Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
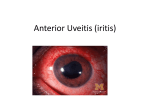
ORIGINAL ARTICLE The Uveo-Meningeal Syndromes Paul W. Brazis, MD,* Michael Stewart, MD,* and Andrew G. Lee, MD† Background: The uveo-meningeal syndromes are a group of disorders that share involvement of the uvea, retina, and meninges. Review Summary: We review the clinical manifestations of uveitis and describe the infectious, inflammatory, and neoplastic conditions associated with the uveo-meningeal syndrome. Conclusions: Inflammatory or autoimmune diseases are probably the most common clinically recognized causes of true uveo-meningeal syndromes. These entities often cause inflammation of various tissues in the body, including ocular structures and the meninges (eg, Wegener granulomatosis, sarcoidosis, Behçet disease, Vogt-Koyanagi-Harada syndrome, and acute posterior multifocal placoid pigment epitheliopathy). The association of an infectious uveitis with an acute or chronic meningoencephalitis is unusual but occasionally the eye examination may suggest an infectious etiology or even a specific organism responsible for a meningeal syndrome. One should consider the diagnosis of primary ocular-CNS lymphoma in patients 40 years of age or older with bilateral uveitis, especially with prominent vitritis, that fails to respond to treatment or who has associated neurologic findings. A paraneoplastic disorder has been described in patients who have combined optic neuritis and retinitis defined serologically by the presence of a paraneoplastic IgG autoantibody CRMP-5-IgG. These patients may have an inflammatory vitritis and may have signs of cerebrospinal fluid inflammation. Key Words: uveitis, vitritis, choroiditis, retinitis, uveo-meningeal syndrome, meningitis (The Neurologist 2004;10: 171–184) T he uveo-meningeal syndromes are a heterogeneous group of disorders that share involvement of the uvea, retina, and meninges. Etiologies of this syndrome include infectious, inflammatory, and neoplastic disorders (Table 1) with the From the *Department of Ophthalmology, Mayo Clinic—Jacksonville, Jacksonville, Florida; and the †Departments of Ophthalmology, Neurology, and Neurosurgery, University of Iowa Hospitals and Clinics, Iowa City, Iowa. This work was supported in part by an unrestricted grant from Research to Prevent Blindness, Inc., New York, NY. Reprints: Paul Brazis, MD, Department of Ophthalmology, Mayo Clinic— Jacksonville, 4500 San Pablo Road, Jacksonville, FL 32224. E-mail: [email protected]. Copyright © 2004 by Lippincott Williams & Wilkins ISSN: 1074-7931/04/1004-0171 DOI: 10.1097/01.nrl.0000131145.26326.ff The Neurologist • Volume 10, Number 4, July 2004 main clinical features being a meningitis or meningoencephalitis associated with uveitis. The meningeal involvement is often chronic and may cause cranial neuropathies, polyradiculopathies, and hydrocephalus. In this review we define and describe the clinical manifestations of different types of uveitis and discuss the individual entities most often associated with the uveo-meningeal syndrome. We review the distinctive signs in specific causes for uveo-meningeal disease and discuss our evaluation of these patients. The uveo-meningeal syndromes are a heterogeneous group of disorders that share involvement of the uvea, retina, and meninges. UVEITIS The uveal tract consists of the iris anteriorly, the ciliary body (the pars plicata anteriorly and the pars plana posteriorly) in the middle, and the choroid posteriorly (Fig. 1). Inflammation of any of these structures is called uveitis. The anatomic division of the uveal tract serves as the basis for classifying uveitis into anterior uveitis (iritis, anterior cyclitis, iridocyclitis), intermediate uveitis, or posterior uveitis (focal, multifocal, or diffuse choroiditis, retinochoroiditis, or neurouveitis with optic nerve inflammation), and panuveitis.1,2 Anterior uveitis may be subdivided into acute or chronic and granulomatous or nongranulomatous forms. Patients with acute anterior uveitis present with eye pain, redness, tearing, photophobia, and decreased vision in the affected eye. There is a perilimbal flush caused by dilation of the circumcorneal conjunctival vessels and often clumps of inflammatory cells called keratic precipitates on the posterior cornea (Fig. 2). The characteristic finding is inflammatory cells and protein in the aqueous humor (cell and flare). With severe inflammation there may be a layering of inflammatory cells in the inferior anterior chamber (hypopyon). Chronic anterior uveitis often develops in a more gradual fashion with patients experiencing decreased vision. Affected eyes are 171 Brazis et al The Neurologist • Volume 