Heart, Lung, and Blood Institute at the National Institutes of Health (NIH), and Dr. Ann Mullally from the Brigham and Women’s Hospital and the Dana-Farber Cancer Institute. Dr. Abdel-Wahab discusses his lab’s efforts to understand the biology of mutations in components of the spliceosome with the goal of using them as therapeutic targets in the treatment of leukemias. Dr. Young covers his lab’s investigation of an improved treatment protocol for aplastic anemia involving immunosuppressive therapy in combination with eltrombopag. In the third video, Dr. Mullally talks about her lab’s study of mutant calreticulin in patients with myeloproliferative neoplasms. To watch all three videos and for more to come soon, visit www.youtube.com/ASHwebmaster under the playlist titled “The Hematologist: Conversations with Innovators.” N O N M A L I G N A N T H E M A T O L O G Y 36 The Hematologist Compendium 2010-2015 Approach to the Diagnosis and Management of the Anemia of Chronic Inflammation – March/April 2013 UPDATE/COMMENTARY Since the publication of this “Ask the Hematologist” article, the role of inflammation in reduced renal erythropoietin (EPO) production has been better defined. With inflammation, renal EPO-producing fibroblasts have increased transforming growth factor-β1 (TGFβ1) and NFκB expressions that mediate their transformation to myofibroblasts, which no longer produce EPO.U1 The potential restoration of EPO production in these cells by inhibition of specific hypoxic-inducible factor (HIF) prolylhydroxylases suggests a potential treatment for some patients with anemia of chronic inflammation (ACI).U2 In a study of patients with unexplained anemia of the elderly (UAE) and age-matched normal controls, increases in serum ferritin and decreases in serum iron as well as increases in neopterin, a product of activated macrophages, correlated with the degree of anemia.U3 Compared to controls, the UAE group had decreased renal function and similar EPO levels despite decreased hemoglobin values, suggesting that they had the restricted iron flux and decreased EPO found in ACI. diagnostic utility in those patients in whom the differentiation of these two disease states is not clear from the standard clinical evaluation. Lastly, targeting hepcidin has further emerged as a potential treatment in ACI, as direct pharmacological inhibition of hepcidin via an antihepcidin oligoribonucleotide has shown promising results in IL-6–induced anemia in a primate modelU5 and similar activity for a limited period in lipopolysaccharide-treated human volunteers.U6 U1.Souma T, Yamazaki S, Moriguchi T, et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol. 2013;24:1599-1616. U2. Souma T, Nezu M, Nakano D, et al. Erythropoietin synthesis in renal myofibroblasts is restored by activation of hypoxia signaling. J Am Soc Nephrol. 2016;27:428-438. U3. Artz AS, Xue QL, Wickrema A, et al. Unexplained anaemia in the elderly is characterised by features of low grade inflammation. Br J Haematol. 2014;167:286-289. U4. van Santen S, de Mast Q, Oosting JD, et al. Hematologic parameters predicting a response to oral iron therapy in chronic inflammation. Haematologica. 2014;99:e171-e173. In terms of differentiating ACI from combined ACI and iron deficiency, a trial of oral iron in patients with rheumatological diseases demonstrated that increased reticulocytes and increased reticulocyte hemoglobin content after one week predicted subsequent responses in hemoglobin.U4 This one-week response may have U5. Schwoebel F, van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013;121:2311-2315. U6. van Eijk LT, John AS, Schwoebel F, et al. Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood. 2014;124:2643-2646. cytokines suppress EPO production, decreasing plasma EPO concentration and increasing apoptosis of erythroid cells in the EPO-dependent stages of differentiation (Figure).4 Finally, the inflammatory cytokines IL-6 and members of the bone morphogenetic protein (BMP) family diminish serum iron concentration by inducing transcription of hepcidin, the master regulator of iron homeostasis.3 Hepcidin exerts its effects primarily through interaction with the cellular iron exporter, ferroportin, expressed on the basolateral surface of enterocytes and on reticuloendothelial cells. Binding to hepcidin induces endocytosis and degradation of ferroportin thereby restricting gastrointestinal iron absorption and impairing macrophage export and recycling of iron from phagocytosed senescent erythrocytes. The trapping of iron in reticuloendothelial cells accounts for the characteristic iron-laden macrophages observed in ACI bone marrow aspirates stained with Prussian blue. Because iron absorption and iron recycling are impaired, plasma iron concentration is low, and transferrin saturation is often subnormal (as in our case), resulting in a functional iron deficiency state with consequent suboptimal delivery of iron to maturing erythroblasts. Interpretation of transferrin saturation must also take into account the fact that transferrin is a negative acute phase reactant, and as such, the serum concentration is often subnormal or at the lower end of the normal range in patients with ACI (as illustrated in our case). P. BRENT FERRELL, MD, 1 AND MARK J. KOURY, MD 2 1. Instructor of Medicine, Division of Hematology/Oncology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 2. Professor of Medicine, Emeritus, Vanderbilt University School of Medicine, Nashville, TN Patient History A 64-year-old man had a history of two years of weight loss and mild normocytic anemia, but over the preceding two months, worsening polyarthritis, fatigue, and anemia (Hgb 8.6 g/dL, Hct 25%, MCV 91 fL) were noted. The reticulocyte count was 1.4 percent with a normal WBC and normal platelet count. He had normal serum cobalamin, methylmalonate, LDH, RBC folate, and negative hepatitis serologies. Laboratory tests were consistent with normal renal, thyroid, and liver function. Abnormal laboratory tests included an erythrocyte sedimentation rate of 87 mm/h, serum C-reactive protein (CRP) of 145 mg/L, haptoglobin of 447 mg/dL, and serum albumin of 2.5 g/dL. Iron studies showed a serum iron of 23 ug/dL, TIBC of 209 ug/dL, transferrin saturation of 11 percent, and serum ferritin of 299 ng/mL. He was transfused with two units of packed erythrocytes, begun on low-dose prednisone, and referred for evaluation of anemia. Diagnostic considerations In most instances, ACI is normocytic, normochromic, but approximately one-third of cases fall into the microcytic, hypochromic morphological classification. In the latter cases, functional iron deficiency as a consequence of excess hepcidin production dominates the pathophysiology, and review of the peripheral blood film reveals features similar to other processes, such as iron deficiency or thalassemia, that affect production of heme or globin. ACI is typically a mild-to-moderate hypoproliferative process manifested as grade I or grade II anemia. If more severe, (grade III or worse, hemoglobin < 8.0 g/dL), the anemia is likely multifactorial with other processes, such as concurrent gastrointestinal bleeding contributing to the etiology. Blood loss and inflammation may coexist, especially in patients with renal failure on hemodialysis, in patients with gastrointestinal malignancy or inflammation, or in patients with arthritis treated with corticosteroids or non-steroidal anti-inflammation drugs. In those cases, determining the relative contributions of absolute iron deficiency and functional iron deficiency can be challenging without performing a bone marrow analysis. The Question What is your approach to the diagnosis and management of the ACI? Our Response Overview The traditional disease categories associated with ACI are malignancy, infection, and connective tissue disorders. When excess cytokine production is a pathologic manifestation, ACI may be associated with such processes as severe heart failure and poorly controlled diabetes that fall outside of Figure the customary listing of chronic inflammatory diseases. ACI is often subtle and insidious in onset, presenting as a mild, persistent anemia that may worsen over time. Causes of the underlying inflammation such as rheumatoid arthritis may be obvious, or the basis of the inflammation may not be immediately apparent as in cases of occult malignancy or chronic infection. Pathophysiology Multiple mechanisms are responsible for the development of ACI.1-3 Although erythrocytes in ACI have slightly shortened survivals, the following three mechanisms, mediated by inflammatory cytokines, exert major effects in sequential periods of erythropoiesis (Figure): 1) inhibited survival and differentiation of erythroid progenitor cells; 2) suppressed EPO production; and 3) hepcidin-mediated sequestration of reticuloendothelial iron. Chronic inflammatory states increase production of cytokines such as IL-1, TNF-α, and interferon-γ that directly inhibit the survival and differentiation of erythroid progenitor cells.1,2 Inflammatory Inhibited Survival and Differentiation Supressed EPO Production Increased RES Iron Sequestration Stage of Erythroid Differentiation HSC BFU-E CFU-E Baso EB Poly EB Ortho EB RET RBC l — EPO dependence — l l — Hemoglobin synthesis — l KEY BFU-E: Burst-forming unit-erythroid CFU-E: Colony-forming unit-erythroid Pro EB: Proerythroblast Baso EB: Basophilic erythroblast Poly EB: Polychromatophilic erythroblast Ortho EB: Orthochromatic erythroblast RES: Reticuloendothelial system RET: Reticulocyte N O N M A L I G N A N T H E M A T O L O G Y 37 Pro EB Mechanisms of inflammatory cytokine inhibition of erythropoiesis in ACI. Three mechanisms (shown in red) that are mediated by inflammatory cytokines are key elements in the pathophysiology of ACI and represent potential therapeutic targets. The main period of erythropoiesis in which each of the respective mechanisms has its major effect is indicated by arrows. The stages of erythropoiesis begin with the hematopoietic stem cell (HSC) and extend through the mature erythrocyte (RBC). The periods of erythropoietin (EPO) dependency and hemoglobin synthesis are shown below those specific stages of differentiation. The Hematologist Compendium 2010-2015 Serum ferritin concentration is less than 20 ng/mL in uncomplicated iron deficiency anemia. Although as an acute-phase protein, the ferritin concentration can be driven into the normal range by the underlying inflammatory process, a ferritin concentration greater than 150 ng/ mL is rare in ACI patients who have concomitant absolute iron deficiency. As noted above, low serum iron is characteristic of ACI, and low serum transferrin concentration and low transferrin saturation are also observed routinely in ACI. The patient described in the introduction was diagnosed with seronegative rheumatoid arthritis, and two months after beginning therapy with prednisone and methotrexate, his arthritis symptoms had improved and his Hgb was 12.5 g/dL. 1. Weiss G, Schett G. Anaemia in inflammatory rheumatic diseases. Nat Rev Rheumatol. 2013;9:205-215. 2. Davis SL, Littlewood TJ. The investigation and treatment of secondary anaemia. Blood Rev. 2012;26:65-71. 3. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434-1443. Transferrin receptor (TfR) expression is regulated post-transcriptionally by intracellular iron concentration through the iron regulatory element (IRE)/IRE binding protein (IREBP) system. When intracellular iron concentration is low, the IRE/IREBP system stabilizes TfR mRNA, thereby increasing translation and protein expression. The effect of intracellular iron concentration on TfR production led to development of a clinical test of iron status. In this case, the concentration of TfR in plasma (soluble TfR or sTfR) serves as a surrogate marker of iron status (i.e., the concentration of sTfR is elevated in absolute iron deficiency). Subsequent studies suggested that the sensitivity of the assay in distinguishing absolute iron deficient states from inflammatory processes that affect iron metabolism can be improved by calculating the sTfR index by dividing the sTfR concentration by the log of the serum ferritin concentration. Another proposed method for identifying iron deficiency is to measure reticulocyte hemoglobin concentration (CHr) by flow cytometry. As the most recently produced 1 percent of erythrocytes in the blood, the reticulocytes are the subpopulation most affected by iron deficiency at the time the blood sample is obtained, and decreased CHr is a sensitive indicator of iron-restricted erythropoiesis.5 Combining CHr with sTfR index has been used to improve the identification of absolute iron deficiency in patients with inflammation.5 While these studies may have value is some particularly problematic cases, their clinical utility is relatively modest. This interpretation is based on the fact that functional iron deficiency plays an important role in the pathophysiology of ACI, and patients with functional iron deficiency, without absolute iron deficiency, may benefit from supplemental iron. Therefore, the practical value of distinguishing functional iron deficiency from absolute iron deficiency is arguable because iron supplementation can be therapeutic in either case. 4. Miller CB, Jones RJ, Piantadosi S, et al. Decreased erythropoietin response in patients with the anemia of cancer. N Engl J Med. 1990;322:1689-1692. 5. Thomas C, Thomas L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin Chem. 2002;48:1066-1076. 6. Pincus T, Olsen NJ, Russell IJ, et al. Multicenter study of recombinant human erythropoietin in correction of anemia in rheumatoid arthritis. Am J Med.1990;89:161-168. 