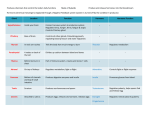
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
PART 19 ENDOCRINE DISORDERS 19.1 Growth and variations in growth J. Batch Growth is the process in our lives that is unique to paediatric and adolescent medicine and distinguishes the health of children and young people from all other branches of clinical medicine. Growth is a multifactorial process and is influenced by the interplay of genetic, nutritional, hormonal, psychosocial and other factors, including the general health of a child. As such, growth mirrors the psychosocial and physical wellbeing of a child and adolescent. The study of normal and abnormal growth is facilitated by a knowledge of the effects of physiological and pathological processes on growth and development at the different stages of life. The three major determinants of growth are: • genetic factors • nutritional factors • hormonal factors. Genetic factors The genetic background of individuals is the basis for the major determinant of growth potential: tall parents tend to have tall children, while short parents have short children. Although children’s heights at maturity resemble those of their parents, little is known about the exact location of the individual height controlling genes, how many genes are involved or how they direct cellular growth. Major genetic disturbances such as occur with chromosomal abnormalities are often reflected in growth patterns. For example, with loss of a sex chromosome in 45,XO Turner syndrome as shown in Figure 19.1.1 (see also Ch. 10.3), adult stature is severely compromised. Other less severe chromosomal abnormalities also may result in abnormalities in stature. Many inherited genetic conditions can also result in growth disturbance. The most striking of these are the skeletal dysplasias, which often follow an autosomal dominant mode of inheritance. The classic example of a skeletal dysplasia is achondroplasia, which is described in Chapter 10.3. Chromosomes may also influence tall stature, as seen in the case of an individual with an extra sex chromosome, for example Klinefelter syndrome XXY, which frequently leads to an adult height above that anticipated from the family pattern. Nutritional factors Nutrition is the second most important factor determining normal growth in childhood and adolescence. In a global sense, malnutrition is the world’s primary cause of poor growth. Both undernutrition and overnutrition may have long-lasting effects on growth patterns. Undernutrition, particularly if it occurs in utero or at significant postnatal periods, may affect both the weight and height growth patterns and also the development of body organs. In utero, undernutrition has also been associated with increased long-term risks of cardiovascular morbidity and mortality in children who are born small for dates. Undernutrition later in life is complicated by the interaction between the quality and the quantity of the diet and the duration of dietary inadequacy. Emotional deprivation also has a profound influence on the growth process and may interact with provision of food. Furthermore, the mechanism of poor growth in many chronic illnesses of childhood is at least in part due to undernutrition or to nutritional influences on the balance of hormones and growth factors. Overnutrition may lead to obesity with advanced linear growth and early pubertal maturation. Overnutrition at a time when linear growth is declining in late adolescence may lead to lifelong obesity with the attendant risks of hypertension, insulin resistance and the development of type 2 diabetes. Hormonal factors Those of significance in growth are: • • • • growth hormone thyroid hormone testosterone and adrenal androgens oestrogens. Growth hormone–insulin-like growth factor I axis The major hormonal influence involved in growth regulation at all ages is the growth hormone–insulin-like growth factor 1 axis (GH–IGF-I). Growth hormone is secreted by the anterior pituitary gland in a pulsatile pattern. Major peaks of secretion occur particularly at night. Growth hormone is bound to a specific growth hormone binding protein and subsequently acts on a broad range of tissues via cell surface receptors, resulting in a range of metabolic and growth-related effects. Many of these effects are mediated via IGF-I, which is produced by the liver and many other tissues (Fig. 19.1.2). The secreted IGF-I then acts either locally on adjacent tissues (paracrine action) or via endocrine mechanisms (i.e. via the circulation). IGF-I levels are age-dependent, being low in the fetus, rising through infancy and childhood, peaking during puberty and then falling to adult levels. IGF-I is very sensitive to nutritional status and in itself is of limited diagnostic value in the assessment of short stature. It circulates bound to one of its major binding proteins (IGFBPs). Thyroid hormone Thyroxine is very important for postnatal growth. Children with untreated hypothyroidism show profound both mental and growth retardation, with very delayed bony development. In many developed countries, neonatal thyroid screening programmes have been set up and treatment of congenital hypothyroidism ensures normal growth and intellectual development. Testosterone and adrenal androgens These hormones are anabolic and growth promoting. In males, testosterone and growth hormone act synergistically to promote the adolescent growth spurt. Excess androgens in childhood due to exogenous treatment, arising from adrenal enzyme disorders (congenital adrenal hyperplasia), precocious puberty or tumours will rapidly cause bone age advancement and may limit the potential for final height. Oestrogens Oestrogens in small doses may synergize with growth hormone to cause growth promotion. In higher doses, oestrogens will inhibit growth and promote early fusion of the bony epiphyses. Phases of growth There are three main phases of growth: fetal and childhood growth, and the pubertal growth spurt. Fetal growth Fetal growth is the most rapid phase of growth. Fetal growth is characterized by rapid differentiation of body organs, while late fetal growth involves continued rapid enlargement in tissues and organs and growth in length. The most rapid linear growth velocity of all ages occurs in the weeks before birth. Factors controlling fetal growth include placental supply of nutrients and oxygen and a range of local growth factors including insulinlike growth factors (IGFs). Pituitary growth hormone probably plays a relatively small part in this phase of growth, while thyroxine is involved in brain and bone growth in the fetus. Pituitary gonadotrophins (luteinizing hormone (LH), follicle stimulating hormone (FSH)) regulate testicular testosterone synthesis in the male fetus, which is essential for normal growth of the male phallus. Thus a male infant with hypopituitarism may have a micropenis at birth. Childhood growth During the first years of life, linear growth velocity is still very rapid (on average 8–12 cm/year) but it plateaus through childhood to an average of approximately 5 cm/year. The growth velocity immediately before the prepubertal growth spurt may be lower than this and represents a transient phase of poor growth. If the onset of the pubertal growth spurt is delayed then this phase of poor growth may be prolonged. During the childhood growth phase the limbs grow faster than the trunk, so that the ratio of the upper to lower body segments (divided at the pubic symphysis) diminishes from approximately 1.7:1 during infancy to 1:1 by age 10. It may fall to around 0.8 by mid puberty. The span:height ratio remains unchanged at approximately 1:1. Factors controlling this phase of growth include genetic determinants, nutrition, absence of chronic disease and hormones, the most important of which are growth hormone and thyroxine. Pubertal growth spurt Puberty is associated with the onset of sex hormone production in boys and girls under the influence of pulsatile release of gonadotrophins (FSH/LH) from the pituitary gland. The earliest sign of puberty in boys, usually occurring at an average age of 11 years, is testicular enlargement (volume greater than 3–4 ml measured with an orchidometer). Penile and scrotal growth follow, with development of pubic and axillary hair in response to testosterone synthesis. In girls, ovarian oestrogen secretion leads to the earliest pubertal sign of breast development at an average age of 10.5–11 years, followed by pubic and axillary hair growth in response to adrenal and ovarian androgens. In boys, testosterone also leads to muscle growth, while in girls, oestrogens cause pelvic broadening and fat redistribution, leading to a female body shape. In both sexes, the onset of puberty is followed by a peak linear growth velocity, at an average age of 12.5 years in girls and 14.5 years in boys. The hormonal events of puberty include an increase in the amplitude of growth hormone pulses, probably due to sex hormone effects. IGF-I levels rise during puberty in association with the high growth hormone levels. The sex hormones (testosterone or oestrogens) also appear to have direct effects at the skeletal growth plate, ultimately leading to fusion of the bony epiphyses and cessation of growth at an average age of 15 years in girls and 17 years in boys. The pubertal growth spurt may be influenced by genetic factors and also may be affected adversely by poor nutrition or chronic disease, both of which can cause pubertal delay. Practical points Puberty • The earliest sign of puberty in females is breast budding at an average age of 10.5–11.0 years • The earliest sign of puberty in males is testicular enlargement, at an average age of 11 years • The pubertal growth spurt occurs at an average age of 12.5 years in females, and 14.5 years in males • The growth spurt in puberty is the most rapid phase of postnatal growth Assessment of growth Percentile charts Any health professional who deals with children must have a working knowledge of normal variations in growth and development and must be able to use a percentile chart. The childhood and pubertal growth patterns can be appreciated by examining growth charts, including linear height and weight charts (Fig. 19.1.3) as well as height velocity charts, indicating annual rate of growth (Fig. 19.1.4). These charts demonstrate the range of normal growth, expressed either as percentiles or as standard deviations (SD) from the mean for age. The percentile curves are derived from the overall distribution (bell-shaped curve) of the data. The median or mid point is the 50th percentile and the normal range of average height or weight on the charts falls between the 3rd and 97th percentiles (Fig. 19.1.3). The median or 50th percentile indicates that 50% of the measurements of a normal group of children are above and 50% are below that point. The 50th centile ‘final’ height value for males is 177 cm and for females is 164 cm. The range between the 3rd and 97th (or approximately 2 SD to 2 SD) includes 94% of all normal children. It must be realized that there will be three normal children in every 100 who will be at or below the 3rd centile and three in every 100 who will be at or above the 97th centile. Assessment of growth velocity (Fig. 19.1.4) is of far greater clinical significance than single measurements of height, and should be based on sequential measurements taken at 3-monthly intervals during a period of 6–12 months. When measured over this time period, a normal child will tend to follow the same height percentile. A child with an organic or endocrine disease will tend to deviate away from the percentile line and may move downwards across percentile lines. Thus serial measurement of children is the key to the assessment of their growth status. Practical points Growth • There is a wide variation of normal growth • 3% of normal children will be above the 97th percentile or below the 3rd percentile • Assessment of growth velocity (growth over time) is of more value than a single growth measurement • The average growth rate during childhood is 5 cm/year Bone age Bone age is an index of physiological maturity, indicating the state of bony epiphysial maturation. A bone age is obtained by performing an X-ray of the left wrist and hand and is interpreted according to an atlas of age- and sex-specific standards. The bone age indicates the average age of children at a similar stage of bony maturation and is a guide to the remaining growth potential of the child. In normal children the bone age can be delayed up to 2 years behind the chronological age but it is rarely advanced a year beyond the chronological age. Height age Height age is the age at which a child’s current height would represent the 50th percentile. This means that the height age indicates the average age of children of a similar height. Midparental height The midparental height (MPH), also known as the target height, allows the height of any individual child to be considered in relation to the heights of his/her biological parents. The midparental height can be calculated using the following formulae: For boys: MPH (father’s height (mother’s height 13))/2, 7.5 cm For girls: MPH (mother’s height (father’s height 13))/2, 6 cm. A child should not be assumed to be short ‘because s/he has a short family’ unless the actual height of the child has been plotted on the growth chart and found to be consistent with the midparental height outlined above. Short stature The management of a child with short stature requires consideration of a number of issues. It is important to realize that the majority of short children will have no pathology but will either be following a familial pattern or have a variant of normal growth. The main causes of short stature in order of frequency of diagnosis are summarized in Table 19.1.1. As can be seen from the table, endocrine causes of short stature are the least common. Practical points Short stature • The majority of short children are normal and healthy • The most common causes of short stature are familial short stature and constitutional delay of growth • Chronic disease is a major cause of short stature and is characterized by a fall off in both height and weight velocity Variations from normal Familial (genetic) short stature Important features of familial short stature are as follows: • these children will be growing on the 3rd centile or below, but the rate of growth is parallel to the 3rd centile • the growth rate (growth velocity) is usually normal. • the adult height percentiles of both parents should be plotted on the child’s growth chart to assess whether the child’s height is appropriate for the heights of the parents • pubertal development usually occurs at the appropriate time • markers of physical maturation such as bone age tend to be consistent with chronological age. Constitutional delay in growth and pubertal development Constitutional delay in growth and pubertal development is a very common variation of growth and leads to short stature during childhood with a height prognosis consistent with the midparental height expectation. Important features are: • affects boys far more commonly than girls • often there is a family history of a parent being short as a child, with delayed puberty and eventual catch-up with peers • these children are the so called ‘slow growers and late bloomers’. The typical growth pattern shows a growth rate that is mostly normal except for a period of 6–12 months in the first 2 years of life, when the growth rate falls transiently • the nadir in the growth velocity prior to puberty may be exaggerated, with the delayed appearance of pubertal development • markers of physical maturation such as bone age are delayed and are consistent with the height age, indicating a true constitutional delay in growth • the delay in puberty and associated delay in closure of bony epiphyses means that both the pubertal growth spurt and the completion of growth will be slowed according to the delay in bone age • these children (most often boys) tend to grow into their late teenage years or early twenties. The remaining causes of short stature are all pathological causes. The differences in growth patterns between genetic short stature, constitutional delay and pathological short stature are summarized in Figure 19.1.5. The relationship between chronological age, bone age and height age for common growth abnormalities is summarized in Table 19.1.2. Pathological causes Intrauterine growth retardation This results in growth-retarded babies of appropriate gestational age. Major causes include a variety of genetic disorders primarily affecting the fetus. An abnormal intrauterine environment due to maternal illness, placental insufficiency or intrauterine infection may also lead to poor fetal growth. If the insult to the fetus occurs early in gestation, cell number is reduced and the potential for catch up is diminished. Chronic disease Chronic disease is a major cause of growth failure: • usually the growth failure is associated with a similar fall off in weight velocity • Chronic infections may also present as failure to grow and gain weight • A hormone or endocrine problem is unlikely to be the cause of poor growth if both the weight and height are affected • Nutritional insufficiency may contribute to the growth failure of chronic disease due to inadequate or inappropriate intake, poor absorption or impaired or excessive tissue utilization. Skeletal disorders Skeletal disorders are usually familial, involving intrinsic cartilage or bone defects. Examples include achondroplasia (the classic dwarf), other milder bony dysplasias (particularly hypochondroplasia) and the mucopolysaccharidoses. The major distinctive features of all of these disorders is body segment disproportion, i.e. increased upper to lower body segment ratios. The limbs are usually short, leading to a reduced span:height ratio. Weight gain is usually normal. These disorders in their mild form are relatively common and often overlooked. More information is given in Chapter 10.3. Iatrogenic Iatrogenic causes include corticosteroid excess, seen in children with steroid-treated chronic illnesses, such as severe asthma, juvenile arthritis and nephrotic syndrome. Marked growth failure is associated with weight gain. Irradiation to the head and spine may result in hypothalamic–pituitary dysfunction and poor spinal growth, which may lead to poor growth of the trunk with increased span:height ratio and a reduced upper to lower body segment ratio. Chromosomal abnormalities and syndromes Turner syndrome and its variants are the most common chromosomal cause of short stature. This condition must be excluded in any girl with short stature, as the typical phenotypic features may not be seen, particularly in the mosaic forms of Turner syndrome. The most common associated abnormality is ovarian dysgenesis resulting in failed pubertal development in 95% of cases. Other commonly seen features include a webbed neck (Fig. 19.1.1), small ears, increased carrying angle, coarctation of the aorta, horseshoe kidney, dysplastic nails and recurrent otitis media. All or none of these dysmorphic and clinical features may be present. The full range of features is summarized in Table 19.1.3. Other chromosomal disorders are less common causes of short stature. Common dysmorphic syndromes presenting with short stature include Noonan syndrome, intrauterine growth retardation (Russell–Silver dwarfism) and Aarskog syndrome. Further details of these conditions are found in Chapter 10.3. Psychosocial Psychosocial causes of short stature cover the spectrum from severe deprivation to overt abuse, and may be associated with nutritional deficiencies. Fall off in weight gain is usually as striking as failure of linear growth. Short stature due solely to psychosocial deprivation is uncommon. Clinical example Tahlia’s parents were concerned about her growth. Tahlia was aged 7 years and had an average birth weight and length but, since she had gone to school, her mother had noticed that she was one of the smaller girls in her class, and she did not seem to be growing as fast as the other children. Tahlia had several urinary tract infections as a toddler, and an ultrasound done after these had shown that she had a single horseshoe kidney. She also had recurrent middle ear infections, and her general practitioner noted that she had a heart murmur. An echocardiogram by a paediatric