Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
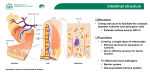
Diabetes & Metabolism 35 (2009) 262–272 Review Intestinal microflora and metabolic diseases M. Serino a,b,∗ , E. Luche a,b , C. Chabo a,b , J. Amar c , R. Burcelin a,b,∗ b a U858, Institut national de la santé et de la recherche médicale (Inserm), Toulouse, France IFR31, institut de médecine moléculaire de Rangueil (I2MR), université de Toulouse Paul-Sabatier, 1, avenue Jean-Poulhes, 31432 Toulouse cedex 4, France c Rangueil Hospital, Department of Therapeutics, Toulouse, France Received 12 March 2009; accepted 15 March 2009 Available online 5 May 2009 Abstract Recent advances in molecular sequencing technology have allowed researchers to answer major questions regarding the relationship between a vast genomic diversity—such as found in the intestinal microflora—and host physiology. Over the past few years, it has been established that, in obesity, type 1 diabetes and Crohn’s disease—to cite but a few—the intestinal microflora play a pathophysiological role and can induce, transfer or prevent the outcome of such conditions. A few of the molecular vectors responsible for this regulatory role have been determined. Some are related to control of the immune, vascular, endocrine and nervous systems located in the intestines. However, more important is the fact that the intestinal microflora-to-host relationship is bidirectional, with evidence of an impact of the host genome on the intestinal microbiome. This means that the ecology shared by the host and gut microflora should now be considered a new player that can be manipulated, using pharmacological and nutritional approaches, to control physiological functions and pathological outcomes. What now remains is to demonstrate the molecular connection between the intestinal microflora and metabolic diseases. We propose here that the proinflammatory lipopolysaccharides play a causal role in the onset of metabolic disorders. © 2009 Elsevier Masson SAS. All rights reserved. Keywords: Diabetes; Obesity; Metagenome; Inflammation; Lipopolysaccharides; Review Résumé Flore intestinale et maladies métaboliques. Les progrès récents des méthodes de séquençage à ultrahaut débit ont permis de répondre à des questions fondamentales concernant la relation entre la flore intestinale, vecteur de la plus grande diversité génétique de notre organisme, et la physiologie de l’hôte. Il est maintenant établi qu’au cours de l’obésité, du diabète de type 1 et de la maladie de Crohn, pour n’en citer que quelques unes, la flore intestinale joue un rôle important. Ce microbiote peut induire, transférer ou prévenir le développement de ces maladies. Certains acteurs moléculaires responsables de ces effets ont été identifiés et seraient relatifs au contrôle du système immunitaire, du développement vasculaire ou de fonctions endocrines et nerveuses principalement et initialement localisées dans l’intestin. Il faut également considérer que la relation entre l’hôte et la flore intestinale est bidirectionnelle. En effet, certaines données démontrent l’impact de l’hôte sur la flore intestinale. Désormais, une écologie mutualisée entre la flore intestinale et l’hôte doit être considérée comme un nouvel acteur de la physiologie et physiopathologie. Cet acteur peut être manipulé par des approches pharmacologiques et nutritionnelles afin de contrôler la physiologie humaine et ses dysfonctionnements. Les mécanismes moléculaires doivent encore être démontrés. Nous proposons dans cette revue que les lipopolysaccharides bactériens, hautement inflammatogènes, sont responsables de l’initiation du développement des maladies métaboliques. © 2009 Elsevier Masson SAS. Tous droits réservés. Mots clés : Diabète ; Obésité ; Métagénome ; Inflammation ; Lipopolysaccharides ; Revue 1. Introduction ∗ Corresponding authors. E-mail addresses: [email protected] (M. Serino), [email protected] (R. Burcelin). 1262-3636/$ – see front matter © 2009 Elsevier Masson SAS. All rights reserved. doi:10.1016/j.diabet.2009.03.003 Our human microflora are an inherited feature resulting from our evolution. With the celebration of 200 years since the birth of Charles Darwin, it makes sense to consider that the development M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 of human physiology could be the consequence of environmental factors such as microflora, which might even be thought of as a selection factor among humans. Indeed, the selection of the species comprising our microflora today could be considered to be the result of evolution, with some strains selected on the basis of our evolving genome. This means that the host–microflora relationship could be the result of a bidirectional selection process that has now established a shared ecology among the different members of the microflora and the host. However, the molecular bases of this ecology and its interaction with the host need deciphering if we are to have access to the potentially vast new spectrum of strategies for the management of human physiology and treatment of diseases. Cancer and the inflammatory and metabolic diseases are among the first in line for the use of microflora-based therapeutic strategies. Given the wide diversity of our entire body’s microflora, this review focuses on the intestinal microflora, and its relationship with metabolic diseases and other related disorders. The choice of this narrow field of discussion is driven by the intuitive concept that feeding our body is also feeding our intestinal flora. Consequently, both the genome and metagenome contribute to the subtle balance of mechanisms involved in metabolism. We discuss here some of the most recent findings on the molecular mechanisms through which the intestinal microflora influence metabolic diseases. 2. Intestinal microflora: a mutual ecology The intestine is inhabited by trillions of microorganisms, including bacteria, fungi and the Archaea. Two sets of microorganisms have been described. In one, the tasks performed by some members are partially known and are most likely beneficial to the host. However, the vast majority of species are neither beneficial nor harmful to human physiology, and their function remains completely unknown. These species are termed “normal flora”, or “microbiota”. It is estimated that 500 to 1000 different species of bacteria live in the human body [1]. The precise composition of the intestinal microflora varies among individuals and animals, with evidence to suggest that its establishment occurs during the first year of life [2], and that host and external environmental factors, as well as changes in the gut environment and in the microbiota itself, can trigger changes from childhood to adulthood [3,4]. The intestinal colonization that follows birth represents the host’s earliest contact with microbes [3]. Indeed, microbes from the mother and surrounding environment colonize the gastrointestinal tract of the infant until the dense, complex microbiota have developed. The succession of microbes colonizing the intestinal tract is most marked during early development, when the feeding mode shifts from breast- and formula-feeding to weaning and the introduction of solid food. In