Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
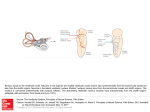
American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 – 371 www.elsevier.com/locate/amjoto Original contributions Large vestibular aqueduct syndrome: audiological, radiological, clinical, and genetic features Stefano Berrettini, MDa,T, Francesca Forli, MDa, Fausto Bogazzib, Emanuele Neri, MDc, Luca Salvatori, MDc, Augusto Pietro Casani, MDa, Stefano Sellari Franceschini, MDa a ENT Unit, Neuroscience Department, University of Pisa, 56126 Pisa, Italy Department of Endocrinology and Metabolism, University of Pisa, 56126 Pisa, Italy c Division of Diagnostic and Interventional Radiology, Department of Oncology, Transplants, and Advanced Technologies in Medicine, University of Pisa, 56126 Pisa, Italy Revised 8 December 2004 b Abstract Purpose: The aim of this study was to analyze the clinical, audiological, radiological, and genetic features of a group of patients affected with large vestibular aqueduct syndrome. Materials and methods: Seventeen patients affected with large vestibular aqueduct syndrome (LVAS), diagnosed by means of high-resolution magnetic resonance imaging of the inner ear, with 3-dimensional reconstructions of the labyrinth and by high-resolution spiral computed tomography of the temporal bone, performed only on the oldest patients, have been submitted to a complete audiological evaluation, a thyroid functional and ultrasonographic study, and a molecular study of the PDS gene. Results: The clinical presentation of LVAS was very variable in our group of patients. The enlarged vestibular aqueduct was bilateral in 15 cases and unilateral in 2; it was the only malformation of the labyrinth in 12 patients, whereas it was associated with other inner ear anomalies in the other 5. The hearing loss was very variable in degree (from mild to profound), age at onset, and progression. Moreover, among the 17 patients, 10 were clinically affected by Pendred’s syndrome (PS), 3 by distal renal tubular acidosis associated with large vestibular aqueduct, whereas in 3 patients the large vestibular aqueduct was not syndromal. Finally, we identified mutations in the PDS gene in 5 of 10 patients with PS. Conclusions: Our data underscore the frequent role of the large vestibular aqueduct syndrome in the pathogenesis of sensorineural hearing loss and the overall wide variability in its audiological features. It is also highlighted that LVAS is often part of some syndromal diseases, most of which are PS, which is often misdiagnosed because of the varying degree of thyroid symptoms. This study also underscores the possible role of hydro-electrolyte and acid-base endolymphatic fluid disorders in the pathogenesis of enlarged vestibular aqueduct syndrome. D 2005 Elsevier Inc. All rights reserved. 1. Introduction The large vestibular aqueduct syndrome (LVAS) is characterized by the presence of an abnormally large vestibular aqueduct (LVA) generally associated with fluctuating, progressive sensorineural hearing loss (SNHL), often T Corresponding author. ENT Unit, Neuroscience Department, University of Pisa, 56126 Pisa, Italy. Tel.: +39 50 992625; fax: +39 50 550307. E-mail address: [email protected] (S. Berrettini). 0196-0709/$ – see front matter D 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.amjoto.2005.02.013 with sudden, stepwise onset or progression secondary to activities involving minor head trauma, large sudden shifts of barometric pressure, the Valsalva maneuver, and so forth [1-11]. Large vestibular aqueduct syndrome is considered to be the most frequent morphogenetic cause of hearing loss in children [12], although its frequency is probably underestimated [3,12,13]. It is often associated with other congenital inner ear anomalies, the most common being an abnormally large vestibule, an enlarged semicircular canal, or a hypoplastic cochlea [9,14,15], although according to some 364 S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 – 371 Fig. 1. Volume rendering of the labyrinth showing the enlarged endolymphatic duct/sac complex (arrow), together with the cochlea, vestibule, and semicircular canals. S.F., m, 12 yrs (case 9) dB HL authors, SNHL associated with LVA as the only radiographically detectable inner ear anomaly constitutes a separate clinical entity [1,15]. Large vestibular aqueduct syndrome may be associated with nonsyndromic [16] as well as syndromic forms of SNHL, such as the Pendred’s syndrome (PS) [17-21] and the branchiootorenal syndrome [22]. Moreover, we have recently reported on 3 consecutive, unrelated pediatric patients in whom LVAS was associated with SNHL and distal renal tubular acidosis (dRTA) [23]. Concerning the genetics of LVAS, it has been postulated to be inherited as an autosomal recessive trait [5,24]. Recently, the locus for nonsyndromic SNHL associated with LVAS has been mapped to the same chromosomal region as the PS locus, 7q31 [25,26], and it has been reported that the gene responsible for the PS, the PDS, is also mutated in patients with LVA associated with nonsyndromic SNHL [16,21,27,28]. In this paper, we describe the clinical, audiological, radiological, and genetic features of a group of 17 patients affected with LVA, studied by means of high-resolution magnetic resonance imaging (HR-MRI) of the inner ear with 3-dimensional (3D) reconstructions of the labyrinth and by high-resolution computed tomography (HR-CT) of the petrous bone, performed only on the oldest patients. patients over 6 years of age, or conditioned audiometry for patients between 1 and 6 years. The hearing loss was considered progressive when a deterioration of more than 15 dB in the PTA (average between 500-1000-2000 Hz) was recorded within a 10-year period [29]. Vestibular function was studied only in the oldest patients (15/17) through evaluation of spontaneous, positional, and head-shaking nystagmus, and