Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
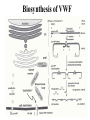
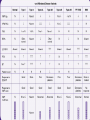
Von Willebrand Disease Ali Akalin Resident in Pathology UCHSC Von Willebrand Factor • The gene for vWF subunit is located on chromosome 12p. • Homomultimeric glycoprotein ranging in size from 600,000 to 20 million daltons • The carbohydrate component is estimated to add approximately 10-15% to the molecular mass. • Synthesized in endothelial cells and megakaryocytes and stored in Weibel-Palade bodies and platelet alpha granules, respectively. • vWF is initially formed in the ER as a pre-pro VWF molecule, which would assembly into homomultimeric protein after glycosylation, dimerization and multimerization in the golgi organelle and storage places in the cells. Biosynthesis of VWF Biosynthesis of VWF Structure of vWF Function of vWF • Serves as the carrier protein for factor VIII (probably factor VIII and vWF are brought together in storage granules). • Serves as the ligand that binds to glycoptrotein Ib receptor on platelets to initiate platelet adhesion to damaged blood vessel walls. • vWF needs to be activated to be able to bind to GP 1b receptor on platelets (Ristocetin, high shear force, collagen, etc) Variations in vWF Activity in Health and Disease • There is a continium of vWF levels between normal subjects and patients with vWD. • In patients with borderline results, it is necessary to repeat diagnostic testing 2 or 3 times in 4-6 weeks intervals. • vWF level varies with blood type. Overlap of vWF levels Variations in vWF by blood type Variations in vWF Activity in Health and Disease • In patients with type O blood and moderate reductions in plasma vWF, the presence or absence of a bleeding history and family studies are needed to confirm or exclude a diagnosis of vWD. • Thyroid hormone and estrogen promote vWF synthesis. • Deficiency of thyroid hormone reduces vWF in both normal subjects and patients with vWD. • Patients with mild vWD who take birth control pills or estrogen replacement therapy may increase their slightly low vWF levels into normal range. • Both factor VIII and vWF are acute phase reactants and their levels increase 1-3 times during exercise, inflammatory conditions, pregnancy, adrenergic stimulation, etc… Von Willebrand Disease • First described by Erik von Willlebrand in individuals living on the Aland Islands, an archipeloga between Sweden and Finland in 1926. • Characterized by mutations that lead to an impairment in the synthesis or function of vWF. Acquired forms are caused by different pathophysiologic mechanisms. • Manifests as mucocutaneuos bleeding (epistaxis, menorrhagia, GI bleed, ecchymosis). • Most common inherited bleeding disorder affecting up to 1% of population. • Autosomal inheritance Classification A- Quantitative deficiency of VWF Type 1: Partial quantitative deficiency of vWF Type 3: Virtually complete deficiency of vWF B- Qualitative deficiency of VWF Type 2A: Qualitative variants with decreased platelet dependent function associated with the absence of high and intermediate molecular weight vWF multimers Type 2B: Qualitative variants with increased affinity for platelet GPIb Type 2M: Qualitative variants with decreased platelet dependent function not caused by the absence of high-molecular weight vWF multimers Type 2N: Qualitative variants with markedly decreased affinity for factor VIII Type I VWD • Accounts for ~ 70 % of patients with VWD. • Autosomal dominant inheritance. • Clinical presentation ranges from mild to moderately severe bleeding. • Some patients are asymptomatic and detected incidentally in studies investigating a relative with type 3 disease. • Result from increased clearance of vWF from the circulation as well as from decreased synthesis. • Lab findings: – Concordant reduction in VWF antigen and activity (ristocetin cofactor activity), and factor VIII activity. – All vWF multimers are present, although in decreased concentration. – RIPA is impaired, if VWF levels are sufficiently low. – Platelet VWF activity and antigen are usually within the normal range, but may be reduced. Type 3 vWD • Very rare (estimated incidence 1/million). • Autosomal recessive inheritance. • Characterized by marked decrease or absence of vWF as a result of mutations in both alleles. • Presents with severe bleeding into soft tissue and joints, in addition to mucocutaneous bleeding. • Lab findings: – – – – vWF antigen