Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Machine Learning in
Computational Biology
CSC 2431
Lecture 9: Combining biological datasets
Instructor: Anna Goldenberg
What kind of data integration is there?
What kind of data integration is there?
SNPs
and gene expression
Networks and gene expression (and
mutations)
ENCODE data. Combining different
epigenetic signals and binding info
Ontologies and genome annotations
Now: integrating
patient data
Data is available
E.g. The Cancer Genome Atlas (TCGA)
Total of 33 cancers.
9 cancers have over 500+ samples
All publicly available!
Why integrate patient data
Why integrate patient data
To
identify more homogeneous subsets of
patients (that might respond similarly to a
given drug)
To
help better predict response to drugs
mRNA
mutations
more
genes
CNV
clinical
more
genes
p-value = {0.2,0.6,0.5}
(Verhaak et al, Cancer Cell, 2010)
mRNA
mutations
more
genes
CNV
clinical
more
genes
p-value = {0.2,0.6,0.5}
What about methylation data?
(Verhaak et al, Cancer Cell, 2010)
More recent GBM study (Sturm et al, 2012)
Methods used in Verhaak 2010
Factor analysis – a dimensionality reduction method –
used to integrate mRNA data from 3 platforms
Consensus clustering (consensus average linkage
clustering) (Monti et al, 2003)
SigClust – cluster significance (Liu et al, 2008)
Silhouette to identify core of clusters (Rousseeuw,1987)
ClaNC – nearest centroid-based classifier to identify
gene signatures (Dabney, 2006)
More recent GBM study (Sturm,
2012)
Missing
values – imputed using k-NN
(Troyanskaya, 2001)
Unsupevised consensus clustering (R:
clusterCons) (Monti, 2003, Wilkerson and
Hayes, 2010)
Consensus matrix was calculated using
the k-means algorithm
Number of clusters is decided by visual
assessment
Breast Cancer Analysis (TCGA,2012)
Integrated pathway analysis using PARADIGM
Significantly mutated genes were identified using MuSiC
package
NMF for unsupervised clustering of somatic and CNV
data, protein expression
RPMM – recursively partitioned mixture model (RPMM
Bioconductor package)
ConsensusClusterPlus (R-package) to combine
clustering based on single data type
MEMo (Mutual Exclusivity Modules) – identifies mutually
exclusive alterations targeting frequently altered genes
that are likely to belong to the same pathway
PARADIGM
Infers Integrated Pathway Levels (IPLs) for genes, complexes,
and processes using pathway interactions and genomic and
functional genomic data from a single patient sample.
Data:
◦ mRNA relative to normal samples
◦ CNVs mapped to genes
◦ Networks: Biocarta (Biocarta, NCIPID, Reactome) –
Superimposed into SuperPathway
Approach: belief propagation to maximize likelihood
(hear more next class!)
Vaske, C. J. et al. Inference of patient-specific pathway activities from multidimensional cancer genomics data using PARADIGM. (2010) Bioinformatics 26
Silhouette statistic
Subtype
1
2
3
−0.2
0
0.2
0.4
0.6
Silhouette Value
0.8
1
Silhouette statistic
a.
b.
c.
d.
Three clusters in 2 dimensions
Three clusters in 10 dimensions, each cluster has 50 observations
4 clusters in 10 dimensions with randomly chosen centers
Six clusters in 2 dimensions
(a)
(d)
Silhouette statistic
a.
b.
c.
d.
Three clusters in 2 dimensions
Three clusters in 10 dimensions, each cluster has 50 observations
4 clusters in 10 dimensions with randomly chosen centers
Six clusters in 2 dimensions
Hossein Parsaei. Finding a number of clusters
NMF – non-negative matrix
factorization
Matrix factorization: NMF(V) = WxH
W and H are non-negative
Current methods (many – gradient descent,
alternating non-negative least squares, etc)
Arora et al (2012) – exact NMF method runs in
polynomial time under separability condition of
W
Consensus Clustering
Resampling based method for class discovery and
visualization of gene expression microarray data
Goal: assessing stability
Method:
◦ For a 1000 iterations
1.