7. Papadaki HA, Kritikos HD, Valatas V, et al. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: improvement following anti-tumor necrosis factor-α antibody therapy. Blood. 2002;100:474-482. 8. Sasu BJ, Cooke KS, Arvedson TL et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115:3616-3624. 9. Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood. 2011;117:4915-4923. 10. Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118:4977-4984. Dr. Ferrell indicated no relevant conflicts of interest. Dr. Koury is a consultant with Keryx Biopharmaceuticals, Inc. and the Pharmaceutical Division of Japan Tobacco, Inc. Treatment Although many patients have mild anemia that does not require treatment, establishing a diagnosis of ACI is important as doing so implies an ongoing inflammatory process, the etiology of which should be investigated. Furthermore, effective treatment of the underlying disease results in improvement or resolution of the anemia. In some instances, however, the underlying disease may be resistant to therapy (e.g., poorly responsive malignancy or refractory connective tissue disease). In such cases, red cell transfusion support and therapy that targets the pathophysiologic mechanisms that underlie ACI (Figure) are the mainstays of management. ACI may respond to recombinant human EPO (rhEPO),6 but the response may be blunted by concomitant functional iron deficiency. Iron supplementation, with a goal transferrin saturation of > 20 percent, may increase the effectiveness of rhEPO, and some patients respond to iron supplementation in the absence of rhEPO support. Infused iron rather than oral iron supplementation is often needed as hepcidin restricts gastrointestinal iron absorption. The desired response (increase in transferrin saturation) to iron supplementation, however, is relatively short lived, and repeated iron infusion puts patients at risk for iatrogenic hemochromatosis1,2 because the high hepcidin prevents the recycling of this iron. AntiTNF-α agents have been shown to increase hemoglobin concentration in patients with ACI independent of an effect on EPO concentration.7 Looking forward, suppression of hepcidin activity has been examined in animal models. In a mouse model of ACI, antibodies to hepcidin also required the concurrent administration of EPO to reverse the anemia,8 whereas inhibition of hepcidin transcription, by blocking BMP receptor activation or downstream signaling, prevented and reversed ACI without the need for exogenous EPO administration.9,10 As more therapeutic options become available, well-designed clinical studies will be needed to determine the optimal pharmacologic approach to the management of ACI. Watch ASH Webcasts and Webinars on ASH On Demand View a wide range of online ASH educational materials including webcasts of ASH live meetings and informative webinars. Visit ashondemand.org for access. N O N M A L I G N A N T H E M A T O L O G Y 38 The Hematologist Compendium 2010-2015 Approach to the Diagnosis and Management of Transfusion-Related Acute Lung Injury – July August 2013 The authors have indicated that there has been no update regarding the content of this article since the original publication date in 2013. HUY P. PHAM, MD, 1 AND STEVEN L. SPITALNIK, MD 2 1. Assistant Professor of Pathology, UAB Medicine, Baptist Health, Birmingham, AL 2. Professor of Pathology and Cell Biology; Vice Chairman, Laboratory Medicine, College of Physicians and Surgeons, Columbia University, New York, The Question What is your approach to the diagnosis and management of transfusion-related acute lung injury (TRALI)? Our Response Epidemiology The blood supply in the United States is safe. Although non-life-threatening adverse events such as allergic and febrile transfusion reactions are encountered regularly by clinicians, transfusion-related fatalities are rare. From 2007 through 2011, 212 fatalities following blood collection and transfusion were reported to the FDA.1 Non-infectious complications pose the greatest mortality risk to the transfused patient with TRALI accounting for 43 percent of deaths and hemolytic transfusion reactions due to ABO (10%) and non-ABO (13%) incompatibility accounting for 23 percent.1 In comparison, 11 percent of transfusion-related deaths were due to microbial infections. Although usually associated with infusion of blood products containing high volumes of plasma (e.g., fresh-frozen plasma and platelets), TRALI has also been linked to red blood cell transfusions. Estimates of the incidence of TRALI may be influenced by factors such as transfusion policy. For example, the incidence fell from 2.57 to 0.81 per 10,000 transfusions concurrent with reduction in the use of plasma from female donors.2 Clinical Presentation and Diagnosis The diagnosis of TRALI is made on clinical grounds, and no single laboratory or radiologic test definitively identifies or excludes this entity. We currently use the criteria and case definitions proposed by a Canadian Consensus Conference (Table).3 Thus, TRALI is an acute event presenting during a transfusion or within six hours of its completion. Characteristic signs and symptoms include fever, chills, dypsnea, hypoxemia, hypotension (or possible transient hypertension), and the new onset of bilateral non-cardiogenic pulmonary edema (e.g., chest x-ray showing bilateral alveolar and interstitial infiltrates in the absence of cardiomegaly). TRALI is often associated with transient leukopenia or neutropenia. A diagnosis of “Possible TRALI” is made based on the same criteria as TRALI except that an alternative risk factor for acute lung injury is present concurrently (Table). The differential diagnosis of TRALI includes the following: transfusion-associated circulatory overload (TACO), anaphylaxis, and sepsis.4 Distinguishing TACO from TRALI may be challenging because some of the signs and symptoms of the two entities overlap; in addition, the two processes can occur concurrently in a given patient. The key difference between these two conditions is the pathophysiologic origin of the pulmonary edema (i.e., cardiogenic in the case of TACO and non-cardiogenic in the case of TRALI). Thus, clinical improvement after treatment with a diuretic and/or an inotropic agent is characteristic of TACO, but not TRALI. Other findings suggestive of TACO include persistent hypertension, a post-transfusion brain natriuretic peptide (BNP) level of at least 100 pg/mL, and a post-transfusion:pre-transfusion BNP ratio of >1.5.5 Although anaphylaxis can present with hypotension, cyanosis, and hypoxia due to bronchospasm and laryngeal edema, the absence of fever and pulmonary edema distinguish this process from TRALI. Although transfusion-induced sepsis, particularly after a platelet transfusion, typically presents with pyrexia and hypotension, respiratory distress is an infrequent complication. Other entities, such as myocardial infarction, pulmonary embolism, and other causes of acute lung injury share clinical features with TRALI and should be considered in the differential diagnosis. The diagnosis of TRALI is particularly challenging in complex inpatients such as those encountered in the intensive care unit setting, given that such patients often have multiple medical problems and may exhibit some symptoms of TRALI even before transfusion. of the donor product could induce the second hit. Together, these processes induce capillary endothelial damage, resulting in vascular permeability and pulmonary edema.6-8 Based on this proposed mechanism, one might hypothesize that the incidence of TRALI would be higher for plasma-rich transfusion products and that the incidence would be lower when plasma derived from female donors (who have a higher prevalence of antiHLA antibodies) is restricted; available data support these two hypotheses.2 In addition, recent studies indicate that circulating platelets are involved in TRALI pathophysiology, suggesting that anti-platelet therapy may be beneficial clinically.7,9 Management The first step in managing any suspected transfusion reaction is to stop the transfusion. Once the patient is stabilized, the episode should be reported to the transfusion medicine service so that a transfusion reaction evaluation can begin. Because hemolytic transfusion reactions are associated with significant morbidity and mortality, the transfusion medicine service will first perform a “clerical check” to ensure that the correct unit was transfused into the correct patient. Next, the ABO type of the patient and the transfused unit will be confirmed, the post-transfusion blood sample will be inspected for visible evidence of hemolysis, and an indirect and direct anti-globulin test will be performed on the posttransfusion sample to determine if circulating and/or red blood cell-bound antibodies are present. Additional laboratory tests to investigate for hemolysis, including a complete blood count, urinalysis, and plasma concentration of bilirubin, lactate dehydrogenase, and haptoglobin, are often needed. To diagnose TRALI, physical exam, chest x-ray, and arterial blood gas studies are recommended. In distinguishing TRALI from TACO, an echocardiogram may be useful in determining whether the observed pulmonary edema is of cardiogenic origin. Other causes of adverse events that share clinical features with TRALI (e.g., sepsis, myocardial infarction, and pulmonary embolus) should be promptly investigated. Current management of TRALI consists of respiratory and circulatory support based on clinical severity. Oxygen supplementation is required in almost all patients; in severe cases, mechanical ventilation may be necessary. Hypotensive episodes can be treated with pressors. Corticosteroid treatment has not improved outcome.10 In theory, the noncardiogenic pulmonary edema of TRALI should not respond to diuresis. Most patients improve within two to three days, but those who do not improve over this period typically have a protracted clinical course or a fatal outcome.8 Patients who have experienced an episode of TRALI are not at greater risk for a second episode, assuming that the initial event was a consequence of infusion of donor antibodies that were present in the transfusion product and that subsequent blood products do not Table 1: Canadian Consensus Conference on the Definition of TRALI TRALI: 1) Acute lung injury a. Acute onset b. Hypoxemia i. Research setting: PaO2/FiO2 < 300 and/or SpO2 < 90% on room air ii. Non-research setting: PaO2/FiO2 < 300 and/or SpO2 < 90% on room air and/or other clinical symptoms of hypoxia c. Bilateral infiltrates on frontal chest x-ray d. No evidence of left atrial hypertension (i.e., no circulatory overload) 2) No pre-existing acute lung injury before transfusion 3) During or within 6 hours of transfusion 4) No temporal relationship to an alternative risk factor for acute lung injury Possible TRALI: Pathophysiology 1) Acute lung injury Although the exact mechanism of TRALI is uncertain, a “two-hit” process has been proposed.6-8 According to this hypothesis, the first “hit” is induced by an underlying condition, such as trauma or sepsis, which primes granulocytes and/or activates endothelial cells, thereby causing neutrophils to become sequestered in the pulmonary vasculature. The second “hit” results from passive infusion of donor antibodies in the blood product that recognize either human leukocyte antigens (HLA) on recipient endothelial cells or human neutrophil antigens (HNA) on recipient neutrophils. Alternatively (or in addition), infusion of biologic response modifiers (e.g., CD40 ligand) in the plasma portion 2) No pre-existing acute lung injury prior to transfusion N O N M A L I G N A N T H E M A T O L O G Y 39 3) During or within 6 hours of transfusion 4) A clear temporal relationship to an alternative risk factor for acute lung injury Kleinman S et al. Transfusion. 2004;44:1774-1789. The Hematologist Compendium 2010-2015 come from the initial donor. However, notification of the transfusion medicine service about a possible TRALI episode has important implications for the donor and the safety of the blood supply. If TRALI is suggested by both clinical presentation and laboratory results (e.g., finding anti-HLA and/or anti-HNA antibodies in the transfused blood product that match the corresponding antigens in the patient), the donor must be evaluated for consideration of their continued eligibility to donate. Indeed, many centers choose to exclude such donors permanently. That the product from a particular donor caused an episode of TRALI may be difficult to prove unequivocally, however, because patients often receive products from multiple donors. Prevention Approaches to reducing the risk of TRALI have included avoiding the use of plasma from female donors, using plasma derived only from males or from never-pregnant females, and testing female donors for anti-HLA antibodies.1,2 Although these measures reduce the incidence, they do not completely eliminate risk because TRALI can be induced by other blood products (e.g., red blood cells, platelet concentrates, cryoprecipitate). In addition to avoiding the use of high-risk blood products, conservative transfusion strategies and interventions that address the “first hit” could also help reduce TRALI incidence. Nonetheless, TRALI continues to be the most common cause of transfusionrelated mortality, making rapid recognition and institution of appropriate supportive care imperative.1,7 1. Fatalities reported to FDA following blood collection and transfusion: annual summary for fiscal year 2011. www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/ReportaProblem/ TransfusionDonationFatalities/ucm302847.htm. 2012. Accessed March 31, 2013. 