Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
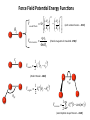
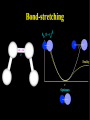
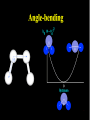
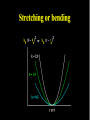
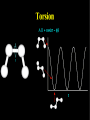
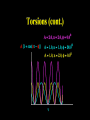
Molecular Mechanics Force Fields Basic Premise If we want to study a protein, piece of DNA, biological membranes, polysaccharide, crystal la;ce, nanomaterials, diffusion in liquids,… the number of electrons (i.e. the number of energy calculaAons) make quantum mechanical calcula-ons impossible even with present-‐day computers. Instead, we replace the nuclei and electrons, and their interacAons, by new potenAal funcAons: ”Classical” atoms. Based on simple physical concepts Enables the systems under study to be VERY large (100,000 atoms). Molecular mechanics force fields The molecular interacAons, also known as the potenAals, together form a force field, A force field is a mathema8cal descrip8on of the classical forces or energies between par8cles (atoms). Energy = func%on of atomic posi%ons (x,y,z) The force field equa%on consists of several func%ons that describe molecular proper%es both within and between molecules The force field also contains parameters (numbers) in the potenAal funcAons that are tuned to each type of molecule (protein, nucleic acid, carbohydrates) There are many different force field equaAons and parameter sets A force field must be simple enough that it can be evaluated quickly, but sufficiently detailed that it reproduces the key features of the physical system being modelled. Force field classifica8on In general, force fields can be classified as either: Specific (many parameters, limited applicability, high accuracy) O>en developed in academic labs for study of specific molecular classes or Generic (fewer parameters, more generaliza-ons, wide applicability, poor accuracy) Easiest to use in point-‐and click so>ware Force Field Parameters can come from: Experimental sources (mainly from x-‐ray diffrac-on) or Theore-cal calcula-ons (mainly from QM) Many force fields employ similar mathemaAcal equaAons but differ in the parameters used in the equaAons. It is therefore extremely dangerous mix to parameters between force fields. Different Force Fields: AMBER ( Assisted Model Building with Energy Refinement). CHARMM (Chemistry at HARvard using Molecular Mechanics). GROMOS (GROenigen Molecular SimulaAon) OPLS (OpAmized Parameters for Large-‐scale SimulaAons) MMFF (the Merck Molecular Force Field) DREIDING Generic force field due to Mayo et al. (1990) UNIVERSAL (UFF)Generic force field due to Rappeet al. (1992) CVFF/PCFF Force fields for fluorinated hydrocarbons MM2, MM3, MM4 Developed by Allinger et al. for calculaAons on small molecules COMPASS Commercial force field marketed by Accelrys Inc. Different Force Fields: AMBER ( Assisted Model Building with Energy Refinement). CHARMM (Chemistry at HARvard using Molecular Mechanics). GROMOS (GROenigen Molecular SimulaAon) OPLS (OpAmized Parameters for Large-‐scale SimulaAons) MMFF (the Merck Molecular Force Field) DREIDING Generic force field due to Mayo et al. (1990) UNIVERSAL (UFF)Generic force field due to Rappeet al. (1992) CVFF/PCFF Force fields for fluorinated hydrocarbons MM2, MM3, MM4 Developed by Allinger et al. for calculaAons on small molecules COMPASS Commercial force field marketed by Accelrys Inc. Force Field Poten8al Func8ons The potenAal funcAons may be divided into bonded terms, which give the energy contained in the internal degrees of freedom, and non-‐bonded terms, which describe interac8ons between molecules. E pot = ∑V + ∑Vθ + ∑Vτ + ∑V r bonds vanderWaals angles torsions atoms Poten8als between bonded atoms Total poten8al Energy, Epot or Vtot + ∑Velectrostatics atoms Poten8als