infants, the microbiota develops rapidly and remains unstable during the first year of life, but becomes more stable after that. This initial stage of microbiota establishment could be the key modulating moment in the establishment of a healthy microbiota in the individual. It is thought that food is one of the major determinants of the intestinal microbiota and, therefore, has important consequences for the development 263 of adult physiology and, hence, pathology. The indigenous gut microbiotas are considered to be among the first stimuli to trigger mechanisms leading to the generation of immunophysiological regulation [3,5,6]. They provide crucial signals for the development of the immune system in infancy and set the immunological “tone” in adulthood. This finding has also been observed in germ-free mice, where oral administration of ovalbumin antigen in neonate mice blocked the inflammatory Th1-mediated response, which usually triggers the production of interferon (IFN)-gamma and IgG2a, in favour of a Th2 tolerogenic response, characterized by IgE, IgG1 and interleukin (IL)-4 in the adult mice [7]. Part of the intestinal mucosal barrier function is formed by a common mucosal immune system that communicates with the various mucosal surfaces of the body. Local T-cell immunity is an important aspect of the specific intestinal immune system [8]. T-cell reactivity is programmed during its initial stages of activation by mucosal dendritic cells, which are thought to play key roles in regulating immune responses in the antigen-rich gastrointestinal environment [8,9]. Defects in this regulatory system are believed to lead to the two main forms of inflammatory bowel disease (IBD)—Crohn’s disease (CD) and ulcerative colitis (UC) [9,10]. This means that the programming of dendritic cell function by intestinal microflora might be involved in the occurrence of inflammatory diseases [8]. A detailed description of the role played by the intestinal microflora in the immune system is beyond the scope of this review. In brief, the first intuitive function attributed to the intestinal microflora is their capacity to break down certain nutrients, such as plant carbohydrates, that the host would otherwise be unable to digest [11], thus allowing the host to recover more energy from ingested food and, hence, better development. Another function is the synthesis of bioactive molecules, such as phytoestrogens [12], that are able to deeply interact with human physiology [13] (Fig. 1). Similarly, the microflora could benefit from the intestinal environmental conditions of a healthy host, ensuring longerlasting dissemination. This idea has been described for many years and is now well established [14]. Some bacteria carry out fermentation of the complex, indigestible carbohydrates of plants, and synthesize vitamins such as folic acid, vitamin K and biotin [15,16]. Other beneficial bacteria in the normal flora include the Lactobacillus species, which convert milk protein to lactic acid in the gut [17]. The presence of such bacterial colonies also inhibits the growth of potentially pathogenic bacteria (usually through competitive exclusion), which is why some of these “good” bacteria are sold as probiotic dietary supplements [18]. This shows that a mutually beneficial ecology is being shared among different members of the microflora and the host. Furthermore, some bacterial functions are involved in host functions such as intestinal epithelial turnover, immune modulation, drug metabolism, gastrointestinal tract motility regulation, mucosal barrier fortification, angiogenesis, intestinal homoeostasis and postnatal intestinal maturation [19–25]. The vast majority of these commensal bacteria are anaerobic, able to survive and grow in an environment lacking oxygen, and some normal flora bacteria can act as opportunistic pathogens in 264 M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 Fig. 1. Intestinal microflora ecology and its functions. The human intestine harbours roughly 1014 microorganisms, comprising bacteria, fungi and protozoa. Bacteria represent the majority of the gut flora, accounting for 60% of the total. The gut microflora has multiple functions, as represented here. cases of lowered immunity. Escherichia coli, for example, lives in the colon and, as an extensively studied model organism, is probably the best understood bacterial species [26]. Mutated strains of this gut commensal, such as E. coli O157:H7, can cause disease. However, it remains “dormant” in a healthy host. Microflora composition varies along the gastrointestinal tract (from the stomach to the small and large intestines), according to factors such as nutrient availability, pH and oxygen concentrations. The bacterial count increases from the upper to the lower parts of the gastrointestinal tract, ranging from 102 colony-forming units (cfu)/mL in the stomach to 1012 cfu/mL in the colon, with species–specific metabolic characteristics that vary according to their localization. The upper portion of the gastrointestinal tract is mostly colonized by aerobic bacteria, while the lower portion is colonized by anaerobic species; this is because oxygen concentration decreases from the duodenum to the colon. The pH also influences the numbers of bacteria found in the different parts of the gastrointestinal tract: the stomach has the smallest number due to its low pH of 1–2, whereas the pH increases within the small and large intestines from 5 (colon) to 7. Our understanding of the complex gut microflora ecology (Fig. 1) has been improved by molecular biology tools that extend the study of microorganisms to those that are not cultivable [27–29]. Sequencing of the gene encoding for 16S ribosomal RNA in nucleic acids extracted from faeces or other luminal samples is a useful tool for the phylogenetic identification of bacteria [30]. Using this molecular approach, it is now possible to study the complexity of all genes of the gut microflora, an analysis termed “metagenomics”. This approach provides a better understanding of the entire genome of the intestinal microflora, including the relationship between the commensal and pathogenic strains. The human colonic microbiota is composed of Bacteroidetes (Bacteroides species) and Firmicutes (Clostridium, Lactobacillus, Enterococcus), which together account for more than 90% of the gut bacterial phyla [29]. Molecular technologies such as fluorescent in situ hybridization (FISH) and DNA microarray chips allow the identification of specific bacterial species. However, besides a qualitative analysis, there is also the possibility of designing strain-specific real-time polymerase chain reaction (PCR) primers for performing quantitative analyses [31]. The advent of these molecular methods has generated enough data to demonstrate that, in a given individual, the composition of the intestinal microflora remains virtually the same throughout life, with no dramatic changes [32]. It has been suggested that this stability is conferred by the intestinal immune system long-term exposure to microbial antigens from childhood to adulthood, allowing it to identify bacterial antigens as normal [19]. 