cardiologist showed that Tahlia had a bicuspid aortic valve. Tahlia had no other medical problems. Tahlia’s height at 7 years of age was 6 cm below the 3rd percentile and well below her midparental height range. Unfortunately despite having seen several doctors over the years, no past height measurements had been recorded. Tahlia’s growth progress was monitored during a 6-month interval, during which time she only grew 1.6 cm (annualized growth velocity 3.2 cm). Her position on the growth chart moved further away from the 3rd percentile. Tahlia’s poor growth was investigated when she was 7.5 years of age. A full blood count and erythrocyte sedimentation ratio (ESR) and a biochemical screen were normal. She was euthyroid and had a negative coeliac screen. A bone age X-ray showed a bone age of 4 years, well below her chronological age. Several weeks later the chromosome result became available, and showed that Tahlia had mosaic Turner syndrome. Tahlia and her parents were referred to a paediatric endocrinologist, who advised them that Tahlia’s growth could be improved with growth hormone injections, and that it was very likely that she would need hormonal induction of puberty and would have later infertility due to the presence of gonadal (ovarian) dysgenesis as part of Turner syndrome. They were also informed that Tahlia would need ongoing medical care into adult life to monitor for other possible health problems, including hypertension, dyslipidaemia and type 2 diabetes, all of which occur more commonly in individuals with Turner syndrome. Endocrine Endocrine causes of short stature are the least common pathological cause and include hypothyroidism, growth hormone deficiency (possibly associated with other pituitary hormone deficiencies), Cushing syndrome (hypercortisolism) and adrenal insufficiency. In all of these cases the fall-off in linear growth exceeds the fall in weight gain. Assessment Issues to determine Assessment of short stature involves determination of the following issues: • • • • • • • is s/he short? is s/he growing slowly (could this be pathological)? what is the underlying cause? what is the adult height prognosis? how is s/he coping with the short stature? is any specific therapy warranted? is any supportive therapy indicated? The approach to the assessment of short stature should include history, examination, investigations if necessary, therapy and follow-up. History When taking the history, the following should be sought: • what is the height compared to peers? How long has the child been short? Who is concerned about the short stature and is there teasing at school? What is the school performance? • what are the birth details and past medical history? Was there unexplained neonatal hypoglycaemia (suggesting pituitary hormone deficiency) or early illnesses? Determine the dental and milestone development, specific disease symptoms and nutritional status. Has puberty commenced? Are previous growth measurements available (child health record or measurements from local doctor or school)? • what are the heights and ages of pubertal onset of the parents and siblings? Is there a family history of specific diseases? Examination On examination ensure/look for: • accurate height and weight (using a reliable measuring device, particularly for height); body proportions (span, upper and lower segments) • assessment of pubertal status. Pubertal stages for boys and girls are summarized in Table 19.1.4. The characteristic pubertal changes in males and females are illustrated in Figures 19.1.6 and 19.1.7. Further issues regarding puberty will be considered later in this section • general physical examination including evidence of chronic disease, nutritional state and dysmorphic features suggesting a syndrome • any sign of goitre or clinical signs of hypothyroidism, including dry hair and skin, bradycardia and delayed reflexes • evidence of ‘midline brain development syndromes’ which may result in hypopituitarism. This includes cleft palate, single central incisor and small male genitalia (associated with gonadotrophin deficiency in utero). The combination of neonatal hypoglycaemia and small genitalia suggests hypopituitarism • examination of visual fields and optic fundi to exclude the possibility of a pituitary lesion, in particular craniopharyngioma. Management The single most important aspect of the management of short stature is to plot the current and previous heights and weights and parental heights on a percentile chart in order to answer the following questions: • is the child short and below the 3rd height centile? Is this appropriate for midparental height? • is the child growing slowly and is there evidence that the height is falling across the percentile lines? This can be further plotted on a height velocity chart (Fig. 19.1.4). A velocity below the 25th centile for bone age is potentially abnormal in a short child. A reliable height velocity requires at least 6 months of growth data, and preferably 12 months with consistent measurements at 3–4monthly intervals over that time. Examination of the growth data plus the points obtained in history and examination should allow distinction between a variation of normal or a pathological cause of short stature. Investigations Investigations should be performed if there is any evidence of specific chronic disease, if there is a suggestion of chromosomal abnormality or if the growth velocity is subnormal. The following investigations may be performed: • • • • • • • • bone age X-ray full blood count and ESR urea, creatinine and electrolytes urinalysis microscopy and culture calcium and phosphate thyroid function tests chromosomes (girls only) screening test for coeliac disease (total IgA and endomysial antibodies or tissue transglutaminase antibodies). It is important to note that all girls with unexplained short stature should have a karyotype performed to exclude the possibility of Turner syndrome. If puberty is markedly delayed it may be worthwhile measuring gonadotrophins (FSH/LH) and testosterone or estradiol. These investigations provide a screen for underlying chronic disease, infection or nutritional deficiency as well as hypothyroidism and Turner syndrome. Other investigations may be indicated by specific physical findings. In a child with unexplained combined weight and height fall-off, a malabsorptive disorder should be excluded and consideration should be given to measuring endomysial antibodies as a possible indicator of the presence of coeliac disease. If these are elevated, a small bowel biopsy may be necessary. Investigation for growth hormone deficiency Growth hormone deficiency may be suggested by association with midline defects or if other pituitary hormone deficiencies, including hypothyroidism or gonadotrophin deficiency, are present. The presence of biochemical growth hormone deficiency suggests a need for magnetic resonance imaging (MRI) of the central nervous system (CNS) and biochemical testing for other pituitary deficits. Specific underlying causes of growth hormone deficiency include idiopathic (most common), pituitary or hypothalamic tumours/structural abnormalities, cranial irradiation and genetic growth hormone deficiency (e.g. gene defects of pituitary transcription factors). Specific tests to determine the presence of growth hormone deficiency include: Clinical example Ben was a 14-year-old boy whose mother complained that he did not seem to be growing at all. She said to her general practitioner that she had not needed to buy Ben any new clothes for the past 3 years. She remembered that her older son was always growing out of his clothes at this age. She was also concerned because Ben did not seem to have as much stamina as other boys his age, and certainly he ate much less than she would have expected at this age, based on her experience with her older son. Further history revealed that Ben had always been a very healthy boy in the past, with no significant medical problems. He had become a fussy eater during the past couple of years. He said that many foods he had previously eaten now made him feel sick in the stomach and gave him stomach cramps. When asked if he had diarrhoea, Ben said ‘no’, but when asked how many times he went to the toilet he admitted that he needed to go at least 6 times per day, and that this pattern had only developed in the last year. Ben said he thought this was normal. Physical examination confirmed that Ben’s height and weight were almost the same as when he was 11 years old. Apart from being a little bit pale, there were no other abnormal physical findings on general examination. Ben was prepubertal. Baseline investigations included a full blood count and ESR. These showed that Ben had a low haemoglobin and his ESR was 79. All the other baseline tests including electrolytes, liver function tests, renal function, screening tests for coeliac disease and thyroid function tests were normal. Ben’s bone age was very delayed. At 14 years of age his bone age was only 10 years. The history of poor growth and the abdominal symptoms together with the low haemoglobin and raised ESR suggested the possibility of a chronic gastrointestinal problem such as inflammatory bowel disease. Ben was referred to a paediatric gastroenterologist and had an endoscopy and colonoscopy, which demonstrated that he had Crohn disease. Ben was treated with steroids and salazopyrine for his Crohn disease and his symptoms improved. He started to gain weight and over the next 12 months his growth also improved. • physiological tests of growth hormone sufficiency, including: • exercise growth hormone (GH) tests: performed in the fasting state on a bicycle ergometer with blood for GH levels taken before and 30 minutes after exercise. This is a screening test with 20% of normal children failing to reach cut off levels • overnight sleep studies of growth hormone secretion • measurement of IGF-I and IGFBP-3 levels. This test is used for screening for GH deficiency prior to proceeding to more involved pharmacological tests. As yet the data are not sufficiently reliable to recommend that these tests be performed as a single diagnostic entity in a clinical setting. They may, however, be useful when considered in combination with other clinical information, results of MRI brain scans and growth hormone tests • pharmacological tests of growth hormone sufficiency, including: • glucagon stimulation test • arginine–insulin hypoglycaemia test • clonidine stimulation test. Although no one test is superior to another, the glucagon stimulation test is at present the preferred pharmacological test of GH secretion. The glucagon stimulation test also provides information about the adrenocorticotrophin–cortisol axis. The arginine–insulin hypoglycaemia test is used rarely in paediatric practice as hypoglycaemia is an obligate part of this test and is potentially dangerous. For the purposes of defining GH deficiency and assessing eligibility for hGH as a pharmaceutical benefit in Australia, biochemical growth hormone deficiency is defined as failure to achieve a peak serum GH concentration of more than 10 mU/l in response to two stimulation tests, at least one of which would be a pharmacological test, or in response to one test in the presence of other evidence suggestive of GH deficiency, such as structural CNS abnormalities or low plasma IGF-I and IGFBP-3 levels. Treatment Short stature is considered by some children and their families to be a physical and psychosocial disability. Extreme short stature can certainly be considered as a handicap in both a social and medical sense. Many paediatricians and paediatric endocrinologists consider that, if the estimated final height of a female will be less than 152.4 cm (5 ft) or a male less than 162.6 cm (5 ft 4 in), then consideration should be given to the use of a growth promoting agent. The major growth promoting agent used in the treatment of short stature is biosynthetic growth hormone. Biosynthetic growth hormone has been available commercially in Australia since 1985. In 1988, the Guidelines for the Use of Growth Hormone in Australia were liberalized, allowing the use of growth hormone in short children who are growing poorly but are not growth-hormone-deficient. Growth hormone can be administered only by subcutaneous injections, usually given 6–7 days per week. Despite the availability of biosynthetic growth hormone, the annual cost of growth hormone therapy remains very high. Growth hormone should therefore only be used for children who have short stature and who could potentially benefit from the therapy. The Commonwealth Department of Health and Family Services has provided guidelines for the use of growth hormone in Australia. These can be summarized as follows: A child must have abnormally short stature (height less than the 1st percentile) with an abnormally low growth rate, measured over a minimum period of 1 year at intervals of not greater than 6 months. The child should be growing at a height velocity below the 25th centile for skeletal age and sex. Variations to these guidelines exist for particular groups of patients, such as Turner syndrome, growth hormone deficiency, combined growth hormone deficiency and precocious puberty, chronic renal insufficiency and those patients with growth retardation secondary to an intracranial lesion or cranial irradiation. Growth hormone treatment has been shown to be of benefit in the following conditions: • growth hormone deficiency • Turner syndrome (final height can be improved by up to 8–10 cm) • growth retardation secondary to renal insufficiency. In general, results of growth hormone therapy for short stature associated with constitutional delay, intrauterine growth retardation, glucocorticoid-induced growth failure, chromosomal and genetic disorders and skeletal dysplasias have been less promising. Although the definite indications for growth hormone treatment of short stature are limited, any child who is short and growing slowly with a poor ultimate height prognosis should be referred for assessment. After assessment it may be appropriate to offer the child a trial of growth hormone therapy. Other specific agents used in the treatment of short stature associated with constitutional delay have included oxandrolone (a biosynthetic testosterone analogue) and low dose oral testosterone preparations. Psychological support and counselling Psychological support and counselling are undoubtedly the most important part of the management of short stature and can be provided by either health professionals or lay support groups. Often reassurance regarding the normality of the child and the reassurance of a reasonable height prognosis is all that is required. If the height prognosis is poor, then counselling and support should be used in conjunction with growth promoting agents. Families should be advised to encourage self esteem in the child by promoting the child’s strengths, for example, sporting, musical or academic, rather than concentrating on the perceived limitations imposed by short stature. Lay support groups associated with growth hormone deficiency or Turner syndrome are now quite common in most large cities, and have active programmes for children and their families. On occasions it may be necessary to seek formal psychological or psychiatric help for a child or family who are very distressed by the problem of short stature. Tall stature Tall stature is a relatively infrequent problem compared with the number of children who present because of short stature. In general, very few tall children have an organic disease process as a basis for their disease. The most common reason for tall stature is genetic tall stature. Compared to 20 years ago, relatively few teenagers and their parents are concerned about tall stature, as it is now more socially acceptable for girls to be tall. However, girls may be presented for assessment of their final height if it is thought that they will be in excess of 178 cm (5 ft 10 in). A final height of 183 cm (6 ft) may be acceptable for a girl if her parents and other siblings are also tall. As with short stature, there is no clear demarcation between normal and tall stature. A child whose height is above the 97th percentile for age should be considered tall. Causes The following may be causes of tall stature: • • • • familial or normal variant precocious puberty syndromes: Marfan, Klinefelter, triple X, homocystinuria, Sotos endocrine causes: hyperthyroidism and pituitary gigantism. Familial/normal variant tall stature Most tall children are normal in all respects and their height is genetically determined. Some children with early but otherwise normal pubertal development (8–10 years in girls, 10–12 years in boys) will appear tall in relation to their peers and family during adolescence, but will have a predicted final height within the accepted normal range. These early developers have an advanced bone age and are at the opposite end of the spectrum to the short children with delayed maturation, who have delayed puberty but who will also reach a normal adult height commensurate with their midparental height. Precocious puberty Precocious puberty is defined as pubertal development at less than 8 years of age in girls and less than 9.5 years in boys. Because of rapid acceleration of bone maturation and early epiphysial closure, many children with precocious puberty are excessively tall in early to mid childhood but finish up as relatively short adults (Fig. 19.1.8). It is important to recognize precocious puberty, as treatment can be provided to switch off the premature activation of the hypothalamic–pituitary–gonadal axis. It is also important to ascertain whether the appearance of precocious puberty may be due to an aberrant source of androgen or oestrogen production, such as an adrenal or ovarian tumour. Occasionally the appearance of precocious puberty may be due to iatrogenic causes, such as oestrogen cream application or excessive administration of anabolic steroids to improve appetite and growth. Syndromes causing tall stature Marfan syndrome may present with the classical picture of arachnodactyly, ligamentous laxity, chest deformity, cardiac abnormalities, high arched palate and subluxation of the lenses. Often, however, one sees tall, thin children with some marfanoid features who are difficult to classify. Children with homocystinuria have a marfanoid phenotype but are retarded intellectually. Homocystinuria may be diagnosed by a study of urinary and serum amino acids. Tall girls with intellectual retardation should also be screened for the triple X syndrome, and tall boys with disorders of pubertal maturation, with small testes, gynaecomastia and sometimes behavioural disturbance, should have chromosomal analysis. They may have either XYY syndrome or Klinefelter syndrome (XXY). Endocrine causes of tall stature Hyperthyroidism is relatively uncommon in childhood and is an infrequent cause of tall stature. The clinical features of hyperthyroidism are discussed in Chapter 19.2. Pituitary gigantism is extremely rare but should be suspected if the history and examination suggest pituitary involvement in association with tall stature. Children with pituitary gigantism will have an abnormally rapid growth rate as well as tall stature, in contrast to genetically tall children who grow above but parallel to the 97th centile but have a normal growth velocity. Approach to diagnosis and treatment The approach to diagnosis in a tall child consists of a full history, including a history of family heights and pubertal maturation patterns. In addition, it is important to ascertain whether there are associated abnormalities or developmental delay which may suggest one of the chromosomal or syndromal disorders causing tall stature. A full physical examination is essential, with emphasis on accurate height measurement, body build, limb proportions and pubertal status. Neurological assessment should include funduscopy, assessment of visual fields and intellectual function and should determine any evidence of hyperthyroidism. In most instances a tall child will in fact have a normal growth velocity above but parallel to the 97th centile. An accurate height measurement and bone age assessment by an experienced radiologist or paediatric endocrinologist will enable an adult height prediction to be made. Frequently this is found to be at or below 178 cm (5 ft 10 in) for a girl or 193 cm (6 ft 