by the caloric stimulation test, following the procedures of Fitzgerald and Hallpike [30], with video-nystagmograph recording. The diagnosis of LVA was made by means of HR-MRI at high field strength (Signa 1.5 T; GE/Medical Systems, Milwaukee, Wis), with 3D reconstructions of the labyrinth (Figs. 1 and 2), performed on each patient and confirmed via spiral HR-CT of the temporal bone in the oldest patients (15/17 patients). High-resolution MRI was performed with a Signa 1.5 T machine (GE/Medical Systems). An acquisition was performed on each ear using a 3-in circular surface coil for signal reception, with a 3D, T2-weighted, fast spinecho sequence in the axial plane (TR 5000, TE 275, echo train length 64, partition thickness 0.5 mm, 72 partitions, field of view 12 cm, matrix 512 512). A single excitation was acquired and the imaging time was 7 minutes and 22 seconds [31]. Images were transferred to a high-end graphics workstation (Dextroscope; Volume Interactions/Bracco, Singapore), and volume analysis was performed with a dedicated software. The software embedded with the workstation allows a rapid segmentation and provides a fast and intuitive method to generate 3D models with a volume rendering algorithm. The 3D models of the membranous labyrinth were displayed with a stereoscopic view. Using hand-tracking devices, it was possible to isolate the vestibular duct and sac from the membranous labyrinth. A volume is calculated by counting bvisibleQ voxels and multiplying them by the voxel dimensions as obtained from the DICOM (Digital Imaging and Communication in Medicine) 3.0 data source. The user needs to specify a rectangular box that completely includes the volume of interest and a voxel 2. Material and methods We report on 17 patients, 9 males and 8 females, affected by LVAS, with a mean age of 24.7 years, ranging from 4 to 50 years. Each patient underwent a complete audiological and clinical evaluation: apart from family and clinical history evaluation and an otomicroscopic examination, the tests included impedance audiometry, the stapedius reflex threshold test, and brainstem auditory-evoked potentials, for all, and pure-tone audiometry (PTA, 0.5-1-2-4-8 kHz) for -20 -10 0 10 20 30 40 50 60 70 80 90 100 110 120 250 500 1000 2000 4000 8000 Hz Fig. 2. Pure tone audiogram of patient 9, showing the presence of an airbone gap bilaterally. S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 –371 threshold value. Voxels inside the volume of interest, and voxel values above the specified threshold, will be counted toward the final volume value. High-resolution CT was performed with a 4-row CT (Light Speed Plus; GE Medical Systems) with a collimation of 1.25 mm, section thickness 0.63 mm, pitch 3, 0.8 s rotation tube, 120 kVp, 200 mAs, in the axial and coronal planes. Images were reconstructed at 0.63-mm interval with a bone algorithm [32]. The diameters of the vestibular aqueduct (VA) and the endolymphatic duct (ED) and sac (ES) were measured by axial CT and axial MRI scans, respectively. A VA was considered to be enlarged if its caliber, measured midway between the common crus and external aperture, was greater than or equal to 1.5 mm [15]. The HR-MR investigations of the inner ear were repeated in 6 patients some years after the diagnosis of LVAS (range 4–10 years, mean 5.6 years) to assess any variations in the volume of the ED and ES; 4 of 6 patients presented a bilateral LVA, whereas 2 of 6 patients presented a unilateral LVA. The MRI data sets of those patients were coregistered, and the volume of the vestibular aqueduct was then calculated by using a 3D workstation (Dextroscope; Volume Interactions/Bracco), to see whether the volume had changed over time. Image coregistration was performed with a semiautomatic registration method (multipoint plus freehand fusion). Three anatomical landmarks were visually identified by the radiologist in the MRI 3D images corresponding respectively to the middle tract of the superior and lateral semicircular canals, and the initial part of the basal turn of the cochlea. A marker was manually placed on each of the identified points. Once the markers were placed in both datasets the software produced a preliminary combined coregistered 3D model. The 3D model was then displayed as a fused 3D image incorporating the magnetic resonance data. An assessment was made by the radiologist by visually inspecting the fused 3D images to determine the accuracy of image coregistration. In the case of incorrect superimposition of labyrinth components, a manual adjustment of the data was made. By using the volume rendering of the MRI data, the membranous tract of the vestibular aqueduct acquired at different time intervals was isolated and displayed both in the same 3D model. The radiological study of all the patients, both HR-CT and HR-MR investigations, has been performed and evaluated by the same radiologist, expert in the imaging study of the petrous bone. Also, the measurements of the diameters, the 3D reconstructions, and the analysis of the volumes of the ED/ ES complexes have been performed by the same radiologist. Each patient also underwent a functional and ultrasonograph evaluation of the thyroid and a perchlorate (KClO4) discharge test to reveal the presence of the PS, as previously described [33], apart from 2 patients: 1 patient was not submitted to perchlorate discharge test, because she was too young (4 years old) and 1 patient, previously submitted to 365 thyroidectomy, was not submitted to the evaluation of thyroid function nor to perchlorate discharge test. Finally, each patient underwent mutation analysis of the PDS gene, as described in previous studies [33]. 