Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
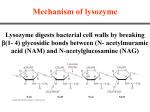
Enzymatic Catalysis Instructor: Dr. Tsung-Lin Li Genomics Research Center Academia Sinica Source book: Biochemistry (3rd) by Voet and Voet Catalytic mechanisms 1. 2. 3. 4. 5. 6. The specificity of substrate binding combined with the optimal arrangement of catalytic groups (as a result of eons of evolution) Acid-based catalysis Covalent catalysis Metal ion catalysis Electrostatic catalysis Proximity and orientation effects Preferential binding of the transition state complex Acid-base catalysis • General acid catalysis is a process in which partial proton transfer from a Brønsted acid (a species that can donate protons) lowers the free energy of a reaction’s transition state. • General base catalysis is a process if the rate is increased by partial proton abstraction by a Brønsted base (a species that can combine with a proton). • Some reactions may be simultaneously subject to both processes: a concerted general acid-base catalyzed reaction A Mechanisms of keto–enol tautomerization. (a) Uncatalyzed. (b) General acid catalyzed. (c) General base catalyzed. RNase A–catalyzed hydrolysis The bovine pancreatic RNase A–catalyzed hydrolysis of RNA is a two-step process with the intermediate formation of a 2,3 -cyclic nucleotide. RNase A–catalyzed hydrolysis The pH dependence of Vmax/KM in the RNase A–catalyzed hydrolysis of cytidine-2,3 -cyclic phosphate. Covalent catalysis • Covalent catalysis involves rate acceleration through the transient formation of a catalyst-substrate bond. The decarboxylation of acetoacetate. Covalent catalysis has both nucelophilic and electrophilic stages • The nucleophilic reaction between the catalyst and the substrate to form a covalent bond • The withdrawal of electrons from the reaction center by the now electrophilic catalyst • The elimination of the catalyst, a reaction that is essentially the reverse of stage 1 Certain amino acid side chains and coenzymes can serve as covalent catalysts • Residues: Lys, His, Cys, Asp, Ser • Cofactors: TPP, PLP Metal ion catalysis • About 1/3 of all known enzymes require the presence of metal ions for catalytic catalytic activity • Two classes of metal ion-requiring enzymes that are distinguished by the strengths of their ionprotein interactions Metalloenzymes Contain tightly bound metal ions (transition metal ions) such as Fe2+, Fe3+, Cu2+, Zn2+, Mn2+, or Co2+ Metal-activated enzymes Loosely bind metal ions from solution, (usually alkali and alkaline earth metal ions) such as Na+, K+, Mg2+, or Ca2+ Metal ions participate the catalytic process in three major ways 1. By binding to substrates so as to orient them properly for reaction 2. By mediating oxidation-reductions through reversible changes in the metal ion’s oxidation state. 3. By electrostatically stabilizing or shielding negative charges Metal ions promote catalysis through charge stabilization • The metal ion acts in much the same way as a proton to neutralize negative charge, that is, it acts as a Lewis acid. • Metal ions are often much more effective catalysts than protons because metal ions can be present in high concentrations at neutral pH’s and can have charges greater than +1. • Metal ions have been dubbed “superacids”. Metal ions promote nucleophilic catalysis via water ionization • A metal ion’s charge makes its bound water molecules more acidic than free H2O and therefore a source of OHions even below neutral pH’s. • For example, (NH3)5Co3+(H2O) (NH3)5Co3+(OH-) + H+ CO2 + H2O HCO3- + H+ X-Ray structures of human carbonic anhydrase. (a) Its active site in complex with bicarbonate ion. X-Ray structures of human carbonic anhydrase. (b) The active site showing the proton shuttle. •Through general base catalysis by His64 (but too far away from Zn2+bound water), they are two intervening water molecules to form a hydrogen bonded network that act as a proton shuttle. •The resulting Zn2+-bound OH- ion nucleophilically attacks the nearby enzymatically bound CO2, thereby converting it to HCO3-. •The Zn2+-bound OH- group donates a hydrogen bond to Thr199, which in turn donates a hydrogen bond to Glu106. These interactions orient the OH- group with the optimal geometry for nucleophilic attack on the substrate CO2. Metal ions promote reactions through charge shielding • The actual substrates of kinases are Mg2+-ATP complexes rather than just ATP. Electrostatic catalysis • Charge distributions about the active sites of enzymes are arranged so as to stabilize the transition states of the catalyzed reactions. • Such a mode of rate enhancement, which resembles the form of metal ion catalysis, is termed electrostatic catalysis. • These charge distributions serve to guide polar substrates toward their binding sites so that the rates of these enzymatic reactions are greater than their diffusion-controlled limits catalysis through proximity and orientation effects • Enzyme catalytic mechanisms are far more efficient than those of organic model reactions due to effects of proximity and orientation. • Reactants must come together with the proper spatial relationship for a reaction to occur It is 24-fold more effective • Proximity along contribute relatively little to catalysis; factors that will increase this value other than proximity alone clearly must be considered. • Properly orienting reactants and arresting their relative motions can result in large catalytic rate