4 in) for a boy and no further action is necessary. In the past, sex steroids have been used to reduce the final height in both males and females where the estimate of final height was thought to be excessive. High-dose oestrogen treatment was used in tall girls in the past. However, recent studies have suggested impaired fertility in women treated with high-dose oestrogen for tall stature in adolescence, and this treatment is now rarely advocated. It should only be considered after paediatric endocrinology input and fully informed consent of the both the parent and young person. Variations of pubertal development Early normal puberty In many countries, including Australia, children appear to be going through puberty at an age which is much younger than children in previous generations. This is called the secular trend in growth and development. The earlier age of puberty is probably due to effects of improved nutrition and living circumstances and absence of chronic disease. This seems to be particularly true for girls, with many girls showing early signs of breast development just before 8 years of age and starting to have menstrual periods while still in primary school. In most cases this early puberty is just a variation of normal. After assessment by a specialist, no specific treatment is usually required. The girl and her family need to have the situation explained. Enlisting the help of the teacher is also very helpful. Delayed puberty Delayed puberty is very common and occurs in approximately 2% of the adolescent population. Delayed puberty is defined as the absence of pubertal changes over the age of 14 years for girls and over the age of 14–15 years for boys. In general, adolescents have a heightened awareness of body image and are often preoccupied with the normality or otherwise of their pubertal development. Boys in particular may suffer major psychological effects resulting from delayed puberty as they may experience bullying, may be left out of sporting teams and may be less generally able to compete with their peers due to poor muscular development. The most common causes of delayed puberty are familial or constitutional delay in puberty for which there is often a family history, particularly in the father of a teenage boy. Puberty may also be delayed in the presence of any chronic illness of childhood or adolescence. The causes of delayed puberty are usually considered on the basis of the serum gonadotrophins (LH/FSH) and are outlined in Table 19.1.5. Clinical example Sophie Jones was taken to see her general practitioner when she was 10 years old. Mrs Jones was very concerned that Sophie was too young to cope with menstrual periods, and was worried that the periods would commence very soon. Mrs Jones insisted on speaking to the doctor privately, before Sophie came into the consulting room, and she became very upset about Sophie’s development when speaking to the doctor. She recalled when she was an adolescent and had ‘terrible period pain’, which prevented her going to school and being in the netball team. Mrs Jones wanted the doctor to give Sophie medication to ‘put puberty on hold’ until Sophie was much older. When asked, Mrs Jones did say that Sophie did not seem to be upset about her development and that a few other girls in Sophie’s class at school appeared to be at a similar stage of development. Sophie was a very healthy girl who was in grade 5 at school. She had a group of friends at school, and enjoyed being part of the local Little Athletics Team. She was the eldest child in her family and had two younger brothers aged 6.5 and 4 years. She had no significant medical problems. Sophie had developed some ‘breast budding’ when she was 9.5 years old, and in the past 12 months had started using a deodorant. She also reported that she had developed some pubic hair and had a few facial blackheads. Examination showed that Sophie’s height was on the 75th percentile and her weight was on the 50th percentile for her age, and confirmed the presence of breast stage 2 and pubic hair stage 2, with some facial seborrhoea. The rest of the physical examination was normal. The doctor advised Mrs Jones that Sophie’s pubertal development was completely normal for her age and that the usual interval from the beginning of breast budding to menarche was usually 18–24 months. She also advised Mrs Jones that it would not be the best option for Sophie to suppress an otherwise normal developmental process. She advised Mrs Jones that Sophie would experience her pubertal growth spurt in the time leading up to menarche, and that Sophie’s growth would then slow down over the following 2 years until Sophie was fully grown. Given the current growth pattern, the doctor advised Mrs Jones that Sophie’s height would end up being consistent with family expectations. She also advised Mrs Jones that the best thing she could do as a mother was to calmly talk to Sophie about the physical changes of puberty, so that Sophie would understand and be prepared for her menstrual periods when they started. During the next 2 years, Sophie and Mrs Jones saw their GP every few months. Mrs Jones took her GP’s advice and, when Sophie started her periods at the age of 11.5 years, she coped very well. Diagnosis and management Assessment of delayed puberty requires a complete history, including a family history of pubertal maturation patterns. Also, it is important to look carefully for the possibility of occult chronic disease, such as inflammatory bowel disease, which may become apparent initially as a delay in the onset of puberty. If the history is suggestive of familial or constitutional delayed puberty, and this is confirmed by physical examination, no further investigation may be necessary. If the diagnosis is not clear, then the following investigations may need to be performed: • full blood count and ESR • urea/creatinine and electrolytes • • • • • • • • liver function tests screening test for coeliac disease (total IgA and endomysial antibodies or tissue transglutaminase antibodies) thyroid function tests chromosomes bone age X-ray serum FSH and LH, testosterone or estradiol serum prolactin growth hormone studies. Treatment Indications for treatment of delayed puberty are primarily psychological. Induction of pubertal development in boys through the judicious use of intramuscular or oral testosterone preparations may be very useful in alleviating the psychological stress caused by delayed puberty. Recent research also suggests that treatment of delayed puberty is important, as puberty is the time when peak bone mass is accumulated. Failure to achieve peak bone mass in adolescence may place individuals at risk of osteoporosis in adult life. Treatment of delayed puberty should only be carried out by paediatricians and endocrinologists experienced in this area, as excessive administration of sex steroids can adversely accelerate bony epiphysial maturation and affect long-term height outcome (Fig. 19.1.9). Psychological support and counselling Psychological support and counselling are an extremely important part of the management of pubertal delay and in some instances may be all that is required while waiting for the onset of spontaneous pubertal development. It is very important to reassure the adolescent and his/her family that s/he is normal and that appropriate pubertal and sexual development will occur or can be relatively easily assisted with hormonal intervention. Such reassurance and support can profoundly improve an adolescent’s self-esteem and can help reverse problems such as truanting from school, oppositional behaviour and bullying. Clinical example Andrew was a 15.5-year-old boy whose father was concerned that Andrew was growing poorly, and that he was underdeveloped for his age. Andrew’s father had also been a late developer, and he remembered being bullied at school by some of the other boys and also being left out of the rugby team because of his size. He reported that he was still growing when he left school and became an apprentice mechanic. The mechanics he worked with used to call him ‘Shorty’. Further history revealed that Andrew was a healthy young man who had always grown along the 3rd percentile, but from 14 years of age his height had fallen away from the 3rd percentile line. He was a very keen sportsman and had always been a very fast runner but could no longer compete successfully with boys his age. Because of this he had recently taken up golf, which he and his father played together every Saturday. Andrew’s physical examination confirmed that he was completely prepubertal, with an otherwise normal physical examination. The most likely diagnosis was familial delayed puberty. Andrew was not distressed at all by his delayed puberty, and his father was reassured when the diagnosis was explained. During the next 3 years, Andrew’s growth rate increased and he went through a delayed but otherwise normal puberty, eventually being the same height as his father. Precocious puberty Precocious puberty is a rare problem and occurs much less frequently than delayed puberty. Precocious puberty is defined as pubertal development before age 8 years in girls and 9.5 years in boys. True or central precocious puberty is associated with raised gonadotrophins. True precocious puberty is much more common in girls than boys, and girls are less likely to have an identifiable underlying pathological cause than boys. Girls with this disorder will have accelerated growth and development of both breasts and pubic hair. Boys with true precocious puberty have evidence of enlargement of both testes as well as accelerated linear and genital growth and pubic hair development. The most common cause of central precocious puberty is a hypothalamic hamartoma, but many tumours involving the hypothalamic–pituitary area can be associated with an increased prevalence of precocious puberty. Gonadotrophin-independent (pseudo) precocious puberty may be seen with congenital adrenal hyperplasia, adrenal, testicular or ovarian neoplasms and tumours that secrete non-pituitary gonadotrophin such as chorionic gonadotrophin. The McCune–Albright syndrome is also a cause of gonadotrophin-independent precocious puberty. If precocious puberty is suspected, referral should be made to a paediatric endocrinologist who will organize appropriate investigations, which will include measurement of serum FSH and LH, testosterone or estradiol, dynamic tests of gonadotrophin secretion such as luteinizing hormone releasing hormone (LHRH) testing, bone age assessment and cerebral imaging including computed tomography (CT) and/or MRI head scanning. Treatment of precocious puberty should be managed by a paediatric endocrinologist experienced in this area. Treatment options include LHRH superagonists, medroxyprogesterone acetate or cyproterone acetate. Indications for treatment for precocious puberty will include factors such as the age of the child and the rate of progression of the pubertal development. Practical points Delayed puberty • Delayed puberty is a very common pubertal problem • Delayed puberty is defined as absence of pubertal development in girls older than 14 years and boys older than 14–15 years • Familial delayed puberty and constitutional delayed puberty are the most common causes, particularly in boys, often with a positive family history • Chronic disease may cause delayed puberty Conditions resembling precocious puberty Premature thelarche Isolated breast development, either unilateral or bilateral, is relatively common in girls older than 2 years of age and may occur at any time throughout childhood. By definition, premature thelarche has no other features of precocious puberty. All cases should be referred for assessment by a paediatrician or endocrinologist; however, in most cases observation and follow-up is all that is required. Clinical example Amy was a 12-month-old girl whose parents were very worried that she was going into early puberty. They reported that Amy had bilateral breast development, which had been present from birth. Amy was the first child of her parents. The pregnancy had been uncomplicated and she born 2 weeks post-term via a normal vaginal delivery. Her birth weight was 3570 g. At birth she was noted to have swollen breasts, which the midwife had said could occur in newborn babies because of the mother’s hormones and would go away. However, the breast development had persisted, and now the family were very worried. Further history revealed that Amy was a very healthy baby who had not had any significant medical problems and had grown and developed normally for her age. At 12 months she was already walking and could say several words. Her weight, height and head circumference were all progressing along the 25th percentile for age. She was not on any medications, health foods or tonics and ate a normal toddler diet. Physical examination revealed that Amy did have breast enlargement (1.5 cm diameter), but there was no sign of any pubic hair development or other pubertal changes. General examination was normal. The most likely diagnosis was premature thelarche (isolated premature breast development), which may persist after neonatal breast engorgement has subsided. Premature thelarche may be unilateral or bilateral, and the breast size may wax and wane. There are no associated pubertal changes in this condition and no growth acceleration. The condition may persist through infancy and childhood, and true puberty and menarche occur at a normal time, consistent with familial patterns. Investigations including measurement of LH, FSH and oestradiol, bone age assessment and pelvic ultrasound are normal for age. Premature adrenarche The appearance of isolated pubic hair development under the age of 8 years in a girl may occur as a variant of normal but may also be associated with an adrenal disorder such as a non-classical form of congenital adrenal hyperplasia. Careful assessment for any associated signs of virilization, such as clitoral enlargement, hirsutism or acne, should be performed. The appearance of pubic hair in a boy before the age of 9 years rarely occurs as a normal variant and should always be investigated. In all cases of premature pubic hair development, referral should be made to a paediatrician or paediatric endocrinologist so that appropriate investigation of adrenal androgens can be performed. Isolated premature menarche At the beginning of normal puberty in a girl, the small amounts of oestrogen made by the ovary switch ‘on and off’. If enough lining of the womb is made with each ‘switch on’, there may be a small vaginal bleed when the ‘switch off’ occurs. This may happen several months in a row, then disappears as total oestrogen increases and normal puberty progresses. It is a normal variant and usually needs no treatment. Before the diagnosis of premature menarche is accepted, all other causes of premature oestrogen secretion and/or any local causes of vaginal bleeding must be eliminated by the specialist. Pubertal gynaecomastia Gynaecomastia is a very common finding in adolescent boys, occurring in 40–70% of 14-year-olds. In most instances the breast development is minor, transient and regresses. Rare causes of marked pubertal gynaecomastia include Klinefelter syndrome, adrenal and gonadal tumours, and drugs such as cimetidine, digoxin, spironolactone and marijuana use. If significant breast enlargement is causing psychosocial difficulties it may be necessary to refer a teenage boy to a plastic surgeon for consideration for subareolar mastectomy. Hormonal therapy does not influence the natural history of pubertal gynecomastia. Asymmetrical breast development Asymmetrical breast development can occur in both males and females. In males, it is clearly a variant of pubertal gynaecomastia. In females, breast development can be asymmetrical at the beginning of breast budding or subsequently through breast development. The degree of asymmetry can be quite marked. Consideration should be given to the possibility of an underlying chest wall or pectoral muscle abnormality and examination should be conducted appropriately. However, in most cases asymmetrical breast development is just a physiological variant of puberty. Clinical example Adam was a 15-year-old boy with bilateral breast enlargement. The breast enlargement had begun not long after his 14th birthday, and had progressed during the next 12 months. Adam was a healthy adolescent, with no significant medical problems. His parents reported that he was growing rapidly and had developed a large appetite. Physical examination revealed a male teenager who had stage 3 pubic hair development and 12 ml testes. He had palpable breast tissue bilaterally (2.5 cm diameter), with normal pectoral muscles and underlying chest wall. The breast tissue was uniform and nontender, and the associated lymph nodes were not enlarged. Adam had gynaecomastia, which is a very common pubertal problem in teenage males. In the presence of a normal past history and examination, no investigations are usually necessary. Adam and his family were reassured that the gynaecomastia was a common pubertal condition in males and, over the following 3 years, as Adam completed his growth and pubertal development, the gynaecomastia resolved. In rare cases, an underlying vascular abnormality or lipoma may cause one breast to appear larger than the other. This can usually be readily determined by a physical examination and confirmed by ultrasound. Reassurance and monitoring are all that are usually required. For self-esteem and cosmetic reasons, advice should be given to teenage girls about temporary use of breast prostheses or even shoulder pads to equalize the breast form. Most girls cope with the situation by wearing loose fitting garments and T shirts over swimwear. In most situations, the asymmetry resolves with full pubertal development. On rare occasions, however, referral to a reconstructive surgeon for breast reduction or augmentation may need to be considered. 19.2 Thyroid disorders in childhood and adolescence F. Cameron, J. Brown Normal thyroid function throughout infancy, childhood and adolescence is essential for a normal developmental and physiological outcome. Thyroid disease is one of the most common groups of endocrine disorders in childhood and adolescence, with approximately 1–2% of all children having a thyroid disorder at some time. Therefore, knowledge of thyroid disease and its management is fundamental to paediatric medicine. Thyroid physiology The thyroid gland removes iodide from the blood stream, combines it with tyrosine and releases iodinated tyrosine to the peripheral tissues. The thyroid gland is able to trap iodide and synthesize iodothyronine from 70 days gestation. Release of thyroxine, however, does not occur until 18–20 weeks gestation. Thyroid gland growth is regulated by thyroid stimulating hormone (TSH), released from the anterior pituitary gland, which is in turn regulated by thyrotropin regulating hormone (TRH), released from the hypothalamus. These regulating hormones are in turn controlled by negative feedback from triiodothyronine (T3), the active metabolite of the major thyroid hormone thyroxine (or tetraiodothyronine, T4). The thyroid gland is extremely effective at trapping serum iodide, with a concentration gradient from thyroid to serum of 30–40. This gradient increases in times of iodide deficiency. Once trapped, iodide is oxidized to iodine and organification occurs. Organification is the iodination of thyroglobulin-bound tyrosyl residues to form monoiodotyrosine (MIT) and diiodotyrosine (DIT). Organification and iodide oxidation (to iodine) are catalysed by thyroid peroxidase. Thyroid peroxidase couples the iodotyrosines to form iodothyronines within the thyroglobulin molecule, resulting in T 4 and T3. In the absence of iodine deficiency the T4 to T3 synthesis ratio is 10–20:1. In adults, the release rate of T4 to T3 is 3:1. Once released, both hormones bind to thyroxine binding globulin (TBG). 