10, Number 4, July 2004 TABLE 1. Etiologies of the Uveo-Meningeal Syndrome Infectious Bacterial Cat-scratch disease Whipple disease Mycobacterial—tuberculosis Syphilis Lyme disease Fungal Toxoplasmosis Viral Gnathostomiasis Inflammatory Wegener granulomatosis Sarcoidosis Behçet disease VKH syndrome APMPPE Neoplastic Lymphoma Metastatic disease Paraneoplastic syndrome of combined optic neuritis and retinitis defined serologically by CRMP-5-IgG often not gray (ie, they are “quiet”) but may have pigmented keratic precipitates, aqueous cells and flare, and areas where the iris has become adherent to the anterior lens capsule (posterior synechiae). Iris nodules may occur on the pupillary margin (Koeppe nodules) or away from the pupillary margin (Busacca nodules). When large keratic precipitates (“mutton fat” precipitates) are present with nodules on the iris, the inflammation is said to be granulomatous. If these iris nodules or mutton fat keratic precipitates are absent, the uveitis is called nongranulomatous. Posterior subcapsular cataracts often occur with chronic anterior uveitis. Inflammatory mediated scarring between the iris and peripheral cornea (anterior synechiae) may lead to chronic glaucoma. Inflammation of the ciliary body (cyclitis and pars planitis) presents with the gradual onset of blurred vision or floaters in a quiet eye. The anterior chamber may have a few cells but there are diffuse cells in the vitreous. Clumps of vitreous inflammatory cells may cause vitreous “snowballs.” Mounds of inflammatory white exudates may accumulate because of gravity on the inferior pars plana (“snow banks”). The release of vasoactive agents lead to macular edema which may cause decreased or distorted vision. Patients with posterior segment inflammation (choroiditis and retinitis) present with reduced vision, distorted vision, and floaters of acute or insidious onset. Pain, redness, tearing, and photophobia are often absent. Inflammation of the retinal 172 FIGURE 1. This cross-section drawing of the eye shows the uveal tract (highlighted in brown) with its 4 anatomic components: iris, ciliary body (pars plicata), pars plana, and choroid. FIGURE 2. Peri-limbal flush, seen here as fine red lines on the sclera just outside the iris, indicates dilated capillaries and small arterioles resulting from iritis. The multifocal accumulation of protein and inflammatory cells on the corneal endothelial surface forms keratic precipitates (KPs). arterioles and venules causes sheathing, focal vascular narrowing and irregularity. Exudates may cluster along vessels appearing as “candle wax drippings” (tache d’bougie). With © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 choroidal and retinal inflammation, cells are often noted in the vitreous (vitritis). The macula and optic nerve may also become swollen or ischemic. Retinitis is characterized by areas of retinal whitening and thickening, while choroiditis is characterized by deeper yellow or cream-colored lesions beneath the retinal pigment epithelium. Vascular leakage from choroidal inflammation may cause exudative retinal detachment. The association of retinitis or choroiditis with a swollen optic disc is referred to as neuroretinitis. The evaluation of uveitis thus requires thorough ophthalmologic examination including assessment of visual acuity and color vision, external examination (for conjunctival or episcleral injection), slit-lamp biomicroscopy (cell and flare, keratic precipitates, iris atrophy, iris nodules, syneciae, cataract, etc.), measurement of intraocular pressures, and dilated fundus examination.2,3 Fluorescein angiography is often useful to confirm retinal vasculitis, macular edema, and areas of retinitis, choroiditis, ischemia, or retinal detachment. It is important to identify the predominant location of the uveitis (anterior, intermediate, posterior) because the differential diagnosis differs according to location. ETIOLOGIES The causes of uveitis include traumatic, infectious, inflammatory, or neoplastic etiologies. It is important to identify the predominant location of the uveitis (anterior, intermediate, posterior) because the differential diagnosis differs according to location. The most common causes of nontraumatic anterior uveitis are idiopathic (38 to 56% of cases), the seronegative spondyloarthropathies (21 to 23%), juvenile rheumatoid arthritis (9 to 11%), and herpetic keratouveitis. Most of intermediate uveitis cases are idiopathic (pars planitis). Toxoplasmosis is the most common cause of posterior uveitis and the most common causes of panuveitis are idiopathic (22 to 45%) and sarcoidosis (14 to 28%).4,5 INFECTIOUS ETIOLOGIES OF THE UVEOMENINGEAL SYNDROME Almost any infectious