Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
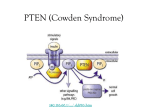
172 Biochemical Society Transactions (2007) Volume 35, part 2 Role of PTEN/PI3K pathway in endothelial cells A. Suzuki*1 , K. Hamada*, T. Sasaki†, T.W. Mak‡§ and T. Nakano¶ *Department of Molecular Biology, Akita University School of Medicine, Akita 010-8543, Japan, †Department of Microbiology, Akita University School of Medicine, Akita 010-8543, Japan, ‡The Campbell Family Institute for Breast Cancer Research, Princess Margaret Hospital, Toronto, ON, Canada M5G 2M9, §Departments of Immunology and Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2C1, and ¶Department of Pathology, Medical School and Graduate School of Frontier Biosciences, Osaka University, Suita 565-0871, Japan Abstract PTEN (phosphatase and tensin homologue deleted on chromosome 10) is an important tumour-suppressor gene that encodes a 3-phosphatase. The major substrate of PTEN is PIP3 (phosphatidylinositol 3,4,5trisphosphate) generated by the action of PI3Ks (phosphoinositide 3-kinases). Hereditary mutation of PTEN causes tumour-susceptibility diseases such as Cowden disease. We used the Cre-loxP system to generate an endothelial cell-specific mutation of PTEN in mice. Heterozygous mutation of PTEN in endothelial cells enhances postnatal neovascularization, including tumour angiogenesis necessary for tumour growth. This observation suggests that Cowden disease patients are not only at risk for additional tumorigenic mutations due to complete loss of PTEN function, but may also experience accelerated growth of incipient tumours due to enhanced angiogenesis. Homozygous mutation of Pten in murine endothelial cells impairs cardiovascular morphogenesis and is embryonic lethal due to endothelial cell hyperproliferation and impaired vascular remodelling. Additional homozygous mutation of p85α, the regulatory subunit of class IA PI3Ks, or p110γ , the catalytic subunit of the sole class IB PI3K, led to a partial rescue of all phenotypes in our PTEN-deficient mice. Thus inhibition of the PI3K pathway, including the targeting of PI3Kγ , may be an attractive therapeutic strategy for the treatment of various malignancies. Introduction PTEN (phosphatase and tensin homologue deleted from chromosome 10) is a ubiquitously expressed tumour–suppressor gene [1]. PTEN is mutated in human sporadic cancers of various tissues, and in hereditary cancer disorders such as Cowden disease, Bannayan–Zonana syndrome, Lhermitte– Duclos disease and Proteus syndrome [2,3]. Patients with these latter disorders suffer from multiple hamartomas and an increased risk of cancer. PTEN is a multifunctional phosphatase whose lipid phosphatase activity is associated with tumour suppression [4]. The major substrate of PTEN is PIP3 (phosphatidylinositol 3,4,5-trisphosphate) [5], a lipid molecule generated by the action of PI3Ks (phosphoinositide 3-kinases). The PI3Ks are evolutionarily conserved lipid kinases [6]. To date, eight distinct PI3K isoforms have been reported in mammals, four of which belong to the class I PI3Ks. PI3Kα, PI3Kβ and PI3Kδ constitute the class IA PI3Ks that are mainly activated by RTK (receptor tyrosine kinase) engagement, while the single class IB PI3K, PI3Kγ , is activated by the βγ Key words: cardiovasculogenesis, endothelial cell, p85α, phosphatase and tensin homologue deleted on chromosome 10 (PTEN), phosphoinositide-3-kinase (PI3K), tumour angiogenesis. Abbreviations used: bFGF, basic fibroblast growth factor; E9.5 (etc.), embryonic day 9.5 (etc.); Foxo1, forkhead box O 1; GPCR, G-protein-coupled receptor; LOH, loss of