80% of circulating T3 results from deiodinatination of T4 in peripheral tissues. Thyroid hormones bind to a nuclear receptor. T 3 binds to this receptor with 10 times the affinity of T4. Once bound, thyroid hormones regulate gene transcription, increasing cytoplasmic proteins, which stimulate mitochondrial activity, thus increasing metabolic rate. Disorders of thyroid function in childhood can be divided into the following categories: • hypothyroidism • hyperthyroidism • thyroid masses. Hypothyroidism Congenital Screening for congenital hypothyroidism has been performed in most developed countries for the last 15–20 years. In Australia, screening for congenital hypothyroidism, phenylketonuria and cystic fibrosis occurs on day 3–5 of life. Because of such screening the clinical picture of ‘cretinism’ (the later effects of congenital hypothyroidism, including severe intellectual disability) thankfully is now rarely seen. Incidence One in 3000–5000, with some geographical variation. Aetiology and genetics Some 75% of cases are due to dysgenesis (agenesis, ectopia), 10% to dyshormonogenesis, 5% to hypothalamic– pituitary deficiency (central hypothyroidism) and 10% to transient hypothyroidism (iodine exposure, maternal antithyroid antibodies, etc.). While thyroid disease appears to be sporadic in the majority of children born with hypothyroidism, evidence for a genetic component is increasing. In up to 2% of cases, hypothyroidism is familial, and children with congenital hypothyroidism have a higher incidence of associated abnormalities (cardiac, renal, hip dysplasia) than the general population. Studies in mouse models with congenital defects of thyroid development have provided the basis for molecular genetic studies in humans with congenital hypothyroidism. Mutations have been described in a number of genes, resulting in absent, misplaced, hypoplastic or unresponsive glands (Table 19.2.1). In some instances, a specific phenotype can be recognized, and prognosis is affected. For example, in individuals with the NKX2.1 mutation, neurological outcome is poor despite early thyroxine treatment. Hypothyroid patients with normally located and normally sized glands have defects in thyroid hormone biosynthesis. Recessive mutations in thyroglobulin, thyroid peroxidase, pendrin (causing Pendred syndrome – sensorineural deafness and hypothyroidism) and sodium/iodide symporter (NIS) genes have been described. Mutations in the TRH receptor gene, TSH beta subunit and transcription factors regulating pituitary development have also been described in some individuals with central hypothyroidism. Clinical picture Often the condition is subclinical and is detected on routine screening. Clinical features that should be looked for are jaundice, dry skin, a hoarse cry, puffy face, prominent tongue, listlessness, umbilical hernia, hypothermia, bradycardia and failure to thrive. Investigation results An unconjugated hyperbilirubinaemia (due to glucuronyl transferase deficiency) is common. An elevated TSH detected on testing of a heelprick drop of blood collected on filter paper on day 3–5 of life is seen in primary hypothyroidism. Management Confirmatory investigations are needed if the screening tests suggest an abnormality: repeat T4 and TSH; thyroid scan (showing absent, lingual or increased uptake of radioisotope), X-ray distal femoral epiphysis (absence implying prolonged/prenatal hypothyroidism), and assessment and imaging of the pituitary gland if indicated. Treatment involves commencement of therapy (thyroxine replacement at 8–10 g/kg/d). Thyroid imaging results for congenital hypothyroidism of varying causes are shown in Figure 19.2.1. Prognosis Normal intellectual and physical development is likely if treatment is commenced promptly and monitored closely. Overtreatment may result in craniosynostosis. Acquired Acquired hypothyroidism in the child or adolescent is relatively uncommon. In iodine-sufficient regions of the world the most common cause is autoimmune thyroiditis. Accordingly, acquired hypothyroidism is seen twice as commonly in females than in males, usually manifesting in early puberty. Prevalence Between the ages of 1 and 18 years the prevalence is 1.2%. Acquired hypothyroidism is rare prior to 4 years of age. Aetiology The causes in order of frequency are as follows: primary hypothyroidism – chronic lymphocytic, autoimmune (Hashimoto) thyroiditis, late-appearing congenital dyshormonogenesis, exogenous factors (high-dose iodine exposure (Wolff–Chaikoff effect), radiation) and severe iodine deficiency; and central hypothyroidism – congenital and acquired hypopituitarism. Clinical picture The most common presentation is growth retardation and goitre. In addition, the triad of short stature, obesity and mental dullness indicates hypothyroidism until proven otherwise. Growth impairment usually mainly affects the limbs so that body proportions predominantly remain infantile. Other features include hypothermia, bradycardia, slow reflex relaxation, constipation, dry hair and skin, pallor, facial puffiness (‘myxoedema’) and dental delay. The onset is often insidious, with delays of up to 4–5 years being reported between the onset of growth retardation and the diagnosis of hypothyroidism. While delayed puberty usually occurs, some cases of precocious puberty have been reported. Autoimmune thyroiditis may be associated with other autoimmune diseases (autoimmune polyglandular syndromes) such as type 1 diabetes, autoimmune adrenalitis (Addison disease), vitiligo and pernicious anaemia. Occasionally, autoimmune hypothyroidism can be preceded by a period of transient hyperthyroidism. Investigation results Goitre (detected either clinically or sonographically) is common in acquired hypothyroidism. Blood tests show a low circulating T4 and (usually) a high circulating TSH. Bone age is delayed and there may be positive thyroid autoantibodies (in autoimmune thyroiditis). There is patchy uptake of isotope on thyroid scan (in autoimmune thyroiditis). Hypothalamic or pituitary anomalies may be seen on computed tomography (CT) or magnetic resonance imaging (MRI) (in tertiary or secondary disease). Management Replacement thyroxine (usually 50–100 g/day in a single dose) is required. An appropriate individual dose is determined by measuring serum TSH at 2 or more weeks after commencing therapy. Prognosis Severely hypothyroid children often show dramatic clinical changes with treatment. These include: weight loss, rapid growth, loss of primary teeth, some transient hair loss and increased energy/alertness. The long-term neurodevelopmental outcome is good, given that the rapid growth phase of the brain in the first 2 years of life has usually been protected. Despite short-term rapid catch-up growth, restoration of full growth potential often does not occur, because of rapid advancement of bone age in the first 18 months of treatment. Long-term treatment is usually required. Hyperthyroidism Congenital This is always due to maternal thyrotoxicosis and is a rare clinical event. However, if unrecognized and untreated, neonatal hyperthyroidism may be fatal. Incidence Maternal thyrotoxicosis is uncommon (1–2 cases per 1000 pregnancies). Neonatal disease occurs in 1 per 70 cases of pregnancies affected by thyrotoxicosis. Aetiology Neonatal hyperthyroidism is the result of the transplacental passage of TSH receptor stimulating antibodies from a mother with either active or inactive Graves disease. Measurement of maternal antibody status, rather than thyroxine levels, is predictive of the likelihood of neonatal hyperthyroidism. Clinical example Sarah had been struggling at school ever since she started year 7. At the age of 13 years, her parents had become concerned that she was having difficulty settling into her new school as she was always complaining of feeling tired and having ‘tummy pains’. Sarah’s parents were at a loss to explain why she was behaving this way as she was a good student at primary school and had lots of friends. More recently Sarah’s parents had noted that her weight had increased dramatically (they attributed this to her lack of activity, as she didn’t seem to eat all that much), so much so that she now had a very ‘fat’ neck. She was also complaining of feeling cold all the time and was constantly wearing extra clothes even when the weather was warm. Her parents were concerned that she had a body image problem as a consequence of her recent weight gain. The school counsellor felt that Sarah might be depressed and her parents were most concerned. On examination Sarah was moderately overweight with cool hands and she had a resting pulse rate of 50. She had dry hair and skin. She had a smooth, uniformly enlarged thyroid gland. Her reflexes showed a markedly delayed relaxation phase. Investigations revealed that Sarah had a serum TSH level of 35 IU/ml (high) associated with a T4 of 5 nmol/l (low). Her bone age was equivalent to 10 years and a plain X-ray of her abdomen showed faecal loading. Her antithyroid microsomal antibody titre was positive. Sarah was commenced on 100 g of thyroxine per day. Within 2 weeks she was reporting much improved energy levels and affect. At 1 month review she had lost 5 kg in weight, her school performance had improved and her goitre was showing some signs of shrinkage. Clinical picture Neonates may present with any of the following: irritability, poor weight gain, tachycardia, cardiac arrhythmias, flushing, hypertension, goitre, exomphalos, jaundice and hepatosplenomegaly. While presentation soon after birth is more common, if the mother has been taking antithyroid drugs, presentation may be delayed until day 8– 9 after birth, when the antithyroid medication has been eliminated from the neonate’s circulation. Investigation results High circulating T4 or T3 levels and low TSH levels are detected in the neonatal blood sample. Management Immediately after diagnosis, sedation and treatment with either beta-blockade or digoxin may be required. Subsequent treatment with antithyroid medication (carbimazole, methimazole or propylthiouracil) is usually required. A therapeutic response should be seen within 24–36 hours after commencing treatment. Prognosis Mortality rates of up to 25% have been reported. The half life of thyroid stimulating antibodies in the fetal circulation is approximately 12 days; however, the clinical course may extend for a period of up to 12 weeks. Acquired Hyperthyroidism in childhood and adolescence is less common than either euthyroid goitre or hypothyroidism. As with acquired hypothyroidism, it is most commonly due to autoimmune disease and is usually seen in young adolescent females. Incidence Females are affected 6–8 times more commonly than males. Some ethnic groups (such as Asian females) have a greater reported incidence of autoimmune hyperthyroidism. Aetiology Autoimmune hyperthyroidism (Graves disease) is the most common form of acquired hyperthyroidism in childhood and adolescence. Less frequent causes include the acute toxic phase of autoimmune hypothyroidism (Hashimoto disease; hashitoxicosis) and a toxic thyroid nodule (rare). There is often a family history of autoimmune thyroid disease (either Graves or Hashimoto disease). The primary defect is the presence of stimulating autoantibodies (thyroid receptor antibodies) which mimic the action of TSH. Overstimulation of the TSH receptor leads to thyroid growth and excess thyroxine production. There is often an associated ophthalmopathy due to the deposition of proteoglycans in the extraocular muscles and retro-orbital spaces. Clinical picture The clinical features of hyperthyroidism result from sympathetic drive causing a hypermetabolic state. Many of the florid signs of thyrotoxicosis that are seen in adults are less pronounced in children. Symptoms include deteriorating school performance, weakness/fatigue, restlessness/sleeplessness, polyuria, hunger, heat intolerance, excessive sweating, anxiety and diarrhoea and weight loss. Clinical signs include: goitre or localized thyroid mass, tremor, tachycardia and brisk reflexes. Approximately 30% of children will have associated proptosis and other signs of thyroidal ophthalmopathy (lid lag, lid retraction and ophthalmoplegia). Investigation results Suppressed serum TSH levels are seen and are associated with elevated T4 or T3 levels. There will also be elevated levels of thyroid autoantibodies (TSHreceptor-antibody-positive in Graves disease). The bone age is advanced and there is sonographic evidence of thyromegaly. Generalized and localized increased uptake of isotope is seen in Graves disease and toxic adenoma respectively. Management In the setting of autoimmune hyperthyroidism there are three treatment options. The first of these, antithyroid medication, is the most commonly used. Carbimazole and methimazole have traditionally been used most commonly in Australia and Europe, whereas propylthiouracil has been more commonly used in North America. Both types of medication block organification and are similarly efficacious, with comparable side effect profiles. Beta-blockade (with propranolol) may also be used in the first 2–4 weeks of therapy to gain symptom control. This is contraindicated in children who suffer from asthma. Treatment with organification blocking drugs is continued for 2 years in the first instance. Other treatment options include thyroidectomy (subtotal or total) and radioactive iodine. In the setting of toxic adenoma, surgery is usually the preferred treatment option. Prognosis After 2 years of medical therapy, approximately 20–50% of patients can be expected to enter spontaneous remission, with resolution of thyroid autoantibody status. Among those patients who do not remit spontaneously, long-term drug therapy is both safe and effective. In the advent of poor compliance with medical therapy, lack of control or increasing thyromegaly, a second treatment option – surgical subtotal or complete thyroidectomy – is considered. This results in 20% of patients becoming euthyroid, 50% of patients becoming hypothyroid and 30% of patients becoming thyrotoxic in the long term. Other paediatric and adult centres use a third treatment option, that of radioactive iodine. This treatment results in total thyroid ablation and requires subsequent lifelong thyroxine replacement therapy. Clinical example Tina, aged 15, had noticed increasing anxiety levels recently. She was quite bright academically and had set high standards for herself at school. Her parents were concerned that her anxiety was associated with some recent difficulties in concentrating during classes. Her teachers complained that she ‘fidgeted’ all the time and was quite restless. Despite a healthy appetite (‘she eats more than anyone else in the family’) Tina had been losing weight and had frequent loose bowel actions. Her mother also reported that, despite it being winter, Tina refused to wear appropriate cold weather clothing, preferring a T shirt most of the time. On examination, Tina appeared quite anxious and had very prominent eyes (proptosis). She had difficulty in sitting still on the examination couch and squirmed around quite a lot. Her resting pulse was 110 and she had a fine tremor when her hands were held out. Her palms were very sweaty. She had a firm, smooth goitre with an audible bruit. Her reflexes were very brisk and she had difficulty standing from a squatting position. A provisional diagnosis of Graves disease was made. This was confirmed by finding that Tina’s serum TSH levels were unrecordably low in the face of a T4 level of 52 nmol/l (high). Her anti-TSH receptor antibody titre was elevated. A thyroid ultrasound demonstrated a uniformly enlarged thyroid with no focal changes. Tina was commenced initially on both carbimazole and propranolol. Her symptoms had largely abated within 3 weeks and her propranolol was ceased at this time. Ophthalmological review confirmed the presence of proptosis, with no other thyroidal eye signs being present. Over the following year Tina’s goitre diminished in size; however, her proptosis remained unchanged. She was initially treated with carbimazole for 2 years. At this time she was still TSH-receptor-antibody-positive and it was decided to continue treatment for a further 2 years. Thyroid masses Goitre The commonest cause of goitre on a worldwide basis remains iodine deficiency. In developed countries this had become rare until recent times, with the iodisation of table salt and some infant milks. Recently, iodine deficiency has been reported again in Australian populations, presumably due to low-salt diets encouraged for cardiovascular health reasons. Incidence Goitres or diffuse enlargement of the thyroid gland occur in 4–5% of all children. They are more common in girls during puberty and are often not detected. Aetiology In Australia the main causes in order of frequency are: Hashimoto thyroiditis (majority are euthyroid), Graves disease, mild dyshormonogenesis, tumour (benign/malignant), acute/subacute thyroiditis and iodine deficiency. Foods that inhibit thyroxine synthesis and can lead to goitre (goitrogens) include cabbage, soybeans and cassava. Clinical picture Most often the goitre is asymptomatic and is frequently detected on routine examination undertaken for other reasons. Thyroid hypofunction or hyperfunction will present with the signs and symptoms described above. Occasionally, pressure symptoms related to the enlarged thyroid (dysphagia, stridor or neck discomfort) may be the presenting feature. Thyroidal tenderness is seen in acute/subacute thyroiditis. Regional lymphadenopathy associated with a goitre or thyroid nodule is suggestive of malignancy and is an ominous sign. Investigation results Sonography, serum thyroid function tests and serum thyroid antibody levels will distinguish most causes of goitre. Fasting urinary iodine levels will also help to define iodine status. Thyroid scanning will show increased uptake with mild dyshormonogenesis and patchy distribution in Hashimoto thyroiditis. Management Smoothly enlarged goitres with normal thyroid function can be managed simply by observation and iodine supplementation if required. Goitres with functional consequences will require either thyroxine supplementation or suppressive medication. Prognosis This depends on the cause of the goitre. As most cases of asymptomatic goitre result in no disturbance of thyroid function, the prognosis is usually good. Thyroid nodules Nodules within the thyroid gland are palpable, localized swellings. They may be single or multiple. The Chernobyl nuclear reactor disaster in 1986 led to a markedly increased incidence of benign and malignant thyroid nodules in children from the surrounding iodine-deficient areas. Incidence Fewer than 2% of children have thyroid nodules. Of these, approximately 2% are malignant. If the nodule is single the risk of malignancy increases to 30–40%, higher than in adults. Aetiology Benign nodules include cysts, cystic adenomas and variations of Hashimoto thyroiditis. Malignant nodules are carcinomas and occur in the following order: papillary/mixed, follicular, medullary and anaplastic. In one series of children with thyroid cancers reported in the 1950s, 80% had a history of having received head/neck radiotherapy. However, head/neck irradiation is now less commonly used and the aetiology of most thyroid cancers remains obscure. Radiation-exposed children need close follow-up, and regular ultrasound surveillance substantially increases the detection of thyroid malignancy. Medullary carcinomas may be sporadic, familial (autosomal dominant mode of inheritance) or part of a multiple endocrine neoplasia (MEN2) complex. Patients with MEN2 have been found to have mutations in the RET proto-oncogene. Clinical picture The most common presentation is the lobular, irregular thyroid gland seen in Hashimoto thyroiditis. Nodules are usually asymptomatic and are often detected coincidentally upon routine examination. Rarely, nodules may be hyperfunctional (‘toxic adenoma’). Medullary carcinomas may be associated with phaeochromocytoma (in later life) and parathyroid hyperplasia (MEN2A), or multiple mucosal neuromas, Marfan-like habitus and phaeochromocytoma (MEN2B). It is very rare for a child with MEN2 to present with clinical disease. They are usually detected as part of a kindred subjected to genetic screening. Investigations Nodules may be detected both sonographically and by thyroid scanning. The finding of multiple hot nodules associated with positive thyroid antibody titres and/or disturbed thyroid function is against a diagnosis of malignancy. Alternatively, a single cold nodule with or without serum calcitonin levels (associated with medullary carcinomas) is suggestive of malignancy. Fine-needle aspiration is not widely used in the diagnosis of thyroid nodules in children. Management If there is any doubt as to the nature of any thyroid nodule it is appropriate to proceed to open biopsy. Solitary benign nodules are usually excised. Papillary thyroid cancers are treated with total thyroid excision, with subsequent radioactive iodine therapy if metastases are thought to be present. Medullary thyroid cancers are unresponsive to radioactive iodine and early total thyroidectomy remains the treatment of choice. In individuals with RET proto-oncogene mutations from families with a strong history of medullary carcinomas, prophylactic thyroidectomy is considered. Prognosis Most thyroid nodules are benign and have an excellent prognosis. In the case of papillary carcinomas, serial thyroid scans for the first 3 years after surgery will detect any residual thyroid tissue or tumour recurrence. In patients suffering from medullary carcinomas, serial measures of serum calcitonin levels are the monitoring strategy of choice. Practical points • Normal thyroid function is essential for normal growth and development • Thyroid disorders are common and affect up to 2% of children and adolescents • Neonatal screening for congenital hypothyroidism allows early detection and treatment, resulting in normal development in the majority of affected infants • Symptoms of acquired hypothyroidism may be subtle in childhood and adolescence. Short stature may be the only presenting feature of hypothyroidism • Hyperthyroidism is much less common than hypothyroidism and non-specific associated symptoms may result in delay in diagnosis • Thyroid malignancy in isolated thyroid nodules is much more common in children than in adults In summary, disorders of the thyroid gland can have many manifestations: hypofunction, hyperfunction, pressure symptoms and incidental tumours. Given the importance of normal thyroid function for both neurological and physical development, and the potential for malignancy in thyroid nodules, a clinical awareness of potential thyroid problems in paediatrics is essential. Once detected, most thyroid problems can be successfully managed, with excellent clinical outcomes. 