Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
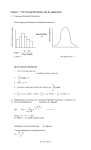
Electronic structure and optical properties of Transparent Conducting Oxides Hemant Dixit Electron Microscopy for Materials Science – EMAT, Universiteit Antwerpen , Campus Groenenborger, Groenenborgerlaan 171, B-2020 Antwerpen, Belgium. Transparent Conducting Oxides (TCO’s) are technologically important materials and have wide range of application in transparent electronics, solar cells, flat panel displays, touch screens and so on. These materials combine the unique material property of being optically transparent (property of an insulator) and electrically conductive (property of a metal). The systematic study of the correlation between the structure of the material with its electronic and optical properties, using firstprinciples electronic structure calculations, is important to understand the physics of these materials and to design new materials cost effectively. We have applied Density-functional theory (DFT) successfully onto basic TCO materials like zincoxide, indiumoxide and cadmium oxide. DFT results show that the structural properties can be accurately described. However the electronic structure, using DFT, shows some discrepancies compared to experiment due to intrinsic complexities involved in the theoretical description of oxides. It is an open area of research and an accurate theoretical description is still not available in literature. To calculate the band gap of these oxides we have used the GW approximation. The GW technique involves the ejection or injection of electrons that links the N particle system with the (N+/-1)-particle system. Thus, the GW approximation offers a strong physical basis to correlate the band energies obtained using Green's function with the experimental band gap measured using photoemission spectroscopy techniques. The GW approximation to many body perturbation theory represents the state of the art technique to calculate the quasiparticle correction to the band gap of solids and has been successfully applied to many materials. Although the GW approximation often works well with pseudopotentials and plane wave basis sets used within Density Functional Theory – Local density approximation (DFT-LDA), the II-VI materials are particularly challenging due to the strong p-d hybridization between the cation 'd' and anion 'p' states. We discuss the modification required in the pseudopotentials for correct description of the GW gap. In this talk we will show the GW results on systems like ZnO, ZnX2O4 (X=Al, Ga and In) spinel oxides and SnO2.