2.
Resample data
Cluster with fav. clust. method (hier, k-means)
◦ Compute consensus matrix
◦ Partition D based on Consensus Matrix
Monti, S., Tamayo, P., Mesirov, J., Golub, T. (2003) Consensus Clustering: A ResamplingBased Method for Class Discovery and Visualization of Gene Expression Microarray
Data. Machine Learning, 52, 91-118.
SigClust
Goal: assess statistical signficance of clustering
H0: data comes from a single Gaussian
H1: not from a single Gaussian
Statistic: Cluster Index (CI) - sum of within-class
sums of squares about the mean of the cluster
divided by the total sum of squares about the
overall mean (mean-shift and scale invariant)
Liu,Yufeng, Hayes, David Neil, Nobel, Andrew and Marron, J. S, 2008, Statistical
Significance of Clustering for High-Dimension, Low-Sample Size Data, Journal of the
American Statistical Association 103(483) 1281–1293
Patient Specific Data Fusion (Yuan
et al, 2011)
Nonparametric
Bayesian model (gene
expression and CNV)
◦ Feature selection (each feature is drawn from
a multinomial distribution with unknown class
proabilities
◦ MCMC inference
Kernel
LearningLearning
10.3 Multiple
Multiple
Kernel
Mostly
used in supervised cases, but
Multipleexists
Kernel
Learning
(MKL)
learn
in unsupervised scenario (Chuang,
improvesCVPR,
the2012)
performance of classifier
Linear
of function
kernels
1, . . . , m,
thecombination
objective
is
to
l
Pm
kernel Kcombine = v=1 ↵v Kv more sui
MKL is used in supervised setting bec
optimal ↵. Recently, an unsupervised M
with spectral clustering framework. Ei
h-dimensional patient-by-feature matrix was then used as input into a
orithm available as part of matlab distribution that yielded a set of cl
ion as the distance metric and ‘average’ as the linkage function. The
hosen to be the same as the result of clustering of the SNF fused matrix.
iCluster (Shen et al, 2009)
latent variable model
Sparsity
regularization
(Lasso-type)
Gaussian latent variable
model
with sparsity regularization
in Lasso-type o
riefly, the main
assumption
behind this
approach is that
the sets of m ge
Latent
variables
(embedding
is
shared)
m
ter
k=1
Gaussian
shared a common set of latent variables zi using the following linear
xik = Wk zi + ✏ik , i = 1, . . . , n, k = 1, . . . , m
notes the loading matrix associated with the k-th genomic data and n is
on variables zi represent the underlying driving factors on patient i that c
ase subtype assignment. iCluster uses the Expectation-Maximization (EM
arameteres due to the assumption that the error in the model follows a G
arsity in the estimated Wk is enforced by adding an `1 norm regulariza
uggested in the method’s manual.
Drawbacks of existing methods
A
lot of manual processing
Many steps in the pipeline
Integration mostly done in the feature
space – if there is signal in a combination
of features, it’ll be lost
Focusing on consensus – what if there is
complementary information?
Similarity Network Fusion (Wang et al, 2014)
Integrate
1.
2.
data in the patient space
Construct patient similarity matrix
Fuse multiple matrices
than
patients
that, N
have
di↵erent subtypes. We denote ⇢(xi , x
where
mean(⇢(x
i
i )) is the average value of the correlations bet
between
xi and xj . We then use a scaled exponent
each of patients
its neighbours.