19.3 The child of uncertain sex J. Fairchild The child of uncertain sex has a genital appearance that does not permit gender assignment. This includes infants with bilateral undescended testes, perineal hypospadias with a bifid scrotum, clitoromegaly, posterior labial fusion, a phenotypical female with a palpable gonad and those with discordant genitalia and sex chromosomes. The birth of a child of uncertain sex presents a psychosocial crisis for the family and may indicate an underlying medical condition such as congenital adrenal hyperplasia, which could be life-threatening if undiagnosed and untreated. These disorders are rare, often complex and always require urgent expert consultation. Normal prenatal development An understanding of normal prenatal development is essential in the evaluation of the child of uncertain sex. Normal sexual development in the embryo consists of three related sequential processes: 1. establishment of chromosomal sex at fertilization, with XY as male and XX as female 2. determination of gonadal sex when the indifferent gonad develops into a testis or an ovary 3. development of phenotypic sex as a result of gonadal differentiation and gonadal hormone production. Internal genitalia Up until about 7 weeks, male and female embryos develop in an identical fashion, with bipotential gonads and both wolffian and müllerian internal genital ducts present (Fig. 19.3.1). In males, the presence of the sex determining region on the Y chromosome (SRY gene) directs the bipotential gonad to become a testis. At least four other genes are also required for normal testicular development. By 7–8 weeks the testis has recognizable tubules and starts producing androgens, including testosterone from the Leydig cells and müllerian-inhibiting substance (MIS) from the Sertoli cells. Circulating hormone levels are low and masculinization of the internal genital ducts occurs by locally acting (exocrine) secretion of these hormones down the wolffian duct. High levels of testosterone promote the ipsilateral development of the wolffian duct to become the epididymis, vas deferens and seminal vesicle. High levels of MIS lead to ipsilateral müllerian duct regression. This process occurs during a critical period between 8 and 12 weeks. The Leydig cells also produce a relaxin-like factor that, together with MIS and androgens, masculinizes the gubernaculum. The gubernaculum holds the testis near the inguinal ligament during early development and from 25 weeks it begins to elongate, steering the testis towards the scrotum. Clinical example A healthy, full-term infant was born with ambiguous genitalia. On examination there was an enlarged clitoris (2 cm in length), posterior fusion of the labia, with a single opening visible, and no gonads were palpable. The genitalia were noted to be hyperpigmented. There was no history of consanguinity but a previous male sibling had died at 2 weeks of age after a vomiting illness. Initial investigations confirmed that the baby’s chromosomes were 46XX and a pelvic ultrasound showed the presence of a normal uterus and ovaries. The 17-hydroxyprogesterone level at 48 hours of age was markedly elevated, confirming the diagnosis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Treatment with hydrocortisone was commenced. Daily electrolytes were normal until the 6th day of life, when hyperkalaemia developed, confirming the salt-wasting form of the condition. Fludrocortisone therapy was then added. The child was referred to a paediatric surgeon for corrective genital surgery. She will require lifelong replacement therapy and monitoring. The hyperpigmentation was due to the adrenocorticotrophic hormone (ACTH) excess and resolved with adequate replacement therapy. It was likely that the previous male sibling also had the condition, as it is inherited as an autosomal recessive trait. The genitalia of affected males are usually normal at birth apart from some hyperpigmentation and affected male infants typically present with a salt-wasting crisis in the first few weeks of life. It is often confused with pyloric stenosis, which also presents at this age with vomiting and dehydration. In females, the absence of testosterone and MIS leads to wolffian duct regression and müllerian duct preservation. The müllerian ducts develop into the uterus, fallopian tubes and upper vagina. External genitalia Up until about 8 weeks, the external genitalia of male and female embryos also have an identical appearance. Both sexes have a genital tubercle, two genital swellings and two genital folds (Fig. 19.3.2). In males, between 8 and 12 weeks, androgen action on the external genitalia results in the development of normal male genitalia. Circulating levels of testosterone are insufficient for this action but testosterone is converted in the periphery to dihydrotestosterone (DHT) by the enzyme 5-reductase. Dihydrotestosterone binds to the androgen receptors much more strongly than testosterone, thereby amplifying its effect. The genital tubercle develops into the glans penis, the genital swellings fuse to form the scrotum and the genital folds elongate and fuse to become the shaft of the penis and the penile urethra. The prostate forms in the wall of the urogenital sinus. With the exception of the phallus, circulating androgens masculinize the external genitalia only during a critical period between 8 and 12 weeks. In females, in the absence of androgen, the genital tubercle forms the glans clitoris and the short female urethra. The genital swellings remain unfused and become the labia majora. The genital folds become the labia minora and the vaginal plate, a thickening of the posterior wall of the urogenital sinus, canalizes to form the lower vagina. Evaluation of the child of uncertain sex The initial evaluation of the child of uncertain sex should include a history, clinical examination, assessment of adrenal secretion, sex chromosomes and internal anatomy. History A careful history should include: • • • • • any prenatal exposure to androgens maternal virilization during pregnancy family history of intersex disorders, females who are childless or females who have amenorrhoea family history of unexplained infantile deaths consanguinity. Clinical examination The clinical examination should include: • inspection of the external genitalia • palpation to determine the presence, location and symmetry of the gonads: • if both gonads are palpable and symmetrical they are almost always testes • gonadal asymmetry implies one testis present, suggesting a mixed chromosomal pattern • if both gonads are impalpable the gonadal and duct status is unknown • general examination looking for: • dysmorphic features or non-genital abnormalities that may point to the diagnosis of a specific disorder • hyperpigmentation and/or systemic illness, which may suggest congenital adrenal hyperplasia. Clinical example A full-term infant with normal birth weight (3.1 kg) was noted to have a small penis (stretched penile length 1.5 cm, normal 2.5 cm) and undescended testes. The infant had had two episodes of hypoglycaemia treated with intravenous dextrose and on general examination was noted to have a small cleft in the palate. Hypoglycaemia is unusual in a full-term infant of normal birth weight and in the presence of a midline defect raises the possibility of hypopituitarism. A diagnostic blood sample taken at the time of the hypoglycaemia revealed low levels of cortisol, growth hormone and free thyroxine, confirming the diagnosis. The baby was commenced on hydrocortisone and thyroxine with resolution of the hypoglycaemia. He was given a short course of testosterone at 6 months of age, which resulted in an increase in his penile length to 2.5 cm. Growth hormone therapy was commenced when his growth rate slowed at 12 months of age. Micropenis and cryptorchidism can occur with either gonadotrophin deficiency or growth hormone deficiency and is a useful clinical sign of congenital hypopituitarism in male infants. Initial investigations The initial investigations that should be undertaken are: • urgent karyotype: • permits classification of the infant into one of three diagnostic categories and determines further evaluation • imaging of internal anatomy: • ultrasound to determine the presence of gonads, uterus and vagina • urogenital sinogram to visualize the urethral and vaginal anatomy • exclude congenital adrenal hyperplasia: • serum electrolytes and blood glucose series • serum 17-hydroxyprogesterone and other adrenal steroids Further evaluation This may include: • serum profile of adrenal steroids, gonadal androgens and their precursors • human chorionic gonadotrophin (hCG) stimulation test to confirm a normal rise in gonadal hormones with stimulation, with the testosterone/DHT ratio reflecting 5-reductase activity • surgical procedures: genital skin biopsies for androgen receptor assay, panendoscopy and/or laparotomy to delineate internal genitalia gonadal biopsy. Practical points Initial evaluation of the child of uncertain sex • History: androgen exposure, family history of intersex or infant deaths, consanguinity • Examination: determine the presence of palpable gonads, hyperpigmentation, systemic illness, non-genital abnormalities • Investigations: include an urgent karyotype, assessment of adrenal secretion, ultrasound urogenital sinogram to assess internal anatomy • Assignment to one of the three diagnostic categories will determine further evaluation: virilized XX, undervirilized XY or mixed chromosome pattern Diagnostic categories The results of the karyotype, in particular the sex chromosomes, will allow the infant to be classified into one of three diagnostic categories which will determine further evaluation: • virilized XX • undervirilized XY • mixed chromosome pattern. Virilized XX In the virilized XX child, the gonads are ovaries and the internal genitalia are female, therefore no gonads are palpable (Fig. 19.3.3). The external genitalia are virilized to a variable degree, from mild clitoromegaly to complete labial fusion with urethral tubularization to the tip of the enlarged phallus (Fig. 19.3.4). If the exposure occurred after 12 weeks there will be isolated clitoromegaly without labial fusion. Causes of the virilized XX state may be: • androgen excess from the fetal adrenals: • congenital adrenal hyperplasia (most common cause) • androgen excess from the mother: • maternal ingestion of androgens • androgen-producing tumour • placental aromatase deficiency. Undervirilized XY The undervirilized XY child usually presents with a small phallus, posterior hypospadias, poorly developed, bifid scrotum and testicular maldescent. The causes of the undervirilized XY state may be: • inadequate androgen production: • hypoplastic testes (luteinizing hormone (LH) deficiency) • dysplastic testes • testosterone synthesis defects • inability to convert testosterone to DHT (5-reductase deficiency) • impaired response to androgen: • androgen insensitivity syndrome. Clinical example A 16-year-old girl presented for investigation of primary amenorrhea. On examination she was found to be a tall girl for her family. She had breast development at stage 5 but sparse pubic hair (stage 2–3) and no axillary hair. She also had an inguinal mass, which was occasionally painful when knocked during sport. Investigations revealed a 46XY karyotype, no uterus on pelvic ultrasound and serum levels of testosterone characteristic of normal men. A diagnosis of complete androgen insensitivity was made. Gonadectomy was performed as there is a high incidence of germ cell cancer and oestrogen replacement therapy was commenced. Psychological support was important in helping her come to terms with the diagnosis. Mixed chromosomal pattern True hermaphroditism In true hermaphroditism, both testicular and ovarian tissues coexist. The gonads are usually ovary and testis or ovary and ovotestis. The chromosomes are usually 46XX, although 46XY mosaicism can occur. Asymmetry of the gonads, internal and external genitalia is the hallmark of this condition. Mixed gonadal dysgenesis In mixed gonadal dysgenesis there is also gonadal asymmetry with a testis on one side and a streak gonad on the other. The testis may be dysgenetic and the streak gonad usually contains ovarian stroma without oocytes. The chromosomes are commonly 45XO/46XY, and these infants may have the phenotypic features of Turner syndrome, but other mosaic patterns can occur. The risk of gonadoblastoma is high in dysgenetic testes and gonadectomy in the first decade of life is recommended. Intersex disorders with unambiguous genitalia It is important to note that some patients with intersex disorders will have unambiguous genitalia. These patients have complete sex reversal with a phenotype the reverse of what would be expected from their genotype. Examples of this would be an XY child with complete gonadal dysgenesis or complete androgen insensitivity syndrome. Intersex abnormalities may be missed if the diagnosis of hypospadias is made without due care. This diagnosis should never be made without full investigations, unless both testes are descended in a fused scrotum. Management The birth of a child of uncertain sex presents a unique set of challenging and often difficult management issues. A multidisciplinary team approach involving the paediatrician, paediatric endocrinologist, surgeon, geneticist, psychologist, social worker and adolescent gynaecologist is required for optimal management. Initial management The initial management of the child of uncertain sex requires attention to medical and psychosocial issues. Evaluation of the child must be carried out urgently, so that a decision about the sex of rearing can be made as quickly as possible and life-threatening medical conditions can be identified. Urgent expert consultation should always be sought. The birth of a child of uncertain sex is a psychosocial crisis for the family and must be handled sensitively. The parents should be honestly informed that you do not yet know the sex that their baby is meant to be, but reassured that this will be determined as soon as possible. Instruct all staff to refer to the infant as ‘your baby’, not he, she or it. The parents will require guidance as to how to deal with family and friends and the support of an experienced social worker or psychologist is invaluable. Every effort should be made to encourage the parents to bond with their baby. The parents should be advised not to name the baby or register the birth until sex assignment is decided. The urgent medical issue is the exclusion of congenital adrenal hyperplasia and the attendant risk of adrenal crisis. Congenital adrenal hyperplasia is the most common cause of XX virilization and one of the causes of XY undervirilization. Serum electrolytes and blood glucose should be monitored closely and serum sent for urgent measurement of 17-hydroxyprogesterone and other adrenal steroids. Congenital adrenal hyperplasia refers to a group of autosomal recessive disorders resulting from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. The most common enzyme deficiency is 21-hydroxylase deficiency, accounting for more than 90% of cases. The deficiency of cortisol results in adrenocorticotrophic hormone (ACTH)-mediated adrenal hypertrophy and excessive production of cortisol precursors, which are diverted to the synthesis of androgens. Concomitant aldosterone deficiency leads to salt wasting in 75% of cases. Symptoms and signs of a salt-wasting adrenal crisis include vomiting, diarrhoea, hypovolaemia, hyponatraemia with hyperkalaemia, hypoglycaemia and cardiovascular collapse and can occur within the first few days to weeks of life. If symptoms or signs of adrenal crisis are present, stress doses of hydrocortisone and intravenous fluid therapy with normal saline and 5% dextrose should be started immediately. The stress dose of hydrocortisone is 100 mg/m2/day and is given intravenously. Practical points Management goals for the child of uncertain sex • An accurate and expeditious diagnosis is essential – and requires urgent expert consultation • Rational sex assignment is important • Therapy is directed towards achieving successful sex assignment • Psychological support for the family is required • Genetic counselling is an integral part of management Gender issues The issue of timing and approach to genital reconstruction is controversial and evolving. An evidence-based approach is hampered by the lack of long-term outcome studies involving large numbers of patients. Gender assignment and sex of rearing should be based on the most probable adult gender identity and potential for adult function. It involves consideration of the diagnosis, degree of virilization, capacity to respond to androgen, potential for adult sexual function and fertility as well as the parent’s social and cultural background. There are no data demonstrating a link between early genital surgery and psychosexual orientation and, although early surgery may make life easier for the parents and the child, these irrevocable decisions may complicate the lives of adults. Decisions about sex of rearing and genital reconstruction should only be made by an informed family after careful evaluation and counselling by an experienced multidisciplinary team. Long-term management Once gender is assigned, every effort should be made to encourage the child’s sex of rearing. Both the child and parents will benefit from long-term psychosocial support and many individuals and their families derive benefit from support groups. Specific management of the gonadal tissue in infants of uncertain sex may be required to reduce the risk of gonadal malignancy, torsion and infertility. Abdominal gonads bearing Y material should be brought into the scrotum or removed surgically. The age of surgery will vary according to the underlying condition. Sex steroid replacement should be provided consistent with the sex of rearing and age of the child to ensure adequate growth and pubertal development and prevent osteoporosis. 