2. Toy P, Gajic O, Bacchetti P, et al. Transfusion-related acute lung injury: incidence and risk factors. Blood. 2012;119:1757-1767. 3. Kleinman S, Caulfield T, Chan P, et al. Toward an understanding of transfusion-related acute lung injury: statement of a consensus panel. Transfusion. 2004;44:1774-1789. 4. Mazzei CA, Popovsky MA, Kopko PM. Noninfectious complications of blood transfusion. In: Roback JD, Grossman BJ, Harris T, et al. (Eds). Technical Manual, 17th ed. AABB. 2011:272-762. 5. Zhou L, Giacherio D, Cooling L, et al. Use of B-natriuretic peptide as a diagnostic marker in the differential diagnosis of transfusion-associated circulatory overload. Transfusion. 2005;45:1056-1063. 6. Silliman CC, Boshkov LK, Mehdizadehkashi Z, et al. Transfusion-related acute lung injury: epidemiology and a prospective analysis of etiologic factors. Blood. 2003;101:454-462. 7. Gilliss BM, Looney MR. Experimental models of transfusion-related acute lung injury. Transfus Med Rev. 2011;25:1-11. 8. Popovsky MA, Moore SB. Diagnostic and pathogenetic considerations in transfusion-related acute lung injury. Transfusion. 1985;25:573-577. 9. Caudrillier A, Kessenbrock K, Gilliss BM, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661-2671. 10. Steinberg KP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354:1671-1684. Dr. Pham and Dr. Spitalnik indicated no relevant conflicts of interest. Make Use of ASH’s Consult-a-Colleague Service This service for ASH members helps facilitate the exchange of information between hematologists and their peers. Members can seek consultation on clinical cases relating to categories including anemias, hematopoietic cell transplantation, hemoglobinopathies, hemostasis/thrombosis, lymphomas, lymphoproliferative disorders, leukemias, multiple myeloma & Waldenström Macroglobulinemia, myeloproliferative disorders, myelodysplastic syndromes, and thrombocytopenias. Email your inquiries to [email protected], or visit www.hematology.org/Clinicians/Consult.aspx for more information. N O N M A L I G N A N T H E M A T O L O G Y 40 The Hematologist Compendium 2010-2015 Criteria for Selecting Patients with Sickle Cell Anemia for Allogeneic Hematopoietic Stem Cell Transplantation – September/October 2013 The author has indicated that there has been no update regarding the content of this article since the original publication date in 2013. CHARLES T. QUINN, MD, MS Associate Professor of Pediatrics and Medical Director, Pediatric Sickle Cell Program; Medical Director, Erythrocyte Diagnostic Laboratory, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH The Question What criteria do you use for selecting patients with sickle cell anemia (SCA) for allogeneic hematopoietic stem cell transplantation (HSCT)? My Response About 40 years ago, only half of the children born with SCA in the United States were expected to live long enough to reach adulthood. Now, however, as a result of the high quality of care available in comprehensive sickle cell centers, disease-related mortality during childhood has been reduced to 1 to 2 percent.1,2 This remarkable improvement in survival stems mainly from interventions aimed at preventing early deaths from infection and splenic sequestration, as well as the greater use of disease-modifying therapies (e.g., hydroxyurea and chronic transfusions). No current U.S. statistics are available, but in a study published in 2001, the median lifespan for individuals with SCA in a Jamaican cohort was estimated to be more than 50 years.3 Not reflected in these improving survival data, however, is the burden of SCA-related chronic organ injury acquired in early childhood that becomes especially manifest in young adulthood, adversely affecting the quality and duration of the remaining life of the patient. The burden of morbidity and mortality in SCA has now simply shifted to adults, and the period after transition to adult medical care is associated with a particularly high risk of death, especially from acute chest syndrome and multi-organ failure syndrome.1 HSCT is the only available cure for SCA, and more than 500 transplants for SCA have been reported to international registries. Although usually successful and curative, the widespread use of HSCT is still limited by the lack of sufficient suitable donors and concerns about regimenrelated morbidity and mortality. HSCT is safest when an HLA-matched sibling donor is available, but only about 10 percent of transplant candidates will actually have such a donor. Current estimates place regimen-related mortality for myeloablative transplantation using an HLAmatched sibling donor at about 5 percent, with a concomitant 9 percent risk of graft rejection and a 15 percent risk of chronic graft-versus-host disease (GVHD) (Table).4,5 There are additional late effects of transplantation not tabulated here, including infertility, endocrinopathies, premature cardiovascular disease, and possibly cancer. One can argue that SCA-related mortality during childhood (in patients cared for in comprehensive centers in developed nations) is now lower than the regimen-related mortality of HSCT, but this comparison does not integrate the lifelong risk of progressive morbidity, impaired quality of life, and early mortality due to SCA. Of course, there is no randomized comparison of transplantation versus no transplantation that accurately quantifies the relative lifetime risks of morbidity and mortality from SCA and HSCT, but life after a successful transplant — the event-free survival (EFS) of a full-sibling-donor myeloablative HSCT is approaching 90 percent (Table) — is arguably better than a life with SCA. An important caveat, however, is that the definition of EFS differs by study and often does not include chronic GVHD, which may be worse than SCA, so EFS rates that are meaningful to patients are usually lower than reported (70-80% vs. 90%, respectively). chronic transfusions. If there is continued interest in pursuing HSCT, then I arrange formal consultation and counseling with the HSCT team. This multi-step process does not lead to HSCT for most patients, but they are at least aware and knowledgeable about the option, and I support them with decision-making. The second scenario is a direct inquiry about HSCT by a patient or parent, oftentimes as a result of something heard or read about on the Internet. Recent immigrants from Africa also tend to ask commonly about transplantation. The third scenario occurs when a patient suffers a particularly serious complication, such as overt stroke. In these instances, I follow the same general process outlined above in counseling patients and their families about HSCT. In essence, I consider every patient with SCA (here I include both HbSS and sickle-β0thalassemia) who has an HLA-matched sibling donor to be a potential candidate for myeloablative HSCT. Honest discussions about the risks, benefits, and alternatives are needed, regardless of the “severity” of a patient’s disease, and the decision to pursue HSCT is a lengthy and patient- and family-centered process. Once a decision to perform HSCT is made, I recommend that it be done during early childhood. My opinions and practices are based upon the assumption that my patient’s HSCT will be performed in a center with substantial experience in transplantation for SCA and, ideally, as part of a clinical research study. The use of alternative donors (unrelated, mismatched, or haploidentical) and stem cell sources (unrelated umbilical cord blood) for children carries a much higher risk of transplant-related morbidity, mortality, and graft rejection and should be performed only as part of a carefully designed investigational study. Adults with SCA tolerate myeloablative conditioning regimens poorly, likely because of the cumulative burden of chronic organ injury acquired during childhood. However, there is very promising early experience with a non-myeloablative conditioning regimen (including total-body irradiation, alemtuzumab, and sirolimus) in severely affected adults with SCA that can produce stable, mixed donor-recipient chimerism that effectively cures the disease.6 These outcomes have been achieved without chronic GVHD or mortality and with a low rate of graft rejection. The mixed chimerism appears to persist after discontinuation of immunosuppression.7 In the future, adults may no longer be summarily excluded from HSCT. Until then, all transplants in adults should be performed as part of a clinical research study with the indications being dictated by the eligibility criteria of the particular study. While the experience with adult recipients and alternative donors matures, research needs to focus on reducing transplant-associated morbidity, especially sterility, in young patients undergoing reduced-intensity conditioning regimens. Ongoing studies will continue to expand access to HSCT for both children and adults, and the next “Ask the Hematologist” question on this topic might also include the option of gene therapy. 1. Quinn CT, Rogers ZR, McCavit TL, et al. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447-3452. 2. Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92:905-912. 3. Wierenga KJ, Hambleton IR, Lewis NA, et al. Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study. Lancet. 2001;357:680-683. 4. Locatelli F, Pagliara D. Allogeneic hematopoietic stem cell transplantation in children with sickle cell disease. Pediatr Blood Cancer. 2012;59:372-376. 5. Lucarelli G, Isgrò A, Sodani P, et al. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med. 2012;2:a011825. Expert panels and professional societies provide differing sets of clinical indications for HSCT as a treatment option for SCA, but common recommendations include recurrent 6. Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361:2309-2317. vaso-occlusive complications (acute chest syndrome and painful events), usually despite 7. Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic hematopoietic stem cell transplantation for sickle cell hydroxyurea therapy, and overt cerebrovascular disease. Other indications have been disease: the time is now. Blood. 2011;118:1197-1207. proposed and used, such as abnormal transcranial Doppler velocities and silent cerebral infarction, despite much less agreement among experts and very limited data. Some hematologists now also advocate that a diagnosis of SCA itself is an indication for HSCT, Dr. Quinn indicated no relevant conflicts of interest. justified by the knowledge that complications of SCA are often unpredictable, and most individuals with SCA will have progressively severe morbidity and early mortality. In my clinical practice, I do not compare a fixed list of indications for HSCT with an ongoing tabulation of the number and type of SCA-related complications experienced by the patient. Table. International Studies of Myeloablative HSCT for SCA Using Matched-Sibling Donors†§ Rather, I initiate (or continue) the discussion of HSCT in three main clinical scenarios. Vermylen Walters Locatelli Bernaudin Panepinto Lucarelli Weighted The first usual scenario is planned. I ensure that all patients with SCA and their parents know about the possibility of cure by HSCT. I include discussion of the option of HSCT as part of the comprehensive, ongoing education about SCA starting with the very first visit after diagnosis (usually the result of newborn screening). I also recommend tissue typing of all unaffected full siblings, including those with sickle trait, to know if there is an HLA-matched donor. If there is a matched-sibling donor, I offer dedicated clinic visits for frank discussions about the risks and benefits of myeloablative HSCT compared with life with SCA, as well as the risks and benefits of hydroxyurea therapy and OS¶ 1998 N=50 2000 N=50 2003 N=11 2007 N=87 2007 N=67 2011 N=11 Average N=276 93 94 100 92 97 90 94 RRM 7 6 0 7 2 10 5 Rejection 10 10 9 5 13 0 9 cGVHD 20 12 6 13 22 36* 17 EFS 82 90 86 85 90 85 84 †Data compiled from tables in Locatelli et al. and Lucarelli et al. § Values are shown as the percentage of the N value for the corresponding study. ¶ Abbreviations: OS, overall survival; RRM, regimen-related mortality; cGVHD, chronic graft-versus-host disease; EFS, event-free survival. *Four of 11 patients had mild, chronic GVHD of the skin that resolved completely after steroid therapy. N O N M A L I G N A N T H E M A T O L O G Y 41 4 5 The Hematologist Compendium 2010-2015 Diagnosis and Management of Platelet Alloimmunization – November/December 2013 UPDATE/COMMENTARY Platelet refractoriness remains a clinical challenge associated with an increased risk of bleeding, prolonged hospital stays, and decreased survival. Poor one-hour post-transfusion increments typically represent immune platelet refractoriness, whereas an adequate one-hour increment followed by a suboptimal 18- to 24-hour count suggests peripheral consumption or sequestration. In the absence of a direct prospective comparison of the three strategies commonly used to support patients who have platelet alloimmunization – human leukocyte antigen (HLA) matching, HLA antibody avoidance, and platelet cross-matching – two recent reviews have concluded there is no definitive evidence that any of these strategies improves hemorrhage control or mortality.U1,8 The provision of HLA-matched platelets can be very efficient in centralized transfusion systems as it has been in the United Kingdom.U2 Apheresis platelets stored in additive solution, which contain 2/3 less donor plasma, have become available in the United States, with a longer track record in Europe. They are associated with lower platelet increments at one hour, but are less likely to cause allergic transfusion reactions or hemolysis, compared with standard platelets suspended in 100 percent donor plasma.U3 Platelets that have undergone psoralenUVA treatment to inactivate bacteria (and other infectious agents) are also known to produce lower post-transfusion increments. These considerations need to be taken into account when evaluating patients who seem to be refractory to platelet transfusion. A recent study in an animal model suggests that immune-mediated clearance of MHC mismatched platelets can occur in the absence of anti-platelet alloantibodies, in mice that are deficient in B cells. Allo-reactive CD8+ T cells appear to mediate antibodyindependent platelet clearance.U4,U5 If this finding is corroborated in humans, immune platelet refractoriness may actually account for some refractory cases now presumed to be non-immune. A putative T-cell—mediated mechanism would also favor HLA matching over cross-matching as a management strategy. U1. Vassallo RR, Fung M, Rebulla P, et al. Utility of cross-matched platelet transfusions in patients with hypoproliferative thrombocytopenia: a systematic review. Transfusion. 