1.determine
to
the weight
the edge eofijthe
: patient network is tw
The advantage
of our of
construction
Construct similarity networks
augments the correlation between patients which facilitates the cl
2 the data.
afterwards; 2) it reduces the e↵ect of scale and
noise
in
⇢(x
,
x
)
i
j
W
(i,
j)
=
exp(
),
Patient
similarity:
A natural kernel acting on functions on V can
be
defined
by n
2
⌘⇠ij
of the weight matrix as follows:
W (i,be
j) empirically set
where ⌘ isAdjacency
a hyperparameter
that
can
P (i, j) = P
,
matrix:
eliminate the scale problem. In our paper,
wek)define
k2V W (i,
P
Patients
Patients
so that j2V P (i, j) = 1.
mean(⇢(x
mean(⇢(x
i , Ni )) +
j , Nj ))
Given amRNA
graph,
G,
we
construct
another
graph
G:
the
vertices
Patients
expression
⇠ij =
genes
same as in G, and the similarities between non-neighboring
points
2
the pairwise similarity values) are set to zero. Essentially we make
tion that local similarities (high values) are more reliable than
and we thus assign similarities to non-neighbors through graph di↵
network. This is a mild assumption widely adopted by other mani
algorithms.
Using K nearest neighbors (KNN) to measure local affinity, we
Patients
Construct similarity networks
Patients
1.
and we This
thus assign
similarities
to non-neighbors
through
graphmanifold
di↵usion
network.
is a mild
assumption
widely adopted
by other
network. This is a mild assumption widely adopted by other manifold le
algorithms.
algorithms.
Using K nearest neighbors (KNN) to measure local affinity, we const
Usingmatrix
K nearest
similarity
as: neighbors (KNN) to measure local affinity, we constru
⇢
similarity matrix as:
W (i, j) if x 2 KN N (x )
W(i, j) = ⇢ W (i, j) if xjj 2 KN N (xi )i
0 otherwise
1) W(i, j) =
0 otherwise
ThenSparsification
the corresponding kernel becomes:
Then the corresponding kernel becomes:
W(i, j)
2)
P(i, j) = P
W(i, j)
P(i, j) = Pxk 2KN N (xi ) W(i, k)
xk 2KN N (xi ) W(i, k)
Note that P carries the full information about the similarity of each da
Note that P carries the full information about the similarity of each data
to all others whereas P only encodes the similarity top2nearby data poi
to all others
whereas P only encodes
the similarity to nearby data poin
Patients
expression
clarity,mRNA
wegenes
call P the status matrix
and P the kernel
matrix. Our al
p1
clarity, we call P the status matrix and P the kernel
matrix. Our algo
always starts from P as the initial status using P as the kernel matri
always starts from P as the initial status using P as the kernel matrix
di↵usion process for computational efficiency.
di↵usion process for computational efficiency.
p9
3 3 Cross
Di↵usion
Process
(CrDP)
with
m
=
2
Simi
Cross Di↵usion Process (CrDP) with m = 2 Simila
Matrices
Matrices(Views)
(Views)
p8
Given
mm
views
can construct
constructsimilarity
similaritymatrice
matric
Given
viewsfrom
fromdi↵erent
di↵erentdomains,
domains, we
we can
(j)(j)
(j)
(j)
(j)
(j)
and
W
using
Eq
4
for
the
j-th
view,
j
=
1,
.
.
.
,
m.
P
and
P
areobo
and W using Eq 4 for the j-th view, j = 1, . . . , m. P and P are
3
Cross Di↵usion Process (CrDP) with
Matrices (Views)
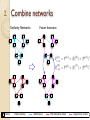
2. Combine
networks
Given
m views from di↵erent domains, we can construct
and W (j) using Eq 4 for the j-th view, j = 1, . . . , m. P
from Eqs 3Fusion
and 5Iterations
respectively.