3. Results A summary description of each patient’s case is provided in Tables 1 and 2. An enlargement of the membranous ED/ ES complex was disclosed by HR-MR bilaterally in 15 cases (30 ears) and unilaterally in the other 2 (right-sided in both). In the patients who underwent HR-CT, it confirmed the presence of an enlargement of the bony VA in every ear in which HR-MR had revealed an enlarged ED and ES. The mean volume of ED and ES complex, measured on 3D reconstructions of the membranous labyrinth as described, was 0.67 mL, ranging from 0.09 to 1.78 mL. In 12 patients, LVA was the only radiographically detectable inner ear anomaly, whereas in 5 patients it was accompanied by other inner ear abnormalities: bilateral dilatation of the vestibule, the apical turn of the cochlea, and the lateral semicircular canal in patient 9, bilateral dilatation of the vestibule in patients 10 and 17, and bilateral classical Mondini deformity in patients 12 and 13. The volume of the ED/ES complex remained stable in the 6 patients who underwent serial HR-MR of the inner ear, and particularly the 2 cases of these 6 with unilateral LVA also exhibited unilateral LVA upon the second HR-MR. The degree of SNHL in the affected ears (32 ears with LVA) ranged from moderate to profound, with a PTA from 47 to 113 dB HL and a downsloping (29 ears) or U-shaped (3 ears) hearing threshold curve; only 2 ears (patients 1 and 6) presented a moderate hearing deficit, 4 ears (patients 1, 2, 10, and 17) exhibited a severe hearing deficit, whereas in the remaining 26 ears the deficit was profound. A conductive component was present in 10 of our patients, 19 ears (cases 1– 3, 5, 8 –11, 16 and 17), whereas in 1 patient it was not tested (case 7: the patient was too young). The average of the air-bone gap was calculated between the frequencies of 500-1000-2000 Hz and ranged between 3 and 26.6 dB HL, with a mean value of 15.2 dB HL. Two of our patients (cases 3 and 4) received a cochlear implant in adult life without intraoperative perilymph gusher and with good postoperative outcomes, 14 patients are traditional hearingaid users, and 1 patient (case 6), with a unilateral moderate hearing deficit, does not use hearing aids. We did not find a clear correspondence between volumes of ED/ES complex and the degree of the hearing deficit; we only found that all the patients with ED/ES complex volume greater than 1 mL exhibited a profound hearing loss. The age at which hearing loss was diagnosed varied widely, although it was reached in childhood or adolescence in all patients: from 7 months to 15 years, with a mean age of 3.9 years. It should be underscored that in 8 (cases 7, 9, 10, 12 – 15, and 17) the hearing loss was probably congenital and diagnosed at different ages (13 months in case 7; 366 Case Side of LVA 1. SL (f, 50 y) Bilateral 15 y left, 45 y right Bilateral 5 y Bilateral 3 y Bilateral 6 y Right 7y 2. 3. 4. 5. BI (f, 17 y) BM (f, 43 y) EA (m, 30 y) SG (m, 18 y) 6. PM (m, 8 y) Right Age of PTA (dB HL) hearing loss onset/diagnosis 8y 7. MC (f, 4 y) Bilateral 13 mo (congenital?) 8. IM (f, 30 y) Bilateral 5 y 9. SF (m, 12 y) Bilateral 7 mo (congenital?) 10. DVS Bilateral 2 y (m, 21 y) (congenital?) 11. SP (m, 22 y) Bilateral 3 y 12. DMA Bilateral 2 y (m, 35 y) (congenital?) 13. DMB Bilateral 2 y (f, 33 y) (congenital?) 14. PF (m, 27 y) Bilateral 18 mo (congenital?) 15. PN (f, 30 y) Bilateral 18 mo (congenital?) 16. KI (f, 22 y) Bilateral 4 y 17. DPF (m, 18 y) Bilateral 8 mo (congenital?) R: 47, L: 81 R: R: R: R: 101, L: 105, L: 113, L: 103, L: Conductive component Sudden hearing Fluctuations Progression Vestibular function (average between 500impairment of hearing of hearing 1000-2000 Hz) (dB HL) (after triggers) function loss Syndromic aspects + (R: 6.6, L: 11.6) Nonsyndromic LVA Absent 81 + (R: 25, L: 13.3) 108 + (R: 16.6, L:20) 108 48 + (R: 26.6) + + R: 55, L: normal hearing 98 (COR) Not tested R: 95, L: 100 + (R: 18.3, L: 23.3) R: 103, L: 103 + (R: 15, L: 23.3) R: 103, L: 91 + Left hypofunction + + + + + Bilateral hypofunction Right hypofunction Bilateral hypofunction Normal + + + + + + + + (R: 5, L: 8.3) R: 105, L: 100 + (R: 13.3, L: 18.3) R: 105, L: 108 + + + R: 103, L: 107 Mutations of PDS gene Nonsyndromic LVA Nonsyndromic LVA Pendred Distal renal tubular acidosis Not executed Distal renal tubular acidosis Not executed Distal renal tubular acidosis Left hypofunction (16 y) Pendred syndrome Bilateral hypofunction Pendred syndrome Absent Absent Absent Absent Bilateral hypofunction Pendred syndrome T410M/L465W Bilateral areflexia Bilateral areflexia Pendred syndrome Pendred syndrome G497R/wt T505N7glV1001 + 1A Bilateral areflexia Pendred syndrome T505N7glV1001 + 1A Absent Absent Absent R409H/L465W R: 103, L: 98 + Bilateral hypofunction Pendred syndrome Absent R: 100, L:95 + Bilateral hypofunction Pendred syndrome Absent + Bilateral hypofunction ? (patient previously Not completed submitted to thyroidectomy) Pendred syndrome Not completed R: 96, L: 98 + (R: 18.3, L: 20) R: 92, L: 95 + (R: 3, L: 3.3) + Pure-tone audiometry average is 0.5-1-2 kHz. COR indicates conditioned-oriented responses. R, right; L, left; wt, wild type. Normal S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 – 371 Table 1 Audiological, clinical, and genetic features of our group of patients S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 –371 367 Table 2 Radiological findings in our group of patients and correlations with hearing loss Case Endolimphatic duct/sac complex volume (mL) PTA (dB HL) Association with other inner ear anomalies 1. 2. 3. 4. 5. 6. 7. 8. 