3. Intestinal microflora and inflammatory diseases The role of the intestinal microflora has been largely described in inflammatory diseases such as IBD, CD and UC. Although beyond the scope of this review, some concepts are helpful for understanding the origin of metabolic disorders. Indeed, it is now thought that obesity and diabetes are associated with a poor inflammatory status, leading to impaired insulin action and adipose tissue plasticity (Figs. 2 and 3) [33,34]. This means that the origin of metabolic inflammation could have some traits in common with inflammatory diseases, albeit of lesser intensity. We briefly review here some of the main concepts. 3.1. Intestinal microflora, CD and UC IBDs such as CD and UC represent abnormal immune responses of the gastrointestinal tract leading to adverse clini- M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 265 Fig. 2. Intestinal microflora changes and metabolic implications. Ingested components such as high-fat foods, antibiotics, dietary fibre (prebiotics) and bacterial additives (probiotics) can change the intestinal microflora ecology, leading to an unbalanced Firmicutes–Bacteroidetes ratio. This imbalance can affect metabolism, leading to a pathological state (obesity and diabetes). cal outcomes. CD is associated with excessive IL-12, IL-23 and IFN-gamma production that affects the wall of the small intestine, leading to inflammation and, often, granulomas. Patients report gastrointestinal symptoms such as abdominal pain, diar- rhoea and rectal bleeding, as well as systemic symptoms of weight loss, fever and fatigue; they can also develop fistulae [35]. UC is associated with excess IL-13 production that primarily affects the colon, with chronic inflammation of the mucosa Fig. 3. Nutrition-induced intestinal microflora changes and their metabolic consequences. A change in nutrition induces changes in the intestinal microflora. In particular, a fat-enriched diet decreases the Gram-positive–Gram-negative ratio, which is associated with increased lipopolysaccharide (LPS) absorption via mechanisms involving lipoproteins and paracellular translocation. LPS then activates cells from the immune system in the liver, adipose tissues and muscles. The increased inflammation eventually triggers insulin resistance and adipose tissue plasticity. 266 M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 [36]. More important, an atypical Th2 response mediated by non-classical natural-killer T (NKT) cells producing the IL-13 is characterized by an exacerbated cytotoxic potential in epithelial cells, altering the integrity of the intestinal wall and, thus, leading to ulceration. Certainly, metabolic diseases are not associated with this degree of inflammation. However, an impaired innate immune system, in which hypersensitive macrophages or lymphocytes can generate an over immune response, could lead to excess cytokine production, thereby increasing systemic inflammation. An increased inflammatory status is now accepted to be at least partly responsible for impaired insulin action [33,34]. Recent data have shown that, when differential gene expression was determined by microarray along the longitudinal axis of the small intestine of C57BL/6J mice fed a diabetogenic highfat diet, after 2, 4 and 8 weeks of the dietary intervention, the most pronounced effects were observed in the middle section of the small intestine [37]. Numerous genes coding for cell cycles, lipid metabolism and inflammation were identified. From this, the authors concluded that high-fat-feeding modulates biological processes, especially those related to lipid metabolism and inflammation. In addition, they found that the differential expression of potential signaling molecules could provoke systemic effects in peripheral organs by influencing their metabolic homoeostasis. Another mechanism common to intestinal inflammatory and metabolic diseases is related to changes in the intestinal mucosa. Using conventional culture techniques, Swidsinski et al. [38] demonstrated greater amounts of mucosa-associated bacteria (those in the mucous layer and at the epithelial surface) in tissue biopsies obtained from IBD patients vs controls. Indeed, concentrations of mucosa bacteria increased progressively with the severity of disease. The authors suggested that changes in the mucosal flora in IBD are not secondary to inflammation, but the result of a specific host response. They further hypothesized that a healthy mucosa is capable of blocking faecal bacteria and that this function is profoundly disturbed in patients with IBD. This was demonstrated by the fact that most germ-free (sterile) rodents have no intestinal inflammation or immune activation, but rapidly develop disease and pathogenic immune responses after colonization by specific pathogen-free enteric bacteria [39]. Although the processes of intestinal inflammatory diseases and diabetes are different, the mucosa hypothesis offers a mechanism that increases contact between the intestinal microflora and cells of the innate immune system, thereby increasing inflammation. The same reasoning could apply as regards an impaired anti-inflammatory mechanism. Sellon et al. [40] showed that mice with targeted deletion of the IL-10 gene spontaneously developed enterocolitis when maintained under conventional conditions, but developed only colitis when kept in specific pathogen-free environments. This study confirmed the hypothesis that enteric bacteria are necessary for the development of spontaneous colitis and immune system activation in IL-10-deficient mice. As for hypotheses based on what is known of the role of intestinal microflora in the development of intestinal inflammatory diseases, there are indeed mechanisms in common between metabolic diseases and IBDs, albeit with considerably less inflammation in metabolic diseases. Taken together, the innate and acquired immune systems are good candidates, as both are involved in the interplay between intestinal microflora and the inflammatory/anti-inflammatory cytokine balance. A good example of this idea is the role of the intestinal microflora in the control of type 1 diabetes (T1D). 4. Intestinal microflora and T1D T1D is a T-cell-mediated autoimmune disease that results in destruction of the insulin-producing beta cells of the pancreas. T1D is known as “childhood”, “juvenile” or “insulin-dependent” diabetes, although it is not exclusively seen in childhood, as demonstrated by its growing incidence in adults. So far, there are no preventative measures against T1D, and the initiating cause of the immune-system damage is still not fully understood, although diet and exercise can help. However, a provocative and growing body of evidences show that the intestinal microflora is involved in the development of T1D. Previous hints showed that the spontaneous incidence of T1D in an animal model—non-obese diabetic (NOD) mice—could be due to the animal microbial environment [41] or microbial stimuli [42,43]. Recently, the interaction between the gut microflora and innate immune system showed that the microflora could be considered a T1D-predisposing factor [44]. In their first series of experiments, the authors found that specific pathogen-free NOD mice lacking the MyD88 