19.4 Childhood diabetes J. Couper Diabetes mellitus Diabetes mellitus is caused by a deficiency in the production or action of insulin. It is the second most common chronic disease of childhood and causes considerable morbidity due to acute metabolic derangements and longterm microvascular and macrovascular complications. In childhood, the majority of diabetes is type 1 diabetes (previously known as insulin-dependent diabetes). However, in parallel with the increasing incidence of type 2 diabetes in childhood in North America, this is also the trend in Australia and New Zealand, especially in Aboriginal and Polynesian populations. The prevalence of type 1 diabetes mellitus in Australia is approximately 1 in 750 for children under the age of 15 years, with an annual incidence of 12–13 per 100 000 population. Prospective registers show a continuing increase in incidence, especially in the under-5 age group. Similar incidences are reported for New Zealand, USA and the UK. In Asia the incidence is low, whereas in parts of Scandinavia it exceeds 30 per 100 000. The reasons for these variations are presumed to be both genetic and environmental. The sex ratio in diabetes is equal. Diabetes is uncommon in infancy. In childhood the incidence shows two peaks, at ages 4–6 years and 10– 14 years. Pathogenesis of type 1 diabetes There are two major factors that contribute to the pathogenesis of diabetes: • genetic predisposition • environmental triggers or protectors. Autoimmune destruction of beta cells Type 1 diabetes is caused by autoimmune destruction of the beta cells (insulin-producing cells) of the islets of Langerhans. T-cell infiltration of the islets and circulating autoantibodies precede the development of diabetes for months to years. Target antigens are insulin, glutamic acid decarboxylase and a tyrosine phosphatase. This preclinical phase, when blood glucose is normal and circulating antibodies are present, provides clues for prevention or postponement of the onset of clinical diabetes. There is an increased frequency of certain HLA types (HLA DR3/DQ2 and DR4/DQ8) in children with diabetes. The HLA genes are located on chromosome 6 and encode HLA molecules on the beta cells. Environmental factors that are potential candidates in the initiation of autoimmunity or that might act as progression factors are viruses (particularly enteroviruses) and dietary factors (cereals and cow’s milk). However, only congenital rubella is a proven environmental trigger and this is a rare cause of type 1 diabetes. The increase in childhood obesity may also account for an earlier presentation of type 1 diabetes due to insulin resistance and beta cell exhaustion but the extent of this contribution to the increasing incidence of type 1 diabetes is unresolved. Metabolic effects of insulin Insulin is the hormone of energy storage and anabolism. It allows glucose to enter cells to be stored as glycogen in the liver and muscle and as triglyceride in fat. Insulin deficiency prevents glycogen and triglyceride storage and causes their breakdown as well as that of protein. In addition, insulin deficiency promotes hepatic gluconeogenesis. The combined effects of glycogen breakdown, enhanced gluconeogenesis and failure of glucose entry into cells results in a rise in blood glucose. When the renal threshold is exceeded, glycosuria occurs. The osmotic effect of the glycosuria causes polyuria and dehydration. The breakdown of triglyceride (lipolysis) releases free fatty acids into the circulation. In the liver these are converted to ketoacids (ketogenesis), with eventual development of ketoacidosis. When the autoimmune process has destroyed approximately 90% of the beta cell mass, persistent hyperglycaemia indicates the onset of clinical diabetes. The diagnosis can be made before symptoms when subjects at risk are followed prospectively in natural history and intervention trials. In routine practice, symptoms are usually present for several weeks before the diagnosis is made; however, a child with a suspected diagnosis of diabetes should be investigated immediately. As the insulin deficiency proceeds, diabetic ketosis and then ketoacidosis develop and, if not treated, result in death. Ketoacidosis causes vomiting and, later, rapid deep breathing (Kussmaul respiration). The hyperventilation is a compensatory response for the metabolic acidosis by removing carbon dioxide. Chemical breakdown of acetoacetic acid in the body yields acetone, which can be detected on the patient’s breath. Breakdown of fat and protein and dehydration lead to weight loss. Abdominal pain mimicking an acute surgical abdomen may occur. Dehydration (shock) occurs with continuing massive urinary losses caused by the osmotic diuresis. The acidosis, dehydration and changes in plasma osmolality cause initial irritability, then confusion, drowsiness and eventually coma. Because immune function becomes compromised, the possibility of serious infection should always be considered, although it is rarely present. A summary of the clinical features and useful investigations at the time of presentation of type 1 diabetes is presented in Table 19.4.1. Clinical example For the previous 2 weeks, a mother had noted her 4-year-old son, Tom, to be irritable, thirsty and wetting the bed at night, having previously been dry. After being generally less well for 24 hours he became lethargic and began vomiting. On presentation at casualty, he was noted to be drowsy, he was dehydrated and he had deep sighing respirations (Kussmaul breathing). The diagnosis of diabetic ketoacidosis was confirmed when he was found to have a blood glucose of 22 mmol/l, blood ketones were 5.2 mmol/l, serum sodium 126 mmol/l, potassium 5.2 mmol/l, bicarbonate 10 mmol/l, pH 7.15 and the base deficit 26. Therapy consisted of intravenous isotonic fluids, intravenous potassium and intravenous insulin. Tom was discharged 4 days later, on two injections of insulin per day, a diabetes food plan and a programme of home blood glucose testing. During the hospital admission his family received education from the diabetes educator and dietitian, and the education programme was continued as an outpatient. A community diabetes educator visited Tom’s kindergarten to educate the staff about hypoglycaemia. Differential diagnoses The diagnosis of type 1 diabetes in childhood is not usually difficult, provided that the physician is aware that this condition occurs even in the very young. The most common misdiagnoses are to mistake: • polyuria for a urinary tract infection • the overbreathing of metabolic acidosis for a respiratory tract infection or asthma • vomiting and abdominal pain for gastroenteritis or an acute abdomen. Children with intercurrent infections, acute asthma or hypernatraemic dehydration may have transient hyperglycaemia and glycosuria that resolves with the intercurrent illness. Only very rarely do these children develop type 1 diabetes. Islet autoantibodies can be tested to determine whether the child is developing type 1 diabetes. Treatment of diabetic ketoacidosis The aims of therapy are: • emergency fluid replacement (10–20 ml/kg/h), using volume expanders, if shock is present so that the circulation is restored • correction of dehydration slowly over 24–48 hours, using normal (isotonic or 0.9%) saline • replacement of electrolyte losses and slow correction of acidosis. Supplemental potassium of 40–60 mmol/l in intravenous fluids is required to maintain normal serum potassium levels once insulin therapy has begun. Higher levels of supplementation will require ECG monitoring • correction of the insulin deficiency, with an infusion of soluble insulin. Treatment should be undertaken only in a centre equipped with paediatric intensive care facilities; the child may need to be transported there by an expert retrieval team. Frequent biochemical monitoring of the blood glucose, electrolytes and blood gases is required. The initial rate of insulin infusion of 0.1 unit/kg per hour is adjusted to produce a slow fall in the blood glucose. Rapid reductions in the blood glucose level and/or a fall in serum sodium concentration alter the plasma osmolality too quickly and increase the risks of life-threatening cerebral oedema. When ketones disappear, subcutaneous insulin is begun with regular food intake. Most children are stabilized on two to four daily injections of ultra-short and intermediate-acting insulins. Within days to weeks of the introduction of insulin, some recovery of the remaining viable beta cells may occur. During this period of partial remission (also known as the ‘honeymoon’ phase), insulin requirements fall. This phase may last for weeks or months but, as the underlying autoimmune destruction of beta cells is still in progress, more complete insulin deficiency occurs, so that blood glucose levels and insulin requirements eventually rise permanently. There is recent evidence that the process of islet destruction and regeneration may continue for years, providing an exciting research avenue to increase viable beta cells. Management Aims of management Type 1 diabetes is a permanent disorder. By definition, insulin treatment is necessary and in childhood at present it can only be given by injection (inhaled insulins can supplement long-acting subcutaneous insulin in adults). The long-term aims are for the child to achieve normal physical and emotional development, to lead a fulfilling life, with as little restriction on lifestyle and occupation as possible, and to minimize the risk of long-term microvascular and macrovascular complications. Management principles The attainment of these aims depends largely on maintaining good diabetic (metabolic) control. This is often difficult to achieve and is especially difficult in the under-5-year age group and for the adolescent. The aim is to keep the blood glucose levels as close to normal as possible pre- and postprandially. To measure the blood glucose, a spring-loaded lancet is used to prick a finger, the blood is placed on a reagent strip and the glucose can be measured accurately by a variety of meters designed for home use. Most children measure their blood glucose two to four times a day. The key elements in achieving stability in the blood glucose levels are: • insulin • diet • exercise. Insulin Insulin therapy is individualized. Generally, prepubertal children require 0.8–1 unit/kg body weight per day, given as two to four subcutaneous injections daily. This requirement increases with more variability during puberty. Preschool children may be very sensitive to short-acting insulin and therefore receive predominantly intermediate-acting insulin. Regimens using bolus doses of ultra-short-acting insulin before meals and an intermediate or longer-acting insulin before bed, or insulin pump regimens (basal insulin infusion subcutaneously and bolus doses of insulin with meals and snacks) are especially suitable for the adolescent needing more flexibility in the treatment schedule. These regimens may improve metabolic control and reduce hypoglycaemia and, in the case of pump therapy, weight gain, provided compliance is excellent and there is accompanying blood glucose monitoring. The dose of insulin is adjusted according to blood glucose measurements, anticipated diet and exercise. Glycosylated haemoglobin (HbA1C) levels indicate the level of control during the preceding 6–8 weeks and often provide the most meaningful guide when blood glucose levels are erratic. The target range for HbA 1C is less than or equal to 7.5%, and has fallen considerably during the last 5 years. Target blood glucose levels are generally 4– 8 mmol/l but may need to be higher in preschool children or children with a history of recurrent severe hypoglycaemia. Practical points Poor metabolic control with high HbA1c • • • • • • Is the prescribed insulin dose adequate? Is insulin omission a problem? Is food intake excessive or inappropriate? Consider education/dietary/psychological review Consider an eating disorder, especially in girls Consider increasing doses, changing insulin types and intensifying the insulin schedule if insulin omission is not a problem Diet Food raises the blood glucose level and this must be balanced by the glucose-lowering effect of insulin and exercise. This balance is usually achieved by having the diabetic diet supply carbohydrate throughout the day with three meals (breakfast, lunch, dinner) and three snacks (morning tea, afternoon tea and supper). However, toddlers will usually have a grazing pattern of food intake and older adolescents may not need three snacks per day. Children receiving insulin pump therapy have more flexibility, with lower carbohydrate intake if desired. The usual diabetic diet is relatively high in complex carbohydrates, low in simple carbohydrates (sugar) and low in saturated fats. Dietitians frequently use the glycaemic index of foods to guide the child and family to appropriate food choices. The glycaemic index is a classification of foods based on their postprandial blood glucose response and, as such, is a more predictable guide than merely measuring the carbohydrate content of foods. The diet must be adequate nutritionally for normal growth and must be acceptable and satisfying to the child. Individual and changing food plans are essential if the diet is to be adhered to. It is also necessary to account for pre-existing family and cultural traditions. Frequent review by an experienced dietitian is necessary to cope with changing requirements as the child grows. Extra food may need to be taken with exercise to prevent hypoglycaemia. Exercise Exercise increases the uptake of glucose by the exercising muscles and lowers the blood glucose level. This effect is seen only if the diabetes is in good control and adequate serum levels of insulin are present (exercise undertaken during poor control and with low insulin levels may paradoxically raise the blood glucose levels). To combat the hypoglycaemic effect of exercise, the child may need extra carbohydrate food. Regular exercise schedules in the older child may be better managed by a small reduction in the preceding insulin dose. Regular exercise should be encouraged. It does not, per se, improve metabolic control in children but it may do so indirectly in some individuals by modifying appetite and improving wellbeing and self esteem. Outpatient management For most children with diabetes, the initial hospitalization or day stay admission at diagnosis for stabilization and education is their only hospital admission. Much or all of the early education programme can be provided by the diabetes educator and dietitian as an outpatient, provided there are adequate resources for this and no contraindications exist, such as severe parental distress or communication difficulties. Follow-up visits are usually every 3 months once the child is stabilized to assess: • • • • • • • • • • general wellbeing history of hypoglycaemic episodes home blood glucose monitoring insulin schedule food plan school progress and absenteeism height and weight injection sites size of the thyroid gland presence of skin infections. The blood glucose profile is examined in the logbook kept by the patient, or is downloaded from the patient’s blood glucose monitor, and the adequacy of the dietary and insulin regimen is assessed. Continuous blood glucose monitoring (Fig. 19.4.1) is particularly useful when fine tuning of the insulin schedule is required, after a change to a new insulin schedule or insulin pump, or when there are concerns of undetectable nocturnal hypoglycaemia. The glycosylated haemoglobin (also called HbA 1C) measures the degree of glycosylation of haemoglobin, while the serum fructosamine measures the extent of non-enzymatic glycosylation of serum proteins. These parameters give an indication of the glycaemic control during the life span of the red cells (120 days) and the serum proteins (3 weeks). HbA1C levels can be measured in capillary blood within minutes in the outpatient clinic setting, which is ideal. With the combined efforts of the parents, the child and the diabetes management team (physician, dietitian, diabetes educator, social worker and psychologist) most children grow and develop normally, achieve their educational potential and have a satisfying childhood and adolescence. However, fewer than 50% of children in Australia achieve target HbA1c levels below 7.5%. Long-term microvascular and macrovascular complications Long-term microvascular and macrovascular complications which may occur in children with diabetes are: • • • • • nephropathy retinopathy neuropathy cardiovascular disease peripheral vascular disease. It is extremely rare for children and adolescents to show clinical signs of these complications but subclinical signs can be detected, particularly from adolescence onwards. These form the basis of complication screening programmes. It is recommended that, from 5 years duration of type 1 diabetes, the patient has an annual review including: • measurement of resting blood pressure • assessment of urinary albumin excretion, either by an overnight urine collection or an early morning sample • fundoscopy on dilated pupils by an ophthalmologist. Serum lipids also are relevant, particularly when there is a family history of a lipid disorder or of premature cardiovascular disease. The major risk factors for the development of complications are: • • • • • degree of metabolic control duration of type 1 diabetes smoking hypertension. family history. It should be reinforced for the patient and their family that any improvement in their glycaemic control, even if it is still not ideal, will reduce their risk of developing complications. The early introduction of angiotensin converting enzyme inhibitors, in patients with persistent microalbuminuria before blood pressure rises, delays the onset of chronic renal failure. Special problems in management Hypoglycaemia For parents, the occurrence of severe hypoglycaemia in their child (loss of consciousness or a convulsion) is one of the most distressing aspects of diabetes management. Usually, no long-term harmful effects result from a severe hypoglycaemic episode. However, the concern remains of possible neurological damage in the very young child, or in the adolescent with a cocktail of other drugs such as alcohol, and the possible development of hypoglycaemic unawareness with recurrent episodes of hypoglycaemia. Practical points Recurrent hypoglycaemia • Is it severe, moderate or mild; is it daytime or nocturnal? • Is there hypoglycaemic unawareness? • What is the HbA1c level? • Consider a change of insulin dose, insulin type (e.g. ultrashort analogues) or insulin schedule • Consider continuous glucose monitoring, particularly if nocturnal hypoglycaemia is suspected • Consider continuous subcutaneous insulin infusions (insulin pump therapy) Minor hypoglycaemic episodes are relatively frequent and reflect the difficulties in achieving stable control with current insulin delivery to the systemic circulation, particularly as an improving HbA 1C increases the risk of hypoglycaemia. However, the advent of insulin analogues (ultra-short and peakless basal) and insulin pumps has reduced the risk of hypoglycaemia. All patients with type 1 diabetes should carry some rapidly absorbable carbohydrate (e.g. glucose tablets or jelly beans) for immediate treatment of hypoglycaemic symptoms. Hypoglycaemia may occur if the insulin dose is excessive, if insufficient food is eaten or if extra exercise is undertaken. Severe hypoglycaemia is most common during the night when the glucose threshold for counterregulatory hormone responses is lower. The clinical features of hypoglycaemia can be divided into: • stimulation of the sympathetic nervous system: anxiety, palpitations, tachycardia, pallor, perspiration, headaches and abdominal pain • effects on the central nervous system: lethargy, dizziness, ataxia, weakness, confusion, personality changes, visual disturbance, unconsciousness, localized and generalized convulsions. These clinical features appear rapidly in a previously well child and there is no difficulty in differentiating hypoglycaemic coma from the coma of diabetic ketoacidosis. The emergency treatment for the unconscious hypoglycaemic child is to lie them on their side and check the airway. Oral fluids must not be given. Intramuscular or subcutaneous glucagon (0.5 mg for children under 5 years of age and 1 mg for older children and adults) or intravenous glucose (2.5 ml of 20% dextrose per kilogram of body weight) is administered. All families with a diabetic child should have glucagon at home and should be able to give it subcutaneously. The response to therapy is seen within minutes. For the less severely affected child, so called mild hypoglycaemia can be treated with oral