Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
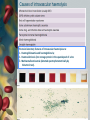
HAEMOLYTIC ANAEMIAS I • • • • • Dr. Zeki Ali Mohamed MRCP UK ( Edinburgh & London ) FIBMS - Haematology Consultant Physician & Haematologist Azadi Teaching Hospital • Lecturer / Departmeent of Medicine • Duhok University – Faculty of Medical Sciences • School of Medicine Normal Red Cell Destruction • • Being anucleate, red cell metabolism gradually deteriorates as enzymes are degraded and not replaced >> non-viable. After a mean lifespan of about 120 days, red cell destruction normally occurs by the macrophages of the reticuloendothelial system (RES): (Bone Marrow , Liver and Spleen) , the breakdown of haem from red cells liberates: • Iron: for recirculation via plasma transferrin mainly to BM erythroblasts. • Protoporphyrin: is broken down to bilirubin. • Bilirubin: circulates to the liver where it is conjugated to glucuronides which are excreted into the duodenum via bile and converted to stercobilinogen and stercobilin (excreted in faeces) • Stercobilinogen and stercobilin: are partly reabsorbed and excreted in urine as urobilinogen and urobilin. • Globin chains: are broken down to amino acids which are reutilized for general protein synthesis in the body. (a) Normal (RBC) breakdown. Extravascularly in the macrophages of the RES (b) Intravascular haemolysis occurs in some pathological disorders. Haemolytic anaemias - Definition Haemolytic anaemias are defined as: Anaemias resulting from increased rate of RBC destruction >> Shortened life span. After full expansion, the normal adult marrow, is able to produce red cells at 6–8 times its normal rate. Because of this (Erythropoietic hyperplasia) and anatomical extension of bone marrow, red cell destruction may be increased several-fold before the patient becomes anaemic (compensated haemolytic disease). Haemolytic anaemia may not be seen until the red cell lifespan is less than 30 days. CLASSIFICATION •Hereditary haemolytic anaemias are the result of ‘intrinsic’ red cell defects •Acquired HAs are usually the result of an ‘extracorpuscular’ or ‘environmental’ change. •(PNH) is exception : an acquired disorder, with intrinsic defect. Clinical features Patients with HA may commonly show: pallor of the mucous membranes, mild fluctuating jaundice and splenomegaly. urine may turn dark on standing because of excess urobilinogen not bilirubin. Pigment (bilirubin) gallstones may complicate the condition some patients (particularly with sickle cell disease) develop ulcers around the ankle. Aplastic crises may occur, usually precipitated by infection with parvovirus which ‘switches off erythropoiesis, and are characterized by a sudden increase in anaemia and drop in reticulocyte count folate deficiency may cause a megaloblastic crisis in the bone marrow . Laboratory findings: The laboratory findings are divided into three groups. 1- Features of Increased red cell breakdown : Serum bilirubin raised, unconjugated ( Indirect ) and bound to albumin Urine urobilinogen increased Serum haptoglobins absent because the haptoglobins become saturated with haemoglobin and the complex is removed by RE cells. 2 - Features of increased red cell production: Reticulocytosis; Bone marrow erythroid hyperplasia; the normal marrow myeloid: erythroid ratio of 2:1 to 12:1 is reduced to 1:1 or reversed. 3 – Features of damaged red cells: Morphology (e.g. microspherocytes, elliptocytes, fragments); Osmotic fragility, autohaemolysis, etc.; Specific enzyme, protein or DNA tests. Intravascular and extravascular haemolysis There are two mechanisms whereby red cells are destroyed in haemolytic anaemia: There may be excessive removal of red cells by macrophages of the RES (Extravascular haemolysis) or they may be broken down directly in the circulation (Intravascular haemolysis) Whichever mechanism dominates will depend on the pathology involved. In intravascular haemolysis, free haemoglobin is released which rapidly saturates plasma haptoglobins and the excess free haemoglobin is filtered by the glomerulus. If the rate of haemolysis saturates the renal tubular reabsorptive capacity, free haemoglobin enters urine and, as iron is released, the renal tubules become loaded with haemosiderin. Methaemalbumin is also formed from the process of intravascular haemolysis. Causes of