protein (a key component for recognition of microbial stimuli by innate immune system receptors) were able to resist T1D, suggesting that the lack of MyD88 protein affected the autoimmune T cells systemically, preventing the destruction of the pancreatic beta cells. Excess activation by microbial antigen could lie at the origin of the autoimmunity. However, a surprising conclusion came with the finding that MyD88 knockout mice without intestinal microflora still developed T1D. This paradoxically suggested that immunization by microflora prevented the overt activity of the immune system, leading to autoimmunity. In fact, colonization of the NOD germ-free mice with a specific microbial combination, comprising bacterial phyla normally present in the human intestine, attenuated T1D. This meant that, first, a given flora combination was responsible for the occurrence of T1D by a mechanism requiring the MyD88 inflammatory signaling molecule and that, second, a different intestinal flora group could protect against T1D by a mechanism that prevented overt activation of the immune system. In addition to these surprising data, a new finding was that, in addition to the capacity of intestinal microflora to control host functions such as the innate immune system or nutrient absorption (Fig. 1) [45], the host itself was able to control the metagenome. This was again demonstrated in mice lacking the MyD88 protein with different intestinal microflora, characterized by a reduced Firmicutes–Bacteroidetes ratio, and an increase in Lactobacillaceae, Rikenellaceae and Porphyromonadaceae families of bacteria in the caecal contents. This change in flora suggested that the host could select a particular intestinal flora to enable regulation of various new bodily functions. M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 For example, T1D is associated with increased intestinal permeability [46], lymphocytic infiltration in the mucosa [47] and morphological changes [48] in the BioBreeding (BB) rat, a model of spontaneous T1D. In humans, Bosi et al. [49] showed that, following the oral administration of two probes—lactulose and mannitol—urinary excretion over the following 5 h was dramatically increased, suggesting augmentation of intestinal uptake of the probes. All subjects with islet autoimmunity analyzed by the investigators showed increased intestinal permeability to the disaccharide lactulose, indicative of intestinal barrier damage. This suggests that numerous other proinflammatory substances might be able to cross the epithelial barrier and reach the immune system to generate and increase an inflammatory response. Substances such as the highly proinflammatory lipopolysaccharides (LPS) and peptidoglycans (PGN) are excellent candidates. 5. Intestinal microflora, obesity and type 2 diabetes (T2D) In the USA, roughly one million diabetic patients are newly diagnosed each year. Given this growing number of cases, as well as the numbers of undiagnosed cases (one-third of patients are not aware of their disease), the treatment of diabetes remains a major therapeutic challenge that the entire population needs to address. Obesity has now been classified as the newest epidemic, as its occurrence is on the increase in Western countries. The World Health Organization (WHO) has estimated that 600 million people will be obese by 2025 [50]. This corresponds to a doubling of the current obese population. The rise in excessive weight gain in the developed countries is associated with metabolic and cardiovascular diseases due to combined genetic and environmental factors. Although numerous hypotheses have been proposed to link these factors with obesity, no clear unifying concept has yet emerged. In terms of environmental factors, a noteworthy point is that reducing dietary fibre induces a cascade of metabolic impairments, leading to excess body weight, and cardiovascular and diabetic complications. Recent evidence also shows that the gut microflora is determinant of body weight and the amount (size) of adipose tissue, and is involved in intestinal permeability (Fig. 2). This suggests that the microflora play an important role in the development of T2D and obesity, two linked pathologies that highlight an epidemiological progression [45,51,52]. A novel hypothesis was recently proposed wherein the intestinal microflora could be considered a causative factor of obesity [52–54]. Briefly, it proposes that a specific intestinal microflora combination is responsible for increased energy storage. Two mechanisms are then involved: first, the microflora increase the bioavailability of energy intake by transforming non-digestible fibre into absorbable nutrients; and second, the microflora regulate intestinal gene expression. Specifically, gut bacteria could reduce the intestinal expression of fasting-induced adipocyte factor (FIAF), a protein that inhibits lipoprotein lipase activity. Its inhibition would favour the release of free fatty acids (FFA) from lipoprotein particles, making FFA more readily available 267 to the liver and other tissues, which would then store them more efficiently [55]. Similarly, a change in the intestinal microflora could favour the production of short-chain fatty acids by means of their own metabolism. Such molecules are activators of triglyceride synthesis. These two arguments could explain why axenic (or germfree) mice are leaner than their conventional counterparts [53]. In contrast, colonization of axenic mice by conventional microflora rapidly increased their body weight [53]. This was associated with decreased expression of FIAF and modulation of other genes involved in glucose and lipid metabolism [54], thus supporting the involvement of intestinal microflora in the control of energy metabolism. Intestinal microflora composition itself is changed in metabolic diseases (Fig. 2) [45,51,56–60]. Genetically ob/ob mice are characterized by a 50% reduction in the number of Bacteroidetes, which is balanced by an increased number of Firmicutes, compared with lean mice. This was not due to a change in the quality of food, but may have been induced by the large amount of food consumed by the obese mice [61]. However, independent of food quantity, the quality of food is certainly a regulating factor in metagenomic profiles (the intestinal microflora). Recently, we found that feeding mice a high-fat diet induces a dramatic change of the metagenomic profile of the intestinal microflora. This was associated with a general reduction in the total amount of intestinal bacteria and a reduction in Gram-positive bacteria, while the proportion of Gram-negative bacteria increased (Fig. 3) [57–60]. Changes in the intestinal microflora have also been reported in human studies (Fig. 2). Obese patients are characterized by a greater disturbance of the Firmicutes–Bacteriodetes ratio [45,51,56,61], which was subsequently reduced and almost normalized when the patients underwent several weeks of body weight loss [51]. In addition, this showed that the intestinal microflora can be adapted by nutritional therapy, which means that metagenomic imprinting is reversible. 