agent may cause a meningoencephalitis and many agents may cause uveitis. The association of an infectious uveitis with an acute or chronic meningitis or meningoencephalitis is unusual. However, occasionally the eye examination may suggest an infectious etiology or even a specific organism responsible for a meningeal syndrome. Infectious agents commonly associated with different types of uveitis are © 2004 Lippincott Williams & Wilkins Uveo-Meningeal Syndromes outlined in Table 2. Only infectious agents known to cause a meningeal syndrome possibly associated with uveitis will be discussed. BACTERIAL INFECTIONS The major bacterial etiologies for uveo-meningeal syndrome include cat scratch disease, Whipple disease, syphilis, tuberculosis, and Lyme disease. Cat scratch disease is usually manifested by a regional lymphadenopathy following the scratch or bite of a kitten or adult cat and is a result of infection by Bartonella henselae. A common ophthalmologic manifestation of cat scratch disease is optic disc edema (Fig. 3) with a macular star (ODEMS).6 This neuroretinitis may be associated with anterior uveitis. Cat scratch disease may cause a focal, multifocal, or diffuse retinitis or choroiditis (Fig. 4).7–9 In a study of 24 patients (35 eyes) with choroidal, retinal, or optic disc manifestations of cat scratch disease, discrete white retinal or choroidal lesions were the most common posterior segment finding (46% of eyes, 63% of patients) followed by macular star (43% of eyes, 63% of patients).9 Some patients with cat scratch disease develop signs of meningeal irritation and TABLE 2. Types of Uveitis Associated with Specific Infectious Agents Anterior uveitis Bacteria (endogenous), mycobacteria, and spirochetes—Listeria Monocytogenes, Neisseria meningitidis, Haemophilus Influenzae, Yersenia, Klebsiella, leprosy, tuberculosis, mycoplama pneumoniae, Whipple disease, syphilis Viral—Herpes zoster, herpes simplex, cytomegalovirus, HIV, hepatitis B Fungi—Candida albicans, coccidiomycosis Protozoa—Toxoplasmosis Rickettsiae—Rocky Mountain spotted fever Intermediate uveitis and vitritis Bacteria, mycobacteria, and spirochetes—cat scratch disease, Whipple disease, tuberculosis, Lyme disease Viral—Cytomeglovirus, EB virus Protozoa—Toxoplasmosis Posterior uveitis Bacteria, mycobacteria, and spirochetes—Neisseria meningitidis, Leptospira, Brucella, Nocardia asteroides, Whipple disease, cat scratch disease, tuberculosis, Lyme disease, syphilis Viral—Cytomegalovirus, herpes simplex, herpes zoster, Rubella, (German measles), Rubeola, HIV Fungi—Candida, Histoplama capsulatum, Cryptococcus neoformans, Aspergillus, Coccidiodes immitis, Blastomyces dermatidis Parasite—Toxoplasmosis, Toxocara canis, Cysticercus cellulosae, Onchocerca volvulus 173 Brazis et al FIGURE 3. Optic disc swelling in cat scratch neuroretinitis. FIGURE 4. Focal choroiditis in cat scratch disease. fever beginning 1 to 6 weeks after the onset of lymphadenopathy or about 3 to 8 weeks after initial animal contact. Other neurologic manifestations include encephalitis, radiculitis, myelitis, or a combination of these findings.10 The diagnosis is confirmed by serologic testing for Bartonella henselae and antibiotic therapy with azithromycin, ciprofloxacin or doxicycline may be useful. Most cases improve over time though some patients experience permanent visual loss. Whipple disease is characterized by arthralgia, abdominal pain, and weight loss. The causative organism is Troph- 174 The Neurologist • Volume 10, Number 4, July 2004 eryma whippelii. Ocular manifestations include chronic anterior uveitis, vitreous opacities (described as “clumps of mulberries”), diffuse chorioretinal inflammation, and varying degrees of retinitis and vitritis.11–16 Although diarrhea and other gastrointestinal signs and symptoms are common, they may be absent in central nervous system Whipple disease. Neurologic manifestations are usually because of parenchymal rather than meningeal involvement and include drowsiness, confusion, amnesia, behavioral abnormalities, intellectual deterioration, ataxia, vertical gaze impairment, and supranuclear ophthalmoplegia. A distinct and presumed pathognomonic condition called oculomasticatory myorhythmia may occur in Whipple disease. It is characterized by constant, 0.5 Hz rhythmic convergence of the eyes associated with synchronous contractions of the eyelids, mouth, face, and neck.17–19 The diagnosis of Whipple disease can be made by small bowel biopsy. Polymerase chain reaction (PCR) testing of tissue or cerebrospinal fluid might be diagnostic. Appropriate and often long-term antibiotic therapy with penicillin, tetracycline, or trimethaprim/sulfamethoxazole may be required in central