heterozygosity; PC, pericyte; PKB, protein kinase B; PI3K, phosphoinositide-3-kinase; PIP3 , phosphatidylinositol 3,4,5trisphosphate; PTEN, phosphatase and tensin homologue deleted on chromosome 10; RT, reverse transcriptase; RTK, receptor tyrosine kinase; αSMA, anti-smooth-muscle antigen antibody; VCAM1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; VGF, vascular growth factor; VSMC, vascular smooth-muscle cell. 1 To whom correspondence should be addressed (email [email protected]). C 2007 Biochemical Society subunit of G-proteins and acts downstream of GPCRs (Gprotein-coupled receptors) [7,8]. PI3K activation occurs in response to the binding of numerous kinds of growth factors, cytokines, chemokines and hormones to their receptors, as well as antigen receptor engagement. PIP3 generated by PI3K activation in turn activates PKB (protein kinase B)/ Akt, a serine/threonine kinase involved in anti-apoptosis, proliferation and oncogenesis. By dephosphorylating the D3 position of PIP3 , PTEN negatively regulates the PI3K pathway and PKB/Akt activation and thus tumorigenesis. To investigate the functions of PTEN in vivo, we and others generated null mutations of Pten in mice [9–13]. Animals heterozygous for a null Pten mutation develop a broad range of tumours, including cancers of the breast, thyroid, endometrium and prostate as well as T-cell lymphomas. This spectrum of neoplasias closely resembles that seen in humans with PTEN mutations. Pten+/− mice also develop signs of auto-immune disease [14]. Homozygosity for a null Pten mutation in mice leads to embryonic lethality at approx. E9.5 (embryonic day 9.5) [9,10], indicating that PTEN is indispensable for embryogenesis. Because this early embryonic lethality precludes the analysis of PTEN functions in adult organs, we and others created conditional mutant mouse strains in which the Pten gene is ‘floxed’ (Ptenflox mice). To determine the role of PTEN in a particular cell type, the Ptenflox mice are crossed with transgenic mice expressing Cre recombinase under the control of a tissue-specific promoter. Analyses of these various conditional mutant mice have revealed that tissue-specific deletion of PTEN causes a wide range of malignancies, including T-cell lymphoma [15], skin 3rd Focused Meeting on PI3K Signalling and Disease cancer [16], teratoma [17], hepatoma [18], breast cancer [19], pancreatic cancer [20], prostatic cancer [21–23], bladder cancer [24] and acute myeloid leukaemia [25,26]. These mutant mice also develop various non-cancerous ailments such as auto-immune disease [15], cardiac failure [27] and non-alcoholic steatohepatitis [18]. Increased angiogenesis and accelerated tumour growth in Tie2CrePtenflox/+ mice Embryonic cardiovascular development and postnatal neovascularization (including tumour angiogenesis) are complex processes that share a dependence on shear stress and particular signalling molecules [28]. Co-ordinated interactions between endothelial VGFs [vascular growth factors; e.g. VEGF (vascular endothelial growth factor), Ang-1, Ang-2, bFGF (basic fibroblast growth factor), PDGF-B (plateletderived growth factor-B), ephrin-B2 and TGF-β (transforming growth factor-β) superfamily members], intracellular signalling molecules [e.g. Notch1 and COUP-TFII (chicken ovalbumin upstream promoter-transcription factor 2)], and intercellular contacts [e.g. VCAM-1 (vascular cell adhesion molecule-1)] are required for both prenatal and postnatal vasculogenesis, and mutations of these molecules cause defects in cardiovascular development [29–33]. Significantly, all of the above VGFs also activate the PI3K–PKB/Akt