Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
XV–74
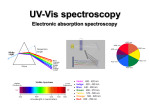
UV-vis (Electronic) Spectra-2014 -Ch.13 Atkins, Ch.19 Engel
Most broadly used analytical tech / especially bio-applic.
inexpensive optics / solvent & cell usually not problem
intense transitions sensitive, low concentrations
broader transitions – mix in vibrational excitation / low res.
Optical Spectroscopy Processes diagram
But some molecules “don’t absorb” in UV-region >200nm
all absorb in vac. UV (<200nm) e.g. salts, ions, saturated
molecules: hydrocarbons, sugars, alcohols, etc.
UV - -systems, open shells, broad – less detail structure
many do not fluoresce, compete paths for energy transfer
XV–75
Basic idea – excite electrons to a new state
Thus - new potential surface, i.e. vibrations will differ
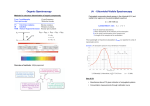
Franck-Condon Principle
“vertical transitions”
Nuclear motion slow compared
to transition time
effectively “frozen” nuclei
In excited state, molecule first
relaxes to new equilibrium
structure, then fluoresces
Vibrational energy goes to solvent
“vibrational relaxation”
Mirror image spectra
A – absorbance
F – fluorescence
broad bands many component
“Vibronic” transitions:
ge g exe ex
vibronic overlap often
unresolved
Born-Oppenheimer, separate
integrals: elec(r) and nucl(R)
Intensity (A or F) ~ De-g = ex* g d 2
(Dipole strength)
= (exe ex* ge g d 2
=(exe*ge dr2(ex*g dR2
integrated
distribution over vib
intensity
F-C factor-vertical trans
XV–76
F-C allowed transitions
“Vertical” excitation of electrons,
means nuclei stay near minimum of
originating surface. Favor vibrations
at turning point reference to minimum
of other state. Multiple vibrations get
excited but with different frequencies,
relative intensity given by square of
overlap of vibrational functions, initial
and final states F-C envelop
Potential energy surfaces shapes Atkins above, Engel (p.459-60)
Top, left: Vibration Spacing reflect: A excited, F ground state
Bottom: bigger potential shift, more distribution,
eventually get continuum (right, structureless—dissociate)
XV–77
Shift of potential surfaces
reflected in F-C bandshape, excitation
to continuum, broad structureless,
dissociating state
Gap - Absorb and Fluor
shift, different geometry
vibs closer, bond strength
Molecule - electronic energies change with nuclear positions,
and gives rise to different
vibrational levels
Ex. Potential energies of I2
electronic statesMany states, not all transitions
seen – selection rules
Plus each has own vibration
energies
XV–78
Absorbance
A = -log10 I/I0 = b c {b – path, c – conc.
– molecular property relate to dipole strength D
QM link: Intensity - A ~ D10 = 1* 0 d 2
Electronic Spectra – Broad - vibrations couple electronic
Spectra reflect: h = E
a) change electronic energies
Eel = E1 – E0
b) change of vibration
(note: frequencies differ)
Evib = (e+½)he – (g+½)hg
initial state – typically g = 0
but small g or high T “hot band”
absorb from g 0
most probable “vertical transition”
(Franck-Condon)
Fluorescence – if relax to e = 0 then can emit photon
Can be mirror image of Absorption, but fluorescence
Vibrational progression reflects lower state
Intensity - IF ~ D01 same probability as absorbance
vibronic pattern differ – spacing g
linear: measure IF ~ Iexcite (if excite by absorption)
but measure fluor. signal against null background
extremely sensitive / can even do single molecule
[Problem – other relaxation limit quantum yield]
XV–79
Ex. absorption/fluorescence spectra –vertical surface
Selection rules
—less simple than for rotations and vibrations
a. Molecule must change dipole moment, normally
change electronic states where charge is dislocated
(if center of symmetry gu allowed, polyatomic use symmetry)
b. Spin not affected by E-field (light) – S = 0
c. Between states, vibrations change - v = 0, ±1, ±2, . .
But rotations restricted: J = 0, ±1
XV–80
What kind of molecules have measurable Absorbance?
a. All absorb vacuum UV ( < 200 nm , > 50,000 cm-1)
everything eventually (shorter ) absorbs
Closed shell, saturated, light atoms only at higher (vacUV)
e.g.:
H2O , MeOH
-- closed shells, saturated
CnH2n+2 , CnH2n-m Fm+2
-- light atoms
LiF , CaF2
-- salts
He, Ne, Ar
– rare gas
b. UV (ultraviolet) (: 200-400 nm, = 50-25,000 cm-1)
big contribution are -systems
aromatics, polyenes, conjugated
O
O
hetero atom:
O
+ lone pair delocalize
N
O
H
plus heavier atom systems
S S
C I
… (also Cl-, Br - . . .)
c. in Visible (: 400-700 nm , = 25,000-14,000 cm-1)
need very delocalized system (-electron)
N
N +
N
N
N
retinal
(off a bit)
dyes are like this-aromatic
porphyrin
_
or open shells – radicals
transition metal Fe(CN)6-3 , CuII(SH)2(NH3)2 etc.
complexes :
red
blue
d. near-IR (: 700-2500 nm , = 14,000-4,000 cm-1)
mostly transition metals (d-d), open shells, NO, 1O2
N O
XV–81
Benzene electronic spectra – * -displaced surfaces
vibronic progressions, vi = ±1, ±2,… totally sym. modes,
for first trans. forbidden, build on four asym modes vj = ±1
allowed transition A1gE1u at <200nm (intense, ~105),
Triplet trans. at ~330 nm, S=1 forbidden (very weak, ~10-3)
XV–82
XV–83
Comparison of porphyrin and hemoglobin absorb. with O2 & CO
Rhodopsin visible
absorbance in
dark and changes
after exposure
and adding
11-cis-retinal
XV–84
Transition metal complexes – open shells, visible absorb
dd transitions are weak because l ~ 0
Mn+2 - d5, ground state - 6A1g (6S related) –
absorbance very weak, S≠0