Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
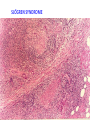
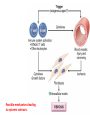
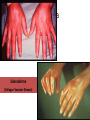
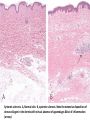
Auto Immune Diseases Ch 4 (P 120 - 134 ( March. 7. 2016 Auto Immune Diseases Autoimmunity is hard to classify as strictly a B cell or T cell mediated disease as multiple arms of the immune system are involved Auto Immune Diseases Symptoms • Initial diagnosis may be missed in patients as diseases present with general symptoms – Fever, muscle ache, fatigue, joint pain • Disease specific manifests – SLE – rash – Sjogren’s – dry mouth, dry eyes Diagnosis • General tests – C - Reactive Protein – Autoantibody titers (anti DNA, anti phospholipids, etc) – Presence of Rheumatoid Factor • Disease specific tests – Neurological exam – MS – Fasting glucose - Diabetes SLE • A multisystem autoimmune disease. • Most organ involve in the body; skin, kidneys, serosal membranes, joints, & heart. • Remitting and relapsing. • autoantibodies, including (ANAs). • Dx; demonstration of four or more of the criteria • (F / M = 9 : 1) • 2nd or 3rd decade of life. SLE • • • • • • Genetic family HLA-DR2, HLA-DR3 Low complement. Environmental factors; UV, Smoking, hormones, Drugs The diagnosis is established by demonstration of four or more of the criteria during any interval of observation. SLE, SKIN SLE, Glomerulous Libman-Sacks vegetations Libman-Sacks vegetations, (Libman-Sacks endocarditis), are on BOTH sides of the leaflet LUPUS (SLE) • Etiology: Abs directed against the patient’s own DNA (ds DNA), HISTONES, NON-histone RNA, & NUCLEOLUS (Sm), & phospholipids. • Abs against Bl. Cells • Abs against phospholipids. • Pathogenesis: Immune complex disease, in skin, joints, kidneys, vessels, heart, CNS • & Type II HR in Blood cells. • Morphology: “Butterfly” rash, skin deposits, GN. • Clinical expression: Progressive renal and vascular disease, POSITIVE A.N.A. Antinuclear Abs in Autoimmune Diseases Antigen Ab System Many nuclear antigens Generic ANA (DNA, RNA, proteins) (indirect IF) SLE DrugInduced LE SS—Diffuse Sjögren S—CREST Syndrome >95 >95 70–90 70–90 50–80 Native DNA Anti–doublestranded DNA 40–60 <5 <5 <5 <5 Histones Antihistone 50–70 >95 <5 <5 <5 Core proteins of small nuclear RNP particles (Smith antigen) Anti-Sm 20–30 <5 <5 <5 <5 RNP (U1RNP) Nuclear RNP 30–40 <5 15 10 <5 RNP SS-A(Ro) 30–50 <5 <5 <5 70–95 RNP SS-B(La) 10–15 <5 <5 <5 60–90 <5 <5 28–70 10–18 <5 DNA topoisomerase I Scl-70 RNP, ribonucleoprotein; Chronic Discoid Lupus Erythematosus. • • • • • • disease with skin manifestations mimic SLE, face and scalp are usually affected, Only 5% to 10% of patients develop systemic dis. 35% of patients show a positive ANA test, Abs to ds DNA are rarely. skin biopsy show deposition of Ig and C3 at the dermoepidermal junction similar to that in SLE. Drug-Induced Lupus Erythematosus • SLE –like syndrome may develop in patients receiving hydralazine, procainamide, isoniazid, and D-penicillamine, • Positive ANAs, • Negative ds DNA Abs are rare, • HLA-DR4 allele are at a greater risk • The disease remits after withdrawal of the offending drug. MORE SYSTEMIC AUTOIMMUNE DISEASES • RHEUMATOID ARTHRITIS • SJÖGREN SYNDROME • SCLERODERMA (SYSTEMIC SCLEROSIS) ↑ Destructive Rheumatoid Synovitis NORMAL Bi-Layered Synovium • Sjögren syndrome • chronic auto imm. disease, • dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia) • CD4+T & Ab mediated reaction against epith. Cells of lacrimal and salivary glands. • primary form • secondary form, associated with rheumatoid arthritis SLE, polymyositis, scleroderma, vasculitis, mixed connective tissue disease, or thyroiditis. • Genetic & Environmental factors. SJÖGREN SYNDROME SJÖGREN SYNDROME • SYSTEMIC SCLEROSIS (SCLERODERMA) • 1. chronic inflammation • 