2014;54:1180-1191. U2. Stanworth SJ, Navarrete C, Estcourt L, et al. Platelet refractoriness – practical approaches and ongoing dilemmas in patient management. Br J Haematol. 2015;171:297-305. U3. Tobian AA, Fuller AK, Uglik K, et al. The impact of platelet additive solution apheresis platelets on allergic transfusion reactions and corrected count increment (CME). Transfusion. 2014;54:1523-1529. U4. Arthur CM, Patel SR, Sullivan HC, et al. CD8+ T cells mediate antibody-independent platelet clearance in mice. Blood. 2016;127:1823-1827. U5. Manis JP, Silberstein LE. Platelet refractoriness: it’s not the B-all and end-all. Blood. 2016;127:1740-1741. HLA alloimmunization is prevented by the transfusion of leukoreduced red blood cells and platelets. KAREN QUILLEN, MD, MPH Medical Director, Blood Bank, Boston Medical Center; Professor of Medicine and Pathology, Boston University School of Medicine, Boston, MA The Question What is your approach to the diagnosis and management of platelet alloimmunization? My Response Platelet refractoriness occurs in 5 to 15 percent of patients who receive chronic platelet transfusions.1 The patient’s size and the number of platelets transfused should be factored into the assessment of refractoriness. For example, one measure, the corrected count increment (CCI), is computed as follows: CCI = (platelet increment after transfusion/µl) x (body surface area in m2) ÷ (platelet dose x 1011). For the purposes of this calculation, assume that each single-donor apheresis unit contains 3 x 1011 platelets or that each wholeblood-derived platelet concentrate contains 5.5 x 1010 platelets. Using this formula, if the platelet count increased by 20,000/µL in a patient who had a body surface area of 2.0 m2 and who received one apheresis unit of platelets, the CCI is 20,000/µL x 2 ÷ by 3 = 13,333/µL. Refractoriness is defined as a CCI value below 2,500 platelets/μL at 18 to 24 hours posttransfusion or a value below 4,500 platelets/μL at 10 to 60 minutes post-transfusion2 on two to three consecutive platelet transfusion episodes. A less rigorous approach is to assume that, for an average size, non-refractory adult, the platelet count increment will be at least 10,000/µl to 20,000/μL one hour after the transfusion of either one unit of apheresis platelets or an equivalent dose of pooled platelets (5-6 combined random-donor platelet concentrates are equivalent to 1 apheresis unit), recognizing that the number of platelets per component unit may vary widely. Non-immune causes of platelet refractoriness predominate. Non-immune factors are present in the majority (72-88%) of transfusion-refractory patients, and immune causes are present in a minority (25-39%).2 Non-immune factors, most of which are prevalent in a high proportion of hematology-oncology patients requiring prolonged platelet transfusion support, include splenomegaly, fever, infection, DIC, bleeding, and drugs such as vancomycin and amphotericin, are associated with platelet refractoriness.2,3 The mechanism for refractoriness associated with various drugs is partially mediated by drugdependent platelet antibodies, although specific testing for such is not widely available and unlikely to be of practical help. Antibodies to HLA antigens account for the overwhelming majority of cases of immune platelet refractoriness, with antibodies to platelet-specific antigens being much less common. HLA alloimmunization may occur in response to prior pregnancies or to transfusions, although only a subset of alloimmunized individuals demonstrates immune platelet refractoriness. The TRAP study4 showed that filtration-removal of leukocytes and ultraviolet B irradiation to inactivate leukocytes were equally effective in preventing the development of platelet transfusion refractoriness, which occurred in 16 percent of control patients, compared with 7 to 10 percent of patients who received leuko-reduced or irradiated platelets. On the other hand, HLA antibodies developed in 45 percent of control patients compared with 17 to 21 percent of intervention patients. Outcomes were equivalent for filtered apheresis platelets and for filtered pooled platelets. In Canada, universal prestorage leuko-reduction of platelets has lessened the incidence of alloimmune platelet refractoriness from 14 to 4 percent.5 Almost all apheresis platelet units and more than 80 percent of packed red blood cell units in the United States are leuko-reduced by filtration either at the time of collection or immediately prior to storage. HLA typing and antibody testing are complementary approaches. Platelets express only HLA Class I antigens. For patients who are candidates for allogeneic stem cell transplantation, HLA typing results may already be known. Most HLA laboratories have adopted high-throughput molecular methods for genotyping HLA Class I and II antigens. A sequence-specific oligonucleotide probe method requires only small amounts of DNA and therefore can be performed on samples from neutropenic patients. Lowresolution HLA-A and B typing (Class I antigens) is adequate for the management of platelet alloimmunization, while high-resolution typing, including sequencing, is reserved for HLA Class II typing to select stem cell donors. HLA antibody detection can be performed using a variety of methods.3 Multiplex flow cytometric bead assays are more sensitive than traditional ELISA.6 In the former assay, patient serum or plasma is incubated with color-coded microbeads that are coated with HLA antigens. Flurochrome-labeled anti-human IgG is added, and a flow analyzer is used to determine the color code of the reactive beads with a computer algorithm determining the specific antigens to which the antibody is reactive. The panel-reactive antibody (PRA) represents the percent of HLA targets to which the patient has made antibodies. PRA can be determined by using the traditional lymphocytotoxic assay, by ELISA, or by fluorescence-based microbead method. Serial assays are useful in assessing candidates for organ transplantation, but less so for management of platelet-refractory patients because a numerical PRA result (the percentage of the population to which the patient has HLA antibodies) does not provide actionable information to guide platelet selection. Use of fresh ABO-matched platelets can improve transfusion response. Strategies to select platelets for refractory patients include HLAmatching, avoidance of known HLA antibody specificities, and platelet cross-matching. Platelets express blood group A and B antigens, but they are often transfused without ABO matching. Major ABO incompatibility (such as group A platelets transfused into a group O recipient) can decrease post-transfusion increments. A trial of fresh ABO-matched platelets can be a valuable temporizing measure while investigation of the basis of platelet transfusion refractoriness is ongoing. For the purpose of platelet donor selection, a grading system (with designated categories A, B1, B2 C, D) is employed. A perfect four-antigen match (2 loci each at HLA-A and HLAB) is grade A. In a B1 match, all of the donor’s HLA antigens are present in the recipient, but the donor lacks one of the recipient’s HLA antigens; in a B2-match, all of the donor’s HLA antigens are present in the recipient, but the donor lacks two of the recipient’s HLA N O N M A L I G N A N T H E M A T O L O G Y 42 The Hematologist Compendium 2010-2015 antigens; in a C-match, the donor has one HLA antigen that is not present in the recipient; in a D-match, the donor has more than one HLA antigen that is not present in the recipient. High-grade matched donors (grade A, B1, or B2) are specifically recruited to donate platelets for a particular patient, but transfusion with grade C or D “matched” units is unlikely to produce a clinically meaningful incremental increase in the platelet count. This grading approach to matching also allows for categorization of antigens into cross-reactive groups (CREG). For example, HLA-A1 and A36 are within the same CREG, so if a patient has the A36 antigen and no available donor platelet is A36 positive, then an A1 donor platelet – typically more prevalent in the Caucasian population – can be used in a grade B match. HLA Matchmaker is an epitope-based computer algorithm used in some centers to identify permissible donor platelets that are more likely to yield adequate platelet increment increases without being HLA matched.7 Despite the resources invested in the management of patients who are refractory to platelet transfusion, a recent review of the literature identified no studies that were adequately powered to detect an effect of transfusion of HLA-matched platelets on mortality or hemorrhage.8 Prospective studies utilizing current technology and examining clinical outcomes are needed to evaluate the effectiveness of HLA-matched platelet transfusion.8 HLA-matched platelets Advantages Disadvantages HLA type may already be on file for allogeneic SCT candidates Testing takes up to 1 week to complete. One-time test per patient Some haplotypes are relatively common such as A1 B8 in Caucasians. Perfect matches are rare. Grade B matches or HLA Matchmaker increase the number of donor possibilities. Avoidance of known HLA-antibody specificities Many more donor possibilities compared with HLA matching. Antibody testing takes several days. Antibody testing should be repeated periodically. Platelet cross-matching For management of the transfusion-refractory patient, available data argue that selection of donors with HLA antigens against which the recipient does not have antibodies is a better strategy than HLA matching. In one observational study involving 29 refractory patients and a database of more than 7,000 HLA-typed donors, a mean of 39 donors were HLA grade A or B matched, but a mean of 1,426 donors were identified as permissible by antibody exclusion.9 Post-transfusion platelet count increments were comparable for the two strategies. HLA antibody testing should be repeated at periodic intervals because antibody specificities may evolve over time. Platelet cross-matching tests the patient’s serum against samples of available donor apheresis platelets using a solid-phase adherence assay or an ELISA. A recent study found a mean CCI of 7,000 at one hour in more than 400 cross-matched platelet doses transfused to 71 refractory patients.10 Platelet cross-matching can be done within a few hours compared with several days for HLA testing, but it does involve frequent repeat testing because the shelf-life of platelets is five days. Automated platforms are invaluable in making this approach efficient and practical.1 A comparison of some of the advantages and disadvantages of the three strategies for dealing with refractoriness to platelet transfusion is contained in the table. Choice of method depends on local resources, and communication between the hematologist and the transfusion service is critical to ensure that donor selection is appropriate and that valuable resources are not wasted or used inappropriately. Testing takes several hours Testing is repeated for each platelet search, labor-intensive unless automated. Applicable to apheresis or pooled whole-blood-derived platelets Commercial kits have been temporarily unavailable in the United States. Anti-fibrinolytic agents can be a useful adjunct for mucosal bleeding. Other approaches to ameliorating the consequences of alloimmune platelet refractoriness include infusion of IVIgG, citric acid treatment of platelets to remove Class I HLA epitopes, and infusion of recombinant activated FVII in actively bleeding patients. Despite anecdotal reports of success, none of these approaches has been validated for clinical use. Use of family members as platelet donors for patients who are potential allogeneic SCT candidates is controversial, based on a theoretical concern for inducing alloimmunization that may jeopardize engraftment. Anti-fibrinolytic agents such as epsilon-aminocaproic acid and tranexamic acid, however, can be useful in platelet-refractory patients with oral mucosal bleeding. 1. Rebulla P. A mini-review on platelet refractoriness. Haematologica. 2005;90:247-253. 2. Stroncek DF, Rebulla P. Platelet transfusions. Lancet. 2007;370:427-438. 3. Hod E, Schwartz J. Platelet transfusion refractoriness. Br J Haematol. 2008;142:348-360. 4. The Trial to Reduce Alloimmunization to Platelets Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med. 1997;337:1861-1870. 5. Seftel MD, Growe GH, Petraszko T, et al. Universal prestorage leukoreduction in Canada decreases platelet alloimmunization and refractoriness. Blood. 