pathway. PTEN is important for normal cardiovascular homoeostasis. In vitro, PTEN inhibits vascular sprouting and endothelial tube formation induced by VEGF, and dominantnegative mutation of PTEN abolishes these effects [34]. In vivo, Su et al. [35] have shown that tumour angiogenesis and tumour growth in mice are blocked by PTEN overexpression in tumour cells or by administration of PI3K inhibitors. However, it was not clear in this study whether the inhibition of tumour growth was caused by the angiogenesis defect or by a direct effect on the tumour cells. To resolve this issue and to investigate the function of PTEN in endothelial cells in vivo, we generated endothelial cell-specific PTENdeficient mice by mating Ptenflox mice and Tie2Cre transgenic mice [36]. Homologous recombination disrupting the Pten gene occurred in approx. 95% of endothelial and endocardial cells in Tie2CrePtenflox/+ mice but not in the PCs (pericytes) or VSMCs (vascular smooth-muscle cells) of these mutants. Histological analyses of systemic vessels and the heart revealed no significant structural differences between Tie2CrePten+/+ and Tie2CrePtenflox/+ mice. We then subcutaneously implanted Matrigels impregnated with bFGF, bFGF+VEGF, bFGF+Ang-1, or PBS into Tie2CrePten+/+ and Tie2CrePtenflox/+ mice and quantified blood vessel infiltration of the implants by immunostaining with anti-CD31 antibody. In contrast with implanted Tie2CrePten+/+ mice, implanted Tie2CrePtenflox/+ mice showed a significant increase in vascularization in response to VGF administration. To determine whether heterozygous Pten deficiency in endothelial cells affected adult tumour angiogenesis, Tie2CrePtenflox/+ and Tie2CrePten+/+ mice were injected subcutaneously with either melanoma (B16BL6) or LLC (Lewis lung carcinoma) cells. Both cell lines induced significantly larger tumours in Tie2CrePtenflox/+ mice than in Tie2CrePten+/+ mice, and microvessels were more abundant and of larger size in the Tie2CrePtenflox/+ mice. In vitro experiments revealed that total numbers of PCNA+ (proliferating-cell nuclear antigen-positive) cells and their migration in response to stimulation with Ang-1 or VEGF-A were significantly increased in Tie2CrePtenflox/+ cultures compared with controls. Our study was the first to demonstrate that the loss of PTEN specifically in mouse endothelial cells makes them hypersensitive to VGFs and thus is responsible for enhanced angiogenesis leading to accelerated tumour growth. An identical mechanism may be operating in humans with Cowden disease, since this cancer susceptibility disorder is caused by heterozygous mutation of PTEN. Our results suggest that an individual who inherits a mutated PTEN allele is not only at risk for additional tumorigenic mutations due to the LOH (loss of heterozygosity) of PTEN, but may also experience accelerated growth of incipient tumours due to enhanced angiogenesis (Figure 1A). Death of Tie2CrePtenflox/flox mice by E11.5 due to endothelial cell hyperproliferation and impaired vascular remodelling The intercrossing of Tie2CrePtenflox/+ and Ptenflox/+ mice generated homozygous mutant embryos that were present at the expected Mendelian frequency up to E9.5. However, resorption of Tie2CrePtenflox/flox embryos commenced at E10.5 and embryonic loss occurred at E11.5. Histologically, E8.25 Tie2CrePtenflox/flox embryos were essentially normal with respect to gross appearance of the central vascular tree, including the rostral-caudal aorta and the anterior and posterior cardiac veins. PTEN is thus dispensable for the differentiation of angioblasts from the ventral mesoderm, their appropriate