6. Intestinal microflora and increased metabolic endotoxaemia A raised hepatic low-density lipoprotein (LDL)–high-density lipoprotein (HDL) ratio defines dyslipidaemia, an important feature of metabolic diseases. The LPS, the principal component of the outer membrane of Gram-negative bacteria and the most powerful of the proinflammatory molecules, is also a component of lipoprotein particles [62,63]. The latter have been proposed to buffer LPS in response to septic shock [63], and to prevent an overt inflammatory response and fatality. As for the proinflammatory characteristics of LPS and its association with a fat-enriched diet and dyslipidaemia, we have already demonstrated that this bacterial component is a triggering factor in metabolic diseases induced by a high-fat diet [57]. When mice were fed a high-fat diet for 4 weeks, their levels of LPS in plasma increased two- to three fold. It further increased throughout the duration of the treatment 268 M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 before reaching a plateau. The increased endotoxaemia—called “metabolic endotoxaemia”—is a pathological condition associated with quantitative and qualitative changes in the intestinal microflora. A high-fat diet increased the Gram-negative–Grampositive ratio in the caecum by reducing the Gram-positive count (Fig. 3). The mechanisms for this remain unknown, but might be related to the reduced dietary fibre in the fat-enriched diet. Indeed, dietary fibre supplementation can reverse changes in the intestinal microflora induced by a fat-enriched diet [60]. Furthermore, by implanting a subcutaneous osmotic minipump to deliver a continuous subcutaneous infusion of LPS, we causally linked LPS to metabolic disease. All features characteristic of the metabolic syndrome—fasting hyperinsulinaemia, glucose intolerance, visceral weight gain, hepatic lipid overload and insulin resistance—were induced by continuous subcutaneous LPS infusion alone. The link between high-fat-feeding in mice and metabolic endotoxaemia has also been demonstrated in humans. Amar et al. [64] showed that fat intake is associated with endotoxaemia in apparently healthy people. We conducted a dietary survey that also assessed other metabolic parameters such as total and HDL cholesterol, and plasma triglyceride levels, in 1015 randomly recruited subjects. Plasma was collected in the morning after overnight fasting. Plasma LPS levels were also measured in a subsample of 201 men. We found that energy intake was associated with increased LPS plasma levels. In contrast, carbohydrate and protein intakes were not associated with increased endotoxaemia. In mice, the proportion of fat in the diet controlled the extent of metabolic endotoxaemia independent of the amount of food ingested [64]. Thus, the authors demonstrated that fat is a highly efficient transporter of LPS from the intestinal lumen to the bloodstream. A molecular demonstration of the role of LPS in the onset of metabolic diseases was made by studying CD14 (LPS receptor)-deficient mice, in which the development of obesity, insulin resistance, hepatic steatosis and most of the metabolic features associated with metabolic diseases were delayed [57]. Either LPS or a high-fat diet can induce adipose tissue infiltration and inflammation, which was totally blunted in the mutant mice. In addition, antibiotherapy can reverse the metabolic phenotype [58]. For this reason, it is now thought that metabolic endotoxaemia is a triggering factor in the onset of metabolic diseases induced by a high-fat diet. However, many questions regarding the role of LPS still need to be addressed. The tissues targeted by LPS, for instance are as yet unknown, and the cells of the innate immune system involved in the inflammatory process also need to be determined. 6.1. A linear concept and other hypotheses So far, this review has found that increased fat ingestion or changes in the intestinal microflora can modulate host gene expression (Fig. 1) and corresponding metabolic functions (Fig. 2). The nutritional and microbiota signal for this could be LPS. The role of lipoprotein synthesis and release by the intestines, and the role of paracellular ingestion of microbiota particles, could be the mechanisms involved in LPS absorption. A change in the structure of the gut barrier, altering the architecture of the modulating tight junctions, would affect intestinal paracellular permeability, resulting in metabolic endotoxaemia. Other processes by which the intestinal microflora could control tight junctions—and, hence, LPS absorption—might involve molecular factors in the control of metabolic diseases. The subsequent local inflammation could also induce abnormal stimulation of the intestinal immune system, leading to localized inflammatory reactions in the intestinal barrier that, in turn, could trigger a systemic inflammatory reaction against white adipose tissue, muscle and the liver, finally resulting in insulin resistance and T2D (Fig. 3). 7. Changing the intestinal microflora 7.1. Antibiotic treatments As the intestinal microflora could be a causal triggering factor of metabolic diseases, Cani et al. [58] treated control, highfat-fed and ob/ob mice with broad-spectrum antibiotics. They used ampicillin and neomycin as examples of antibiotics that are poorly absorbed (or unabsorbed in the case of neomycin) by the organism to reduce the occurrence of systemic effects [65]. The results showed that changes in the gut microbiota induced by antibiotic treatment reduced metabolic endotoxaemia and the caecal content of LPS in both the high-fat-fed and ob/ob mice. The reduced endotoxaemia correlated with improved glucose tolerance and decreases in body weight gain, fat-mass development, inflammation, oxidative stress and macrophage infiltration marker mRNA expression in visceral adipose tissue. In addition, the use of ob/obxCD14–/– mutant mice showed that the lack of CD14 reduced inflammatory markers and metabolic impairments [58]. Likewise, Membrez et al. [66] tested a similar antibiotherapy against the gut microbiota in two different mouse models with insulin resistance. In this study, the investigators used a combination of norfloxacin and ampicillin, as this treatment is known to result in maximum suppression of the numbers of caecal aerobic and anaerobic bacteria in ob/ob mice [67]. After 2 weeks of intervention with the antibiotic combination, both the ob/ob and the diet-induced obese and insulin-resistant mice showed significant improvements in fasting glycaemia and oral glucose tolerance. The authors found that this effect was independent of food intake or adiposity because pair-fed ob/ob mice were as glucose intolerant as the control ob/ob mice. Reduced liver triglycerides and increased liver glycogen concentrations were also associated with the improved glucose tolerance. In addition, the antibiotic treatment in ob/ob mice showed antidiabetic effects by reducing endotoxaemia and increasing adiponectin levels. In this study, the investigators discovered that modulation of the gut microbiota improved glucose tolerance in mice by altering the expression of the hepatic and intestinal genes involved in inflammation and metabolism, and changing the hormonal, inflammatory and metabolic status of the