enhancements Reactants react most readily only if they have the proper relative orientation The geometry of an SN2 reaction. Relative Rates of Anhydride Formation for Esters Possessing Different Degrees of Motional Freedom in the Reaction Above. Catalysis by preferential transition state binding • The binding of the transition state to an enzyme with greater affinity than the corresponding substrates or products. • Rack mechanism: enzymes mechanically strained their substrates toward the transition state geometry through binding sites into which undistorted substrate did not properly fit. • The strained reactant more closely resembles the transition state of the reaction than does the corresponding unstrained reactant. Reaction coordinate diagrams for a hypothetical enzymatically catalyzed reaction (single substrate - blue; corresponding uncatalyzed reaction red). The effect of preferential transition state binding Transition state analogs are competitive inhibitors • Transition state analogs, stable molecules that resemble S or one of its components, are potent competitive inhibitors of the enzyme. X O Lysozme The alternating NAG–NAM polysaccharide component of bacterial cell walls. Enzyme structure Primary structure of HEW lysozyme. X-Ray structure of HEW lysozyme. (c) A computer-generated model showing the protein’s molecular envelope (purple) and Ca backbone (blue). X-Ray structure of HEW lysozyme. (b) A ribbon diagram of lysozyme highlighting the protein’s secondary structure. Table 15-2 Rates of HEW Lysozyme-Catalyzed Hydrolysis of Selected Oligosaccharide Substrate Analogs. X-Ray structure of HEW lysozyme. (a) The polypeptide chain is shown with a bound (NAG)6 substrate (green). The fourth NAG residue (D) appeared unable to bind to the enzyme because it C6 and O6 atoms too closely contact Glu35, Trp108, and the acetamido group of NAG residue C. This steric interference could be relieved by distorting the glucose ring from its normal chair conformation to that of a half-chair. Chair and half-chair conformations. Interactions of lysozyme with its substrate. •This distortion moves the –C6H2OH group from its normal equatorial position to an axial position where it makes no close contacts and can hydrogen bond to the backbone carbonyl group of Gln57 and the amido group of Val109 •The lactyl side chain of NAM cannot be accommodated in the binding subsites of either residues C or E. Hence, NAM residues must bind to the enzyme in substies B, D and F. •The probable cleavage site is between residues D and E. (Or, between second and third residues from its reducing terminus). Catalytic mechanism Mechanism of the nonenzymatic acid-catalyzed hydrolysis of an acetal to a hemiacetal. The Phillips mechanism for the lysozyme reaction. 1. Asp52 is surrounded by polar residues and thus predicted to have a normal pK ( unprotonated) 2. Glu35 is nestled in a nonpolar pocket, and is thus likely to remain protonated (C1, C2, C5, and C5 are coplanar) The D-ring oxonium ion intermediate in the Phillips mechanism is stabilized by resonance. Testing the Phillips mechanism 1. Identification of the catalytic residues (site-directed mutagenesis). 2. Role of strain The d-lactone analog of (NAG)4. Binding Free Energies of HEW Lysozyme Subsites. 3. The lysozyme reaction proceeds via a covalent intermediate For all b-glycosidase but not lysozyme Probing the adduct by incubating the analogue with E35Q mutant (ESI/MS) (X-ray) The HEW lysozyme covalent intermediate (a double displacement mechanism). Serine Proteases Chymotrypsin, trypsin and elastase A. Kinetics and catalytic groups a. Ester hydrolysis as a kinetic model Burst phase (stoichiometric) Steady state phase (rate is constant and independent with substrate conc. Ping pong Bi Bi A Selection of Serine Proteases. b. Identification of the catalytic residues Chemically labeling Chymotrypsin Ser195 (a serine esterase) (insecticides) Reaction of TPCK with chymotrypsin to alkylate His 57. Affinity labeling (a substrate analog bearing a reactive group) Chymotrypsin His57 B. Structure X-Ray structure of bovine trypsin. (a) A drawing of the enzyme in complex. (b) A ribbon diagram of trypsin. X-Ray structure of bovine trypsin. (c) A drawing showing the surface of trypsin (blue) superimposed on its polypeptide backbone (purple). The active site residues of chymotrypsin. The catalytically essential His57, and Ser195 are located at the substrate binding site together with the invariant Asp102, which is buried in a solvent-inaccessible pocket. These three residues form a hydrogen bonded constellation referred to as the catalytic triad. Serine Protease Catalysis • Serine proteases are enzymes that catalyze the hydrolysis of peptide bonds. In each case, the enzymes have a serine residue that plays a critical role in the catalysis. The enzymes cuts preferentially in distinct sites. The active site regions of all of the serine proteases have a number of common factors. For example, an aspartate residue, a histidine residue, and a serine residue are always clustered about the active site depression. •The shape and charge of the "pocket," however, vary between different serine proteases. Thus, it is the nature of the pocket that gives a serine protease its specificity. For example, in chymotrypsin, the pocket is wide and lined with hydrophobic residues to accommodate a hydrophobic side chain, such as phenylalanine. Substrate specificity b. Evolutionary relationships among serine proteases • The sequence and structural homologies among proteins reveal their evolutionary relationships. • The greater similarities among chymotrypsin, trypsin, and elastase indicate that these proteins evolved through gene duplications of an ancestral serine protease followed by the divergent evolution of the resulting enzymes. Relative positions of the active site residues in subtilisin, chymotrypsin, serine carboxypeptidase II, and ClpP protease. These proteins apparently constitute a remarkable example of convergent evolution: nature seems to have independently discovered the same catalytic mechanism at least four times. Catalytic mechanism The catalytic mechanism of chymotrypsin. These steps include the following: 1. Polypeptide substrate binding. 