Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
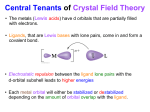
F. ELECTRONIC SPECTRA OF COORDINATION COMPOUNDS Introduction: Transition metal compounds display a wide variety of colors and many of these compounds are used as artist's pigments (e.g., copper phthalocyanine, prussian blue, and Thénard's blue are all various shades of blue). A compound is colored if it absorbs part of the visible light. When a sample absorbs visible light, the color we observe is the sum of the remaining colors that are reflected or transmitted by the sample and strikes our eyes. Remaining colors can conveniently be remembered as the color pairs on opposite sides of the color wheel (Newton Wheel) shown in Figure. 1. Figure.1 Newton Wheel (It was first created by Sir Isaac Newton in 1666.) The color of these compounds is due to electronic transitions induced by the incident light, causing absorption at certain wavelengths, depending on the structure and bonding in the molecule, which is also known as Crystal Field Theory (CFT) in Figure 2. UV–Visible spectroscopy is used to evaluate the absorption properties of molecules. The electronic transitions of transition metal compounds can be categorized into three types: charge transfer, d–d spin–allowed, and d–d spin–forbidden. Charge transfer bands occur when the excited electron moves from a metal centered orbital to a ligand–centered orbital (metal–to–ligand charge transfer – MLCT) or when the excited electron moves from a ligand–centered orbital to a metal centered orbital (ligand–to–metal charge transfer – LMCT). MLCT bands are more common than LMCT. Spin–allowed and spin–forbidden d–d transitions find the excited electron moving from one d orbital to another d orbital. In the spin–allowed transition the spin quantum number (or more accurately, the spin angular momentum) in the ground and excited states are the same while in the spin–forbidden transition the spin state changes during the excitation. These three types of transitions are distinguished by the molar absorption coefficient (in the Beer–Lambert law: A=bc): charge transfer bands have large , generally in the range of 1000 to 10000 L/mol–cm; spin–allowed d–d transition have values of 10–100 L/mol–cm for octahedral complexes and 500–1000 for tetrahedral complexes; and spin–forbidden bands have values less than 5 L/mol–cm. Spin–forbidden bands are often not observed. UV–Vis spectroscopy can be used to distinguish octahedral from tetrahedral geometries (Figure. 3). It is important to report both peak positions and molar extinction coefficients for electronic spectra. Crystal Field Splitting of d- orbitals is arranged differently by changing the environment around the central metal, which can be seen in Figure. 3. Figure 2. Summary of Crystal Field Theory The primary use of UV–Vis spectra for transition metal complexes is the elucidation of electronic structure parameters. Generally this means the crystal field parameter ∆0 and sometimes the Racah inter-electron repulsion constant B. These values are easy to find for most of the electron configurations of octahedral complexes. For d1, d4 (high spin), d6 (high spin), and d9 complexes a single spin–allowed d–d transition is observed with energy ∆0. High spin d5 complexes have no spin– allowed d–d bands so are lightly colored unless charge transfer bands dominate the spectrum. For d 3 and d8 octahedral complexes, three spin–allowed transitions are predicted at energies given by: v1 ∆0 v2 7.5B 1.5∆0 1/2 [225B2 10∆02 18B∆0] v3 7.5B 1.5∆0 1/2 [225B2 10∆02 18B∆0] where all the parameters are in units of cm–1. If all three transitions are observed, ∆0 can easily be found from ν1 and B can be found from 15B=ν3+ν2–3ν1. Often a charge transfer band will obscure 3 so that B must be found from v2 2v1v2 v1 3B 5v2 9v1 If the complex has a d2 or d7 (high spin) configuration, three bands are also expected and are given by: v1 7.5B 0.5∆0 1/2 [225B2 10∆02 18B∆0] v2 7.5B 1.5∆0 1/2 [225B2 10∆02 18B∆0] v3 [225B2 10∆02 18B∆0] where