between non-‐bonded atoms Force Field Poten8al Energy Func8ons i Rij VvanderWaals j ⎡⎛ σ ⎞12 ⎛ σ ⎞6 ⎤ ij ij = 4ε ⎢⎜ ⎟ − ⎜ ⎟ ⎥ ⎢⎜⎝ Rij ⎟⎠ ⎜⎝ Rij ⎟⎠ ⎥ ⎣ ⎦ VElectrostatic = i rij j Vbonds qi q j (Charles Augus-n de Coulomb -‐1785) 4πεRij 1 ij = k r rij − rij0 2 ( 2 ) (Robert Hooke -‐ 1660) j k i θijk 1 Vangles = kθijk θ ij − θ ij0 2 ( (John Lennard-‐Jones – 1931) k τijkl 2 ) j l Vtorsions = i 1 ijkl (1 − cos(nτ )) k ∑ n n 2 (Jean Bap-ste Joseph Fourier – 1822) AlternaAvely, a power-‐series expansion of the Morse potenAal can be used Graphical comparison of Morse and power law potenAals Problem with harmonic approximaAon: Bonds cannot break (essence of Molecular Mechanics; no bonds are broken or formed, cannot be used for chemical reacAons). Torsion Angle or Dihedral Angle Energy The torsional energy is defined between every four bonded atoms (1-‐4 interacAons), and depends on the torsion (aka dihedral) angle ϕ made by the two planes incorporaAng the first and last three atoms involved in the torsion Torsion terms account for any interacAons between 1-‐4 atom pairs that are not already accounted for by non-‐bonded interacAons between these atoms For example: they could be used to describe barriers to bond rotaAon from electron delocalizaAon (double bonds or parAal double bonds), or stereo-‐electronic effects Torsion Example – The Single Bond Using the standard cos3φ potenAal, there are three equilibrium posiAons: ϕ = 180° (trans state) and ± 60° (gauche states). In pracAce, the energies of the gauche states are slightly different than that of the trans state, depending on the atoms involved in the torsion. To introduce a difference between the stabiliAes of the gauche and trans conformaAons, the torsion funcAon can be expanded with addiAonal terms, each with it’s unique contribuAon to the rotaAonal energy: Electrosta8cs Difference in electronegaAvity between atoms generates unequal charge distribuAon in a molecule Omen electronegaAvity differences are represented as fracAonal point charges (q) within the molecule (normally centered at the nuclei (parAal atomic charges) ElectrostaAc interacAon energy is calculated as a sum of interacAons between parAal atomic charges, using Coulombs law Naturally, this equaAon is also used for modeling interacAons between integral charges, such as between ions VElectrostatic = qi q j 4πεRij The problem with this approach is that there is no such thing as a fracAonal electron, therefore there is no perfect method to derive the parAal atomic charges Van der Waals Interac8ons Non-‐bonded interacAon that are not electrostaAc (e.g. between atoms in noble gas) are labeled van der Waals interacAons Contains dispersion and short-‐range components Dispersion interacAons always aoracAve. Arise from instantaneous dipoles that occur during fluctuaAons within the molecular electron cloud Short-‐range interacAons are always unfavorable. Also labeled exchange, or overlap, forces. They occur between electrons with the same spin so they do not occupy same region in space (Pauli exclusion principle) SUMMARY – Force Field Terms ElectrostaAc energy is represented using a set of parAal atomic charges van der Waals energy has both weakly aoracAve and strongly repulsive components and arises from represents electron correlaAon The dispersion term is always negaAve whereas short-‐range energy is always repulsive. Torsion terms describe bond rotaAonal properAes that arise from non-‐classical effects, such as electron delocalizaAon The remaining bond and angle terms describe covalent bonding Once we have our force field, what can we do with it? –Energy minimisaAon – Molecular Dynamics –ConformaAonal analysis The accuracy of the output from all these techniques will obviously be sensi8ve to a greater or lesser extent on the parameteriza8on of the force field