2. Proton transfer from Ser to His. The substrate forms a tetrahedral transition state with the enzyme. 3. Proton transfer to the C-terminal fragment, which is released by cleavage of the C-N bond. The N-terminal peptide is bound through acyl linkage to serine. 4. A water molecule binds to the enzyme in place of the departed polypeptide. 5. The water molecule transfers its proton to His 57. Again, a tetrahedral transition state is formed. 6. The second peptide fragment is released. The acyl bond is cleaved, the proton is tranferred from His back to Ser, and the enzyme returns to its initial state. A key to the mechanism of serine protease catalysis lies in the stability of the two tetrahedral intermediate states, which are very similar to the essential transition states. They appear to be stabilized by hydrogen bonds from backbone amino protons from residues Ser 195 and Gly 193 to one of the oxygens in the tetrahedral complex (the carbonyl oxygen of the substrate). The hydrogen bonding can occur only with formation of the tetrahedral state and thus stabilizes the intermediates. Serine proteases have a histidine and an acidic residue in their active site. Histidine is very common in active sites, because it readily accepts or donates protons at physiological pH. Catalysis of peptide bond hydrolysis by chymotrypsin. Catalytic mechanism of the serine proteases. Catalytic mechanism of serine proteases D. Testing the catalytic mechanism a. The tetrahedral intermediate is mimicked in a complex of trypsin with trypsin inhibitor (BPTI) Trypsin–BPTI complex. (a) The X-ray structure shown as a cutaway surface drawing indicating how trypsin (red) binds BPTI (green). (b) Trypsin Ser 195, the active Ser, is in closer-than-van der Waals contact with the carbonyl carbon of BPTI’s scissile peptide. b. Serine protease preferentially bind the transition state The conformational distortion that occurs with the formation of the tetrahedral intermediate causes the carbonyl oxygen of the scissile peptide to mover deeper onto the active site so as to occupy a previously unoccupied position, the oxyanion hole. Transition state stabilization in the serine proteases. (a) The Michaelis complex. (b) The tetrahedral intermediate. c. The tetrahedral intermediate and the water molecule attacking the acylenzyme intermediate have been directly observed. X-Ray structures of porcine pancreatic elastase in complex with the heptapeptide BCM7 (YPFVEPI). (a) The complex at pH 5. (b) The complex at pH 9. d. The role of the catalytic triad: low-barrier hydrogen bonds. Implausible? The hydrogen atom becomes more or less equally shared between donor and acceptor, when pK’s of both are nearly equal. (D H---A) In nonaqueous solution or in enzyme active site, LBHB can occur. e. Much of a serine protease’s catalytic activity arises from preferential transition state binding. 1010 106 104 Blocking all catalytic triad As a consequence, a large portion of chymotrypsin’s rate enhancement must be attributed to its preferential binding of the catalyzed reaction’s transition state. Zymogen Activation of trypsinogen to form trypsin. Activation of chymotrypsinogen by proteolytic cleavage. Hypothetical QSAR plots of log(1/C) versus log P for a series of related compounds. (a) A plot that is best described by a linear equation. Hypothetical QSAR plots of log(1/C) versus log P for a series of related compounds. (b) A plot that is best described by a quadratic equation. The combinatorial synthesis of arylidene diamides. X-Ray structure of cytochrome P450CAM from Pseudomonas putida showing its active site region. The metabolic reactions of acetaminophen that convert it to its conjugate with glutathione. The assembly, budding, and maturation of HIV-1. HIV-1 polyproteins. (a) The organization of the HIV-1 gag and gag–pol polyproteins. HIV-1 polyproteins. (b) The sequences flanking the HIV-1 protease cleavage sites (red bonds) indicated in Part a. Renin participation in blood pressure regulation. X-Ray structure of pepsin. (a) Ribbon diagram. X-Ray structure of pepsin. (b) Enlarged view of the active site Asp residues and their bound water molecule indicating the lengths (in Å) of possible hydrogen bonds (gray). Catalytic mechanism of aspartic proteases. X-Ray structure of HIV-1 protease. (a) Uncomplexed. X-Ray structure of HIV-1 protease. (b) In complex with its inhibitor. Arrangement of hydrogen bonds between HIV-1 protease and a modeled substrate. Comparison of a normal peptide bond to several groups (red) that are similar to the tetrahedral intermediate of aspartic proteases. Some HIV-1 protease inhibitors that are in clinical use. Mechanism of carboxypeptidase A.