again, all parameters are in units of cm–1. Here ∆0 is found from the difference of the first two bands, ∆0=ν2–ν1, and B is found from 15B=ν3+ν2–3ν1. If only the first two transitions can be observed, then B can be found from v2 2v1 v1 3B 9v1 4v2 For complexes with other electron configurations no simple formulas exist to find the crystal field parameters. The crystal field strength, ∆0, measures the splitting between the t2g and eg orbitals and depends on both the metal ion and the ligand. The spectrochemical series denotes the variation of ∆0 as a function of ligand with weak ligands such as Cl– or H2O leading to small values of ∆0 and stronger ligands like NH3 or CN– giving larger values of ∆0. The inter-electron repulsion constant B is usually reported as β= Bcomplex/Bfree ion. Bonding reduces the inter-electron repulsion in a complex relative to the free (gas phase) ion (this happens because the metal d electrons expand to bond to the ligands and electrons that are farther apart from each other repel each other less) so that βis always less than 1. The value of βalso varies from ligand to ligand and this series is known as the nephelauxetic series, which is similar to but not identical to the spectrochemical series. Figure 3. Crystal Field Splitting parameters for different symmetrical fields. The energy difference between spherical field and octahedral field is called Crystal Field Stabilization Energy (CFSE), which can be calculated as: CFSE = 0.4 ∆0 . (# of electrons in t2g level) - 0.6 ∆0 . (# of electrons in t2g level) Also, CFSE would be different for high spin and/or low spin complexes. Experimental reports of UV–Vis data are not complete unless both the peak position and the molar absorptivity of each peak are given. A solution should be prepared so that the absorbance is between 1.5 and 2.0 and analytically diluted 4 or 5 times so that the most dilute concentration is at least 10 times smaller than the initial concentration. The spectrum is recorded at each dilution and the absorbance of each peak is plotted as a function of concentration. This plot should yield a straight line with a slope of (for a 1 cm cell) and an intercept of 0. If the slope has curvature, then the either experimental technique needs to be improved or the compound is reacting with solvent, itself, or perhaps air. To calculate ∆0/10 and/or B, the peak positions must be identified as spin–allowed d–d transitions in an octahedral environment and then converted to energy units, usually cm–1. Reference James E. Huheey, Inorganic Chemistry, Harper Collins College Publisher, 1993, p. 387. Experiment 9. Experimental Procedure: 1. Prepare the diluted solutions of i) KMnO4 ii) [Co(NH3)5NO2]SO4 (product material of experiment #4 part-III), iii) [Ni(NH3)6]Cl2 (product material of experiment #3 part-I), iv) FeCl2.4H2O, v) VO(acac)2 (product material of experiment #7), vi) K[Cr(ox)2(H2O)2].2H2O by using 0.1-0.2 g of samples in 50 mL water. If the concentration of the solution is high (dark color), you need to dilute the solution. 2. Scan the UV-Vis range of 300 nm to 900 nm by using UV-Vis Spectrophotometer. The instruction for this instrument is given by the lab intsructors. DO NOT USE THE INSTRUMENT BY YOURSELF. Calculating Crystal Field Splitting Parameter, ∆0: 1. Convert all transition wavelenght values (nm) to energy (kj/mol) by using E=hc/ and multiply by Avagadro’s number to molar energy. 2. Calculate Racah Parameter by using equations given in the introduction section. 3. Calculate Crystal Splitting Parameter, ∆0, by using equations given in the introduction section 4. Calculate Crystal Field Stabilization Energy (CFSE) by using equations given in the introduction section. DATA and RESULTS SHEET Sample i) KMnO4 ii) [Co(NH3)5NO2]SO4 iii) [Ni(NH3)6]Cl2 iv) FeCl2.4H2O v) VO(acac)2 vi) K[Cr(ox)2(H2O)2].2H2O 1 2 3 B ∆0 CFSE


















![[Fe(NH 3 ) 6 ] 2+ Finding optimal parameters for spin](http://s1.studyres.com/store/data/008100713_1-7d638d4cd4cd73a8d1a509d7fe17060e-150x150.png)