nervous system (CNS) Whipple disease. The prognosis is variable but recurrent disease is common in patients with neurologic involvement. Tuberculosis (TB) is a multisystem infectious disorder with protean manifestations. The causative organism is Mycobacterium tuberculosis. TB may cause a chronic granulomatous iridocyclitis that is usually bilateral.20,21 The disease may less often present with high-grade, nongranulomatous anterior chamber inflammation. The most common posterior ocular presentation is a bilateral multifocal choroiditis, with or without overlying retinal necrosis and vitritis.20 Retinal periphlebitis may be seen. Choroidal tuberculosis may present as tubercles or tuberculomas (large solitary masses).22 Intraocular inflammation may be acute, relapsing, or chronic and persistent. Tuberculous meningitis arises as a consequence of bacteremia that occurs after primary infection or may also occur after late reactivation of TB elsewhere in the body. It often starts with a prodrome characterized by malaise, low-grade fever, headache and mentation, or personality changes. Within several weeks, a well-defined meningitic phase develops with headache, meningismus, vomiting, confusion, and multiple cranial neuropathies.23 The diagnosis may require positive skin testing (eg, purified protein derivative), chest radiography, or cerebrospinal fluid (CSF) analysis. PCR for TB may be positive in the CSF or tissue. Choroidal biopsy may establish the presence of acid-fast bacilli. Appropriate long-term multidrug anti-TB therapy with isoniazid, rifampin, ethambutol, and pyrazinamide is generally required for TB meningitis. Syphilis, the great mimicker, may present with anterior or posterior uveitis. The disease is caused by Treponema pallidum. The most common ocular presentation is a nonspecific iritis or iridocyclitis.24 The anterior uveitis © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 may be granulomatous or nongranulomatous, unilateral, or bilateral. Multiple forms of posterior uveitis have been described, including retinal vasculitis, necrosis, and optic disc swelling. Diffuse or multifocal chorioretinitis is the most common type of chorioretinal inflammation in patients with acquired syphilis.25 This chorioretinitis is usually bilateral and associated with vitritis. Other types of chorioretinitis include disseminated choroiditis, areolar choroiditis (characterized by individual foci of choroiditis with subsequent lesions developing further and further peripherally), localized central chorioretinitis, and isolated retinitis.26 Most ocular manifestations occur with secondary syphilis. Meningitis frequently complicates secondary syphilis27 with spirochetes sometimes seen on dark field examination of the CSF. General paresis, tabes dorsalis, and meningovascular neurosyphilis occur in the tertiary stage of this disease. The diagnosis is made by serologic and CSF testing for Treponemal (eg, Fluorescent Treponemal antibody or Microhemagglutination for Treponema pallidum) and non-Treponemal testing (eg, rapid plasma reagin or Venereal Disease Research Laboratory). The diagnosis of neurosyphilis in cases of uveo-meningeal syndromes is important because the treatment of tertiary and neurosyphilis requires high-dose intravenous penicillin therapy (4 million units every 4 hours) and therapy is generally longer duration than primary or secondary syphilis regimens (eg, 2 weeks). Lyme disease is a spirochetal infection that results from tick-borne transmission of Borrelia burgdorferi. The disease progresses through 3 stages.28 The first stage (early localized disease) usually includes a rash (erythema chronicum migrans) characterized by an expanding erythematous margin, often with a central area of clearing, at the site of the tick bite. Associated symptoms include fever, malaise, arthralgias, and a flu-like illness. Conjunctivitis is the most common ocular manifestation of early Lyme disease and occurs in approximately 10% of cases. The secondary and tertiary stages of the disease (early disseminated and chronic disseminated stages) are characterized by neurologic impairment, cardiac disease, and arthritis. Neurologic manifestations include cranial nerve palsies (especially facial nerve impairment), meningitis, encephalitis, myelitis, meningoradiculitis, peripheral neuropathies and, rarely, pseudotumor cerebri.29 Uveitis is a relatively rare manifestation of Lyme disease, but may occur in the later stages. Unilateral or bilateral anterior uveitis, intermediate uveitis, vitritis, and choroiditis have been described, with other ocular manifestations including keratitis, episcleritis, optic neuritis, neuroretinitis, and motility problems.30 –34 The