Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
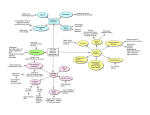
From www.bloodjournal.org by guest on June 18, 2017. For personal use only. chromatin features, types of targeted genes, CpG islands, and other genomic features. Thus, using a standardized bioinformatic platform it is possible to visually compare the AAV vector, lentiviral vector, and ␥RV integration profiles. The strength of the study is that coupling the whole transcriptome analysis to the integration site data obtained from HCCs, it was possible to measure the impact of AAV integration on the expression of genes targeted in these tumors in vivo. Importantly, genes in neighboring AAV vector integrations did not display aberrant levels of expression. Overall, the level of detail reached by this analysis sets a new standard for AAV vector safety studies in mice. It is also possible also to appreciate some peculiar challenges in the study of AAV vector safety that instead do not appear to be a problem with retroviruses. For example, vector integration levels are difficult to estimate as AAV vector genomes coexist both in episomal (mostly) and integrated forms that cannot be easily distinguished. Also, if the integration frequency as determined by Li and colleagues is ⬃ 1/1500, can we expect the HCCs to be marked? It is difficult to answer this question because in the complex tumor microenvironment, expanding tumor cells are mixed with bystander cells marked by integrated and/or episomal AVV genomes and unmarked cells, rendering vector marking measurements problematic. How is it possible, then, to distinguish the integrations in tumor cells (if any) from the bystanders? In the HCCs in this study, 1-2 integrations are represented by high numbers of sequencing reads compared with other integrations from the same sample. Could these be the ones that form tumor cells? Ultimately however, the relative contribution of each integration site should be addressed experimentally. Why in some studies does AAV appear to be genotoxic while in others it does not? The AAV vector from Donsante et al4 contained a viral-derived cytomegalovirus early enhancer/ chicken  actin promoter driving the expression of the human -glucuronidase, while Li et al used a cellular promoter chimera composed of the apolipoprotein E enhancer/ alpha1-antitrypsin promoter driving Factor IX expression. These differences may be an important to considered in genotoxicity assays to confirm that the use of cellular promoters provides an added safety value to retroviral 3250 vectors.11 Other not mutually exclusive variables may be related to an enhanced susceptibility to insertional mutagenesis of the disease model on the mouse strain or environmental factors that may trigger chronic liver damage.12 All these factors play an important role in HCC formation and could play a role in the selection of hepatocytes with genotoxic AAV integrations. It is expected that in the quest for safer AAV vectors, future studies will involve in vivo testing and validation of optimized vector constructs and dissection of the role of genetic and environmental variables in genotoxicity. Conflict-of-interest disclosure: The author declares no competing financial interests. ■ 3. Deyle DR, Russell DW. Adeno-associated virus vector integration. Curr Opin Mol Ther. 2009;11(4):442-447. 4. Donsante A, Miller DG, Li Y, et al. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317(5837):477. 5. Gariboldi M, Manenti G, Canzian F, et al. Chromosome mapping of murine susceptibility loci to liver carcinogenesis. Cancer Res. 1993;53(2):209-211. 6. Dupuy AJ, Rogers LM, Kim J, et al. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009; 69(20):8150-8156. 7. Zhang L, Volinia S, Bonome T, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105(19):7004-7009. 8. Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9(6):389-399. 9. Baum C. Insertional mutagenesis in gene therapy and stem cell biology. Curr Opin Hematol. 2007;14(4):337-342. REFERENCES 10. Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342-347. 1. Li H, Malani N, Hamilton SR, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood. 2011;117(12):3311-3319. 11. Zychlinski D, Schambach A, Modlich U, et al. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16(4):718-725. 2. Mueller C, Flotte TR. Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther. 2008;15(11):858-863. 12. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674-687. ● ● ● IMMUNOBIOLOGY Comment on Liu et al, page 3257 Lineage-specific pleiotropy in immune aging ---------------------------------------------------------------------------------------------------------------Parisa Eshraghi and K. Lenhard Rudolph UNIVERSITY OF ULM In this issue of Blood, Liu et al provide the first genetic evidence that p16 defines a lymphoid lineage intrinsic gatekeeper, which prevents B-lymphocyte transformation but impairs T-lymphocyte function in aging mice.1 magine if we could influence our immune system to produce youthful numbers of highly functional lymphocytes throughout life up to a very advanced age, without increasing the risk of tumors derived from the lymphoid lineage. It is conceivable that such an intervention would reduce the risk of deleterious infections and cancer.2 To therapeutically target the immune system to prevent aging-associated declines in immune function, it is of utmost importance to identify molecular causes of this process. Molecular mechanisms that limit cell proliferation (eg, telomere shortening) have been implemented in cancer protection, but were also shown to contribute to the decline in tissue maintenance and regeneration during ag- I ing.3 The cycling-dependent kinase inhibitor p16 represents one example of a cell-cycle inhibitor, which has not only been implemented to prevent cancer formation at early age, but also to impair stem cell function and tissue maintenance during aging.4 The concept that biologic processes, regulating organismal function (such as the expression of specific genes), can have pleiotropic effects during lifetime (tissue protective during early life but tissue destructive in late life) has led to the theory of “antagonistic pleiotropy” in aging.5,6 It remains an open debate whether genes that exhibit a pleiotropic function during lifetime could represent targets for novel therapies, aiming to prevent the evolution or progression 24 