and activity below the limits of detection RIPA is absent Factor VIII activity is 1-10 % of normal. vWF multimers are not visible on gel electrophoresis. Type 2A vWD • Accounts for ~ 10-15 % of cases of vWD. • Autosomal dominant inheritance. • Affected patients typically present with moderate to moderately severe bleeding. • Results from mutations in A2 domain that – cause a defect in the intracellular assembly and transport of normal VWF multimers (group 1), or – affect a normal cleavage site in VWF and cause increased susceptibility to proteolysis by the VWFcleaving protease after secretion (group 2) Type 2A vWD • Recessive forms of type 2A are caused by mutations within the propeptide, resulting in impaired multimer formation (type 2C) or dimer formation (type 2D). • Lab findings: • Discordant reduction in vWF activity and antigen (Activity/Ag < 0.6). • RIPA is reduced • vWF multimer analysis show an absence of high and intermediate molecular weight multimers. • Factor VIII may be normal or reduced. Type 2B • • • • Accounts for ~ 5 % of cases with vWD. Autosomal dominant inheritance. Presents with moderate to moderately severe bleeding. Caused by mutations in A1 domain that result in readily binding of vWF to GP 1b on platelets (gain of function mutations). • The increase in binding of larger multimers to platelet GP 1b results in their loss from the circulation. • Lab findings: – Discordant reduction in vWF activity and antigen (Activity/Ag < 0.6). – RIPA is increased (aggregation in low concentrations of ristocetin). – vWF multimer analysis show an absence of high molecular weight multimers. – Thrombocytopenia – Factor VIII may be normal or reduced Type 2B • Type 2B vWD should be differentiated from platelet type vWD. • A distinction can be made by modifying RIPA assay as follow: – – – – Patient plasma (vWF) + exagenous fresh normal platelets (1) Patient platelet + exageneous plasma with normal VWF (2) Type 2B: Low dose RIPA present in test 1, absent in test 2. Platelet type vWD: Opposite Type 2M • • • • • • • • • • • Rare Autosomal dominant inheritance. Significant bleeding symptoms Mutations in A1 domain that result in reduced binding of vWF to platelet GP 1b. Vicenza variant show larger than normal multimers in plasma Some variants contain the propeptide in the multimers Lab findings: Discordant reduction in vWF activity and antigen (Activity/Ag < 0.6) vWF multimer analysis shows the presence of full spectrum of multimers. RIPA is reduced. Factor VIII level is reduced if vWF is sufficiently low. Differentiate Between BernardSoulier- Type1- Type 2M vWF:RCo Type 1 vWD RIPA Often Normal Type 2M VwD Bernard-Soulier Syndrome Normal Absent Type 2N (Normandy) vWD • Uncommon • Autosomal recessive inheritance. • Mutations affecting the N-terminus (D’ and D3) of mature vWF monomer within the binding site for factor VIII, causing decreased binding to factor VIII. • Presents with bleeding into soft tissue, joints. • Low levels of factor VIII ( 5-15 %). • Should be suspected who presents with isolated factor VIII deficiency and in families in which an autosomal inheritance pattern for factor VIII is suggested. Type 2N (Normandy) vWD • Lab findings: – Normal vWF activity, antigen, RIPA and vWF multimer analysis. – Factor VIII is usually 5-15 % of normal. – Decreased binding of normal factor VIII to the patient’s VWF. Diagnostic Laboratory Testing for vWD A- Tests to establish the diagnosis 1- PTT 2- VWF antigen 3- vWF:R:Co 4- Factor VIII 5- vWF multimers B- Tests to further define vWD 1- Low dose RIPA 2- Collagen binding 3- vWF- Factor VIII binding (Type 2N) 4- vWF antibodies 5- Platelet vWF 6- vWF AgII 7- DNA analysis vWF Antigen Testing • Immunoelectrophoresis (Laurell) (In the past) • ELISA • Automated latex bead assay (Rheumatoid factor might cause false elevation of vWF antigen level in this assay) vWF Activity • Ristocetin cafactor activity (vWF:RCo): tests the ability of the patient’s vWF to agglutinate formalin fixed commercial platelets by binding to platelet GP 1b receptor in the presence of ristocetin. • (Patient plasma(vWF)+ formalin fixed commercial platelet+Ristocetin) Collagen Binding Assay (vWF:CB) • Patient plasma (vWF) is added onto collagen-coated plates. The amount of vWF bound is proportional to the patient’s plasma vWF level. Ristocetin-Induced Platelet Aggregation (RIPA) • Measures the affinity with which vWF binds to the platelet GP 1b receptor by limiting the concentration of