Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
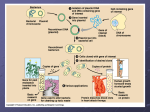
Ch. 19 Part 2 Gene Technology Vectors • Using RE you have cut (or maybe made) your desire gene • How do you get it into organism? • Must use a VECTOR (a go-between) • Viruses • bacteriophages • Liposomes • Tiny spheres of lipids containing DNA • MOST Common vector is a PLASMID • Small, circular piece of double-stranded DNA • Easy incorporation from prokaryotes to eukaryotes • Occur naturally in bacteria • Replicated independently in bacteria • Contain genes for antibiotic resistance • Can be exchanged between bacteria (during conjugation, even with different species) • If your gene is inserted in bacteria, you can use bacteria to make your desired protein from that gene (use the bacterial cell for your benefit) Plasmid Extraction and Gene Insertion • Treat bacteria with your desired plasmid with enzymes to break down cell wall • Naked bacteria centrifuged • Large bacteria chromosomes = heavy = sink to bottom • Small chromosomes (plasmids) = lighter = remain on top • Remove lighter (plasmid) DNA • Cut circular DNA of plasmid using Restriction enzyme • Must USE the same enzyme you used to cut out your desired gene • Creates the COMPLEMENTARY sticky ends (IMPORTANT in integrating your desired DNA into plasma seamlessly) • If blunt ends are created, nucleotides with sticky ends must be added to both gene AND plasmid in order to achieve insertion • Mix open plasmids and desired gene together • Plasmid sticky ends pair up with sticky ends of desired gene • Add DNA ligase to the mixture • Links together the sugar-phosphate backbone od desired gene and plasmid • Produces RECOMBINANT DNA = CLOSED circle of double stranded DNA (plasmid with your desired gene inserted) Why use plasmids? • Plasmids can be modified to produce excellent vectors • Artificial plasmids can be created • Example: pUC group of plasmids • Low molecular mass • Allows easy uptake by bacteria • Origin of replication • Easily copied • Contain polylinker • Several single target sites for different restriction enzymes in one short stretch of DNA • short DNA sequence containing two or more different sites for cleavage by restriction enzymes • Introduced into vectors to make cloning easier • Provide sites that allow cloning DNA, cut with any of a number of different restriction enzymes, into a single plasmid • One or more marker genes • Allows for easy identification of cells that have taken up plasmid Getting Plasmids into Bacteria • Treat bacteria with solution containing HIGH concentration of CALCIUM ions • Cool solution • Heat shock solution • Increases chances of plasmids passing through the surface of he cell membrane • Small proportion of bacteria (1%) TRANSFORMED • They have taken up the plasmid • Remaining bacteria cannot be used because: • They have taken up taken up closed plasmids and have NOT incorporated them into their DNA • They have not taken up plasmids at all Identifying Bacteria with Recombinant DNA • In order to successfully make desired proteins (gene product), you must find and identify the bacteria that have successfully incorporated the plasmid into their DNA • Processes of Identification • Use of antibiotic resistance • Not favored • Use of Gene markers • Favored method Identification with Antibiotic Resistance • Desired gene inserted into bacterial at the point of a gene for antibiotic resistance • Bacteria will no longer be resistant to the antibiotic • Spread the bacteria on agar plate containing antibiotic • Any bacteria that has taken up the plasma will NOT be resistant (will not grow on the plate) • Those are the bacteria with successful incorporation of the plasmid • Bacteria’s DNA polymerase copies plasmids • Bacteria divide by binary fission • Each daughter cell has several copies of plasmid • Each bacteria transcribes the protein (gene product) made by the gene • Problems: • Spread of antibiotic resistant genes to other bacteria (leads to untreatable diseases) Insulin Production • Diabetes Mellitus • Caused by inability of pancreas to make insulin • Treatment • Prior to 1980’sInsulin extract from pigs/cattle and injected into patients • 1983 human insulin made used GM bacteria • Problems Encountered • Locating and isolating gene that produced insulin in human DNA difficult • Cutting out gene for insulin too difficult Production of Human Insulin • mRNA for insulin in pancreatic beta cells extracted • Only cells expressing insulin gene • Contain large quantities of insulin mRNA • mRNA incubated with reverse transcriptase enzyme to produce single stranded DNA • Single stranded DNA converted to double-stranded DNA molecules with DNA polymerase • DNA molecule for insulin now inserted into plasmids of E. coli bacteria • Advantages: • Reliable supply to meet increasing demand for insulin • Not dependent on animals • Nucleotide sequence can be modified to create more effective insulin with different properties • Act faster • Act slower Reverse transcriptase • Enzyme extracted from RETROVIRUSES • Reverses transcription (mRNA single stranded DNA) DNA Polymerase • Enzyme • Assembles nucleotides to make complementary DNA strand Genetic Markers • a gene or DNA sequence with a known location on a chromosome that can be used to identify individuals or species • Use to identify transformed bacteria • Bacteria that contain the plasmid with the inserted gene (that contains the instructions to make our desired protein) • Common Genetic Markers: • Antibiotic resistance genes • Less popular • Lead to antibiotic resistant bacteria (dangerous) • Genes that code for unique enzymes • More popular Useful Enzymes as Genetic Markers • Enzymes that make protein GFP (green fluorescent protein) • • • • • Obtained from jellyfish DNA GFP fluoresces bright green in UV light Gene for GFP is inserted into plasmids Use UV light on bacteria Whichever bacteria fluoresce bright green are the bacteria that contain the transformed plasmid (the genetically modified DNA) • Enzyme B-glucuronidase (GUS) • Obtained from E. coli bacteria • Cells with this enzyme need to be incubated with specific, colorless/non-fluorescent substrates • They will transform this substrate into color of fluorescent products • Color/fluorescence helps identify activity of inserted genes • Color change = genetically modified organism Promoters in Bacteria • Not all bacterial genes are switched on • Environmental conditions determine the genes that bacteria will express • Recall E.coli making the enzyme Blactosidase in presence of lactose medium and no glucose (lac operon) • Promoters control expression of genes • Promoter region of DNA where RNA polymerase binds to begin transcription of DNA • What does this mean for our inserted genes in the bacteria? • We have to insert a promoter as well (to ensure our gene is turned on) • Promoter does TWO things: 1. Allows RNA polymerase to bind to DNA 2. Ensures that RNA polymerase recognizes which of the 2 DNA strands is the template strand Promoters in Bacteria • Promoter does TWO things: 1. Allows RNA polymerase to bind to DNA 2. Ensures that RNA polymerase recognizes which of the 2 DNA strands is the template strand • Promoter region of DNA contains TRANSCRIPTION STRAT POINT • First nucleotide of gene to be transcribed • Promoter: • CONTROLS the expression of the gene • ENSURES high level of gene expression • Eukaryotic Cell Transcription • Transcription factors required to bind to promoter region or RNA polymerase for transcription to start Gel Electrophoresis • method for separation and analysis of macromolecules (DNA, RNA and proteins) and their fragments, based on their size and charge • Used in analysis of DNA and protein • Involves: • Agarose gel with “wells” in it • Molecules placed in wells • Electrical field applied to gel to make molecules move through gel • Movement of molecules depends on factors: • Net (overall) charge • Size of molecules • Composition of gel Gel Electrophoresis Factors • Net (overall) charge • Gel in chamber attached to voltage differential • Negatively charged molecules move towards POSITIVE anode • Positively charged particles move towards NEGATIVE anode • Highly charged molecules move faster than less charged molecules • Size of molecules • Small molecules move faster than large molecules • Composition of gel • Gel contains “pores” tiny openings in which molecules move through • Size of pores determine speed which molecules move through the gel • Gel for DNA electrophoresis AGAROSE • Gel for protein electrophoresis POLYACRYLAMINE Gel Electrophoresis of Proteins • Used to separate the polypeptides made by different alleles of many genes • One gene can have many versions or “alleles” (sections of DNA that code for a specific protein) • Proteins usually have an overall NEGATIVE charge • Proteins need to go through IONIZATION process • R-groups of amino acids become charged • Procedure needs to be carried out at a constant pH using a buffer solution Example of Protein Gel Electrophoresis • Hemoglobin • Many variants • Adult hemoglobin 4 polypeptide chains • 2 alpha chains/ alpha globins • 2 beta chains/ beta globins • Sickle cell anemia • Point mutation has caused a change in the beta globin chain • normal glutamic acid (polar R-group) • after mutation valine (non-polar Rgroup) • Difference in charges of each polypeptide chain means: • Variants of beta globin can be separated with protein electrophoresis • • Sickle cell anemia polypeptide chain less negative charge, protein molecules move SLOWER through the gel = shorter distance traveled Normal polypeptide chain greater negative charge, protein molecules move FASTER through gel = farther distance across gel • Test used to find out if some carries sickle cell allele Gel Electrophoresis of DNA • DNA structure • Negatively charged phosphate group = overall negative charge • SIZE of DNA is important • Small fragments of DNA move fast • Large fragments of DNA move slower • Genetic Profiling/DNA fingerprinting is the application of gel electrophoresis of DNA and forensic science Steps of Gel Electrophoresis • • • • Make and use a gel (agarose for DNA, polyacrylamide for protein) Use a comb when making gel to create “wells” Wells should be made at the negative cathode end of the gel Place samples of DNA into wells, using micropipette, changing tip for each different sample to avoid cross-contamination • Micropipette prevents movement fi DNA sideways/ensure DNA samples stay in their specific well • • • • • Add stain/dye to DNA in each well Add glycerin to sink DNA into well Add buffer solution to gel in chamber (has specific pH) Apply potential difference/voltage difference by turning on power supply Stain and observe DNA • Use UV light & DNA staining • DNA samples are radioactive at beginning • Use autoradiograph or X-ray to visualize • Hazards and safety precautions: • Electrical • Chemical Irritants • UV light DNA Stains: • Methylene blue • Crystal violet • Sybr green • Ethidium bromide • Acridine orange • fluorescien The t-Test • Used to assess whether or not the MEANS of two sets of data with roughly normal distributions are significantly different from each other • Use when you want to know if two sets of continuous data are significantly different from one another • Criteria for using test: • You have 2 sets of continuous, quantitative data • Quantitative numerical data collected from experiment • Continuous each measurement, count, or reading can be any value between two extremes • Not necessarily whole numbers • Discrete each measurement, count, or reading can only be one of a set number of discrete values • MUST be whole numbers • • • • You have more than 10 but less than 30 readings of each set of data Both sets of data come from populations that have normal distribution Standard deviation for two sets of data is very similar How to analyze results: • Get your t-value • If it is GREATER than the t-value for a probability of 0.05 significant difference Standard Deviation Formula 𝑆= 𝑥−𝑥 𝑛−1 2 Step 3: calculate difference between observation and mean Step 1: List each x observation • 𝑥 = the mean • Ʃ = “sum” • x = number of individual values in a set of data • n = total number of observations (individual values, readings, or measurements) in one set of data Step 2: Calculate • s = standard the mean deviation (sum of all x • Create a table!!! values divided by total amount of x values) 𝑥−𝑥 𝑥−𝑥 1.1 -0.2 0.04 1.2 -0.1 0.01 1.3 0.0 0.0 1.4 0.1 0.01 1.5 0.2 0.04 1.4 0.1 0.01 1.1 -0.2 0.04 𝑥 = 9.0 n=7 𝑥 = 1.3 2 Step 4: calculate the square root of each difference Step 5: calculate the 𝑥−𝑥 2 = 0.15 n–1=6 𝑥−𝑥 𝑛−1 2 SUM of all the square differences Step 6: Divide the = 0.025 s = 0.158 SUM of all the square differences by (n-1) Step 7: Find the square root to get the standard deviation The t-Test • Start with a null hypothesis • There is no significant difference between two sample of data • Must have two sets of data • Calculate mean of EACH set of data • Calculate difference from the mean of all observations in each set of data (𝑥 − 𝑥) • Calculate the squares of each of the above (𝑥 − 𝑥)2 • Calculate the sum of the squares (𝑥 − 𝑥)2 • Divide by n1 – 1 for the first set and n2 – 1 for the second set • n = number of individual measurements of sample set • Take the square root of the above answer (standard deviation for each set) • Square the standard deviation and divide by the number of observations in that sample, for both samples • Add values together and take square root of the total • Divide the difference in the two sample means with the value from the previous step • This is your t-value • Calculate degree of freedom for all data (v) • v = (n1 - 1) + (n2 - 1) • Refer to table of t-values for your specific degree of freedom • Probabilities in t-test table are probabilities that the null hypothesis IS CORRECT (there is no significant difference) • Probability you find is the probability that any difference in data is due to chance 𝑡= 𝑥1 − 𝑥2 𝑠12 𝑠22 𝑛1 + 𝑛2 𝑥1 = mean of sample data set 1 𝑥2 = mean of sample data set 1 s1 = standard deviation of sample 1 s2 = standard deviation of sample 1 n1 = number of individual measurements of sample 1 n2 = number of individual measurements of sample 1 t-Test • Probaility of 0.05 is the critical value • 5% confidence level • Differences in data are due to chance if: • T-test value represents a probability 0.05 or MORE • Differences in data are NOT significant • Number of observations IMPORTANT • Total # of observations (both samples added together) is LESS than 30 error due to chance is significant • Table of t makes adjustment to critical values to take this into account • This is why value of degrees of freedom needs to be calculated • Total # of observations (both samples added together) is MORE than 30 number of observations make little or no difference to critical values of t