MARCH 2011 I VOLUME 117, NUMBER 12 blood From www.bloodjournal.org by guest on June 18, 2017. For personal use only. It has been shown that tumor suppressor genes (eg, p16) have pleiotropic effects during lifetime preventing the formation of cancer during early life but contributing to impairments in stem-cell function and organ maintenance during aging. Liu et al provide the first experimental evidence for lineage-specific pleiotropic effects of p16 in the aging immune system. B lineage–specific deletion of p16 leads to B cell–derived neoplasms in middle-aged mice without improving B lymphopoiesis at this age. In contrast, T lineage–specific deletion of p16 does not lead to tumorigenesis but significantly improves T lymphopoiesis and immune functions in aging mice. The data indicate that p16 does not contribute to tumor suppression in T-lymphoid cells but has an important tumor-suppressive role in B-lymphoid cells. In contrast, p16 contributes to impairments in T lymphopoiesis during aging but has no significant effects on B lymphopoiesis in middle-aged mice. Inhibitory effects of p16 on B lymphopoiesis at advanced age cannot be excluded. The data support a new concept indicating that lineage-specific gene-targeting (targeting of p16 in T-lymphoid lineage) could represent a therapeutic option to improve tissue maintenance during aging (T lymphopoiesis) without affecting gene expression and transformation in other compartments (B-lymphoid lineage). of age-associated organ dysfunction. In this regard, it is an important question whether lifecycle-dependent pleiotropic effects of genes occur within a single cell lineage or in separate lineages. If the latter holds true, a pleiotropic tumor suppressor gene may prevent tumor formation in one cell lineage and induce the evolution of age-associated dysfunction in a different lineage. In the case of lineage-specific pleiotropy, cell type–specific gene targeting could improve age-associated impairments in maintenance of one tissue without affecting gene expression and cancer risk in other tissues. To investigate the role of p16 in lymphocyte aging and transformation, Liu et al analyzed aging mouse cohorts carrying B or T lymphocyte–specific deletions of the INK4A gene locus encoding for p16 protein compared with a wild-type cohort. The authors demonstrate that p16 deletion in Tlymphocyte progenitor cells impairs agedependent involution of the thymus and improves the production of naive and memory blood 2 4 M A R C H 2 0 1 1 I V O L U M E 1 1 7 , N U M B E R 1 2 T lymphocytes in aging mice. Importantly, this enhancement in age-dependent T-cell production was associated with improved immune responses in aging mice and did not increase the cancer risk. These results provide a first example of a T cell–specific gene knockout impairing age-dependent declines in thymopoiesis. The findings do not argue against inhibitory effects of the aging environment (thymic niche or systemic blood circulation) on T lymphogenesis. It is conceivable that inhibitory effects of the aging environment on T lymphogenesis could lead to a cell-intrinsic up-regulation of p16 in T-lymphocytic progenitor cells. An important question in future studies is to delineate the molecular pathways that control the up-regulation of p16 in Tlymphocyte progenitor cells during aging. In sharp contrast to the beneficial effects of p16 deletion in the T-lymphocyte lineage, Liu et al demonstrate that the deletion of p16 in B-lymphocyte progenitor cells led to an early formation of B cell– derived neo plasms in middle-aged mice. Positive effects of p16 dele- tion on the prevention of B-lymphocyte aging could not be observed in this time window. The contrasting effects of p16 deletion on aging and transformation in the B-lymphopoietic versus T-lymphopoietic cells represent the most important finding of the current study. To our knowledge, these data provide the first experimental evidence that lineagespecific deletion of a single gene (p16) can have age-dependent, pleiotropic effects, improving maintenance and function in one compartment (T lmphocytes), but promoting tumorigenesis in another compartment (B lymphocytes). The authors conclude from their findings that “this work serves as a cautionary tale for those who would seek to ameliorate aging by globally attenuating tumor suppressor function.”1 This conclusion is important, but to see it more positively we can also conclude from these data that cell type–specific targeting of pleiotropic tumor suppressor genes could represent a novel therapeutic concept for the improvement of tissue maintenance and function during aging (see figure). In addition, it should be noted that the deletion of tumor suppressor genes (p21 or Exonuclease-1) at the level of the whole organism (germline deletion) has been shown to elongate the lifespan of prematurely aging mice with dysfunctional telomeres.7,8 These data indicate that a systemic inactivation of tumor suppressor genes could have beneficial effects at advanced age, when defects in organ maintenance become life limiting. Conflict-of-interest disclosure: The authors declare no competing financial interests. ■ REFERENCES 1. Liu Y, Johnson SM, Fedoriw Y, et al. Expression of p16INK4a prevents cancer and promotes aging in lymphocytes. Blood. 2011;117(12):3257-3267. 2. Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9(1):57-62. 3. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31(1):9-18. 4. Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007; 8(9):703-713. 5. Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398-411. 6. Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120(4):437-447. 7. Choudhury AR, Ju Z, Djojosubroto MW, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet. 2007;39(1):99-105. 8. Schaetzlein S, Kodandaramireddy NR, Ju Z, et al. Exonuclease-1 deletion impairs DNA damage signaling and prolongs lifespan of telomere-dysfunctional mice. Cell. 2007;130(5):863-867. 3251 From www.bloodjournal.org by guest on June 18, 2017. For personal use only. 2011 117: 3250-3251 doi:10.1182/blood-2011-02-332650 Lineage-specific pleiotropy in immune aging Parisa Eshraghi and K. Lenhard Rudolph Updated information and services can be found at: http://www.bloodjournal.org/content/117/12/3250.full.html Articles on similar topics can be found in the following Blood collections Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.