2004;103:333-339. 6. Fadeyi E, Adams S, Peterson B, et al. Analysis of a high-throughput HLA antibody screening assay for use with platelet donors. Transfusion. 2008;48:1174-1179. 7. Nambiar A, Duquesnoy RJ, Adams S, et al. HLAMatchmaker-driven analysis of responses to HLA-typed platelet transfusions in alloimmunized thrombocytopenic patients. Blood. 2006;107:1680-1687. 8. Pavenski K, Rebulla P, Duquesnoy R, et al. Efficacy of HLA-matched platelet transfusions for patients with hypoproliferative thrombocytopenia: a systematic review. Transfusion. 2013;53:2230-2242. 9. Petz LD, Garratty G, Calhoun L, et al. Selecting donors of platelets for refractory patients on the basis of HLA antibody specificity. Transfusion. 2000;40:1446-1456. 10. Wiita AP, Nambiar A. Longitudinal management with crossmatch-compatible platelets for refractory patients: alloimmunization, response to transfusion, and clinical outcomes (CME). Transfusion. 2012;52:2146-2154. WATCH THE EXPERTS Dr. Quillen indicated no relevant conflicts of interest. Adam Cuker, MD, MS Dr. Adam Cuker discusses the latest developments in monitoring and administering oral anticoagulants. He presented on this topic at the 2015 ASH Annual Meeting in a session titled “Evolving Issues in Anticoagulation.” Watch the video and the rest of the ASH Impact Series on ASH On Demand at www.ashondemand.org/FreeContent/Videos. Read an article by Dr. Cuker in this Compendium on page 44. N O N M A L I G N A N T H E M A T O L O G Y 43 The Hematologist Compendium 2010-2015 Recommendation of Light Transmission Aggregometry as Part of the Evaluation of a Patient with a Suspected Bleeding Disorder – March/April 2014 The author has indicated that there has been no update regarding the content of this article since the original publication date of 2014. Figure ADAM CUKER, MD, MS Assistant Professor of Medicine, and of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA The Question When do you recommend light transmission aggregometry as part of the evaluation of a patient with a suspected bleeding disorder? My Response Light transmission aggregometry (LTA) remains the reference method for measurement of platelet function in patients with suspected platelet function disorders (PFDs). Remarkably, the core principles on which LTA is based have not changed since it was first described by O’Brien and Born more than 50 years ago.1,2 Platelet-rich plasma is stirred in a cuvette that is placed between a light source and a photocell. The plasma is cloudy due to suspension of platelets and allows relatively little light to pass through. Upon the addition of an agonist, platelets aggregate, and the sample becomes clearer, permitting greater light transmission. Transmission of light is detected by the photocell and recorded as a function of time (Figure). An enhancement of modern LTA is the capacity to simultaneously monitor ATP secretion from dense granules using a luciferin-luciferase reagent.3 Representative normal and abnormal LTA tracings. Figure A shows normal responses to epinephrine 1.0 µM (curve 1) and collagen 2.0 mg/mL (curve 2). At critical concentrations, weak agonists such as epinephrine and ADP induce a biphasic response. The primary wave of aggregation reflects response to the addition of exogenous agonist. When normal platelets are activated, they secrete an endogenous pool of agonists. The secondary wave represents aggregation in response to this endogenous pool. Figure B is a tracing from a patient with epistaxis and menorrhagia. The response to ADP 5.0 µM (curve 1) is attenuated. A primary wave of aggregation is followed by deaggregation and an absent secondary wave of aggregation. There is no response to epinephrine 5.0 µM (curve 2). The patient’s platelets also demonstrated abnormal ATP secretion in response to ADP and epinephrine. Electron microscopy of the patient’s platelets revealed an absence of dense granules consistent with a diagnosis of dense granule storage pool disorder. When do I recommend LTA? Rare PFDs such as Glanzmann thrombasthenia (GT) and Bernard– Soulier syndrome (BSS) are usually diagnosed early in life because of the severity of the bleeding phenotype. In syndromic PFDs, the presence of associated features (e.g., oculocutaneous albinism in Hermansky–Pudlak syndrome) may facilitate diagnosis. Far more common, however, are mild PFDs without associated syndromic features. Even with a detailed bleeding history, these disorders may be difficult to distinguish from normal variation due to the high frequency of mucocutaneous bleeding symptoms in the general population. In one study of healthy adults, epistaxis, easy bruising, and prolonged bleeding after tooth extraction were reported in 25 percent, 18 percent, and 18 percent of subjects, respectively; and 47 percent of menstruant women reported heavy menses.4 patients with a suspected bleeding disorder were enrolled. A normal basic hemostatic work-up (platelet count, PT, APTT, fibrinogen, and von Willebrand panel) was required for study entry. The median ISTH-BAT score was significantly greater in patients than in controls (12 vs. 0). Among the patients who had a suspected bleeding disorder, however, there was no difference in score between the 41 who were diagnosed with a PFD by LTA and the 38 with a normal LTA (11 vs. 12).5 A bleeding history may be taken using either a conventional approach or a standardized bleeding assessment tool (BAT). Published BATs are effective in discriminating patients with bleeding disorders from healthy controls, but they have not proven effective in predicting the presence of PFDs among patients referred for a suspected bleeding disorder. A recent study investigated the utility of the International Society on Thrombosis and Haemostasis (ISTH)-BAT for this purpose. Twenty-one healthy controls and 79 In my practice, I employ a conventional approach to the bleeding history. I query patients about spontaneous, mucocutaneous bleeding symptoms (location, frequency, severity, treatment); bleeding with hemostatic challenges (e.g., surgery, trauma, childbirth, menstruation); and family history of bleeding. I also ask about prescription and over-the-counter medications (e.g., anti-platelet agents) and comorbidities (e.g., liver disease, uremia) that could promote bleeding. I use data from the history to estimate the pretest probability of a PFD. I recommend LTA when I judge the pretest probability to be intermediate or high (i.e., >5-10%). My approach is likely to be highly operator-dependent and result in Other Features over-testing (only about half of the patients I refer for LTA have an identifiable defect). Although BATs hold promise for ameliorating these deficiencies, I have opted not to use BATs in my practice until they are shown to improve diagnostic accuracy compared with conventional approaches to assessment of bleeding. Macrothrombocytopenia Table. Characteristic LTA Patterns of Selected Platelet Function Disorders Disorder Aggregation Response Primary ADP Secondary ADP Epinephrine AA Collagen Ristocetin Inherited disorders GT Absent Absent Absent Absent Absent Normal BSS Normal Normal Normal Normal Normal Absent Dense granule SPD Normal Decreased or Absent Variable Normal Normal Normal Deficiency of dense granules seen with platelet electron microscopy. May be syndromic or occur in isolation. Acquired disorders Aspirin Normal Decreased or Absent Decreased or Absent Absent Decreased or Absent Normal P2Y12 inhibitors (e.g., clopidogrel) Decreased or Absent Absent Normal Normal Normal Normal Glycoprotein IIb/IIIa inhibitors Absent Absent Absent Absent Absent Normal Uremia Normal Decreased Variable Decreased Normal or Decreased Normal Inherited aspirin-like defects in AA metabolism show a similar pattern GT=Glanzmann thrombasthenia; BSS=Bernard–Soulier syndrome; SPD=storage pool disorder; ADP=adenosine diphosphate; AA=arachidonic acid N O N M A L I G N A N T H E M A T O L O G Y 44 How is LTA interpreted? A panel of seven platelet agonists – ADP, epinephrine, collagen, thrombin receptor-activating peptide, the thromboxane A2 mimetic U46619, arachidonic acid, and ristocetin – is recommended by an international consensus panel.6 Aggregation tracings (Figure) are evaluated with respect to a number of parameters including shape change; length of lag phase; slope of aggregation; presence of a secondary wave of aggregation produced by weak agonists, such as epinephrine; maximal percent aggregation; and presence of deaggregation. The Hematologist Compendium 2010-2015 The pattern of aggregation and/or secretion defects observed with the agonist panel facilitates identification of a variety of hereditary and acquired PFDs (Table) and also helps to localize lesions to specific pathways. Classic examples include GT (caused by mutations in integrin αIIbβ3) and BSS (caused by mutations in the platelet glycoprotein Ib-IX-V complex), both of which have characteristic LTA signatures (absent aggregation to all agonists except ristocetin and absent ristocetin-induced agglutination, respectively). In practice, LTA abnormalities often do not conform to one of the textbook patterns shown in the Table. Many of these “non-specific” abnormalities are of uncertain clinical significance and molecular pathogenesis. However, critical new insights are emerging from genotype–phenotype correlation analyses such as the United Kingdom Genotyping and Platelet Phenotyping (GAPP) study.7 GAPP is a multicenter study of patients with clinically suspected inherited PFDs. Results from the first 111 patients were recently reported.8 A defect by LTA was detected in 64 patients (58%). Most defects (84%) involved Gi (i.e., ADP and epinephrine) receptor signaling, thromboxane A2 receptor signaling, or dense granule secretion. Targeted genotyping subsequently identified novel mutations in the P2Y12 ADP receptor, the thromboxane A2 receptor, and FLI1 and RUNX1, genes encoding transcription factors that participate in megakaryopoiesis and dense granule secretion.8,9 Several limitations must be borne in mind in the interpretation of LTAs. First, some LTA “abnormalities” are observed in a small percentage of healthy volunteers. Clinical correlation is required, but not always conclusive, in distinguishing disease from normal variation. LTA cannot assess platelet function under flow conditions and may be influenced by a variety of preanalytic, analytic, and biologic variables. Which variables can confound LTA? Many drugs affect platelet function in vitro. Drugs with reversible effects on platelets (e.g., NSAIDs) should be held for at least three days and drugs with irreversible effects (e.g., aspirin, clopidogrel) for at least 10 days prior to testing. An international consensus panel recommends a short rest period and avoidance of smoking and caffeine prior to blood collection to mitigate the effects of epinephrine release.6 Because chylomicrons in plasma can interfere with LTA, patients should be counseled to avoid fatty meals shortly before testing. Laboratories must adhere to meticulous specifications for sample collection and processing to avoid in vitro platelet activation. The platelet count in platelet-rich plasma should be measured. Results may be inaccurate when the platelet count is < 150 × 109/L. Studies should be completed within four hours after blood collection.6 Conclusion As they have for decades, a detailed bleeding history and LTA form the backbone of the diagnostic evaluation of patients with suspected PFDs. These approaches are not without shortcomings. The bleeding history has relatively poor specificity because of the frequency of mucocutaneous bleeding symptoms in the general population. LTA is labor-intensive, requires considerable expertise and a fresh blood sample, is not well-standardized, and shows overlap between normal variation and pathology across certain parameters. Continued development of BATs and improved methods for measuring platelet function are needed to overcome these limitations. Genotype–phenotype correlation studies are elucidating novel defects, are providing new insights into platelet function in health and disease, and may ultimately pave the way for more exacting approaches to diagnosis. 1. O’Brien JR. The adhesiveness of native platelets and its prevention. J Clin Pathol. 1961;14:140-149. 2. Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194:927-929. 3. Miller JL. Platelet function testing: an improved approach utilizing lumi-aggregation and an interactive computer system. Am J Clin Pathol. 1984;81:471-476. 4. Mauer AC, Khazanov NA, Levenkova N, et al. Impact of sex, age, race, ethnicity and aspirin use on bleeding symptoms in healthy adults. J Thromb Haemost. 2011;9:100-108. 5. Lowe GC, Lordkipanidzé M, Watson SP; UK GAPP study group. Utility of the ISTH bleeding assessment tool in predicting platelet defects in participants with suspected inherited platelet function disorders. J Thromb Haemost. 2013;11:1663-1668. 6. Cattaneo M, Cerletti C, Harrison P, et al. Recommendations for the standardization of light transmission aggregometry: a consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J Thromb Haemost. 2013;11:1183-1189. 7. Watson SP, Lowe GC, Lordkipanidzé M, Morgan NV; GAPP consortium. Genotyping and phenotyping of platelet function disorders. J Thromb Haemost. 2013;11:351-363. 8. Dawood BB, Lowe GC, Lordkipanidzé M, et al. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonist panel. Blood. 2012;120:5041-5049. 9. Stockley J, Morgan NV, Bem D, et al. Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood. 2013;122:4090-4093. Dr. Cuker indicated no relevant conflicts of interest. ASH-SAP, Sixth Edition The sixth edition of the ASH SelfAssessment Program (ASH-SAP) is available for ordering. This educational resource will help hematologists and others working in hematology-related fields to stay up to date with the latest advances in the rapidly evolving disciplines of adults and pediatric hematology, including malignant and nonmalignant disorders. The program includes multimedia featuring 3D animation and audio, a question-and-answer book, and six tests you can take to earn up to 70 CME/MOC Credits. For pricing, to order your copy, and for any additional information, visit www.ash-sap.org. N O N M A L I G N A N T H E M A T O L O G Y 45 The Hematologist Compendium 2010-2015 Anti-Phospholipid Antibodies and Pregnancy – July/August 2014 The authors have indicated that there has been no update regarding the content of this article since the original publication date in 2014. DEEPA SURYANARAYAN, MD, 1 AND DAVID GARCIA, MD 2 1. Clinical Lecturer, Department of Medicine, McMaster University Medical Center, University of Calgary, Alberta, Canada 2. Professor of Medicine, University of Washington School of Medicine, Seattle, WA The Question What is your approach to a patient with laboratory evidence of anti-phospholipid antibodies who is pregnant or considering pregnancy? Our Response Antiphospholipid antibody syndrome (APS) is a heterogeneous autoimmune disorder characterized by arterial and venous thromboembolic events and obstetric complications (especially recurrent fetal losses) in association with persistent laboratory evidence of anti-phospholipid antibodies (aPL). The aPL associated with the disease include the lupus anticoagulant (LA), anti-cardiolipin (α-CL), and anti-β2 glycoprotein-1 (anti-β2 GP-1). APS and aPL can occur in isolation (i.e., primary) or in association with other disease processes (i.e., secondary) including connective tissue diseases, most commonly systemic lupus erythematosus (SLE). Diagnostic Criteria for APS The presence of aPL alone does not constitute APS. The international classification criteria for APS (the Sapporo criteria) were introduced in 1998 and revised in 2006.1 To make a definitive diagnosis of APS, the presence of at least one clinical feature (thrombosis or pregnancy morbidity) and one laboratory abnormality must be observed, and the laboratory abnormality must be present on two or more occasions at least 12 weeks apart. Laboratory parameters are as follows: IgG and/or IgM α−CL or anti-β2 GP-1 in moderate to high titre (i.e., a titre > 40 for IgG or for IgM or a titre > the 99th percentile), or documentation of the LA. Further details about the diagnostic criteria for APS can be found in the Journal of Thrombosis and Haemostasis.1 Maternal and Fetal Morbidity The pathogenesis of aPL-related pregnancy morbidity is incompletely understood. For a pregnant woman, APS (and perhaps the presence of aPL) raises the possibility that she is at increased risk for thrombotic events and/or obstetric complications such as recurrent pregnancy losses, preeclampsia, eclampsia, intra-uterine growth restriction, and Hemolysis, Elevated Liver enzymes, Low Platelets (HELLP) syndrome. In a European referral center cohort of 1,000 patients with APS (many of whom had a connective tissue disease), 188 pregnancies were documented over 10 years; among patients in this group, early pregnancy loss (16.5%), intra-uterine growth restriction (26.3%), and prematurity (48.2%) were common.2 A systematic review found that lupus anticoagulant was associated with preeclampsia (OR 2.34), intra-uterine growth restriction (OR 4.65), and late (after 10 weeks of gestation) fetal loss (OR 4.73); however, numerous case-control studies were included in the analysis. When the analysis was restricted to cohort studies, late fetal loss was the only outcome associated with the aPL.3 Catastrophic APS (CAPS), a rare but devastating form of this syndrome, has been associated with pregnancy and the puerperium with 0.9 percent of the pregnancies from the aforementioned cohort being complicated by CAPS.2 Laboratory Diagnosis of aPL The laboratory identification of aPL can be problematic because of variability in the sensitivity of assays and reagents, high false-negative and false-positive detection rates, lack of standardization of assays, and lack of adherence to established guidelines. To address these issues, guidelines for laboratory detection of aPL were published in 2009 by the Scientific Standardization Subcommittee (SSC) of the International Society of Thrombosis and Haemostasis (ISTH) and by the British Committee on Standards in Hematology. The Clinical Laboratory Standards Institute (CLSI) has also prepared guidelines for laboratory testing for LAC that are to be published soon. Before counseling a patient about risk or recommending therapy, we suggest that the hematologist ensure that aPL testing is not only confirmed on more than one occasion but also performed by a laboratory that follows approved aPL testing guidelines. Pre-Pregnancy Counseling Management of a woman with APS should start with pre-pregnancy counseling that focuses on the involved risks. Discussion should include risks of maternal thrombosis and fetal loss (both early- and late-term) as well as the increased risk of placenta-mediated complications such as preeclampsia, HELLP syndrome, preterm delivery, and intra-uterine growth restriction. Involvement by a multidisciplinary management team that includes N O N M A L I G N A N T H E M A T O L O G Y 46 a maternal-fetal medicine specialist and a hematologist experienced in the management of thrombophilic conditions is recommended. APS patients on chronic anticoagulation should be informed about the potential teratogenic effects of warfarin, and oral anticoagulation should be switched to a therapeutic dose of low-molecular-weight heparin (LMWH) either before or very shortly after conception. For all women with APS, the American College of Chest Physicians (ACCP) evidence-based guidelines4 recommend commencing low-dose aspirin on confirmation of pregnancy, not only for its antithrombotic effects but also to reduce the risk of preeclampsia.5 In practice, it would be reasonable for a woman with obstetric APS to begin aspirin during the time that she and her partner are trying to conceive, because the risk of ASA is low and the impact of a delay between conception and ASA use is unknown. Pregnancy with aPL or APS – Clinical Management Scenarios 1. Pregnant women with persistent evidence of aPL who do not fulfill the diagnostic criteria for APS There are no robust data to guide management of pregnant women in this cohort. Dr. Anne Lynch et al. measured aPLs in early pregnancy in 451 low-risk nulliparous women and found that 24.4 percent had aPLs.8 The rate of fetal loss in this cohort was higher than those without aPLs (15.8 vs. 6.5%), but the rate of adverse maternal outcomes was similar in both groups. We usually recommend low-dose ASA because it is easy to take and there is a reasonably high quality of evidence that it reduces the risk of preeclampsia; however, the significant uncertainty about the benefit of any antepartum, antithrombotic therapy in women who do not meet criteria for APS means that decisions should be individualized. The role of post-partum thromboprophylaxis in this patient group is not established. 2. Women with adverse obstetric outcomes plus aPL, but no history of thrombosis In three meta-analyses of randomized trials in women with APS, the combination of ASA with prophylactic heparin significantly reduced pregnancy loss (RR 0.46)7 or firsttrimester loss (OR 0.39)9 and increased live births (RR 2.3).10 These analyses, however, have several limitations, including a small number of trials from which to gather data, small sample size within trials, and low quality of trial design. Compared with women who have had recurrent pregnancy losses, but whose thrombophilia testing is negative, women with a history of obstetric APS are at increased risk for both arterial and venous thrombotic events.11 In the NOH-APS observational study among women with prior fetal loss, those treated with LMWH and low-dose aspirin had lower pregnancy loss but higher preeclampsia rates than other women. Therefore, the evidence that combined (LMWH + ASA) therapy improves outcomes is, at best, of modest quality. Nonetheless, we suggest combination LMWH + ASA for pregnant women with APS and adverse obstetric outcomes, not only because there is some evidence that this will decrease the likelihood of pregnancy complications, but also because LMWH + ASA should also reduce the risk of thrombosis during pregnancy. However, because the quality of evidence to support combination therapy over LMWH or ASA alone is not compelling, we suggest that the hematologist consider not only patient preferences, but also the recommendation of a maternal-fetal medicine specialist before choosing LMWH + ASA over LMWH or ASA alone. The optimal doses for LMWH and ASA have not been defined, but we suggest 75 to 100 mg of ASA per day plus prophylactic-dose LMWH (Table). For women with obstetric APS and no history of thrombosis, thromboprophylaxis during the postpartum period should be considered, but the net benefit is not well established. 3. Pregnant women with thrombotic APS If a woman has APS and is anticoagulated for prior thrombosis, we suggest switching to therapeutic-dose LMWH before six weeks gestation. Full-intensity anticoagulation is important during the pregnancy and the postpartum period since these women have a significant, ongoing risk of thrombosis that is likely magnified by pregnancy. If a woman is on therapeutic-dose LMWH, delivery should be planned, and LMWH should be discontinued at least 24 hours prior to the procedure. Because there is no high-quality evidence to define the optimal anticoagulant management of patients who desire regional anesthesia, communication with maternal-fetal medicine and anesthesiology is especially important. For a woman with prior thrombosis who has discontinued anticoagulation, we suggest LMWH be started upon confirmation of pregnancy; whether to use prophylactic/ intermediate or therapeutic-dose LMWH would depend on the perceived risks of thrombosis and bleeding. An elevated risk of thrombosis persists for up to 12 weeks following delivery.12 The absolute risk for thrombosis beyond six weeks, however, is small. Thus, thromboprophylaxis beyond the six-week mark of the postpartum period should be reserved for those patients with an especially high baseline risk. Conclusion Management of pregnant women with APS is challenging. Despite the imperfect data that are currently available, pregnancy outcomes in women with APS who are managed in centers that use a multidisciplinary approach, which includes input from clinicians experienced in thrombophilia management and fetal-maternal medicine, is often favorable. The Hematologist Compendium 2010-2015 Table. Overview of the Therapeutic Options Used in Antiphospholipid Syndrome Pregnancy Drug Use Evidence Safety in pregnancy Safety in breastfeeding Aspirin 75-100 mg daily from conception until ≥ 36 weeks gestation Reduction of fetal loss No randomized controlled trials of aspirin for preventing VTE Will cross the placenta; human data inconsistent, but risk is likely low Does enter breast milk, but at low doses; should be safe Prevention of eclampsia Antiplatelet effect Some evidence for aspirin use in improving pregnancy outcomes, specifically reduction in rates of preeclampsia6 Warfarin Discontinue as soon as pregnancy is confirmed Not recommended in pregnancy unless LMWH may be less effective (e.g., prosthetic heart valves) Teratogenic between 6-12 weeks of gestation; switch to LMWH before 6 weeks of gestation Teratogenic Not excreted in breast milk Unfractionated heparin (UFH) May be used in event of massive pulmonary embolism Most studies using UFH have been superseded by studies using LMWH. Safe Not excreted in breast milk Some first-trimester analyses have shown small increased risk of gastroschisis If rapid reversal of anticoagulation is needed during peripartum period or operative procedures LMWH Drug of choice for women on warfarin, women who have had VTE or arterial thrombosis during pregnancy, women with previous pregnancy complications, or women who require thromboprophylaxis Evidence for its role in preventing firsttrimester loss remains controversial7 Does not cross placenta Does enter breast milk, but of little concern due to low bioavailability Steroids (e.g., prednisone) Little evidence for benefit in APS; used in immune thrombocytopenia associated with APS or SLE Minimal evidence of therapeutic benefit Cleft palate reported with first-trimester use Low concentrations in breast milk Intravenous immunoglobulins Used in a small number of women with APS as therapy for concomitant, immunemediated thrombocytopenia No additional benefit when added to conventional therapy with ASA and LMWH Crosses placenta after 30 weeks of gestation Excretion is unknown Hydroxychloroquine (HCQ) Used when women have coexisting SLE Mild antithrombotic effects No reports of fetal toxicity Considered safe despite excretion in breast milk Plasmapheresis Reserved for treatment of catastrophic APS Rarely used in pregnancy Unknown Benefit outweighed by its adverse effects (e.g., preeclampsia, gestational diabetes, increased risk of preterm deliveries) Decreased risk of congenital heart block in women on HCQ in case-controlled studies Limited to case reports Adapted with permission from Soh, MC and Nelson-Piercy C. Antiphospholipid syndrome in pregnancy http://informahealthcare.com/doi/abs/10.1586/eog.10.57. Expert Rev Obstet Gynecol. 