migration within the embryo, and their alignment to form major vessels. However, E9.5 homozygous mutants exhibited an enlarged capillary plexus featuring a meshwork of interconnected, oversized endothelial celllined tubes that showed increased staining for Ki67. Distinct branches of large vessels such as the anterior cardinal vein in the embryo proper and vitelline vessels in the yolk sac were not formed because of a failure in primary vascular plexus remodelling. By E10.5, most of the mutants showed profound growth retardation as well as pericardial cavity enlargement and frequent bleeding into the pericardial cavity or large trunk vessels. Cross-talk between endothelial cells and PCs/VSMCs is critical for vascular remodelling and maturation. We therefore examined the recruitment of PCs and VSMCs to developing vascular channels using immunostaining with αSMA (antismooth-muscle antigen antibody). At E10.0, αSMA+ cells were observed in perivascular regions of the Tie2CrePten+/+ yolk sac. In contrast, no αSMA+ PCs/VSMCs were present C 2007 Biochemical Society 173 174 Biochemical Society Transactions (2007) Volume 35, part 2 Figure 1 Parallels between Cowden disease in humans and endothelial-cell-specific mutation of Pten in mice (A) Cowden disease patients are heterozygous for a germline mutation of PTEN. Such individuals are not only at risk for additional tumorigenic mutations due to the LOH of PTEN, but may also experience accelerated growth of incipient tumours due to enhanced angiogenesis. (B) Mutant mice homozygous for an endothelial cell-specific mutation of Pten are embryonic lethal at E11.5. The loss of PTEN results in constitutive activation of PKB/Akt, and positively regulates eNOS (endothelial nitric oxide synthase) and negatively regulates Foxo1. Moreover, in the absence of PTEN, endothelial cells experience up- or down-regulation of the indicated molecules (Ang-1, Ang-2 etc.). This mass dysregulation leads to the indicated failures in cardiovasculogenesis that most likely underlie the lethality of Tie2CrePtenflox/flox mice. adjacent to endothelial cells in the vessels or yolk sacs of Tie2CrePtenflox/flox mice. In the embryo proper, the αSMA staining seen in the vessels, heart and dorsal aorta of Tie2CrePten+/+ embryos was dramatically decreased in Tie2CrePtenflox/flox embryos. The bleeding from the large trunk vessels noted in the mutants may have resulted from the impaired recruitment of PCs/VSMCs to these tissues. A reduction in the size of the developing intraventricular septum and cardiac trabeculae, and a thinning of the cardiac wall, were also observed in homozygous mutants. The thinnest cardiac walls resembled membranes such that bleeding in the mutant pericardial cavity could have resulted from cardiac wall rupture or leakage of blood cells from the cardiac lumen. RT (reverse transcriptase)–PCR analyses of gene expression in whole yolk sacs from E8.5 Tie2CrePten+/+ and Tie2CrePtenflox/flox embryos showed that a lack of PTEN significantly decreased expression of Ang-1, ephrinB2 and VCAM-1 but increased expression of Ang-2, VEGFA, VEGFR1(Flt-1) (where VEGFR is VEGF receptor) and VEGFR2(Flk-1). These differences were confirmed by RT– PCR analyses of VEGFR2+ cells from E9.5 Tie2CrePten+/+ and Tie2CrePtenflox/flox embryos, and by RT–PCR and protein analyses of HUVEC (human umbilical vein endothelial cells) in which PTEN expression was decreased by siRNA (small interfering RNA) treatment. These results suggest that C 2007 Biochemical Society PTEN deficiency leads directly to an altered VGF profile that may be responsible for the cardiovascular defects of