9. SL (f, 50 y) BI (f, 17 y) BM (f, 43 y) EA (m, 30 y) SG (m, 18 y) PM (m, 8 y) MC (f, 4 y) IM (f, 30 y) SF (m, 12 y) R: R: R: R: R: R: R: R: R: 0.28, L: 0.3 0.32, L: 0.93 0.21, L: 0.23 0.4, L: 1.55 0.44, L: not detectable 0.68, L: not detectable 0.09, L: 0.78 1.07, L: 0.97 1.01, L: 1.18 R: 47, L: 81 R: 101, L: 81 R: 105, L: 108 R: 113, L: 108 R: 103, L: 48 R: 55, L: normal hearing 98 (COR) R: 95, L: 100 R: 103, L: 103 10. DVS (m, 21 y) 11. SP (m, 22 y) 12. DMA (m, 35 y) 13. DMB (f, 33 y) 14. PF (m, 27 y) 15. PN (f, 30 y) 16. KI (f, 22 y) 17. DPF (m, 18 y) R: R: R: R: R: R: R: R: 0.74, L: 0.72 1.16, L: 1.78 0.76, L: 0.42 0.52, L: 0.6 0.72, L: 0.85 0.65, L: 0.73 0.81, L: 1.64 0.3, L: 0.23 R: R: R: R: R: R: R: R: Absent Absent Absent Absent Absent Absent Absent Absent Dilatation of the vestibule, of the apical turn of the cochlea and of the LSC bilaterally Dilatation of the vestibule bilaterally Absent Mondini deformity bilaterally Mondini deformity bilaterally Absent Absent Absent Dilatation of the vestibule bilaterally 103, L: 91 105, L: 100 105, L: 108 103, L: 107 103, L: 98 100, L:95 96, L: 98 92, L: 95 Pure-tone audiometry average is between 500-1000-2000 Hz. LSC indicates lateral semicircular canal. 7 months in case 9; 2 years in case 10, 12, 13; 18 months in cases 14 and 15; and 8 months in case 17). The hearing loss had a sudden onset in 5 patients (cases 2, 3, 8, 11, and 16), in correspondence with some triggering activity. It was progressive in 12 of 17 patients (cases 1– 8, 11, 14 – 16) and presented fluctuations in 5 of 17 patients (cases 2, 3, 5, 6, and 11). One of the 2 patients with unilateral LVA also presented a hearing deficit in the contralateral ear, which manifested some years later and was less severe than in the affected one (case 5). Among the 15 adult patients who underwent vestibular function tests, only 7 complained of vestibular disturbances, although all but 2 (case 5 and 17) exhibited a marked vestibular deficit when studied with the caloric stimulation test. There was no strict correspondence between the side of the vestibular deficit and the side of enlarged vestibular aqueduct (EVA) in these patients; infarct case 3 with a bilateral LVA presented a right vestibular hypofunction, case 4 with a right LVA presented a bilateral vestibular hypofunction, and case 8 with a bilateral LVA presented a left vestibular hypofunction. Of the 17 patients, 10 were clinically affected by PS; all of them presented goiter and positive perchlorate discharge test, and 8 of 10 presented a mild hypothyroidism, whereas 2 of 10 had a normal thyroid function. In these patients, serum anti-Tg, anti-TPO, and anti–TSH-receptor antibodies were undetectable, whereas in all of them serum Tg levels were increased. Another patient (case 16) had previously undergone thyroidectomy for goiter abroad, and the functional evaluation of the thyroid and the perchlorate discharge test could not be performed. Six patients lacked any pathological alterations of the thyroid: of these, 3 patients presented LVAS associated with dRTA (one of these patients, case 7, was not submitted to perchlorate discharge test, because of very young age), whereas the other 3 showed a nonsyndromic hearing loss associated with LVA. Five of the 10 patients with PS had mutations in the PDS gene. We identified 2 novel mutations (G497R and L465W) in the exon 13 nucleotide 1489 GYC and the exon 11 nucleotide 1226 G YA [34]. The other mutations had already been reported (T410M, R409H, T508N, guanine IVS8 1001 + 1 adenine) [33,35]. Four patients presented compound heterozygosis (cases 9, 10, 12, 13), whereas 1 patient (case 11) had a single heterozygous mutation (G497R). The other patients with a clinical diagnosis of PS did not show any mutation of the PDS gene, whereas molecular analysis of the PDS gene is not completed yet in 1 patient with a clinical diagnosis of PS (case 17) and in the patient who previously underwent thyroidectomy (case 16). No evidence of mutations of the PDS gene was found in the 3 patients with EVA syndrome associated with dRTA or the 3 patients with nonsyndromic LVAS. 4. Discussion Enlarged vestibular aqueduct syndrome is described as the most common imaging finding in individuals with SNHL dating to infancy or childhood [12,36]. The earliest description of the EVA syndrome was made by Mondini in 1971 [37], whereas it was Valvassori and Clemis [15] who, using polytomography, first described the association between EVA and SNHL in 50 cases and coined the term LVAS. They defined the large vestibular aqueduct as having a large antero-posterior diameter (N 1.5 mm), often associated with profound, nonprogressive sensorineural hearing loss [15]. Large vestibular aqueduct syndrome has been reported to be bilateral in 55% to 94% of cases [6,12,38]; some authors claim a slight female preponderance [6,12], whereas others report the inverse [38]. In our group, LVA was found to be bilateral in 15 of 17 cases (88%). The audiological features of SNHL in patients affected by LVAS have been described by many authors: SNHL may 368 S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 – 371 be mild to profound, and patients affected by LVAS without hearing impairment have also been reported [2,8,9,14,39]. In a variable percentage of cases (11%-65%), it has been described to be fluctuant and progressive [1,3-5,7,39], often with sudden, stepwise onset or progression (50% of cases, according to Walsh et al [6]), secondary to trigger activities such as those involving the Valsalva maneuver, minor head trauma, scuba diving, jogging, common colds, and so forth [1-9]. When bilateral, the hearing deficit is generally reported to be asymmetrical [9]. Sensorineural hearing loss onset may be from birth to adolescence, with the highest frequency in childhood [3-5,7,9]. In our group of LVA patients, the degree of the hearing deficit varied among the patients: it was moderate to profound. In accord with the literature data, the hearing loss onset or diagnosis has been dated to childhood in all our patients but one (case 1), in whom SNHL started during adolescence. In 5 (29.4%) of 17 cases, the hearing deficit typically started suddenly after some characteristic triggering event. It was progressive in 12 (70.5%) of 17 patients and presented fluctuations in 5 (29.4%) of 17 patients. It is worth noting that all the patients with nonprogressive hearing deficit were those whose