host. M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 7.2. Prebiotic treatment One key question still remains: why is the intestinal microflora in obese patients and mice associated with an increased Firmicutes bacterial count? The answer may be related to changes in the amount of dietary fibre consumed nowadays. The data show the importance of the intestinal microflora in controlling body weight, confirmed by the observation that dietary fibre—which can also change the gut microflora [60]—is associated with metabolic changes. Dietary fibre is a strong inducer of fermenting bacteria [68–71]. The molecular relationship between dietary fibre, microflora and an improved metabolic status is still unknown. However, our recent work has shown that the intestinal microflora can regulate inflammation, which is closely linked to metabolic diseases [58]. The latter are characterized by a low inflammatory status, which contributes to the impaired insulin action [72]. Recently, it has been shown that high-fat-feeding contributes to the induction of moderate inflammation, characterized by cytokine secretion (TNF-␣, IL-6, IL-1) that strongly impairs insulin action and signaling [33,73]. The result is that the glucose transported by muscle or produced in the liver is no longer controlled by insulin, thereby resulting in hyperglycaemia. Consequently, hyperinsulinaemia develops to compensate for the hyperglycaemia, and glucose is then stored in adipose tissue, leading to obesity. Other recent findings propose that dietary fibre can improve the metabolic status of diabetic mice by reducing inflammation [71]. This effect could be due to improved secretion of glucagon-like peptide-1 (GLP-1), a gut hormone which increases glucose-stimulated insulin secretion [74,75]. This idea is physiologically relevant as GLP-1 is secreted into the hepatoportal vein and controls glucose metabolism (insulin secretion, peripheral glucose utilization and vascular blood flow) [76–81]. Increasing the secretion of this hormone at its physiological site of production may be a therapeutic advantage in light of other strategies that aim to increase GLP-1 secretion in the systemic blood by pharmacological means. Intestinal endocrine function may also be improved when the deleterious effects of a high-fat diet that triggers inflammation is blunted by dietary fibre. The control of GLP-1 secretion by inflammation could have a major impact on human health as this hormone is the basis of three new antidiabetic drugs that are already on the market [82,83]. However, the molecular mechanisms linking high-fat-feeding, inflammation and dietary fibre are still unknown. Prebiotics are non-digestible food oligosaccharides that beneficially affect the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon, thereby improving host health [84]. At present, only two dietary non-digestible oligosaccharides fulfill all the criteria for prebiotic classification: fructooligosaccharides (FOS); and galactooligosaccharides (GOS). Classification as a prebiotic food ingredient is based on the following criteria: 269 • it is neither hydrolyzed nor absorbed in the upper part of the gastrointestinal tract; • it is selectively fermented by one or a limited number of potentially beneficial bacteria in the colon; • it alters the composition of the colonic microbiota toward a healthier composition; • it preferably induces effects that are beneficial to host health [85]. In fact, not all oligosaccharides—such as mannanoligosaccharides (MOS)—fit this definition. They may confer other positive benefits, but are minimally utilized by commensal bacteria (Bifidobacterium and Lactobacillus species). Also, prebiotics are usually carbohydrates (such as oligosaccharides), but other non-carbohydrate molecules can be used. The most common form of prebiotic is soluble fibre. We reported that high-fat-feeding was associated with more endotoxaemia and fewer Bifidobacterium species in the caecal contents in mice [58]. Cani et al. [71] found that oligofructose can restore the bifidobacteria count in high-fat-fed mice, and this finding positively correlated with improved glucose tolerance, glucoseinduced insulin secretion, decreased endotoxaemia, and plasma and adipose tissue proinflammatory cytokines. Indeed, Watanabe et al. [86] have now confirmed the beneficial effect of FOS supplementation in female BALB/c mice fed a synthetic diet for 3 weeks compared with control mice that did not receive FOS. The mice were then epicutaneously immunized with 2,4-dinitrofluorobenzene, after which the mice continued to receive their respective diets. Five days after immunization, the mice were ear-challenged with hapten, but the subsequent ear-swelling was significantly reduced in the FOS-supplemented mice compared with the control-diet-fed mice. Using denaturing gradient gel electrophoresis (DGGE), the investigators assessed changes in the intestinal microbiota and found that the numbers of bifidobacteria (Bifidobacterium pseudolongum, assessed by sequence analysis), but not lactobacilli, were significantly higher in the FOS-supplemented mice compared with the mice fed the control diet. Ear-swelling was also negatively correlated with the numbers of bifidobacteria found in the faeces. The authors concluded that supplementing the diet with prebiotics leads to changes in the intestinal microflora that improve health. This means that strategies involving prebiotics should further improve patients’ well-being and health. 7.3. Probiotic treatment Probiotics are dietary supplements that contain potentially beneficial bacteria or yeasts. According to the current definition of the United Nations’ Food and Agriculture Organization (FAO)/WHO, probiotics are “live microorganisms which, when administered in adequate amounts, confer a health benefit on the host”. Interest in probiotics has recently increased, with studies investigating their beneficial roles in the treatments of various diseases. Rautava et al. [87] showed the beneficial effects of Lactobacillus rhamnosus GG and B. lactis Bb-12 in reducing the risks of early acute otitis media and antibiotic use, and recurrent respiratory infections, in the first year of 270 M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 life. Probiotics can also improve the effects of certain drugs such as gliclazide, a sulphonylurea with beneficial extrapancreatic effects in diabetics. Al-Salami et al. [88] showed a threefold probiotic-induced reduction in gliclazide bioavailability in healthy rats whereas it increased uptake (by twofold) in diabetic rats, leading to a reduction in blood glucose levels by insulin-independent mechanisms. In addition, probiotics can be administered as a food supplement in yogurt, for example [89]. In their study, Bajaj et al. found that probiotic-complemented yogurt was able to reverse minimal hepatic encephalopathy (MHE), the preclinical stage of overt hepatic encephalopathy (OHE), a significant condition that affects up to 60% of cirrhosis patients. Yadav et al. [90] found that supplementing with the probiotics L. acidophilus and L. casei increased the efficacy of dahi (Greek yogurt) in suppressing streptozotocin-induced diabetes in rats by inhibiting insulin depletion, preserving diabetic dyslipidaemia, and inhibiting lipid peroxidation and nitrite formation. In ob/ob mice, a genetic mouse model of obesityrelated non-alcoholic fatty liver disease and T2D, Li et al. [91] found that 4 weeks of treatment with VSL#3, a probiotic combination of bifidobacteria, lactobacilli, and Streptococcus thermophilus, decreased both hepatic total fatty-acid content and serum alanine aminotransferase. These benefits were associated with normalization of hepatic fatty-acid -oxidation and reduction of hepatic NF-B activity and uncoupling protein (UCP)-2 expression, suggesting that VSL#3 therapy improves hepatic insulin resistance and lipid metabolism. The benefits of VSL#3 probiotics were also demonstrated by Ma et al. [92] in another mouse model of high-fat-diet-induced obesity, steatosis and insulin resistance. Oral probiotic treatment significantly improved the depleted numbers of hepatic NKT cells, insulin resistance and hepatic steatosis. The effect was NKT-dependent, resulting from attenuation of inflammatory signalling. 