Similarity Networks
Below we introduce our network fusion Cross-Di
First, we calculate the status matrices P (1) and P (2) a
similarity matrices; then the kernel matrices P (1) and
(1)
(2)
Eq 5. Let P0 = P (1) and P0 = P (2) . The cross-di↵us
(1)
(2)
(2)
(1)
Pt+1 = P (1) ⇥ (Pt ) ⇥ (P (1) )0
Pt+1 = P (2) ⇥ (Pt ) ⇥ (P (2) )0
Patient
Patient similarity:
mRNA-based
DNA Methylation-based
Supported by all data
unknown class probabilities. Multiple MCMC c
and infer the statical uncertainties in PSDF. In o
100 MCMC iterations in each step and fusion we
While PSDF appears to be a powerful frame
are essential
precluding
Similarity Networks
Fusion disadvantages
Iterations
Fusedthe use o
this paper: 1) large number of unknown
Similarity param
computationally expensive; 2) it isNetwork
only suitable
tially be applied to the METABRIC cohort whi
the approach is not scalable to the full size of th
2. Combine networks
6
6.1
Patient
Patient similarity:
Supplementary Methods
Stopping Criteria
SNF is proved to converge, and empirically it co
Wt k
in consecutive rounds Et = kWt+1
. ≤ One
10-6 si
kWt k
✏ = 10 6 and if the relative change is lower t
empirical observations about the convergence ca
mRNA-based
Supported by all data
when
the numberDNAofMethylation-based
iterations exceeds
20, it is
process a patient is always most similar to himself than to other
Given m views from di↵erent domains, we can construct similarity matrice
ensure that our final network is full rank, important
for (j)
the class
(j)
and W (j) using
Eq
4
for
the
j-th
view,
j
=
1,
.
.
.
,
m.
P
and
P
are ob
clustering applications of the final network. Finally, we have found
from Eqs 3 and
5 respectively.
of regularization
leads to quicker convergence of CrDP.
Below we introduce
network
fusion
Cross-Di↵usion
The input our
to our
algorithm
can be
feature vectors,Process
pairwise(C
(2)
First, we calculate
status matrices
P (1) status
and Pmatrix
as in
Eqcan
3 from
pairwisethe
similarities.
The learned
P (c)
then tw
be
(1)
(2)
trieval, clustering,
classification;
in thisand
paper,
focus
on cl
similarity matrices;
then the and
kernel
matrices P
P weare
obtaine
(1)
(2)for more
(2) details.
to P[3]
Eq 5. Let P0refer
= readers
P (1) and
=
P
. The cross-di↵usion process is defi
0
Network Fusion
(1)
(2)
Pt+1 = P (1) ⇥ (Pt ) ⇥ (P (1) )0
4 Extension
to m > 2 (1)
Fusing 2 networks:
(2)
Pt+1 = P (2) ⇥ (Pt ) ⇥ (P (2) )0
We extend the CrDP above to multiple (m > 2) similarity matrice
adjusting Eq (6) as follows
Fusing m networks:
(i)
Pt+1
=
P
(i)
⇥(
1
m
1
X
j6=i
(j)
Pt ) ⇥ (P (i) )0 + ⌘I
where i = 1, . . . , m. The corresponding final status matrix is comput
Pm
(i)
1
P
i=1 t .
m
Experiments
Data:!
"2 simulations"
"5 TCGA cancers"
"METABRIC (Large "
Breast Cancer db)"
"
Comparative Methods:!
"Concatenation"
"iCluster"
"PDSB"
"Multiple kernel learning"
"
Criteria: !!
"
"
-log10(log rank pvalue)"
"
Silhouette score (cluster homogeneity)"
"
Running time"
Simulation 1 – complementarity
Simulation 2 - removing noise
Simulation 2 - removing noise
TCGA Data
Gene pre-selection across cancers
Bo Wang
Clustering of the network
Bo Wang
Patient networks:
advantages and disadvantages
-
-
-
Integrative feature selection
Growing the network requires extra work
Unsupervised – hard to turn into a supervised problem
ü Creates
a unified view of patients based on multiple
heterogeneous sources
ü Integrates gene and non-gene based data
ü No need to do gene pre-selection
ü Robust to different types of noise
ü Scalable
Package on CRAN: SNFtool
Data integration - future
Data integration - future
Simultaneous
feature selection and data
integration
Supervised vs unsupervised approaches –
do we really need unsupervised methods?
Priors on contributions of different types
of data
Automate feature pre-selection if
necessary
Next class
iCluster – joint latent variable model (Shen et
al, 2009) - Ladislav
PARADIGM – Andrew
Next topic: pharmacogenomics (guest lecture
by Dr Benjamin Haibe-Kains)