glucose, e.g. half a glass of sugarcontaining non-diet lemonade, or jelly beans. On improvement after the emergency treatment, the child should receive some complex carbohydrate. Management of sick days Children with well controlled diabetes are no more prone to infections than the non-diabetic child. However, the stress of the infection, especially if associated with fever, causes a temporary insulin resistance and more insulin is required. Blood glucose tends to rise despite a poor oral intake. Ketosis may occur. This is a sign of significant insulin deficiency and, if untreated, diabetic ketoacidosis could develop. Parents are taught to measure blood glucose levels frequently, monitor the presence of ketosis (home measurement of blood ketones is more accurate than for urinary ketones) and give frequent small doses (10–20% of daily requirements) of short-acting insulin every 3–4 hours until the diabetes is stabilized again. Generally regular phone contact with the diabetes consultant or educator will keep the child out of hospital, except when vomiting and persistent ketosis complicates home management. Low doses of glucagon can also help prevent hypoglycaemia in the vomiting child at home, but the parents should be in close contact with a diabetes specialist for advice. Growth and delayed puberty Because insulin is the principal hormone of energy storage and anabolism, growth disturbances and pubertal delay can occur if diabetes control is poor. Mauriac syndrome is an extreme example of this, with short stature and hepatic enlargement due to fatty infiltration of the liver. The possibilities of Hashimoto thyroiditis, coeliac disease or, less commonly, adrenal insufficiency should also be considered because of their association with type 1 diabetes: many clinics routinely screen for thyroid function and coeliac disease. Psychological stresses Major problems arise with diabetic control in the presence of psychological stresses. Easily identifiable acute stresses such as school examinations do not usually cause significant problems. However, family conflicts, parental separation, teenage rebellion and other emotional problems may cause a more profound instability, and psychological counselling or formal psychotherapy may be required for the child and the whole family. The relevance of psychological wellbeing to good diabetes control is of such importance that the social worker or psychologist is an integral part of the management team. Most units also have patient and parent support groups. Diabetes camps also nurture self-esteem and confidence. Adherence Excellent diabetes control in childhood and adolescence is difficult in the best of circumstances. It becomes impossible when the child or the family become non-compliant with diet, monitoring or injections of insulin. Refusal to perform blood tests and to conform to the prescribed diet are not unusual periodically and represent a normal rebellion against the never ending discipline that characterizes diabetes management. The commonest cause of recurrent diabetic ketoacidosis and chronic poor metabolic control in adolescence is insulin omission. Patience and counselling are necessary, especially in adolescence, when normal risk-taking behaviour and growing independence do not combine well with diabetes. The adolescent and family should not be made to feel guilty when diabetes control is less than ideal. It is often useful for the frustrated doctor or educator to consider how well s/he would have coped with the tedious diabetes regimen during adolescence. Clinical example Jacinta, a 14-year-old girl with a 5-year history of diabetes, had had three episodes of diabetic ketoacidosis in 3 months. Her blood glucose logbook showed the values to be 4–10 mmol/l but a glycosylated haemoglobin level (HbA1C) was 10.8% (normal range 4– 6%). Her insulin dose was 0.9 units/kg per day and the recorded blood glucose levels appeared spurious. Admission to hospital for stabilization confirmed elevated blood glucose levels. Management required appropriate increases in insulin dose, education and counselling. The dietitian suspected that Jacinta might also have had an eating disorder complicating her insulin omission, and psychological review was arranged. Sometimes it is necessary for someone else (e.g. the parents or a community nurse) to temporarily take over responsibility for the insulin injections when there is a serious problem with insulin omission. Factors limiting management Factors that are often present when diabetes is proving difficult to manage, and that need to be considered further, are: • difficulties in family functioning • hypoglycaemia, which may be unrecognized • adherence problems and psychological stresses (particularly in adolescence). Future directions Families frequently ask about research advances and it is important that they have access to up-to-date information through regular seminars and reliable websites. New synthetic insulin analogues provide a better range of very short, intermediate and peakless long-acting insulins. Inhaled, oral and transdermal insulins are under investigation. Specific immunotherapy of the autoimmune process is being trialled in subjects with preclinical diabetes in an attempt to prevent diabetes and to prolong life of beta cells after diagnosis. Novel approaches to preventing vascular complications include targeting the intracellular mechanisms by which glucose is toxic, e.g. inhibitors of protein kinase C and agents that interfere with the accumulation of advanced glycation end-products. Transplantation of isolated islets of Langerhans in adults has shown recent promise with less beta-cell toxic immunosuppressive drugs. Whole pancreas transplants can occur successfully in conjunction with renal transplantation. Prevention of rejection of the transplant requires lifelong immunosuppression and donors of the pancreatic graft are not plentiful. Stem cells and islet regeneration are now major research directions. Type 2 diabetes The true incidence of type 2 diabetes is not known, as patients are frequently asymptomatic. Characteristics of type 2 diabetes at diagnosis are shown in Table 19.4.2. The distinction between type 1 and type 2 diabetes in childhood may be difficult to make on clinical grounds alone (for example, many children with type 1 diabetes are overweight and have a family history of type 2 diabetes). Measurement of islet antibodies to show their absence is usually necessary to confirm the diagnosis. The distinction is important, as patients with type 2 diabetes are treated with a weight-reducing diabetes diet and oral hypoglycaemics as first-line measures. Insulin may still be required, especially during intercurrent acute infections, when ketoacidosis can occur. Screening for microvascular and macrovascular complications should begin from the time of diagnosis in type 2 diabetes. There is an association between hyperandrogenism in adolescent females and type 2 diabetes. 19.5 Bone mineral disorders C. Jones Hypocalcaemia, rickets and hypercalcaemia are the most common manifestations of disorders of calcium, phosphate and vitamin D metabolism. Disorders of magnesium metabolism are rare but share many features of calcium disorders. Calcium, magnesium and phosphorus (Table 19.5.1) Calcium and phosphate form the major structural components of bone in the form of hydroxyapatite. The majority of magnesium is also found in bones. A large proportion of each mineral in bone is freely exchangeable with the extracellular fluid (ECF). Calcium and phosphate ions, under normal circumstances, are present in a supersaturated solution. A rise in phosphate will lead to the deposition of more calcium phosphate into bone as hydroxyapatite and cause hypocalcaemia. The distribution of calcium and phosphate between bone and the ECF is determined by hormonal regulation of the concentrations of these minerals. The most important hormones are 1,25-dihydroxyvitamin D3 (activated vitamin D) and parathyroid hormone (PTH). The actions of these hormones are summarized in Figure 19.5.1. The ionized ECF forms of calcium and magnesium are responsible for their physiological effects. The ionized form of calcium should be measured to confirm that true abnormalities in concentration are present because the equilibrium between the ionized and protein bound forms can change. For instance, an increase of 0.1 pH unit decreases the ionized calcium by 10%, and hypoalbuminaemia reduces total serum calcium but not the ionized calcium concentration because 90% of bound calcium is bound to albumin. Hypocalcaemia In the neonatal period, hypocalcaemia is defined as a total serum calcium concentration below 1.8 mmol/l (ionized calcium 1.0 mmol/l). Beyond this age, a total plasma calcium below 2.1 mmol/l (ionized calcium 1.2 mmol/l) constitutes hypocalcaemia. Clinical signs usually only occur when the total serum calcium is below 2 mmol/l (ionized calcium 0.75 mmol/l), although some patients will tolerate much lower levels and will still remain asymptomatic. The signs of hypocalcaemia are due to neuromuscular excitability. Jitteriness, apnoea, laryngeal spasm causing stridor and convulsions are frequent in infants. Tetany, carpopedal spasm and the Chvostek (facial twitch on percussion of the facial nerve near the temporomandibular joint) and Trousseau (tetany produced by inflating the sphygmomanometer above systolic blood pressure for up to 2 min) signs are seen mainly in older children. Intracerebral calcification and cataracts are complications. The ECG may show a prolonged QT interval. The causes of hypocalcaemia are listed in Table 19.5.2. Clinical example Kylie, a 6-year-old girl, had nephritis with a metabolic acidosis (pH 7.32, [HCO3] 15 mmol/l, PCO2 30 mmHg). She had an albumin of 20 g/l, the serum total [Ca2] was 1.0 mmol/l and the serum [phosphate] was 3.6 mmol/l. Would it have been safe to correct the acidosis with intravenous NaHCO3? From Table 19.5.1, if the serum albumin was normal, 46% of the total serum Ca2 would be ionized. However, the serum albumin concentration was reduced by 50%, so the amount of calcium bound to albumin would be reduced by 50% (the proportion of total calcium bound to albumin would be reduced to approximately 20%), leaving the ionized proportion of total serum calcium increased from the normal 46% to approximately 66%, equivalent to 0.66 mmol/l. Increasing the pH to 7.4 could reduce the ionized portion and precipitate overt symptoms of hypocalcaemia. Thus, the calcium concentration needed to be corrected before giving NaHCO3. Early neonatal hypocalcaemia is common in premature infants and in infants of diabetic mothers. In premature infants, it is possibly an exaggerated response to the normal interruption of the maternofetal calcium transfer; the serum calcium falls following delivery to a nadir reached at a few days of age and then increases to normal levels at 1–2 weeks of age. The signs are seen within hours of birth, become most severe about 48 hours after birth and then improve spontaneously. It may be aggravated by early phosphate-rich formula feeding or hypoxic– ischaemic injury. Treatment of symptomatic infants involves intravenous calcium and commencement of an appropriate diet, following the guidelines provided in Table 19.5.3. Late neonatal hypocalcaemia usually presents as tetany after the first few days of life. The main cause is transient hypoparathyroidism, as demonstrated by high plasma phosphate and low serum PTH concentrations in the face of hypocalcaemia. A similar clinical picture is seen in infants of mothers with hyperparathyroidism and in infants with congenital heart disease. Treatment may involve calcium infusion, calcitriol, oral calcium supplementation and a low phosphate formula. The infant can often be weaned from this treatment after a few weeks. Persistence of the hypocalcaemia beyond this time should prompt a search for other causes of hypoparathyroidism such as DiGeorge syndrome (aplasia of the parathyroids, thymic aplasia with T cell immunodeficiency and cardiovascular abnormalities), hypomagnesaemia or idiopathic congenital hypoparathyroidism. An abnormality of the calcium sensing receptor on the parathyroid cells (an activating mutation decreasing PTH release) is also a cause. Treatment is based on the use of calcitriol, often in combination with phosphate restriction. Late onset hypoparathyroidism may occur with destructive injury of the parathyroids, e.g. copper deposition in Wilson disease, iron deposition in haemosiderosis or autoimmune type I polyglandular syndrome. In this latter condition, children, usually girls, present with tetany or convulsions and may have candidiasis and adrenal insufficiency, or other autoimmune disorders such as alopecia, malabsorption, thyroiditis and diabetes. Pseudohypoparathyroidism is due to end-organ resistance to PTH and blood levels of PTH are high. Mental deficiency and skeletal abnormalities (particularly a short fourth metacarpal) may be associated. Treatment is the same as for hypoparathyroidism. Clinical example Stephanie had received induction treatment for T-cell lymphoma. She developed hypocalcaemia associated with a raised serum phosphate in the days following this treatment. How should the hypocalcaemia have been managed? The administration of intravenous calcium would have resulted in metastatic deposition of calcium phosphate, as the solubility product of calcium phosphate, was exceeded. The calcification would occur in blood vessels and soft tissues. Such an approach might, in fact, have been necessary if Stephanie had symptomatic hypocalcaemia (convulsions) but it would have been preferable to lower the serum phosphate first. This was be achieved by implementing a low phosphate diet, and administering oral phosphate binders (such as calcium carbonate), which bind the phosphate in the gut (calcium complexes phosphate and is not absorbed by the intestine). It would be uncommon to use dialysis or haemofiltration but these treatments would also lower serum phosphate concentrations. Hyperphosphataemia can cause hypocalcaemia acutely (as seen in tumour lysis syndrome, where cell death following the initiation of chemotherapy for bulky tumours results in the release of phosphate). Acute renal failure with retention of phosphate is associated with hypocalcaemia. The primary treatment in these conditions is to reduce serum phosphate concentrations through dietary restriction of phosphate, the use of phosphate binders (such as calcium carbonate) and dialysis. Rickets due to vitamin D deficiency or disorders of vitamin D metabolism may cause hypocalcaemia (see below.) Rickets Rickets is impaired mineralization of osteoid tissue in the growing child. The mineralization defect affects the epiphysial growth plates, where cartilage cells proliferate, and unmineralized osteoid tissue accumulates, resulting in widening metaphyses, weak bones and development of deformities, particularly in the weightbearing bones. In established bones there is continued bone resorption but failure of mineralization results in soft, rarefied bones (this is osteomalacia, which is the same disease as rickets, in the adult). Practical points Hypocalcaemia • Suspect symptomatic hypocalcaemia when there is tetany, unexplained stridor, irritability or convulsions • Confirm the diagnosis of hypocalcaemia with total and ionized calcium concentrations and take blood for magnesium, phosphate and parathyroid hormone concentrations • Treat symptomatic hypocalcaemia with intravenous 10% calcium chloride 0.2 ml/kg/dose, given into a well placed intravenous line slowly, and monitor the ECG during the infusion. Causes of rickets are deficiency of the effect of vitamin D and phosphate depletion. Figure 19.5.2 outlines the causes of vitamin D deficiency and abnormal metabolism of vitamin D in relation to the physiological production of the active product, 1,25-dihydroxyvitamin D3. The recommended dietary intake of vitamin D is 400 international units (IU) per day. With exposure to a small amount of afternoon sunlight, vitamin D supplementation is unnecessary. Breast milk contains 20–50 IU/l of vitamin D, yet rickets is uncommon in breastfed infants in the first year of life, provided the mother does not have osteomalacia. Most commercial milk formulas have 400 IU/l of vitamin D. In the last 25 years, the incidence of vitamin D deficiency has increased in Australia. This increase has been seen in the children of migrants from the Middle East, southern Europe and Asia, especially where the mother wears a hejab. Reduced sunlight and darkly pigmented skin combined with a range of social factors, including poor housing, lack of adequate infant welfare services for non-English-speaking migrants and unusual feeding patterns, have combined to make rickets more prevalent. Malabsorption associated with gastrointestinal, hepatic or pancreatic disease can cause vitamin D deficiency. In approximately 50% of cases hypocalcaemia is present at the time of diagnosis. Severe liver disease and prolonged use of anticonvulsant therapy with diphenylhydantoin or phenobarbital (through an increased hepatic turnover of vitamin D) can cause rickets. 1-hydroxylase deficiency (type 1 vitamin-D-dependent rickets, characterized by normal or high 25 dihydroxyvitamin D3 levels and low 1,25 dihydroxyvitamin D3 levels) and peripheral resistance to 1,25 dihydroxyvitamin D3 (type II vitamin-D-dependent rickets, characterized by high concentrations of 1,25 dihydroxyvitamin D3) present as severe hypocalcaemic vitamin D deficiency that fails to respond to treatment with vitamin D. The clinical features of these forms of rickets are quite variable: young children may present with tetany and convulsions; older children may present incidentally when a chest X-ray is performed for an intercurrent chest infection. Early signs in nutritional rickets include weakness of the outer table of the skull (craniotabes), thickening of the costochondral junctions (the ‘rachitic rosary’) and widening of the wrists and ankles. Later, asymmetry of the head with delayed closure of the anterior fontanelle, frontal and occipital bossing of the skull, development of a Harrison groove, scoliosis and lumbar lordosis, delayed dentition and bowing and bending of the legs develop. Muscular weakness, ligament laxity and fractures are common. The radiology of the ends of long bones is characteristic, showing widening of the space between metaphysis and epiphysis (Fig. 19.5.3). The metaphyseal ends of the long bones are widened and appear cupped and frayed. The biochemical changes are a normal or a low serum calcium, a low serum phosphate, a high serum alkaline phosphatase and a high PTH. Treatment of nutritional vitamin D deficiency is with oral vitamin D at a dose of 3000–5000 IU/day for 6 weeks. In some cases, treatment precipitates uptake of calcium into bones (‘hungry bones’) to such an extent that hypocalcaemia is accentuated and large doses of calcium supplement may be required to maintain normocalcaemia for the first week(s). In the longer term, an adequate calcium intake (600–1500 mg/d) needs to be maintained, as does an adequate intake of vitamin D (400 IU/d to age 4 years). In cases where poor compliance may be suspected, ‘stoss therapy’ using 50 000 IU of vitamin D every 6 weeks may be used. Radiological improvement is usually apparent within 4 weeks. Some cases are refractory and require longer treatment periods. If there is no response, tests for 1-hydroxylase deficiency, peripheral resistance to 1,25 dihydroxyvitamin D3 or hypophosphataemic rickets should be undertaken. Hypophosphataemic rickets X-linked dominant hypophosphataemic (vitamin-D-resistant) rickets is caused by failure of phosphate reabsorption in the renal tubule and lack of an appropriate increase in 1-hydroxylase activity (low phosphate concentrations normally increase the activity of this enzyme, which produces 1,25 dihydroxyvitamin D 3). In 50% of cases it is familial; the rest of the cases are due to new mutations. Clinically, the rickets affects the lower limbs predominantly, and investigations show that the calcium and PTH concentrations are normal, while the phosphate concentration is quite low. Males are more severely affected than females. Treatment consists of large amounts of dietary phosphate supplementation and large doses of calcitriol. Practical points Rickets • Suspect rickets when there is bowing of the long bones, exaggerated lumbar