intravascular haemolysis The main laboratory features of intravascular haemolysis are: 1 - Haemoglobinaemia and haemoglobinuria; 2 - Haemosiderinuria (iron storage protein in the spun deposit of urine 3 - Methaemalbuminaemia (detected spectrophotometrically by Schumm’s test). Hereditary haemolytic anaemias Membrane defects Hereditary spherocytosis Hereditary Spherocytosis (HS) is the most common hereditary haemolytic anaemia in northern Europeans. It is usually caused by defects in the proteins involved in the vertical interactions between the membrane skeleton and the lipid bilayer of the red cell. The loss of membrane may be caused by the release of parts of the lipid bilayer that are not supported by the skeleton. In HS, the marrow produces red cells of normal biconcave shape but these lose membrane and become increasingly spherical (loss of surface area relative to volume) as they circulate through the spleen and the rest of the RES. Ultimately, the spherocytes are unable to pass through the splenic microcirculation where they die prematurely. Clinical features The inheritance is Autosomal Dominant with variable expression; rarely it may be Autosomal Recessive. The anaemia can present at any age from infancy to old age. Jaundice is typically fluctuating. Splenomegaly occurs in most patients. Pigment gallstones are frequent Aplastic crises, usually precipitated by parvovirus infection, may cause a sudden increase in severity of anaemia Haematological findings Anaemia is usual but not invariable; its severity tends to be similar in members of the same family. Reticulocytes are usually 5–20%. The blood film shows Microspherocytes that are densely staining with smaller diameters than normal red cells. Eosin-5-maleimide staining in hereditary spherocytosis (HS) showing reduced mean channel fluorescence due to membrane band 3 protein deficiency. Investigation and treatment A rapid fluorescent flow analysis of eosin-maleimide bound to red cells is used as a test for HS and membrane band 3 protein deficiency (This has replaced the classic osmotic fragility test which showed the HS red cells to be excessively fragile in dilute saline solution. The identification of the exact molecular defect is NOT NEEDED for management. The direct antiglobulin (Coombs) test is, Negative, excluding an autoimmune cause of spherocytosis and haemolysis. The principal form of treatment is Splenectomy, preferably laparoscopic. Indications: symptomatic anaemia, gallstones, leg ulcers or growth retardation. NB: (Splenectomy should not be performed Unless cCinically Indicated ) This is because of the risk of post-splenectomy sepsis, particularly in early childhood. There is also evidence for late vascular complications. Cholecystectomy should be performed with splenectomy if symptomatic gallstones are present. Splenectomy should always produce a rise in the haemoglobin level to normal, even though microspherocytes formed in the rest of the RE system will remain. Hereditary elliptocytosis This usually a clinically milder disorder with similar clinical and laboratory features to HS except for the appearance of the blood film. Usually discovered incidentally on a blood film and there may be no evidence of haemolysis. Occasional patients require splenectomy. The basic defect is a failure of spectrin heterodimers to self-associate into heterotetramers. Patients with homozygous or doubly heterozygous elliptocytosis present with a severe haemolytic anaemia with microspherocytes, poikilocytes and splenomegaly (hereditary pyropoikilocytosis). Hereditary elliptocytosis. Peripheral blood smears representative of different hereditary elliptocytosis syndromes are shown. A: Micropoikilocytes and elliptocytes in a neonate with transient poikilocytosis and an α-spectrin gene mutation. B: Same child at 7 months of age, now exhibiting morphology of common hereditary elliptocytosis. C: Compound heterozygous hereditary elliptocytosis due to two α-spectrin self-association–site structural mutations. Note distorted red cell shapes, elliptocytes, and fragments. D: Hereditary pyropoikilocytosis. Red cell abnormalities are similar to those in (A) and (C) with prominent budding and fragmentation. Defective red cell metabolism Glucose-6-phosphate dehydrogenase deficiency Glucose-6-phosphate dehydrogenase (G6PD) functions to reduce