ristocetin in the assay. • Patient’s fresh platelet rich plasma (provides both platelet and vWF) is mixed with sequentially lower concentrations of ristocetin, covering a range of concentrations from 0.4 to 1.2 mg/ml. The absence or presence of aggregation at each concentration is recorded. • Platelet-rich plasma from patients with normal vWF does not show aggregation to ristocetin concentrations below 0.6-0.8 mg/ml. • Platelet-rich plasma from patients with type 2B vWD show aggregation to ristocetin concentrations 0.4-0.5 mg/ml. Ristocetin-Induced Platelet Aggregation (RIPA) • RIPA assay assumes that the patient’s platelet GP 1b is qualitatively and quantitatively normal. • Platelets from patients with Bernard-Soulier syndrome (decrease or abnormality in platelet GP 1b) do not aggregate in the presence of normal vWF and ristocetin. • Gain of funtion mutations in platelet GP 1b cause an increase in the affinity between vWF and GP 1b, resulting in aggregation of platelets at low concentrations of ristocetin, which mimics type 2b vWD. • A distinction can be made by modifying RIPA assay as follow: – Patient plasma (vWF) + exagenous fresh normal platelets – Patient Platelet + exageneous plasma with normal VWF RIPA vWF Multimers • Patient’s plasma vWF multimers (rarely eluted vWF from platelets) are sizeseparated by electrophoresis in low concentration agorose gels to look for the presence or absence of high, intermediate and low molecular weight vWF multimers. • • • • • 1: Type 3 2: Normal 3: Type 2A 4: Type 2B 5: vWF extracted from platelets Acquired vWD • Assocciated with a number of different disease states. Acquired vWD • Multimer analysis reveals a decrease in high molecular weight multimers, suggesting preferential removal of high molecular weight multimers. • Pathophysiologic mechanisms include: – Antibody formation to vWF (SLE) • Mixing studies • ELISA • Immunoblotting – Proteolysis • Plasmin (decompensated cirrhosis, pancreatitis, DIC, thrombolytic tx) • Elastase-lik e enzymes (myeloproliferative disorders) Acquired vWD – Binding to tumor cells with increased clearance (Wilms’ tumor, multiple myeloma, Waldenstrom macroglobulinemia) – Decreased synthesis (hypothyroidism) – High shear force (Noncyanotic congenital heart disease, high grade aortic stenosis) • vWF readily binds to platelets and/or proteolysis by the vWFcleaving protease at sites of high shear force. Treatment • • • • General Considerations Treatment of Inherited vWD Treatment of Acquired vWD Treatment of vWD during pregnancy General Considerations • Accurate and complete diagnosis. • Patient’s – past history of bleeding, – response to treatment, – associated medical conditions – current medications. • Aspirin containing medications • NSAID Treatment of Inherited vWD • • • • • Desmopressin Replacement therapy with vWF Antifibrinolytic therapy Estrogen Topical agents Treatment of Acquired vWD • Depends on underlying disease and pathogenetic mechanism – Treatment of underlying disease – Desmopressin – vWF replacement therapy – IVIG – Corticosteroids – Plasmapheresis – Extracorporeal immunoadsorption – Immunosuppressive medications (cyclophosphomide for SLE) – Factor VIIa Treatment of vWD During Pregnancy • Not needed in the majority of women with vWD because vWF levels rise during second and third trimesters. • If needed during pregnancy, desmopressin should not be used. • Low factor VIII level appears to be most important determinant of increased bleeding during delivery. Recommendation is >50 % factor VIII level. • The average time of onset for postpartum hemorrhage is between 11-23 days post-delivery. • Desmopressin and/or vWF replacement therapy is apropriate during the first 2-4 weeks of post-delivery, if needed. References • • • • • • • Rick M. E. www.uptodate.online.com Ginsburg D., Wagner D. D. Hoffman: Hematology: Basic Principles and Practice, 4th ed., Copyright © 2005 Elsevier Sadler JE. New concepts in von Willebrand disease.Annu Rev Med. 2005;56:173-91. Federici AB. Clinical diagnosis of von Willebrand disease. Haemophilia. 2004 Oct;10 Suppl 4:169-76. Favaloro E. J. et al. von Willebrand disease: laboratory aspects of diagnosis and treatment. Haemophilia. 2004 Oct;10 Suppl 4:164-8. Behrman: Nelson Textbook of Pediatrics, 17th ed., Copyright © 2004 Elsevier Colman R. W. et al (editors). Hemostasis and thrombosis : basic principles and clinical practice. Philadelphia : Lippincott Williams & Wilkins, c2001. 4th Edition.