lordosis, splayed wrists and other rachitic changes in an irritable child • Examine for signs of hypocalcaemia and treat symptomatic hypocalcaemia • Suspect nutritional vitamin D deficiency in breastfed infants of mothers where covering clothing is worn at all times, children of dark skin colour and children with malabsorption or liver disease • Suspect another cause if there is failure to respond to vitamin D, low PTH concentrations and very low phosphate concentrations Chronic phosphate deficiency (e.g. renal Fanconi syndrome, dietary phosphate deficiency) from any cause will result in rickets. Renal osteodystrophy This condition predictably occurs when the glomerular filtration rate in chronic renal failure decreases to less than 25% of normal. It occurs as a result of a combination of events, including an increase in plasma phosphate and decreased 1-hydroxylation of 25 hydroxyvitamin D3 in the kidney, leading to a fall in 1,25 dihydroxyvitamin D3 production. This leads to hypocalcaemia and an increase in PTH. The combination of rickets and secondary hyperparathyroidism can result in gross skeletal deformation. Treatment consists of a graded range of measures beginning with dietary phosphate restriction, use of phosphate binders (calcium carbonate) and addition of calcitriol. Hypercalcaemia Hypercalcaemia is defined as a total serum calcium above 2.7 mmol/l (ionized calcium 1.3 mmol/l). Symptoms of hypercalcaemia include nausea and vomiting, polyuria and polydipsia, hypertension and failure to thrive. Ensuing hypercalciuria may cause nephrocalcinosis and urinary calculi. The ECG may show a shortened QT interval. A list of causes of hypercalcaemia is given in Table 19.5.4. Most of these are rare. Primary hyperparathyroidism occurs with some frequency in the second decade of life and is due to hyperplasia or adenoma of the parathyroid glands. It may occur as part of the multiple endocrine neoplasia syndromes (type I hyperparathyroidism associated with prolactinoma or gastrinoma; type II with hyperfunction of the adrenal, parathyroid and medullary cells of the thyroid). Investigations reveal high PTH concentrations, and X-ray appearances include subperiosteal resorption of bone (particularly the phalanges), ‘salt and pepper’ appearance of the cranium and cyst formation in long bones. Treatment of symptomatic disease usually involves subtotal parathyroidectomy. Familial hypocalciuric hypercalcaemia is an autosomal dominant disorder and is usually an asymptomatic condition caused by an inactivating mutation of the parathyroid calcium sensing receptor. Idiopathic hypercalcaemia of infancy is a condition in which there is increased absorption of dietary calcium. The condition usually resolves by the end of the first year of life. Occasionally it is associated with cardiovascular abnormalities (supravalvular aortic stenosis) and dysmorphic facial ‘elfin’ features (as in Williams syndrome). The genetic defect for Williams syndrome involves the elastin gene and many cases can be diagnosed using fluorescent in situ hybridization (FISH) studies. Treatment is directed at the cause of the hypercalcaemia. Severe symptoms may necessitate initiation of a diuresis using sodium chloride infusions, dietary phosphate supplements to bind calcium, glucocorticoids to reduce intestinal calcium absorption in vitamin D excess and bisphosphonates to prevent calcium release from bone. A low calcium milk formula is used to treat idiopathic hypercalcaemia of infancy. Hypercalciuria The normal upper limit of urinary calcium excretion is 0.15 mmol/kg/d or 0.7 mmol per mmol creatinine. Hypercalcaemia usually causes hypercalciuria (Table 19.5.4), with the notable exception of familial hypocalciuric hypercalcaemia. Normocalcaemic hypercalciuria is caused by furosemide or corticosteroid therapy, immobilization of limb fractures, distal renal tubular acidosis and some rare syndromes. Idiopathic hypercalciuria is common and appears to be an autosomal dominant condition with incomplete penetrance. On a calcium-rich or calcium-sufficient diet there is intestinal hyperabsorption of calcium and the PTH and 1,25-dihydroxyvitamin D concentrations are normal. On a lower calcium diet, ‘renal’ loss of calcium occurs and higher levels of PTH and 1,25 dihydroxyvitamin D are found. Excessive bone resorption occurs in some patients and can cause osteoporosis. Thus, the therapeutic safety of a low calcium diet is limited because of the risks of development of osteoporosis. Hypercalciuria can cause nephrocalcinosis (Fig. 19.5.4) and urinary calculi. Where treatment is necessary, thiazide diuretics have proved useful. Osteoporosis Osteoporosis is a condition where there is a decreased mineral content of the skeleton. Measurement of bone mineral density has improved over the last decade with the use of dual energy X-ray absorptiometry (DEXA). However, DEXA must be carefully interpreted with regard to the size of the bone where measurements are taken and the age of the patient, especially with regard to pubertal development of the child. Osteoporosis is only apparent radiologically when approximately half of the bone mineral content has been lost. Causes of osteoporosis are given in Table 19.5.5. Idiopathic juvenile osteoporosis is a rare condition that occurs in mid childhood with gross demineralization of the skeleton, which can result in extensive fractures (particularly vertebral crush fractures). The disease remits spontaneously with the onset of puberty. Osteopetrosis Osteopetrosis is a rare familial disorder of the skeleton in which there is a defect in bone and cartilage resorption and hence a deficiency of bone remodelling. This leads to a dense but brittle type of bone that fractures easily. Infants usually present early in life with a severe leukoerythroblastic anaemia, symptoms of compression of cranial nerves (particularly optic atrophy leading to blindness) and marked hepatosplenomegaly. The bones show a characteristic dense and poorly modelled X-ray appearance. Magnesium disorders Causes of hypomagnesaemia are given in Table 19.5.6. The symptoms of hypomagnesaemia resemble those of hypocalcaemia, with increased neuromuscular irritability. Severe hypomagnesaemia interferes with the release of PTH and consequently hypomagnesaemia and hypocalcaemia often coexist. Hypermagnesaemia occurs rarely in the absence of renal failure. The exception is the neonate born prematurely to a mother given magnesium sulphate for pre-eclampsia. Fig. 19.1.1 Turner syndrome. Note webbing of the neck and the broadly spaced nipples. Fig. 19.1.2 A model showing the factors regulating pituitary growth hormone (GH) release and its targets in specific tissues as well as the liver, inducing release of insulin-like growth factor I (IGF-I) and its binding proteins (IGFBPs). Fig. 19.1.3 Male height centile chart. A similar chart is available for females. Fig. 19.1.4 Male height velocity centile chart. A similar chart is available for females. Fig. 19.1.5 The typical growth patterns and height outcomes for familial short stature, constitutional delay and pathological short stature are illustrated on the percentile chart. The arrow indicates detection of the cause of the pathological growth pattern and commencement of appropriate therapy, thus allowing catch-up growth. Fig. 19.1.6 A Pubertal pubic hair changes in the female. B Pubertal genital and pubic hair changes in the male. A more detailed explanation is given in Table 19.1.4. Fig. 19.1.7 Pubertal breast changes in the female. A more detailed explanation is given in Table 19.1.4. Fig. 19.1.8 The typical growth pattern and outcome of precocious puberty. The vertical arrow indicates commencement of specific therapy to halt/slow down the precocious puberty. The horizontal arrows indicate the degree of bone age at the given chronological ages, advancement (e.g. bone age 12 years at chronological age 7.5 years) ultimately leading to premature fusion of the epiphyses and early cessation of growth. Fig. 19.1.9 The typical growth pattern of a boy with short stature and familial delayed puberty. The vertical arrow indicates the point at which a short course of testosterone therapy was given with a resulting growth spurt, onset of pubertal development and resumption of normal growth and variations in growth. Fig. 19.2.1 Thyroid uptake scan appearances in congenital hypothyroidism. A Thyroid agenesis. No functioning thyroid tissue present in the neck or in the usual ectopic sites. B Dyshormonogenesis. The radioangiogram reveals increased thyroid perfusion. The uptake of pertechnetate in 20 min is 14% (normal 2–5%). The thyroid scan reveals a diffuse goitre normally located in the neck. C Lingual thyroid. There is no evidence of perfused thyroid tissue in the neck. The thyroid scan reveals a prominent midline lingual thyroid. The uptake of pertechnetate in 20 min is 1% (normal 2–5%). D Normal thyroid. The radioangiogram of the head, neck and upper torso is unremarkable. The uptake of pertechnetate in 20 min is 5% (normal 2–5%). The thyroid scan reveals a normally located bilobular gland. Fig. 19.3.1 Normal prenatal development: internal genitalia. Fig. 19.3.2 Normal prenatal development: external genitalia. Fig. 19.3.3 Schematic illustration of normal and ambiguous female genitalia as in congenital adrenal hyperplasia. Note the urogenital sinus leading to a single opening and the enlarged clitoris induced by androgens (Adapted from Moore and Persaud, 1993). Fig. 19.3.4 Ambiguous genitalia of a female infant with congenital adrenal hyperplasia associated with salt wasting. Fig. 19.4.1 Continuous interstitial glucose monitoring obtained from an indwelling subcutaneous sensor in three patients with type 1 diabetes. In A, the pattern of results shows that the patient is in the remission phase. Fig. 19.4.1, cont’d In B, excellent control is seen, but the a.m. rise in glucose concentration requires an increase in a.m. ultra-short insulin. In C, the results show asymptomatic nocturnal hypoglycaemia and daytime hyperglycaemia. This requires adjustment of night-time long-acting insulin and daytime ultra-short insulin. Fig. 19.5.1 Hormonal control of calcium, magnesium and phosphate. Fig. 19.5.2 Vitamin D metabolism and rickets. Fig. 19.5.3 X-rays of the wrist (A) and knee (C) of a child with nutritional rickets at 18 months of age when the diagnosis was made, and 8 months later (B, D) after treatment was finished. Fig. 19.5.4 Renal ultrasound showing nephrocalcinosis of the medullary pyramids in a 4-year-old child with idiopathic nephrocalcinosis. Table 19.1.1 • • • • • • • • • Causes of short stature Genetic/familial short stature Constitutional delay Intrauterine growth retardation Chronic illness – malnutrition Skeletal dysplasia Iatrogenic (steroids/irradiation) Chromosomal abnormality/syndrome Psychosocial Endocrine Table 19.1.2 The relationship between chronological age, bone age and height age for common growth problems Growth problem Chronological age (CA) (years) Genetic short stature Constitutional delay 10 10 Delayed puberty Precocious puberty 15 6 Table 19.1.3 Height age (HA) Bone age (BA) HA CA BA HA CA BA BA HA CA BA HA CA BA Features of Turner syndrome General • Short stature • Developed puberty • Amenorrhoea Lymphatic abnormalities • Neck webbing • Low posterior hairline • Lymphoedema • Nail convexity/dysplasia Skeletal abnormalities • Micrognathia • High arch palate • Short fourth/fifth metacarpals • Increased carrying angles • Madelung deformity • Kyphoscoliosis • Broad chest • Abnormal upper/lower body segment ratio Metabolic • Thyroiditis • Carbohydrate intolerance Miscellaneous • Recurrent middle ear infections • Decreased hearing • Cardiac: coarctation or aortic stenosis (bicuspid aortic valve) • Renal anomaly • Naevi Table 19.1.4 Stages of puberty CA CA HA CA CA Males: genital (penis) development • Stage 1: Preadolescent, testes, scrotum and penis are of about the same size and proportion as in early childhood • Stage 2: Enlargement of scrotum and testes. Skin of scrotum reddens and changes in texture. Little or no enlargement of penis at this stage • Stage 3: Enlargement of the penis, which occurs at first mainly in length. Further growth of the testes and scrotum • Stage 4: Increased size of penis with growth in breadth and development of glans. Testes and scrotum larger; scrotal skin darkened • Stage 5: Genitalia adult in size and shape Females: breast development • Stage 1: Preadolescent: elevation of papilla only • Stage 2: Breast bud stage: elevation of breast and papilla as small mound. Enlargement of areola diameter • Stage 3: Further enlargement and elevation of breast and areola, with no separation of their contours • Stage 4: Projection of areola and papilla to form a secondary mound above the level of the breast • Stage 5: Mature stage: projection of papilla only, due to recession of the areola to the general contour of the breast Both sexes: pubic hair • Stage 1: Preadolescent: the vellus over the pubes is not further developed than that over the abdominal wall, that is, no pubic hair • Stage 2: Sparse growth of long, slightly pigmented downy hair, straight or slightly curled at the base of the penis in boys, or chiefly along labia in girls • Stage 3: Considerably darker, coarser and more curled. The hair spreads sparsely over the junction of the pubes • Stage 4: Hair now adult in type, but area covered is still considerably smaller than in adult. No spread to the medial surface of thighs • Stage 5: Adult in quantity and type with distribution of the horizontal (or classic ‘feminine’) pattern. Spread to medial surface of thighs but not up linea alba or elsewhere above the base of the inverse triangle (spread up linea alba occurs late and is rated stage 6) Table 19.1.5 Causes of delayed puberty Associated with normal or low serum gonadotrophins Constitutional delay Usually familial: associated with slow growth and a delayed bone age Chronic illness Poor nutrition, e.g. cystic fibrosis, juvenile arthritis, inflammatory bowel disease Endocrine causes Hypopituitarism, isolated gonadotrophin deficiency, Kallmann syndrome, hypothyroidism, hyperprolactinaemia Associated with elevated serum gonadotrophins* Gonadal dysgenesis Turner syndrome, Klinefelter syndrome, Noonan syndrome Anorchia Gonadal damage, Vascular damage, irradiation, vascular events infection (mumps), torsion or autoimmune disease * This usually signifies primary gonadal dysfunction. Table 19.2.1 Mutations in genes involved in thyroid development resulting in congenital hypothyroidism Gene Thyroid gland imaging Clinical features Genetics Comments PAX-8 Hypoplastic and cystic; ectopic Mild to moderate congenital hypothyroidism Familial and sporadic Autosomal dominant heterozygous mutations TTF-2 Thyroid agenesis Developmental delay, cleft palate, choanal atresia, spiky hair Familial, homozygous mutations Rare ‘Bamforth syndrome’ NKX2.1/ TTF-1 Normal, hypoplastic gland or agenesis Congenital hypothyroidism, lung disease, hypotonia, developmental delay and choreoathetosis Sporadic; heterozygous loss of function mutations Poor neurological outcome despite thyroid replacement TSH-R Normal to severely hypoplastic, normally located Compensated hypothyroidism to severe congenital hypothyroidism Recessive, inactivating mutations in compound heterozygotes Heterozygous carrier state may be relatively common in Caucasians Activating mutations cause congenital hyperthyroidism TTF: thyroid transcription factor; TSH-R: thyroid stimulating hormone receptor. Table 19.4.1 Type 1 diabetes: clinical features at presentation and useful investigations at diagnosis Clinical presentation • Polyuria and polydipsia • Enuresis and nocturia • Weight loss and fatigue • Thrush • Vomiting (with increasing ketosis) • Kussmaul breathing and coma (with increasing acidosis) Investigations • Urinalysis for glucose and ketones • Random blood glucose (postprandial) • Blood electrolytes and acid–base when unwell • Blood or other cultures and blood count (if infection suspected) • Islet antibodies (if type 2 diabetes suspected) Table 19.4.2 Characteristics of type 2 diabetes at diagnosis • Obesity • Acanthosis nigricans • Family history of type 2 diabetes • Absent or mild ketosis (although ketosis and ketoacidosis can occur ) • Absence of islet antibodies • Raised C peptide/insulin levels • Microvascular complications, hypertension and lipid abnormalities may be present • Hyperandrogenism maybe present • More common in Aboriginal and Polynesian populations Table 19.5.1 Distribution, serum concentrations, dietary requirements and sources of calcium, magnesium and phosphate Calcium Magnesium Phosphorus Body distribution (%) Bone* Intracellular 99 – 60 40 80 20 Serum status (%) Ionized† 46 55 85 Complexed Protein bound 14 40 25 20 5 10 2.4 2.1–2.7 2.2–2.6 0.7 0.75–1.1 0.75–1.1 1.6 2.0–3.3 1.0–1.3§ 300 550 800 1200 40 60 100 200 150 300 800 1200 300–500 1500 Tinned fish, dairy products 40 130 Green vegetables, seeds, nuts Serum levels (mmol/l) Cord blood‡ Neonatal Adult Dietary intake RDI (mg/d) 0.6 months 6–12 months 1–10 years 11–18 years Milk content (mg/l) Human Cow Other food sources 100–300 1000 Meats, dairy products * Ionic exchange occurs readily between extracellular fluid (ECF) and bone, enabling ECF concentrations to be kept fairly constant. † The ionized form of phosphorus at pH 7.4 is HPO42 (70%) and H2PO4 (20%). ‡ Cord-blood concentrations are higher than maternal blood concentrations, indicating active transport mechanisms are involved in transplacental transfer. Parathyroid-hormonerelated peptide (PTHRP) is probably involved in these processes. § Levels of phosphate slowly decline during childhood and reach adult levels on completion of bone growth. RDI, recommended dietary intake. Table 19.5.2 Causes of hypocalcaemia • Neonatal Early neonatal Late neonatal – prematurity, IDM, IUGR, birth asphyxia – di George syndrome – Phosphate load – Low magnesium – IDM • Pseudohypocalcaemia Hypoalbuminaemia • Vitamin D deficiency Nutritional deficiency Disorders of vitamin D metabolism 1-hydroxylase deficiency Vitamin-D-dependent rickets • Parathyroid-hormone associated Hypoparathyroidism Idiopathic Autoimmune polyglandular disease (with mucocutaneous candidiasis, or Addison disease) Calcium-sensing receptor activating mutations or antibodies Hypomagnesaemia Destructive lesions of the glands Hypoplasia PTH receptor defect – pseudohypoparathyroidism • Hyperphosphataemia Tumour lysis Renal failure • Pancreatitis • Medical treatment Large blood transfusion/exchange transfusion PTH, parathyroid hormone; IDM, infant of diabetic mother; IUGR, intrauterine growth retardation. Table 19.5.3 Treatment of hypocalcaemia Emergency • Patients with hypocalcaemia and symptoms should be treated with intravenous calcium* • Electrocardiography monitor (bradycardia) • Intravenous calcium chloride 10%, 0.2 ml/kg/dose (max 1 g), repeated at 4–6 h or followed by Infusion of 1 mmol ( 1.5 ml of 10% CaCl2)/kg/d • Correct the concurrent hypomagnesaemia (magnesium chloride, 0.2 mmol/kg over 1 h) Maintenance • Treat the underlying condition (see text) * Note: extravasations of intravenous calcium cause skin and subcutaneous tissue necrosis. Table 19.5.4 Causes of hypercalcaemia • Neonatal Hyperparathyroidism – primary – maternal hypoparathyroidism – maternal pseudohypoparathyroidism Idiopathic infantile hypercalcaemia Williams syndrome Familial hypocalciuric hypercalcaemia Subcutaneous fat necrosis • Vitamin D excess Iatrogenic Ectopic production – sarcoidosis, tuberculosis, lymphoma • Parathyroid hormone excess Primary hyperparathyroidism multiple endocrine neoplasia (MEN) syndromes Familial hypocalciuric hypercalcaemia Abnormalities related to PTH receptor or PTH-related peptide (PTHRP) • Other Bone disease, trauma and immobilization Thyrotoxicosis and hypothyroidism Neonatal fat necrosis Table 19.5.5 Causes of osteoporosis • Calcium deficiency Nutritional Malabsorption • Malignancy Leukaemia • Glucocorticoid excess Iatrogenic Cushing disease • Homocystinuria • Osteogenesis imperfecta • Immobilization • Idiopathic juvenile osteoporosis Table 19.5.6 Causes of hypomagnesaemia and hypermagnesaemia Hypomagnesaemia • Malabsorption, prolonged intravenous therapy • Diuretic therapy • Renal tubular acidosis • Hereditary disorders of renal tubular reabsorption Hypermagnesaemia • Neonate of mother given MgSO4 for pre eclampsia • Medications containing magnesium given to patients with renal failure