2. widespread damage to small bl ves. • 3. progressive fibrosis in skin & organs • Two types; a. Diffuse type, SS b. Limited “CREST” • Two ANAs ; a. Anti DNA topoisomerase I (anti-Scl 70), highly specific. systemic sclerosis. • anticentromere Ab, in CREST or limited sclerosis. Possible mechanisms leading to systemic sclerosis. Scleroderma Scleroderma (Collagen Vascular Disease) SYSTEMIC SCLEROSIS (SCLERODERMA) Systemic sclerosis. A, Normal skin. B, systemic sclerosis. Note the extensive deposition of dense collagen in the dermis with virtual absence of appendages &foci of inflammation (arrow). PAN IgG4-RD (Fibroiflammatory disease). • Inflamm. • Fibrosis • Obliterative phlebitis. Amyloidosis Pathogenesis of Amyloidosis • Amyloidosis - abnormal folding of pr., which are deposited as fibrils in EC tissues and disrupt normal function. • The Pr. that form amyloid fall into two general categories: • (1) Normal proteins that have an inherent tendency to fold improperly, and do so when they are produced in increased amounts; and • (2) mutant proteins that are prone to misfolding and subsequent aggregation Physical Nature of Amyloid • nonbranching fibrils, 7.5 - 10 nm. • cross-β-pleated sheet conformation Chemical Nature of Amyloid • 95% of amyloid material consists of fibril pr, • (1) AL (amyloid light chain) is derived from Ig light chains produced in plasma cells; • (2) AA (amyloid-associated) is derived from a unique non-Ig protein synthesized by the liver; and • (3) Aβ amyloid is produced from β amyloid precursor protein and is found in the cerebral lesions of Alzheimer disease. Classification of Amyloidosis • Primary • Secondary • systemic • Localized • Congenital • Acquired Primary Amyloidosis Secondary (Reactive) Amyloidosis • AA protein. • Systemic amyloidosis, • TB, bronchiectasis, ch. Osteomyelitis. • Rh arthritis, other CT disorders such as ankylosing spondylitis, and IBD. • Heroin abusers • renal cell carcinoma • Hodgkin lymphoma. Pathogenesis of amyloidosis Clinicopathologic Category Associated Diseases SYSTEMIC (GENERALIZED) AMYLOIDOSIS (primary amyloidosis) Multiple myeloma and Major Fibril Protein Chemically Related Precursor Protein AL Immunoglobulin light chains, (secondary amyloidosis) Chronic inflammatory conditions AA SAA Hemodialysis-associated amyloidosis Chronic renal failure Aβ2m β2-microglobulin AA SAA Familial amyloidotic neuropathies (several types) ATTR Transthyretin SYSTEMIC SENILE AMYLOIDOSIS LOCALIZED AMYLOIDOSIS Senile cerebral Endocrine Medullary carcinoma of thyroid Islets of Langerhans ATTR Transthyretin Ab A Cal AIAPP APP Calcitonin Islet amyloid peptide AANF Atrial natriuretic factor HEREDITARY AMYLOIDOSIS Familial Mediterranean fever Alzheimer disease Type 2 diabetes Clinical Features. • Renal involvement proteinuria, nephrotic syndrome. renal failure and uremia. • Cardiac amyloidosis ____congestive heart failure. • Gastrointestinal amyloidosis, Amyloidosis of the tongue. • Depositions in the stomach and intestine may lead to malabsorption, diarrhea, and disturbances in digestion. • Diagnosis of amyloidosis • Biopsy: rectal or gingival tissues in patients suspected of having systemic amyloidosis. • Examination of abdominal fat • kidney, when renal manifestations are present • Congo red. • serum and urine protein electrophoresis. • Bone marrow aspirates show monoclonal plasmacytosis, • Radiolabeled serum amyloid P (SAP) binds to the amyloid deposits and reveals their presence. • The prognosis for individuals with generalized amyloidosis is poor. • New therapeutic strategies aimed at correcting protein misfolding and inhibiting fibrillogenesis are being developed. Amyloidosis of the kidney. The glomerular architecture is almost totally obliterated by the massive accumulation of amyloid. Liver Amyloidosis Liver Amyloidosis Liver Amyloidosis Skin Amyloidosis