Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Nonlinear optics wikipedia , lookup
Photon scanning microscopy wikipedia , lookup
Astronomical spectroscopy wikipedia , lookup
Thomas Young (scientist) wikipedia , lookup
Upconverting nanoparticles wikipedia , lookup
Ultrafast laser spectroscopy wikipedia , lookup
Smart glass wikipedia , lookup
Reflection high-energy electron diffraction wikipedia , lookup
Phase-contrast X-ray imaging wikipedia , lookup
Diffraction topography wikipedia , lookup
Surface plasmon resonance microscopy wikipedia , lookup
Retroreflector wikipedia , lookup
Magnetic circular dichroism wikipedia , lookup
Anti-reflective coating wikipedia , lookup
Rutherford backscattering spectrometry wikipedia , lookup
Scanning electrochemical microscopy wikipedia , lookup
Ultraviolet–visible spectroscopy wikipedia , lookup
Gaseous detection device wikipedia , lookup
Low-energy electron diffraction wikipedia , lookup
Powder diffraction wikipedia , lookup















































































































































































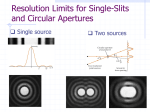



![Scalar Diffraction Theory and Basic Fourier Optics [Hecht 10.2.410.2.6, 10.2.8, 11.211.3 or Fowles Ch. 5]](http://s1.studyres.com/store/data/008906603_1-55857b6efe7c28604e1ff5a68faa71b2-150x150.png)