hearing loss was already severe or profound at the moment of diagnosis, dated in each one to the prelingual period (cases 9, 10, 12, 13, and 17). In such patients at so early an age, it is difficult to establish hearing loss progression. Some authors mention a conductive component in a minority of their patients (17 –38%), probably caused by decreased mobility of the stapes, due in turn to increased perilymphatic or endolymphatic pressure [3,5,6,8,9,12,15]. Ten of our patients (59%) presented a conductive component, a percentage that is slightly high in comparison with data in the literature. The presence of an air-bone gap in patients with EVA syndrome could be the reason why such subjects often present better hearing and communicative results with conventional hearing aids than expected, given the degree of hearing loss. In fact, some of our patients with profound bilateral hearing loss (7 –11 and 16) exhibited satisfactory hearing outcomes with conventional hearing aids, despite the degree of the hearing deficit, whereas 2 of them (patients 3 and 4) received a cochlear implant, without any surgical problem and presenting satisfactory postoperative outcomes, similar to those of patients with normal inner ears; so according to literature data, cochlear implantation is to be considered a viable option for such patients [40-45]. Interestingly, one of the 2 patients with unilateral LVA subsequently developed a progressive hearing deficit in the radiologically normal ear; the hearing deficit in the morphologically normal ear had a later onset and was less severe than in the ear with EVA. This is a novel finding, as the literature contains no reports of cases of unilateral LVA at CT and above all unilateral enlarged endolymphatic duct and enlarged endolymphatic sac (EES) at MRI, with a bilateral hearing deficit; in 1997, Dahlen et al [46] reported on a patient with bilateral SNHL and unilateral LVA at CT scan, in which MRI disclosed the presence of an EES in the CT-normal ear. We can suppose that in such cases the development of an abnormally widened VA is an epiphenomenon of some endolymphatic hydro-electrolytic disorder, which is probably the cause of both the hearing deficit and enlargement of the VA and ES. Vestibular symptoms are reported in less than one third of cases and range from severe episodic vertigo to occasional unsteadiness in adults, whereas incoordination and imbalance predominate in children [24]. Vestibular hypofunction seems to be more frequent [1,3,6,39,47,48], whereas additional otologic symptoms are variable and nonspecific, such as tinnitus and occasional aural fullness [24]. Seven of our patients complained of vestibular disturbances, despite the marked vestibular deficit found in 13 of the 15 patients in whom vestibular function was studied. Diagnosis of LVA is radiological. Computed tomography scan shows the bony labyrinth anatomy, and an axial CT with 1.5-mm sections generally provides the best view of the VA from the vestibule to the posterior surface of the petrous bone, so the VA diameter can be measured midway between the outer aperture and common crus [38,46,49-52]. Magnetic resonance imaging discloses the fluid-filled spaces of the inner ear, and the high contrast of fluid in the membranous labyrinth, especially on T2-weighted images, allows for visualization of the membranous labyrinth with exquisite detail [4,31]. Moreover, MRI is the only imaging technique that enables visualization of the extra-osseous portion of the ES. Three-dimensional reconstructions from MRI data sets are often helpful to detect the sac and other inner ear structures and to better define their morphological features [4,31], so that MRI is considered superior to CT in LVAS evaluation by some authors [49,50]. Not all authors agree on the criteria for defining the vestibular aqueduct as large, but the most accepted criterion is that suggested by Valvassori and Clemis [15]: a vestibular aqueduct is considered large whenever its antero-posterior diameter is 1.5 mm or more, as measured via CT scans midway between the outer aperture and common crus [3,7,15,46,50]. Enlargement of the endolymphatic duct-sac complex is also measured at the midpoint between the posterior margin of the petrous bone and the common crus; it is difficult to measure consistently because of the variability of the extracanalicular portion of the ES [38]. Literature reports of correlations between CT and MRI findings are inconsistent: cases with LVA at CT and normal inner ear at MRI have been reported, as have cases of EES at MRI with normal bony VA at CT [19,46,49]. In our study group, we found a strict correspondence between CT and MRI in those cases for which both had been performed: in fact, the presence of enlarged ED and ED detected via MRI was confirmed by CT in every case it was performed. Literature reports generally describe a correlation between the side of the more enlarged VA and ES, and the side of the more marked SNHL, and conclude that a markedly widened VA is usually associated with profound hearing S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 –371 loss [4,46,49,53], although a strict correlation between the size of the VA and ES and the degree of hearing deficit has not been established [3,4,7,46,53]. In our patients, we could not observe a strict correlation between the size of the ED/ ES complex and the degree of the hearing deficit; at any rate, each of our patients who presented a very large ED/ES complex volumes ( N1 mL) exhibited also a profound hearing deficit. In 2002, Naganawa et al [54] performed serial MRI examinations in 2 patients with bilateral LVAS and unilateral fluctuations of hearing before and during periods of hearing loss exacerbation and found that EES volumes and signal intensity varied dynamically in both patients, independent of degree of hearing loss. In this study, we performed serial