2010;5:748,749. Future studies that expand our understanding of this complex disease, that clearly define obstetric and thrombotic risks, and that identify optimal antithrombotic regimens are needed. 1. Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306. 2. Cervera R, Serrano R, Pons-Estel GJ, et al. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2015;74:1011-1018. 3. Abou-Nassar K, Carrier M, Ramsay T, et al. The association between antiphospholipid antibodies and placenta mediated complications: A systematic review and meta-analysis. Thromb Res. 2011;128:77-85. 4. Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141:e691S-e736S. 10. Mak A, Cheung MW, Cheak AA, et al. Combination of heparin and aspirin is superior to aspirin alone in enhancing live births in patients with recurrent pregnancy loss and positive anti-phospholipid antibodies: a meta-analysis of randomized controlled trials and meta-regression. Rheumatology (Oxford). 2010;49:281-288. 11. Gris J, Bouvier S, Molinari N, et al. Comparative incidence of a first thrombotic event in purely obstetric antiphospholipid syndrome with pregnancy loss: The NOH-APS observational study. Blood. 2012;119:2624-2632. 12. Kamel H, Navi BB, Sriram N, et al. Risk of a thrombotic event after the 6-week postpartum period. N Engl J Med. 2014;370:1307-1315. Dr. Suryanarayan and Dr. Garcia indicated no relevant conflicts of interest. 5. Farquharson RG, Quenby S, Greaves M. Antiphospholipid syndrome in pregnancy: a randomized, controlled trial of treatment. Obstet Gynecol. 2002;100:408-413 6. Empson M, Lassere M, Craig JC, et al. Recurrent pregnancy loss with antiphospholipid antibody: a systematic review of therapeutic trials. Obstet Gynecol. 2002;99:135-144. 7. Laskin CA, Spitzer KA, Clark CA, et al. Low molecular weight heparin and aspirin for recurrent pregnancy loss: results from the randomized, controlled HepASA Trial. J Rheumatol. 2009;36:279-287. 8. Lynch A, Marlar R, Murphy J, et al. Antiphospholipid antibodies in predicting adverse pregnancy outcome. A prospective study. Ann Intern Med. 1994;120:470-475. 9. Ziakas PD, Pavlou M, Voulgarelis M. Heparin treatment in antiphospholipid syndrome with recurrent pregnancy loss: a systematic review and meta-analysis. Obstet Gynecol. 2010;115:1256-1262. N O N M A L I G N A N T H E M A T O L O G Y 47 The Hematologist Compendium 2010-2015 Treatment Approaches to Castleman Disease with the Advent of Anti-Interleukin-6 Therapy – March/April 2015 Due to the recent publication date of this article, an update has not been requested. FRITS VAN RHEE, MD, PhD, MRCP(UK), FRCPath, 1 AND DAVID FAJGENBAUM, MD, MSc 2 1. Professor of Medicine, Myeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences; Co-Founder and Scientific Board Member Castleman Disease Collaborative Network; Little Rock, AR 2. Adjunct Assistant Professor of Medicine, Department of Hematology/ Oncology, University of Pennsylvania; Co-Founder and Executive Director Castleman Disease Collaborative Network; Philadelphia, PA The Question What are your treatment approaches to Castleman disease with the advent of anti– interleukin-6 therapy? Our Response Castleman disease (CD) describes a group of heterogeneous lymphoproliferative disorders that share common histopathological lymph node changes. CD can present with localized (unicentric CD or UCD) or generalized lymphadenopathy (multicentric CD or MCD). MCD should be further divided into human herpesvirus 8 (HHV-8) –positive and HHV-8–negative MCD (Table). The latter is also referred to as idiopathic MCD (iMCD). MCD patients can exhibit a spectrum of clinical features from mild flu-like symptoms to sepsis-like multiorgan failure.1 It is important to distinguish these three entities since they require entirely different therapeutic approaches. UCD UCD presents with lymphadenopathy confined to one lymph node region, and many patients are asymptomatic. Symptoms are usually due to compression of vital structures such as the trachea, blood vessels, or nerves. UCD occurs most frequently in younger females. The diagnosis is made when excisional lymph node biopsy shows characteristic Castleman changes. Approximately 90 percent of UCD cases demonstrate the hyaline vascular (HV) subtype. The other 10 percent of patients with UCD demonstrate mixed cellularity or plasmacytic (PC) pathology and may have constitutional symptoms such as fever, fatigue, and weight loss. UCD is not associated with HHV-8 or HIV infection. There is an increased association with lymphoma, which may have been present all along, or the UCD may “transform” into lymphoma. The etiology of UCD is poorly understood, and there is usually no excess interleukin-6 (IL-6) secretion. Limited studies have demonstrated abnormal cytogenetics or evidence of monoclonality in stromal cells, but the significance of these findings is unknown.2 Surgical extirpation is curative in 95 percent of UCD cases.3 Unresectable cases can be treated with rituximab and steroids, which may induce complete responses or shrink the mass sufficiently to make it resectable. UCD lymphadenopathy is highly vascularized, and embolization is a further therapeutic option. Involution of lymphadenopathy after radiotherapy has also been reported. Difficult cases require a multimodal approach and are best managed at an experienced center. HHV-8–Positive MCD studies. Siltuximab, which is a monoclonal antibody to IL-6, has not been studied in HHV-8– positive MCD because it did not bind to viral IL-6 in preclinical studies. iMCD HHV-8–negative MCD patients present with generalized lymphadenopathy, constitutional symptoms, and can also develop multiorgan failure. The disease can wax and wane, be gradually progressive, or have severe episodic flares resulting in death.8 In a recent literature review, 22 percent of HHV-8–negative MCD patients had died by a median follow-up time of 29 months.9 HHV-8–negative MCD is driven by pro-inflammatory hypercytokinemia, most notably of IL-6, which leads to: anemia; anasarca due to hypoalbuminemia from IL-6–mediated liver dysfunction and VEGF-mediated vascular permeability; and systemic inflammation with elevated ESR, CRP, and fibrinogen.8 Some patients may develop kidney, liver, and bone marrow failure and succumb to their disease. Patients with HHV-8–negative MCD may also have one or more features of POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) syndrome, or a concurrent POEMS syndrome.10 Patients with one or more features of POEMS often have less florid constitutional symptomatology. The etiology of the pro-inflammatory hypercytokinemia in iMCD has not yet been identified. The pathologic cell and the responsible intracellular inflammatory pathway responsible for producing the IL-6 in these patients has also not been elucidated. Hypothesized etiologies include an unknown virus, a small population of malignant cells, or germline genetic mutations in the immune system.1 No single gene causing iMCD has been identified, but systematic sequencing has not been performed. The disease may be influenced by ethnicity and genetic factors such as a polymorphism of the IL-6 receptor.11 Asian patients may demonstrate violaceous skin lesions or interstitial pneumonitis usually not seen in other populations. Currently, a diagnosis of iMCD can be made when patients have 1) histopathology typical of CD on excisional lymph node biopsy, 2) multiple regions of enlarged lymph nodes, 3) negative quantitative PCR for HHV-8 in the peripheral blood or negative LANA-1 staining of the lymph node biopsy, and 4) systematic exclusion of diseases known to demonstrate Castleman-like histopathology (e.g., systemic lupus erythematosus, Epstein-Barr virus, lymphoma, IgG4-associated lymphadenopathy). Hence, both HHV-8–positive and –negative MCD are not purely pathologic diagnoses. Efforts are currently underway to establish international consensus around clinical, pathologic, and exclusion criteria for the diagnosis of iMCD. Although the PC variant predominates in iMCD, HV and mixed pathology have also been reported. HHV-8-negative MCD has been historically managed with corticosteroids, rituximab, and/or chemotherapeutic agents derived from the CHOP regimen. Corticosteroids may temporarily control symptoms, but patients relapse on tapering. Rituximab has not been systematically evaluated in iMCD and a limited number of case reports suggest that patients often relapse. Monoclonal antibodies targeting IL-6 have been recently developed and more rigorously evaluated. A single-arm study of 28 Japanese patients on tocilizumab demonstrated a high response rate in terms of symptoms, laboratory parameters, and reduction in lymphadenopathy.12 Siltuximab was evaluated in a double-blind, placebo-controlled, randomized study using a control arm of best supportive care including up to 60 mg of prednisone. The combined durable symptomatic and tumor response was 34 percent, and 50 percent of patients remained on drug for the duration of the study. This study provided the first placebo-controlled evidence for an iMCD therapy, and siltuximab is the first drug approved for iMCD by the U.S. Food and Drug Administration and European Medicines Agency. Both siltuximab and tocilizumab are safe and well-tolerated.13 For patients that do not respond to anti–IL-6 therapy, immunosuppressants, immunomodulators, biologics, and cytotoxic chemotherapies, including cyclosporine, sirolimus, bortezomib, thalidomide, anakinra, interferon-α, cyclophosphamide, and etoposide, have been reported to have some success in case reports or small series. HHV-8–positive MCD presents with generalized lymphadenopathy and constitutional symptoms and can progress to multiorgan failure leading to death. The disease is driven by the excessive release of viral IL-6, which is encoded by the HHV-8 virus and drives human IL-6, IL-10, and vascular endothelial growth factor (VEGF) secretion. HHV-8–positive MCD was first described during the AIDS epidemic and is classically thought to be due to lytic replication of HHV-8 in the setting of HIV infection.4 However, there are also patients with HHV-8–positive MCD who are HIV negative. These patients may have another cause for immunosuppression that impairs their control of Table. Features of the Different Types of Castleman Disease HHV-8.5 Approximately 10 to 20 percent of individuals have been exposed to HHV-8, and the virus remains dormant in Type of Type of Pathology IL-6–Driven Virologic Status Treatment Castleman Lymphadenopathy Inflammatory B lymphocytes. A diagnosis of HHV-8–positive MCD can Disease Syndrome* be rendered if the patient has 1) a pathologic diagnosis of CD made on an excisional lymph node biopsy and 2) Unicentric Localized 90 percent hyaline Typically not • Negative for Complete excision actively replicating HHV-8 virus detected in the peripheral vascular HHV-8 by QPCR or negative blood by molecular testing (quantitative polymerase chain LANA-1 stain reaction [PCR]) or a positive stain of the lymph node for the HHV-8 latency associated nuclear antigen (LANA-1). HIVMulticentric Generalized ± Plasmacytic or Yes • Positive for HHV-8 • Rituximab ± positive, HHV-8–positive MCD patients may have coexistent HHV-8–Positive hepatosplenomegaly plasmablastic by QPCR etoposide Kaposi sarcoma and are prone to develop HIV-associated • May be positive • Optional valgancifor HIV clovir maintenance lymphomas. Lymph node pathology shows plasmacytic or plasmablastic changes, and the plasma cells may exhibit Multicentric Generalized ± Mostly plasmacytic, Yes, but variable • Negative for • Siltuximab light chain restriction. Intranodal microlymphomas have also HHV-8–Negative hepatosplenomegaly but can be hyaline clinical presentaHHV-8 by QPCR • Tocilizimab been reported. HHV-8–positive MCD is effectively treated with (Idiopathic) vascular or mixed tion from mild to or cellularity very severe Negative LANA-1 • Rituximab rituximab, which depletes the reservoir of HHV-8–positive stain • Chemotherapy in cells and significantly reduces the risk of lymphoma.6 severe cases • Negative for HIV More severely afflicted patients may additionally require etoposide. Some experts recommend maintenance therapy *Symptoms: fevers, night sweats, anorexia, weight loss, fatigue. Laboratory abnormalities: anemia, thrombocytopenia or thrombocytosis, with valganciclovir. HIV-positive patients should receive elevated C-reactive protein, Westergren erythrocyte sedimentation rate, fibrinogen, hypergammaglobulinemia, abnormal renal function, 7 appropriate HAART therapy. The value of tocilizumab, which increased interleukin-6 (IL-6), vascular endothelial growth factor, interleukin-10. blocks the human IL-6 receptor is the subject of ongoing Abbreviations: QPCR, quantitative polymerase chain reaction; LANA-1, latency associated nuclear antigen. N O N M A L I G N A N T H E M A T O L O G Y 48 The Hematologist Compendium 2010-2015 Choice of Therapy for iMCD In the authors’ opinion, patients with iMCD should first be treated with anti–IL-6 therapy approved in that region (siltuximab in North America and the European Union; tocilizumab in Japan). Patients with few symptoms or laboratory abnormalities suggestive of little excess IL-6 may not respond well to anti–IL-6 blockade and should be considered for rituximab and steroids. Patients with severe hypercytokinemia and organ failure may not respond sufficiently to anti–IL-6 targeting monoclonal antibodies, and they require combination chemotherapy or consideration of experimental treatment. Dosing intervals can be spaced out in selected patients responding to anti-IL-6 therapy. Progressive motor polyneuropathy suggesting coexistent POEMS does not respond well to rituximab or to IL-6–targeted therapy, and these patients require autologous stem cell transplantation as part of their treatment plan. 1. Fajgenbaum DC, Van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123:2924-2933. 