8. Microflora as a friend Mazmanian et al. [93] showed that a specific component of the intestinal microflora can directly interact with the innate immune system of the host, resulting in a beneficial effect. In their elegantly written report, the investigators showed that Bacteroides fragilis can protect animals against experimental colitis induced by Helicobacter hepaticus, a potential pathogenic commensal of the intestinal microflora. The benefit depends on the production of a single specific molecule produced by B. fragilis called ‘polysaccharide A’ (PSA). In fact, animals with a mutant variant of B. fragilis lacking PSA cannot resist H. hepaticus colonization, leading to disease and proinflammatory cytokine production in the colon. In addition, the purified molecule of PSA alone is enough to protect against H. hepaticus colonization and even against chemically induced (trinitrobenzene sulphonic acid, TNBS) experimental colitis. PSA protection is mediated by IL-10-producing CD4+ T cells. In conclusion, the present review shows, for the first time, that specific intestinal microflora components can produce molecules that are contributory to host health. Indeed, the intestinal microflora can act, via the production of specific molecules, not only as an enemy of the host, but also—as described above—as a new friend to improve the defense mechanisms of the host (Figs. 1–3). 9. Conflicts of interest The authors have no conflicts of interest to declare in relation to this report. References [1] Sears CL. A dynamic partnership: celebrating our gut flora. Anaerobe 2005;11:247–51. [2] Berg RD. The indigenous gastrointestinal microflora. Trends Microbiol 1996;4:430–5. [3] Mackie RI, Sghir A, Gaskins HR. Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr 1999;69:1035S–45S. [4] Gorbach SL. Intestinal microflora. Gastroenterology 1971;60:1110–29. [5] Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, et al. Feeding our immune system: impact on metabolism. Clin Dev Immunol 2008;2008:639803. [6] Schenk M, Mueller C. The mucosal immune system at the gastrointestinal barrier. Best Pract Res Clin Gastroenterol 2008;22:391–409. [7] Sudo N, Sawamura S, Tanaka K, Aiba Y, Kubo C, Koga Y. The requirement of intestinal bacterial flora for the development of an IgE production system fully susceptible to oral tolerance induction. J Immunol 1997;159:1739–45. [8] Niess JH. Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol 2008;14:5138–48. [9] Strober W, Immunology. Unraveling gut inflammation. Science 2006;313: 1052–4. [10] Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–34. [11] Rouzaud G, Rabot S, Ratcliffe B, Duncan AJ. Influence of plant and bacterial myrosinase activity on the metabolic fate of glucosinolates in gnotobiotic rats. Br J Nutr 2003;90:395–404. [12] Possemiers S, Rabot S, Espin JC, Bruneau A, Philippe C, Gonzalez-Sarrias A, et al. Eubacterium limosum activates isoxanthohumol from hops (Humulus lupulus L) into the potent phytoestrogen 8-prenylnaringenin in vitro and in rat intestine. J Nutr 2008;138:1310–6. [13] Yuan JP, Wang JH, Liu X. Metabolism of dietary soy isoflavones to equol by human intestinal microflora–implications for health. Mol Nutr Food Res 2007;51:765–81. [14] Wong JM, Jenkins DJ. Carbohydrate digestibility and metabolic effects. J Nutr 2007;137(Suppl. 11):2539S–46S. [15] O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 2006;7:688–93. [16] Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol 2006;59:1639–50. [17] Gorbach SL. Lactic acid bacteria and human health. Ann Med 1990;22:37–41. [18] Salminen SJ, Gueimonde M, Isolauri E. Probiotics that modify disease risk. J Nutr 2005;135:1294–8. [19] Ouwehand A, Isolauri E, Salminen S. The role of the intestinal microflora for the development of the immune system in early childhood. Eur J Nutr 2002;41(Suppl. 1):I32–7. [20] Rolfe RD. Interactions among microorganisms of the indigenous intestinal flora and their influence on the host. Rev Infect Dis 1984;6(Suppl. 1):S73–9. [21] Abrams GD, Bishop JE. Effect of the normal microbial flora on gastrointestinal motility. Proc Soc Exp Biol Med 1967;126:301–4. [22] Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A 2002;99:15451–5. M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 [23] Mathan VI, Wiederman J, Dobkin JF, Lindenbaum J. Geographic differences in digoxin inactivation, a metabolic activity of the human anaerobic gut flora. Gut 1989;30:971–7. [24] Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science 2001;291:881–4. [25] Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008;456:507–10. [26] Lee PS, Lee KH. Escherichia coli–a model system that benefits from and contributes to the evolution of proteomics. Biotechnol Bioeng 2003;84:801–14. [27] Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science 2006;312:1355–9. [28] Palmer C, Bik EM, Digiulio DB, Relman DA, Brown PO. Development of the Human Infant Intestinal Microbiota. PLoS Biol 2007;5:e177. [29] Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005;308:1635–8. [30] Macfarlane S, Macfarlane GT. Bacterial diversity in the human gut. Adv Appl Microbiol 2004;54:261–89. [31] Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol 2002;30: 503–12. [32] Zaneveld J, Turnbaugh PJ, Lozupone C, Ley RE, Hamady M, Gordon JI, et al. Host-bacterial coevolution and the search for new drug targets. Curr Opin Chem Biol 2008;12:109–14. [33] Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793–801. [34] Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444: 860–7. [35] Podolsky DK. Inflammatory bowel disease. N Engl J Med 2002;347: 417–29. [36] Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest 2004;113:1490–7. [37] de Wit NJ, Bosch-Vermeulen H, de Groot PJ, Hooiveld GJ, Bromhaar MM, Jansen J, et al. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med Genomics 2008;1:14. [38] Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002;122:44–54. [39] Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm Jr TE, Balish E, et al. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest 1996;98:945–53. [40] Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998;66:5224–31. [41] Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world–recent facts and figures. Immunol Today 1993;14:193–6. [42] McInerney MF, Pek SB, Thomas DW. Prevention of insulitis and diabetes onset by treatment with complete Freund’s adjuvant in NOD mice. Diabetes 1991;40:715–25. [43] Sadelain MW, Qin HY, Lauzon J, Singh B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes 1990;39:583–9. [44] Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 2008;455:1109–13. [45] Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–31. [46] Meddings JB, Jarand J, Urbanski SJ, Hardin J, Gall DG. Increased gastrointestinal permeability is an early lesion in the spontaneously diabetic BB rat. Am J Physiol 1999;276:G951–7. 271 [47] Graham S, Courtois P, Malaisse WJ, Rozing J, Scott FW, Mowat AM. Enteropathy precedes type 1 diabetes in the BB rat. Gut 2004;53:1437–44. [48] Neu J, Reverte CM, Mackey AD, Liboni K, Tuhacek-Tenace LM, Hatch M, et al. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J Pediatr Gastroenterol Nutr 2005;40:589–95. [49] Bosi E, Molteni L, Radaelli MG, Folini L, Fermo I, Bazzigaluppi E, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006;49:2824–7. [50] Formiguera X, Canton A. Obesity: epidemiology and clinical aspects. Best Pract Res Clin Gastroenterol 2004;18:1125–46. [51] Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022–3. [52] Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718–23. [53] Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Hostbacterial mutualism in the human intestine. Science 2005;307:1915–20. [54] Backhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 2007;104:979–84. [55] Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, et al. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem 2000;275:28488–93. [56] Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature 2007;449:804–10. [57] Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–72. [58] Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008;57:1470–81. [59] Cani PD, Delzenne NM, Amar J, Burcelin R. Role of gut microflora in the development of obesity and insulin resistance following high-fat diet feeding. Pathol Biol (Paris) 2008;56:305–9. [60] Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve highfat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007;50:2374–83. [61] Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005;102:11070–5. [62] Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 2009;50:90–7. [63] Vreugdenhil AC, Rousseau CH, Hartung T, Greve JW, van’t Veer C, Buurman WA. Lipopolysaccharide (LPS)-binding protein mediates LPS detoxification by chylomicrons. J Immunol 2003;170:1399–405. [64] Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr 2008;87:1219–23. [65] Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, et al. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 2006;168:1148–54. [66] Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. Faseb J 2008;22:2416–26. [67] Chou CJ, Membrez M, Blancher F. Gut decontamination with norfloxacin and ampicillin enhances insulin sensitivity in mice. Nestle Nutr Workshop Ser Pediatr Program 2008;62:127–37 [137–40 discussion]. [68] Langlands SJ, Hopkins MJ, Coleman N, Cummings JH. Prebiotic carbohydrates modify the mucosa associated microflora of the human large bowel. Gut 2004;53:1610–6. [69] Lesniewska V, Rowland I, Cani PD, Neyrinck AM, Delzenne NM, Naughton PJ. Effect on components of the intestinal microflora and plasma neuropeptide levels of feeding Lactobacillus delbrueckii, Bifi- 272 [70] [71] [72] [73] [74] [75] [76] [77] [78] [79] [80] [81] M. Serino et al. / Diabetes & Metabolism 35 (2009) 262–272 dobacterium lactis, and inulin to adult and elderly rats. Appl Environ Microbiol 2006;72:6533–8. Kleessen B, Hartmann L, Blaut M. Oligofructose and long-chain inulin: influence on the gut microbial ecology of rats associated with a human faecal flora. Br J Nutr 2001;86:291–300. Cani PD, Daubioul CA, Reusens B, Remacle C, Catillon G, Delzenne NM. Involvement of endogenous glucagon-like peptide-1(7-36) amide on glycaemia-lowering effect of oligofructose in streptozotocin-treated rats. J Endocrinol 2005;185:457–65. Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998;41:1241–8. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 2006;116:3015–25. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87: 1409–39. Burcelin R. The incretins: a link between nutrients and well-being. Br J Nutr 2005;93(Suppl. 1):S147–56. Knauf C, Cani PD, Perrin C, Iglesias MA, Maury JF, Bernard E, et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J Clin Invest 2005;115:3554–63. Burcelin R, Cani PD, Knauf C. Glucagon-like peptide-1 and energy homeostasis. J Nutr 2007;137(Suppl. 11):2534S–8S. Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagon-like peptide-1 receptor. Diabetes 2001;50:1720–8. Knauf C, Cani PD, Ait-Belgnaoui A, Benani A, Dray C, Cabou C, et al. Brain glucagon-like peptide 1 signaling controls the onset of high-fat dietinduced insulin resistance and reduces energy expenditure. Endocrinology 2008;149:4768–77. Knauf C, Cani PD, Kim DH, Iglesias MA, Chabo C, Waget A, et al. Role of central nervous system glucagon-like Peptide-1 receptors in enteric glucose sensing. Diabetes 2008;57:2603–12. Cabou C, Campistron G, Marsollier N, Leloup C, Cruciani-Guglielmacci C, Penicaud L, et al. Brain glucagon-like peptide-1 regulates arterial blood flow, heart rate, and insulin sensitivity. Diabetes 2008;57:2577–87. [82] Ahren B. Emerging dipeptidyl peptidase-4 inhibitors for the treatment of diabetes. Expert Opin Emerg Drugs 2008;13:593–607. [83] Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006;368:1696–705. [84] Gibson GR, Roberfroid MB. Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 1995; 125:1401–12. [85] Gibson GR. Dietary modulation of the human gut microflora using the prebiotics oligofructose and inulin. J Nutr 1999;129(Suppl. 7): 1438S–41S. [86] Watanabe J, Sasajima N, Aramaki A, Sonoyama K. Consumption of fructo-oligosaccharide reduces 2, 4-dinitrofluorobenzene-induced contact hypersensitivity in mice. Br J Nutr 2008;100:339–46. [87] Rautava S, Salminen S, Isolauri E. Specific probiotics in reducing the risk of acute infections in infancy - a randomised, double-blind, placebo-controlled study. Br J Nutr 2008:1–5. [88] Al-Salami H, Butt G, Fawcett JP, Tucker IG, Golocorbin-Kon S, Mikov M. Probiotic treatment reduces blood glucose levels and increases systemic absorption of gliclazide in diabetic rats. Eur J Drug Metab Pharmacokinet 2008;33:101–6. [89] Bajaj JS, Saeian K, Christensen KM, Hafeezullah M, Varma RR, Franco J, et al. Probiotic yogurt for the treatment of minimal hepatic encephalopathy. Am J Gastroenterol 2008;103:1707–15. [90] Yadav H, Jain S, Sinha PR. Oral administration of dahi containing probiotic Lactobacillus acidophilus and Lactobacillus casei delayed the progression of streptozotocin-induced diabetes in rats. J Dairy Res 2008;75: 189–95. [91] Li Z, Yang S, Lin H, Huang J, Watkins PA, Moser AB, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37:343– 50. [92] Ma X, Hua J, Li Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol 2008;49:821–30. [93] Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008;453:620–5.