HR-MRI in 6 patients (4 with bilateral and 2 with unilateral LVA): in 5 patients the hearing loss was stable, whereas in 1 patient with a bilateral LVA, the hearing threshold worsened in one ear during the period between the first and the second MRI investigation (case 1). In none of these 6 patients did we find any changes in the volume of the ED/ES complex in the serial HR-MR. Therefore, also in our series it seems that hearing loss progression is not correlated with ED and ES volumes. Generally, LVAS is nonfamilial. A genetic predisposition is evident in a portion of patients affected by LVAS [3-5, 7,24]. Most reported cases are sporadic single cases, but, recently, familial cases have been reported, and some authors suggest an autosomal recessive inheritance in families with members affected by LVAS [4,5,8,24]. Large vestibular aqueduct syndrome may be associated with both nonsyndromic and syndromic forms of SNHL, such as PS [18,19], branchiootorenal syndrome [22,55], and, recently, we have described an association of LVAS with dRTA [23,56]. The relationship between LVAS and the PS has recently stimulated much attention, because it is so common. In fact, the PS is the most frequent form of syndromic SNHL. Pendred’s syndrome is an autosomal recessive disease characterized by goiter, sensorineural deafness, and defective iodide organification. Sensorineural hearing loss is generally prelingual and frequently associated with a wide range of morphological inner ear anomalies, such as the Mondini deformity, LVA, and others [18,57-60]. Recently, the presence of LVA in association with a widened endolymphatic duct and sac has been described as a constant feature of the PS inner ear [19,20,57,61,62]. In 1996, the gene responsible for PS, the PDS gene (the pendrin gene), was mapped to chromosome 7q31 and has subsequently been cloned [27]. Numerous PDS mutations have now been identified in affected patients, suggesting a wide allelic heterogeneity [63-68]. The PDS gene encompasses 21 exons, contains 2343-bp open reading frame, and encodes for a 780–amino acid protein with 11 putative transmembrane domains. The pendrin gene appears to function as an iodidechloride transporter and is expressed at the level of the apical pole of thyrocytes of the distal nephron [69]. Moreover, it has recently been demonstrated to be expressed at the level of 369 the fetal and adult mouse inner ear, in regions involved in the regulation of the endolymphatic fluid composition, such as the endolymphatic duct and sac, distinct areas of the utricle and saccule, and the external sulcus region within the cochlea [70,71]. These observations may be important to our understanding of the mechanisms of inner ear pathology in patients with PDS mutations: abnormal endolymphatic hydro-electrolyte balance and osmotic pressure may be the underlying causes of an abnormal development of the VA, ED, and ES, their enlargement and consequent SNHL. Of these 17 patients affected with LVA, 10 were affected by PS, 3 by LVAS associated with dRTA, and, finally, 3 by nonsyndromic SNHL associated with LVAS, whereas in 1 patient, the one previously submitted to thyroidectomy, it was not possible to study thyroid function nor to perform perchlorate discharge test. All the patients with the nonsyndromic form of LVAS and with LVAS associated with dRTA were single sporadic cases, whereas 7 of the 10 patients with PS were sporadic cases, and the other 4 were 2 pairs of siblings. In the 10 patients affected with PS, clinical involvement of the thyroid was highly variable, and in 1 patient (case 9) it was discovered only after evidence of inner ear malformation was found. The mild degree of thyroid symptoms, so frequently reported in patients with PS, could be the reason why the presence of PS goes undiagnosed in many cases of EVA. In our study group, we were able to find PDS mutations only in patients affected with PS and not in any of the patients with nonsyndromic LVA or in those with LVA associated with dRTA; moreover, 4 of the patients with PS did not present PDS mutations, whereas in 2 patients the molecular analysis has not been completed yet (cases 16 and 17). The 4 patients with PS without PDS mutation had an indistinguishable phenotype from that of patients with PS and PDS mutations. We must suppose that in the patients without PDS mutations the development of EVA, both syndromal (PS) or nonsyndromal, could be due to other factors apart from PDS mutations, such as environmental factors or other genetic dysfunctions. The literature contains some reports of cases of PS with LVA and nonsyndromic LVA in which no PDS gene mutations were found: in these cases, the presence of mutations at the level of intronic or regulatory sequences of the PDS gene, as well as the involvement of other genes, has been supposed [26,28]. Moreover, the role of a single PDS mutation in our patient with PS who was heterozygous for PDS mutations is unclear. Cases of nonsyndromic LVA or PS and heterozygosis for PDS mutations have been reported in the literature, and in such cases mutations of intronic or regulatory sequences of the PDS gene have also been supposed to be involved [26,28]. In conclusion, we wish to emphasize the frequent role of LVAS in the pathogenesis of SNHL and the overall wide variability in its audiological features. In every case of SNHL of unknown origin, the possible presence of LVA should be 370 S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 – 371 investigated via inner ear HR-MRI, especially if dating back to infancy or childhood. Moreover, it should be underscored that LVAS is often part of some syndromic diseases, most of which are PS, which is often misdiagnosed because of the varying degree of thyroid symptoms. Thyroid function and perchlorate discharge tests should be performed on every case of LVAS, together with molecular analysis of the PDS gene, if