2. Chang KC, Wang YC, Hung LY, et al. Monoclonality and cytogenetic abnormalities in hyaline vascular Castleman disease. Mod Pathol. 2014;27:823-831. 3. Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255:677-684. 4. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95:1406-1412. 5. Dossier A, Meignin V, Fieschi C, et al. Human herpesvirus 8-related Castleman disease in the absence of HIV infection. Clin Infect Dis. 2013;56:833-842. 6. Gerard L, Michot JM, Burcheri S, et al. Rituximab decreases the risk of lymphoma in patients with hivassociated multicentric Castleman disease. Blood. 2012;119:2228-2233. 7. Bower M. How I treat HIV-associated multicentric Castleman disease. Blood. 2010;116:4415-4421. The Future 8. van Rhee F, Stone K, Szmania S, et al. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol. 2010;8:486-498. The introduction of rituximab has been a major advance in HHV-8–positive MCD, while therapy with IL-6–targeting monoclonal antibodies is an important innovation in iMCD. However, anti– IL-6 therapy is not effective for all patients, and it is not curative, as cessation of treatment results in relapse. In 2012, we co-founded the Castleman Disease Collaborative Network (CDCN; www.castlemannetwork.org) to accelerate research and elucidate the pathogenesis of MCD. In 2.5 years, we have assembled a 23-member Scientific Advisory Board representing seven countries; built a global community of more than 200 researchers and physicians worldwide; leveraged the community to establish and execute an international research agenda; and engaged patients throughout the entire process. We are currently finalizing plans to establish a global registry/natural history study, which we believe will be crucial for establishing diagnostic criteria and improving patient care. We also plan to launch viral discovery, serum proteomics, intracellular inflammatory pathway identification, and sequencing studies. We invite you to register on our website, attend our annual meeting that occurs during the ASH annual meeting, conduct research, contribute samples for research, and encourage your patients to enroll in our registry. 9. Fajgenbaum DC, Liu A, Ruth J, et al. HHV-8-negative, idiopathic multicentric Castleman disease (iMCD): a description of clinical features and therapeutic options through a systematic literature review [abstr]. Blood. 2014;124:4861a. 10. Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87:997-1002. 11. Stone K, Woods E, Szmania SM, et al. Interleukin-6 receptor polymorphism is prevalent in HIV-negative Castleman disease and is associated with increased soluble interleukin-6 receptor levels. PLoS One. 2013;8:e54610. 12. Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106:2627-2632. 13. van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15:966-974. Dr. Van Rhee and Dr. Fajgenbaum indicated no relevant conflicts of interest. ASH Meetings Visit www.hematology.org/Meetings to learn about upcoming meetings on malignant and nonmalignant hematology. Find out date, location, and program or content information for each meeting online. N O N M A L I G N A N T H E M A T O L O G Y 49 The Hematologist Compendium 2010-2015 Treatment with Direct Oral Anticoagulants – May/June 2015 Due to the recent publication date of this article, an update has not been requested. DAVID GARCIA, MD Professor of Medicine, University of Washington School of Medicine; Assistant Medical Director for Antithrombotic Therapy at the University of Washington Medical Center, Seattle, WA The Question Given the recent U.S. Food and Drug Administration (FDA) approval of edoxaban, the fourth direct oral anticoagulant (DOAC) to come to the U.S. market since 2010, what should I know about this class of medications, and what is their role in patient care? Are there patients for whom these medications are not (or possibly not) appropriate? My Response This is an exciting time for clinicians caring for patients with (or at risk for) thrombosis, but the amount of clinical trial data, not to mention the dosing peculiarities, relevant to the DOACs, can be difficult to digest. Here, I will try both to provide take-home points about safety and efficacy relative to other therapies as well as to address some of the practical management questions I am frequently asked about DOACs. One or more of the DOACs has now been approved for three main indications: 1) the prevention of stroke and systemic embolism in patients with atrial fibrillation (AF), 2) the acute and extended treatment of venous thromboembolism, and 3) the prevention of venous thromboembolism (VTE) after total knee or hip arthroplasty (Table). These medications are small molecules that inhibit their serine protease target by binding to its active site (thrombin in the case of dabigatran and activated factor X [FXa] in the case of rivaroxaban, apixaban, and edoxaban). There are some very important pharmacologic differences with warfarin: All these drugs rely, to varying degrees, on elimination by the kidneys. The prescriber must consult the package insert of a particular agent to determine whether his or her patient’s renal function would be a contraindication to treatment or mandate a dose adjustment. There are no important dietary interactions for the DOACs. A possible exception is that the bioavailability of rivaroxaban seems to be increased if it is taken with a meal; however, the clinical relevance of this is uncertain. Although the DOACs can interact with other medications, the number of potentially clinically important interactions is small compared to the corresponding number with warfarin. My patient taking a DOAC needs to undergo an elective procedure. How far in advance of the procedure should I stop the medication, and do I need to provide “bridge” therapy? Unlike vitamin K antagonists (VKAs), all of the DOACs have both a rapid onset of action and a relatively short half-life. DOACs induce maximal anticoagulant effect within two to three hours of the first dose and, unless renal function is impaired, will essentially disappear from the plasma 24 to 48 hours after the last dose. Therefore, periprocedural anticoagulation (“bridging”) with a parenteral anticoagulant is not necessary for a patient taking a DOAC unless the patient undergoes a surgery that will preclude swallowing or gastrointestinal absorption. Patients with renal impairment will need to interrupt the DOAC longer (see individual prescribing information for more details), but others can safely undergo most interventions as early as 24 to 48 hours after their last DOAC dose. My patient has moderate renal impairment. Does that mean I cannot prescribe a DOAC? Although all DOACs depend on renal clearance more than warfarin does, renal insufficiency is not a contraindication in all cases. Indeed, an analysis of the subgroup of patients with moderate renal impairment (creatinine clearance 30 to 50 mL/min) in the phase III clinical trials indicates that the DOACs (when dosed according to the manufacturers’ recommendations) are at least as safe as warfarin for such individuals.1 The thresholds at which dose-adjustment is required vary depending on the DOAC; most providers would avoid DOACs altogether in patients with severe renal impairment (creatinine clearance < 30 mL/min). That being said, the U.S. FDA has approved a dose adjustment for apixaban in patients on hemodialysis. Apixaban is the DOAC that depends least on renal clearance. On the other end of the spectrum of renal function, the U.S. FDA has recommended that edoxaban not be used in AF patients with excellent renal function (creatinine clearance > 95 mL/min) because of a subgroup analysis suggesting it may be less effective than warfarin to prevent stroke or systemic embolism in this subgroup. When will antidotes for some or all of the DOACs be available? Should I avoid prescribing DOACs until antidotes reach the market? The lack of experience managing DOAC-associated bleeding is an understandable concern. However, the absence of a dedicated antidote should probably not be a major consideration in the decision to use (or not use) this class of medications. Outcome data from randomized phase III clinical trials that included more than 100,000 patients show that, although no antidote was available, the likelihood of fatal bleeding was lower among the patients N O N M A L I G N A N T H E M A T O L O G Y 50 randomized to a DOAC than among the patients randomized to warfarin. Furthermore, taking a DOAC (as opposed to warfarin) was not associated with a higher likelihood of death among patients who experienced a major bleed. These seemingly counterintuitive observations are probably explained by several factors: 1) Despite the availability of vitamin K and plasma products, warfarin-associated major bleeding has a very high associated mortality; 2) compared with warfarin, the DOACs cause 50 percent fewer intracranial bleeds; and 3) the short half-life of the DOACs means that in many cases of major bleeding, no antidote or “reversal agent” is needed. These observations notwithstanding, antidotes and reversal agents in late-phase clinical development are likely to reach the U.S. market within the next two to three years. The DOAC antidotes that are furthest in the development process are andexanet (a factor Xa “decoy” molecule that is intended for patients on a FXa inhibitor) and idarucizumab (an antibody fragment targeting the direct thrombin inhibitor, dabigatran). What should I do if consulted about a patient with DOACassociated bleeding? Many patients with DOAC-associated bleeding will be best managed with supportive care such as red blood cell transfusions and volume support; excellent renal perfusion will maximize the rate at which the anticoagulant effect dissipates. Unless a patient has compromised renal function, the plasma DOAC concentration (and corresponding drug effect) will have decreased by more than 50 percent six to 12 hours after the last dose. In the small minority of patients with DOAC-associated bleeding who require more aggressive efforts to counteract the anticoagulant effect immediately, one could consider prothrombin complex concentrates or recombinant activated factor VII.2 However, the thrombosis risk associated with these interventions is not trivial, and the rationale for their use is based entirely on animal and other preclinical models. Condition Dabigatran Rivaroxaban Apixaban Edoxaban Stroke Prevention in Atrial Fibrillation ü ü ü ü ü* ü - ü ü ü ü ü ü ü ü ü* - Venous Thromboembolism Treatment Acute Extended Knee Arthroplasty Hip Arthroplasty *Edoxaban and dabigatran are approved for the acute treatment of venous thromboembolism only after an initial 5-day course of treatment with a parenteral anticoagulant (e.g., low-molecular-weight heparin, fondaparinux, heparin). In which patients should DOACs be avoided? In general, DOACs should be used only for approved indications. Because patients with mechanical prosthetic heart valves were not included in the phase III studies, and because dabigatran was inferior to warfarin in the only head-to-head comparison that has been done in this population,3 the DOACs should not be prescribed to patients with mechanical prosthetic heart valves. There are also subgroups of patients with (or at risk for) VTE where routine DOAC use should also be avoided pending further evidence. Because of their highly prothrombotic tendencies, patients with active cancer, heparin-induced thrombocytopenia, and bona fide antiphospholipid syndrome are other groups for whom I do not think DOACs should be a first option. Instead, we should work hard to enroll such patients in clinical trials that will provide evidence on which we can base future practice. Conclusion In summary, the DOACs offer hematologists and well-selected patients four oral alternatives to warfarin and other VKAs.4,5 Important ongoing trials will tell us more about how best to manage serious DOAC-related bleeding, whether DOACs can be safely used in patients with cancer-associated VTE, and what is the safest way to combine DOACs with antiplatelet agents (e.g., in patients with previous acute coronary syndromes). In the meantime, many patients for whom these medications are indicated (and for whom cost is not a barrier) will likely choose them over warfarin because of their convenience and impressive safety profile. 1. Harel Z, Sholzberg M, Shah PS, et al. Comparisons between novel oral anticoagulants and vitamin K antagonists in patients with CKD. J Am Soc Nephrol. 2014;25:431-442. 2. Siegal DM, Garcia DA, Crowther MA. How I treat target-specific oral anticoagulant-associated bleeding. Blood. 2014;123:1152-1158. 3. Eikelboom JW, Connolly SJ, Brueckmann M, et al. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med. 2013;369:1206-1214. 4. Yeh CH, Gross PL, Weitz JI. Evolving use of new oral anticoagulants for treatment of venous thromboembolism. Blood. 2014;124:1020-1028. 5. Dzeshka MS, Lip GY. Non-vitamin K oral anticoagulants in atrial fibrillation: Where are we now? Trends Cardiovasc Med. 2014;S1050-1738:00195-00199. Dr. Garcia indicated no relevant conflicts of interest. The Hematologist Compendium 2010-2015 ©2016 The American Society of Hematology All materials contained in this compendium are protected by copyright laws and may not be used, reproduced, or otherwise exploited in any manner without the express prior written permission of The Hematologist: ASH News and Reports. All articles in this compendium were originally published in The Hematologist.