nicotinamide adenine dinucleotide phosphate (NADP). It is the only source of NADPH that is needed for the production of reduced glutathione. Deficiency of G6PD renders the red cell susceptible to oxidant stress Haemoglobin and red blood cell (RBC) membranes are usually protected from oxidant stress by reduced glutathione (GSH). In G6PD deficiency, NADPH and GSH synthesis is impaired. F6P fructose6-phosphate; G6P glucose-6-phosphate; G6PD, glucose-6-phosphate dehydrogenase; GSSG, glutathione (oxidized form); NADP nicotinamide adenine dinucleotide phosphate Epidemiology There is a wide variety of normal genetic variants of the enzyme G6PD, the most common being type B (Western) and type A in Africans. In addition, more than 400 variants caused by point mutations or deletions of the enzyme G6PD have been characterized that show less activity than normal and worldwide over 400 million people are G6PD deficient in enzyme activity Clinical features G6PD deficiency is Usually Asymptomatic. Although G6PD is present in all cells, the main syndromes that occur are as follow: 1 - Acute haemolytic anaemia in response to oxidant stress, e.g. drugs, fava beans or infections. The acute haemolytic anaemia is caused by rapidly developing intravascular haemolysis with haemoglobinuria (The anaemia may be self-limiting as new young red cells are made with near normal enzyme levels. 2 - Neonatal jaundice. 3 - Rarely, a Congenital Non-Spherocytic Haemolytic Anaemia. These syndromes may result from different types of severe enzyme deficiency. Agents that cause haemolysis in (G6PD) deficiency: Infections and other acute illnesses (e.g. diabetic ketoacidosis) Drugs Antimalarials (e.g. primaquine, pamaquine, chloroquine, Fansidar®, Maloprim®) Sulphonamides and sulphones (e.g. co-trimoxazole, sulfanilamide, dapsone, Salazopyrin®) Other antibacterial agents (e.g. nitrofurans, chloramphenicol) Analgesics (e.g. aspirin), moderate doses are safe Antihelminths (e.g. β-naphthol, stibophen) Miscellaneous (e.g. vitamin K analogues, naphthalene (mothballs), probenecid) Fava beans (possibly other vegetables) NB. Many common drugs have been reported to precipitate haemolysis in G6PD deficiency in some patients (e.g. aspirin, quinine and penicillin) but not at conventional dosage. Diagnosis Between crises the blood count is normal. The enzyme deficiency is detected by one of a number of screening tests or direct enzyme assay on red cells. During a crisis the blood film may show contracted and fragmented cells, ‘bite’ cells and ‘blister’ cells which have had Heinz bodies removed by the spleen. Heinz bodies (oxidized, denatured haemoglobin) may be seen in the reticulocyte preparation, particularly if the spleen is absent. There are also features of intravascular haemolysis. Because of the higher enzyme level in young red cells, red cell enzyme assay may give a (False Normal) level in the phase of acute haemolysis with a reticulocyte response. Subsequent assay after the acute phase reveals the low G6PD level when the red cell population is of normal age distribution. Treatment The offending drug is stopped, The underlying infection is treated, High urine output should be maintained Blood transfusion undertaken where necessary for severe anaemia. G6PD-deficient babies are prone to neonatal jaundice and in severe cases phototherapy and exchange transfusion may be needed. The jaundice is usually not caused by excess haemolysis but by deficiency of G6PD affecting neonatal liver function. Pyruvate kinase deficiency This is inherited as an autosomal recessive, the affected patients being homozygous or doubly heterozygous. Over 100 different mutations have been described. The red cells become rigid as a result of reduced (ATP) formation. Clinically: The severity of the anaemia varies widely (haemoglobin 4–10 g/dL) and causes relatively mild symptoms because of a shift to the right in the oxygen (O2) dissociation curve caused by a rise in intracellular 2,3-diphosphoglycerate (2,3-DPG). jaundice is usual and gallstones frequent. Frontal bossing may be present. The blood film shows poikilocytosis and distorted ‘prickle’ cells, particularly post-splenectomy. Direct enzyme assay is needed to make the diagnosis. Splenectomy may alleviate the anaemia but does not cure it and is indicated in those patients who need frequent transfusions.