available. Furthermore, we would like to underscore the role of hydro-electrolyte and acid-base endolymphatic fluid disorders in the pathogenesis of EVA syndrome, as documented by recent studies on PDS expression in the inner ear [70,71] and the association between LVAS and dRTA, which in its recessive form is related to the alteration of the B1 subunit of H+-ATPase (ATP6B1), found in high concentrations in both the distal nephron and the inner ear epithelia [55]. In fact, PDS mutations cause an alteration of iodide-chloride transport (pendrin), and in recessive dRTA, ATP6B1 mutations cause an alteration of a proton pump as well. As they are expressed in the inner ear, both could theoretically lead to hydro-electrolyte and acid-base imbalance of inner ear fluids. The exact mechanism leading to the development of LVA, enlarged endolymphatic duct, and EES is unclear, but future elucidation of this process would furnish an important contribution to our understanding of, and ability to treat, LVAS, and, consequently, SNHL associated with LVAS. References [1] Jackler RK, De La Cruz A. The large vestibular aqueduct syndrome. Laryngoscope 1989;99:1238 - 43. [2] Levenson MJ, Parisier SC, Jacobs M, et al. The large vestibular aqueduct syndrome in children. Arch Otolaryngol Head Neck Surg 1989;115:54 - 8. [3] Irving RM, Jackler RK. Large vestibular aqueduct syndrome. Curr Opin Otolaryngol Head Neck Surg 1997;5:267 - 71. [4] Tong KA, Harsenberger HR, Dahlen RT, et al. Large vestibular aqueduct syndrome: a genetic disease? AJR Am J Roentgenol 1997;168:1097 - 101. [5] Abe S, Usami S, Shinkawa H, et al. Three familial cases of hearing loss associated with enlargement of the vestibular aqueduct. Ann Otol Rhinol Laryngol 1997;106:1063 - 9. [6] Walsh RM, Ayshford CA, Chavda SV, et al. Large vestibular aqueduct syndrome. ORL J Otorhinolaryngol Relat Spec 1999;61:41 - 4. [7] Nowak KC, Messner AH. Isolated large vestibular aqueduct syndrome in a family. Ann Otol Rhinol Laryngol 2000;109:40 - 4. [8] Satoh H, Nonomura N, Takahashi S. Four cases of familial hearing loss with large vestibular aqueducts. Eur Arch Otorhinolaryngol 1999;256:83 - 6. [9] Govaerts PJ, Casselman J, Daemers K, et al. Audiological findings in large vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol 1999;51:157 - 64. [10] Okumura T, Takahashi H, Honjo I, et al. Sensorineural hearing loss in patients with large vestibular aqueduct. Laryngoscope 1995;105: 289 - 93. [11] Belenky WM, Madgy DN, Leider JS, et al. The enlarged vestibular aqueduct syndrome (EVA syndrome). Ear Nose Throat J 1993;72: 746 - 51. [12] Puls T, Van Fraeyenhoven L. Large vestibular aqueduct syndrome with mixed hearing loss: a case report. Acta Otorhinolaryngol Belg 1997;51:185 - 9. [13] Callison DM, Horn KL. Large vestibular aqueduct syndrome: an overlooked etiology for progressive childhood hearing loss. J Am Acad Audiol 1998;9:285 - 91. [14] Emmet JR. The large vestibular aqueduct syndrome. Am J Otolaryngol 1985;6:387 - 415. [15] Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope 1978;88:723 - 8. [16] Abe S, Usami S, Hoover DM, et al. Fluctuating sensorineural hearing loss associated with enlarged vestibular aqueduct maps to 7q31, the region containing the Pendred gene. Am J Med Genet 1999;82:322 - 8. [17] Pendred V. Deaf-mutism and goitre. Lancet 1896;II:532. [18] Cremers CWRJ, Bolder C, Admiraal RJC, et al. Progressive sensorineural hearing loss and a widened vestibular aqueduct in Pendred syndrome. Arch Otolaryngol Head Neck Surg 1998;124: 501 - 5. [19] Phelps PD, Mahoney CFO, Luxon LM. Large endolymphatic sac. A congenital deformity of the inner ear shown by magnetic resonance imaging. J Laryngol Otol 1997;111:754 - 6. [20] Fugazzola L, Mannavola D, Cerutti N, et al. Molecular analysis of the Pendred’s syndrome gene and magnetic resonance imaging studies of the inner ear are essential for the diagnosis of true Pendred’s syndrome. J Clin Endocrinol Metab 2000;85:2469 - 75. [21] Scott DA, Wang R, Kreman TM, et al. Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet 2000;9:1709 - 15. [22] Stinckens C, Standaert L, Casselman JW, et al. The presence of a widened vestibular aqueduct and progressive sensorineural hearing loss in the branchio-oto-renal syndrome. A family study. Int J Pediatr Otorhinolaryngol 2001;59:163 - 72. [23] Berrettini S, Forli F, Sellari Franceschini S, et al. Distal renal tubular acidosis associated with isolated large vestibular aqueduct and sensorineural hearing loss. Ann Otol Rhinol Laryngol 2002;111: 385 - 91. [24] Griffith AJ, Arts HA, Downs C, et al. Familial large vestibular aqueduct syndrome. Laryngoscope 1996;106:960 - 5. [25] Sheffield VC, Kraiem Z, Beck JC, et al. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification. Nat Genet 1996;12:424 - 6. [26] Coyle B, Coffey R, Armour JAL, et al. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4. Nat Genet 1996;12:421 - 3. [27] Everett LA, Glaser B, Beck JC, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 1997;17:411 - 22. [28] Usami S, Abe S, Weston MK, et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet 1999;104:188 - 92. [29] Stephens D. Audiological terms. Definitions, protocols and guidelines in genetic hearing impairment. London7 Whurr Publishers Ltd; 2001. p. 9 - 14. [30] Fitzgerald G, Hallpike CS. Studies in the human vestibular function: observations on the directional preponderance of caloric nystagmus resulting from cerebral lesions. Brain 1942;65:115 - 37. [31] Neri E, Caramella D, Cosottini M, et al. High-resolution magnetic resonance and volume rendering of the labyrinth. Eur Radiol 2000;10:114 - 8. [32] Neri E, Caramella D, Panconi M, et al. Virtual endoscopy of the middle ear. Eur Radiol 2001;11:41 - 9. [33] Bogazzi F, Raggi F, Ultimieri F, et al. A novel mutation in the pendrin gene associated with Pendred’s syndrome. Clin Endocrinol 2000;52: 279 - 85. [34] Bogazzi F, Russo D, Raggi F, et al. Mutations in the SLC26A4 (pendrin) gene in patients with sensorineural deafness and enlarged vestibular aqueduct. J Endocrinol Invest 2004;27(5):430 - 5. S. Berrettini et al. / American Journal of Otolaryngology – Head and Neck Medicine and Surgery 26 (2005) 363 –371 [35] Lopez-Bigas N, Rabionet R, de Cid R, et al. Splice-site mutation in PDS gene may result in intrafamilial variability for deafness in Pendred syndrome. Hum Mutat 1999;14:520 - 6. [36] Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope 1987;97(Suppl 40):2 - 14. [37] Mondini C. Caroli Mundini Anatomica surdi nati sectio. De bononiensi scientiarum et artium. Instituto atque Academia commentarii. 