Tie2CrePtenflox/flox mice. Several lines of evidence support our contention that dysregulation of downstream targets of PKB/Akt due to loss of PTEN function is responsible for the lethal phenotype of Tie2CrePtenflox/flox mice (Figure 1B). Constitutively active PKB/Akt (myrAkt) expression in vivo causes fatal vascular malformations and bleeding due to a failure in vascular remodelling [37]. Fatal vascular remodelling and cardiac defects are also present in mice deficient in Foxo1 (forkhead box O 1), a transcription factor negatively regulated by PKB/ Akt [38]. Mice deficient in the ephrinB2, which is downregulated by PTEN deficiency, die at E10.5 due to defects in vascular remodelling and sprouting that lead to pericardial effusion or bleeding [39]. Similarly, disruption of Ang-1 or its receptor Tie2 in knockout mice causes vascular defects due to impaired recruitment of PCs/VSMCs [29,40]. In contrast, administration of Ang-2 (whose action opposes Ang-1) causes a dose-dependent pericyte ‘dropout’ in the normal retina [41]. VEGF expression, and increased VEGF during lung organogenesis, overstimulates endothelial cell growth that leads to abnormally large capillaries [42]. Consistent with this latter observation, mice overexpressing VEGF die at E14 due to cardiac abnormalities [43]. Finally, VCAM-1 deficiency in mice causes pericardial bleeding and impaired 3rd Focused Meeting on PI3K Signalling and Disease cardiomyocyte development [44]. Taken together, these results highlight the vital role PTEN plays in maintaining normal vascular biology. Partial rescue of Tie2CrePtenflox/+ and Tie2CrePtenflox/flox mice by null mutation of p110γ or p85α p85α is the most abundantly expressed regulatory subunit of the class IA PI3Ks that are activated following RTK engagement by VGFs. p110γ is the catalytic subunit of the sole class IB PI3K that is activated by the βγ subunit of Gproteins and acts downstream of activated GPCRs [7,45]. To clarify the contribution of class IA and class IB PI3Ks to the phenotypes observed in Tie2CrePtenflox/+ and Tie2CrePtenflox/flox mice, we generated p85α −/− Tie2CrePtenflox/+ , p85α −/− Tie2CrePtenflox/flox , p110γ −/− Tie2CrePtenflox/+ and p110γ −/− Tie2CrePtenflox/flox double mutant mice. Homozygous mutation of p110γ partially restored control of angiogenic responses to VGFs and reduced decreased angiogenesis and tumour growth in Tie2CrePtenflox/+ mice. Moreover, loss of p110γ led to a dramatic rescue of all Tie2CrePtenflox/flox phenotypes such that these double mutant mice survived until E18.5–19.5. Homozygous mutation of p85α restored control of angiogenic responses in Tie2CrePtenflox/+ mice to the same degree as mutation of p110γ . However, the defects in cardiovascular morphogenesis seen in Tie2CrePtenflox/flox mice were only partially rescued by p85α deficiency such that these animals survived only until E14.5–15.5. Because most VGF receptors have tyrosine kinase activity, class IA PI3Ks likely play major roles in cardiovasculogenesis and tumour angiogenesis. Consistent with this notion, loss of p85α partially resolved the enhanced angiogenesis and accelerated tumour growth observed in our Tie2CrePtenflox/+ mice, as well as the impaired cardiovascular morphogenesis seen in Tie2CrePtenflox/flox mice. Intriguingly, the enhanced angiogenesis in Tie2CrePtenflox/+ mice induced by RTK agonists (e.g. VEGF and Ang-1) was partially rescued by p110γ deficiency. This result was unexpected because p110γ has been postulated to be activated downstream of GPCRs but not of RTKs. It may be that, in vivo, an unknown VGF (possibly a GPCR ligand) activates p110γ and