1791:419 - 31. [38] Reussner LA, Dutcher PO, House WF. Large vestibular aqueduct syndrome with massive endolymphatic sacs. Otolaryngol Head Neck Surg 1995;113:606 - 10. [39] Yetiser S, Kertmen M, Özkaptan Y. Vestibular disturbance in patients with large vestibular aqueduct syndrome (LVAS). Acta Otolaryngol (Stockh) 1999;119:641 - 6. [40] Aschendorff A, Marangos N, Laszig R. Large vestibular aqueduct syndrome and its implication for cochlear implant surgery. Am J Otolaryngol 1997;18:S57. [41] Au G, Gibson W. Cochlear implantation in children with large vestibular aqueduct syndrome. Am J Otolaryngol 1999;20:183 - 6. [42] Bent JP, Chute P, Parisier SC. Cochlear implantation in children with enlarged vestibular aqueducts. Laryngoscope 1999;109:1019 - 22. [43] Miyamoto RT, Bichey BG, Wynne MK, et al. Cochlear implantation with large vestibular aqueduct syndrome. Laryngoscope 2002;112: 1178 - 82. [44] Bichey BG, Hoversland JM, Wynne MK, et al. Changes in quality of life and the cost-utility associated with cochlear implantation in patients with large vestibular aqueduct syndrome. Otol Neurotol 2002;23:323 - 7. [45] Vescan A, Parnes L, Cucci RA, et al. Cochlear implantation and Pendred’s syndrome mutation in monozygotic twins with large vestibular aqueduct syndrome. J Otolaryngol 2002;31:54 - 7. [46] Dahlen RT, Harnsberger HR, Gray SD, et al. Overlapping thin section fast spin echo MR of the large vestibular aqueduct syndrome: comparison with CT. Am J Neuroradiol 1997;18:67 - 75. [47] Schessel DA, Nedzelski JM. Presentation of large vestibular aqueduct syndrome to a dizziness unit. J Otolaryngol 1992;21:265 - 9. [48] Okumura T, Takahashi H, Honjo I, et al. Vestibular function in patients with large vestibular aqueduct. Acta Otolaryngol (Stockh) Suppl 1995;520:323 - 6. [49] Harnsberger HR, Dahlen RT, Shelton C, et al. Advanced techniques in magnetic resonance imaging in the evaluation of the large endolymphatic duct and sac syndrome. Laryngoscope 1995;105:1037 - 42. [50] Okamoto K, Ito J, Furusawa T, et al. Large vestibular aqueduct syndrome with high CT density and high MR signal intensity. Am J Neroradiol 1997;18:482 - 4. [51] Mafee MF, Charletta D, Kumar A, et al. Large vestibular aqueduct and congenital sensorineural hearing loss. Am J Neuroradiol 1982;13: 805 - 19. [52] Schroeder AA, Kuhn J. Large vestibular aqueduct syndrome. Am J Otolaryngol 2000;21:433 - 4. [53] Okamoto K, Ito J, Furusawa T, et al. MRI of enlarged endolymphatic sacs in the large vestibular aqueduct syndrome. Neuroradiology 1998; 40:167 - 72. 371 [54] Naganawa S, Koshikawa T, Fukatsu H, et al. Serial MR imaging studies in enlarged endolymphatic duct and sac syndrome. Eur Radiol 2002;12:S114 - 7. [55] Chen A, Francis M, Ni L, et al. Phenotypic manifestations of branchiootorenal syndrome. Am J Med Genet 1995;58:365 - 70. [56] Karet FE, Finberg KE, Nelson RD, et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 1999;21:84 - 90. [57] Cremers CWRJ, Admiraal RJC, Huygen PLM, et al. Progressive hearing loss, hypoplasia of the cochlea and widened vestibular aqueducts are very common features in Pendred’s syndrome. Int J Pediatr Otorhinolaryngol 1998;45:113 - 23. [58] Reardon W, Coffey R, Phelps PD, et al. Pendred syndrome — 100 years of underascertainment? Q J Med 1997;90:443 - 7. [59] Reardon W, Coffey R, Chowdhury T, et al. Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet 1999;36:595 - 8. [60] Iwasaki S, Usami S, Abe S, et al. Long-term audiological feature in Pendred syndrome caused by PDS mutation. Arch Otolaryngol Head Neck Surg 2001;127:705 - 8. [61] Reardon W, O Mahoney CF, Trembath R, et al. Enlarged vestibular aqueduct: a radiological marker of Pendred syndrome, and mutation of the PDS gene. Q J Med 2000;93:99 - 104. [62] Masmoudi S, Charfedine I, Hmani M, et al. Pendred syndrome: phenotypic variability in two families carrying the same PDS missense mutation. Am J Med Genet 2000;90:38 - 44. [63] Fujita S, Sando I. Three-dimensional course of the vestibular aqueduct. Eur Arch Otorhinolaryngol 1996;253:122 - 5. [64] Greinwald Jr JH, Wayne S, Chen AH, et al. Localization of a novel gene for nonsyndromic hearing loss (DFNB17) to chromosome region 7q31. Am J Med Genet 1998;78:107 - 13. [65] Li XC, Everett LA, Lalwani AK, et al. A mutation in PDS causes nonsyndromic recessive deafness. Nat Genet 1998;18:215 - 7. [66] Ishinaga H, Shimizu T, Yuta A, et al. Pendred’s syndrome with goiter and enlarged vestibular aqueducts diagnosed by Pds gene mutation. Head Neck 2002;24:710 - 3. [67] Lopez-Bigas N, Melchionda S, de Cid R, et al. Identification of five new mutations of PDS/SLC26A4 in Mediterranean families with hearing impairment. Hum Mutat 2001;16:1 - 4. [68] Kitamura K, Takahashi K, Noguchi Y, et al. Mutations of the Pendred syndrome gene (PDS) in patients with large vestibular aqueduct. Acta Otolaryngol 2000;120:137 - 41. [69] Royaux IE, Suzuki K, Mori A, et al. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 2000;141:839 - 45. [70] Everett LA, Morsli H, Wu DK, et al. Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A 1999;96: 9727 - 32. [71] Everett LA, Belyantseva IA, Noben-Trauth K, et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 2001; 10:153 - 61.