influences RTK signalling initiated by VEGF or Ang-1 that leads to angiogenesis. Our generation of double mutant mice lacking both PTEN and class IB PI3K functions has shed much needed light on the potential role of class IB PI3K in cardiovascular morphogenesis and postnatal angiogenesis. Conclusion Our study has demonstrated that normal function of the PI3K–PTEN–PKB/Akt pathway in endothelial cells is required to control cardiovascular morphogenesis and postnatal neovascularization, including tumour angiogenesis supporting tumour growth. Inhibition of the PI3K pathway, including the targeting of PI3Kγ , may be an attractive therapeutic strategy for the treatment of various malignancies. References 1 Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S.I., Puc, J., Miliaresis, C., Rodgers, L., McCombie, R. et al. (1997) Science 275, 1943–1947 2 Liaw, D., Marsh, D.J., Li, J., Dahia, P.L., Wang, S.I., Zheng, Z., Bose, S., Call, K.M., Tsou, H.C., Peacocke, M. et al. (1997) Nat. Genet. 16, 64–67 3 Marsh, D.J., Dahia, P.L., Zheng, Z., Liaw, D., Parsons, R., Gorlin, R.J. and Eng, C. (1997) Nat. Genet. 16, 333–334 4 Myers, M.P., Pass, I., Batty, I.H., Van der Kaay, J., Stolarov, J.P., Hemmings, B.A., Wigler, M.H., Downes, C.P. and Tonks, N.K. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13513–13518 5 Maehama, T. and Dixon, J.E. (1998) J. Biol. Chem. 273, 13375–13378 6 Toker, A. and Cantley, L.C. (1997) Nature 387, 673–676 7 Stoyanov, B., Volinia, S., Hanck, T., Rubio, I., Loubtchenkov, M., Malek, D., Stoyanova, S., Vanhaesebroeck, B., Dhand, R., Nurnberg, B. et al. (1995) Science 269, 690–693 8 Stephens, L.R., Eguinoa, A., Erdjument-Bromage, H., Lui, M., Cooke, F., Coadwell, J., Smrcka, A.S., Thelen, M., Cadwallader, K., Tempst, P. and Hawkins, P.T. (1997) Cell 89, 105–114 9 Suzuki, A., de la Pompa, J.L., Stambolic, V., Elia, A.J., Sasaki, T., del Barco Barrantes, I., Ho, A., Wakeham, A., Itie, A., Khoo, W. et al. (1998) Curr. Biol. 8, 1169–1178 10 Di Cristofano, A., Pesce, B., Cordon-Cardo, C. and Pandolfi, P.P. (1998) Nat. Genet. 19, 348–355 11 Podsypanina, K., Ellenson, L.H., Nemes, A., Gu, J., Tamura, M., Yamada, K.M., Cordon-Cardo, C., Catoretti, G., Fisher, P.E. and Parsons, R. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1563–1568 12 Stambolic, V., Suzuki, A., de la Pompa, J.L., Brothers, G.M., Mirtsos, C., Sasaki, T., Ruland, J., Penninger, J.M., Siderovski, D.P. and Mak, T.W. (1998) Cell 95, 29–39 13 Stambolic, V., Tsao, M.S., Macpherson, D., Suzuki, A., Chapman, W.B. and Mak, T.W. (2000) Cancer Res. 60, 3605–3611 14 Di Cristofano, A., Kotsi, P., Peng, Y.F., Cordon-Cardo, C., Elkon, K.B. and Pandolfi, P.P. (1999) Science 285, 2122–2125 15 Suzuki, A., Yamaguchi, M.T., Ohteki, T., Sasaki, T., Kaisho, T., Kimura, Y., Yoshida, R., Wakeham, A., Higuchi, T., Fukumoto, M. et al. (2001) Immunity 14, 523–534 16 Suzuki, A., Itami, S., Ohishi, M., Hamada, K., Inoue, T., Komazawa, N., Senoo, H., Sasaki, T., Takeda, J., Manabe, M. et al. (2003) Cancer Res. 63, 674–681 17 Kimura, T., Suzuki, A., Fujita, Y., Yomogida, K., Lomeli, H., Asada, N., Ikeuchi, M., Nagy, A., Mak, T.W. and Nakano, T. (2003) Development 130, 1691–1700 18 Horie, Y., Suzuki, A., Kataoka, E., Sasaki, T., Hamada, K., Sasaki, J., Mizuno, K., Hasegawa, G., Kishimoto, H., Iizuka, M. et al. (2004) J. Clin. Invest. 113, 1774–1783 19 Li, G., Robinson, G.W., Lesche, R., Martinez-Diaz, H., Jiang, Z., Rozengurt, N., Wagner, K.U., Wu, D.C., Lane, T.F., Liu, X. et al. (2002) Development 129, 4159–4170 20 Stanger, B.Z., Stiles, B., Lauwers, G.Y., Bardeesy, N., Mendoza, M., Wang, Y., Greenwood, A., Cheng, K.H., McLaughlin, M., Brown, D. et al. (2005) Cancer Cell 8, 185–195 21 Backman, S.A., Ghazarian, D., So, K., Sanchez, O., Wagner, K.U., Hennighausen, L., Suzuki, A., Tsao, M.S., Chapman, W.B., Stambolic, V. and Mak, T.W. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 1725–1730 22 Wang, S., Gao, J., Lei, Q., Rozengurt, N., Pritchard, C., Jiao, J., Thomas, G.V., Li, G., Roy-Burman, P., Nelson, P.S. et al. (2003) Cancer Cell 4, 209–221 23 Chen, Z., Trotman, L.C., Shaffer, D., Lin, H.K., Dotan, Z.A., Niki, M., Koutcher, J.A., Scher, H.I., Ludwig, T., Gerald, W. et al. (2005) Nature 436, 725–730 24 Tsuruta, H., Kishimoto, H., Sasaki, T., Horie, Y., Natsui, M., Shibata, Y., Hamada, K., Yajima, N., Kawahara, K., Sasaki, M. et al. (2006) Cancer Res. 66, 8389–8396 25 Zhang, J., Grindley, J.C., Yin, T., Jayasinghe, S., He, X.C., Ross, J.T., Haug, J.S., Rupp, D., Porter-Westpfahl, K.S., Wiedemann, L.M. et al. (2006) Nature 441, 518–522 26 Yilmaz, O.H., Valdez, R., Theisen, B.K., Guo, W., Ferguson, D.O., Wu, H. and Morrison, S.J. (2006) Nature 441, 475–482 27 Crackower, M.A., Oudit, G.Y., Kozieradzki, I., Sarao, R., Sun, H., Sasaki, T., Hirsch, E., Suzuki, A., Shioi, T., Irie-Sasaki, J. et al. (2002) Cell 110, 737–749 28 Daniel, T.O. and Abrahamson, D. (2000) Annu. Rev. Physiol. 62, 649–671 C 2007 Biochemical Society 175 176 Biochemical Society Transactions (2007) Volume 35, part 2 29 Suri, C., Jones, P.F., Patan, S., Bartunkova, S., Maisonpierre, P.C., Davis, S., Sato, T.N. and Yancopoulos, G.D. (1996) Cell 87, 1171–1180 30 Lindahl, P., Johansson, B.R., Leveen, P. and Betsholtz, C. (1997) Science 277, 242–245 31 Lindblom, P., Gerhardt, H., Liebner, S., Abramsson, A., Enge, M., Hellstrom, M., Backstrom, G., Fredriksson, S., Landegren, U., Nystrom, H.C. et al. (2003) Genes Dev. 17, 1835–1840 32 Dickson, M.C., Martin, J.S., Cousins, F.M., Kulkarni, A.B., Karlsson, S. and Akhurst, R.J. (1995) Development 121, 1845–1854 33 Larsson, J., Goumans, M.J., Sjostrand, L.J., van Rooijen, M.A., Ward, D., Leveen, P., Xu, X., ten Dijke, P., Mummery, C.L. and Karlsson, S. (2001) EMBO J. 20, 1663–1673 34 Huang, J. and Kontos, C.D. (2002) J. Biol. Chem. 277, 10760–10766 35 Su, J.D., Mayo, L.D., Donner, D.B. and Durden, D.L. (2003) Cancer Res. 63, 3585–3592 36 Hamada, K., Sasaki, T., Koni, P.A., Natsui, M., Kishimoto, H., Sasaki, J., Yajima, N., Horie, Y., Hasegawa, G., Naito, M. et al. (2005) Genes Dev. 19, 2054–2065 37 Sun, J.F., Phung, T., Shiojima, I., Felske, T., Upalakalin, J.N., Feng, D., Kornaga, T., Dor, T., Dvorak, A.M., Walsh, K. and Benjamin, L.E. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 128–133 C 2007 Biochemical Society 38 Furuyama, T., Kitayama, K., Shimoda, Y., Ogawa, M., Sone, K., Yoshida-Araki, K., Hisatsune, H., Nishikawa, S., Nakayama, K., Ikeda, K. et al. (2004) J. Biol. Chem. 279, 34741–34749 39 Wang, H.U., Chen, Z.F. and Anderson, D.J. (1998) Cell 93, 741–753 40 Dumont, D.J., Gradwohl, G., Fong, G.H., Puri, M.C., Gertsenstein, M., Auerbach, A. and Breitman, M.L. (1994) Genes Dev. 8, 1897–1909 41 Hammes, H.P., Lin, J., Wagner, P., Feng, Y., Vom Hagen, F., Krzizok, T., Renner, O., Breier, G., Brownlee, M. and Deutsch, U. (2004) Diabetes 53, 1104–1110 42 Zeng, X., Wert, S.E., Federici, R., Peters, K.G. and Whitsett, J.A. (1998) Dev. Dyn. 211, 215–227 43 Miquerol, L., Langille, B.L. and Nagy, A. (2000) Development 127, 3941–3946 44 Kwee, L., Baldwin, H.S., Shen, H.M., Stewart, C.L., Buck, C., Buck, C.A. and Labow, M.A. (1995) Development 121, 489–503 45 Dimmeler, S., Assmus, B., Hermann, C., Haendeler, J. and Zeiher, A.M. (1998) Circ. Res. 83, 334–341 Received 28 November 2006