diagnosis is made by serologic testing and appropriate antibiotic therapy may be curative. © 2004 Lippincott Williams & Wilkins Uveo-Meningeal Syndromes FUNGAL INFECTIONS Various fungi may cause chronic granulomatous or nongranulomatous uveitis or other intraocular infections. The fungi may reach the eye via penetrating trauma (exogenous) or through hematogenous spread of infection (endogenous). Immunocompromised hosts (eg, cancer patients, transplant recipients on immunosuppresive medications, intravenous drug abusers, patients with chronic indwelling catheters, AIDS patients, etc.) are especially susceptible to systemic fungal infections. Fungal organisms that may cause uveitis in the setting of menigoencephalitis include Candida albicans, Coccidiodes immitis, Cryptococcus neoformans, Histoplasmosis capsulatum, Blastomyces dermatitidis, and Aspergillus. Candida albicans may cause an endogenous endophthalmitis presenting with ocular pain, decreased vision, and floaters.35,36 The typical lesion of Candida endophthalmitis is a fluffy, white chorioretinal lesion with overlying vitreous haze. The infection often extends into the vitreous, causing cotton ball-like opacities so characteristic in appearance as to be almost pathognomonic of Candida infection.37 Progressive retinochoroiditis with satellite lesions may occur. Other manifestations include anterior uveitis with hypopyon, scleritis, keratitis (Fig. 5), papillitis, or even panophthalmitis. CNS candidiasis usually develops in the setting of disseminated candidiasis and manifests as meningitis, sometimes associated with cerebral granulomas. Coccidiodes immitis infection is endemic in certain areas of the southwestern United States as well as Central and South America. It may cause iridocyclitis, iris granuloma, choroiditis, chorioretinitis, or diffuse endophthalmitis.38,39 Pulmonary infection is the most common manifestation of FIGURE 5. Fungal corneal abscess. 175 Brazis et al symptomatic infection, but subacute or chronic meningitis, usually without abscess or granuloma formation, may develop. Cryptococcal retinochoroiditis occurs in immunosuppressed patients (especially those with AIDS, lymphoma, sarcoidosis, or diabetes mellitus) and may be associated with meningitis or meningoencephalitis.40 Cryptococcus is the most common cause of clinically recognized fungal meningitis or meningoencephalitis in adults or children. The onset of this meningitis is often insidious but acute meningitis may also occur. Patients may complain of headache, somnolence, irritability, and clumsiness that may be present for weeks or months before the patient seeks medical attention. Progressive visual loss because of papilledema and fungal optic nerve sheath invasion may occur.41 Most cases of intraocular Cryptococcus infection manifest as posterior segment infection with initial small white spheres in the superficial retina or vitreoretinal interface. These initial lesions are often asymptomatic when diagnosed. These lesions may be associated with adjacent retinal hemorrhage and most patients eventually develop more extensive whitish lesions that may or may not be associated with hemorrhages in the retina, choroid, or both.40,42 Overlying vitritis may also be present. Histoplasmosis capsulatum is responsible for 3 different forms of ocular involvement: Histoplasma endophthalmitis with diffuse uveal and retinal infection from disseminated histoplasmosis; solitary histoplasmic chorioretinal granuloma; and the presumed ocular histoplasmosis syndrome (POHS).43 The presumed ocular histoplasmosis syndrome is endemic in the Ohio and Mississippi River Valleys of the Eastern United States and consists of peripapillary chorioretinal atrophy and scarring, peripheral punched-out chorioretinal scars, choroidal neovascularization with hemorrhage into the macula, and absence of anterior segment or vitreous inflammation. Solitary histoplasmic granuloma is extremely rare and consists of a variable ill-defined white choroidal lesion. Neither the POHS nor solitary histoplasmic granuloma is associated with meningeal involvement. Histoplasmic endophthalmitis usually occurs in immunosuppressed individuals, especially in patients with AIDS, as part of a disseminated histoplasmosis infection. Affected patients complain of decreased vision, floaters and eye pain. Ophthalmic manifestations include conjunctival injection, anterior uveitis, vitritis, and multiple white retinochoroidal foci of infection (Fig. 6).43,44 Although meningitis with disseminated histoplasmosis is usually mild and subclinical, some patients may develop severe meningitis, focal or diffuse encephalitis, or myelitis.45 Disseminated histoplasma infection is frequently fatal. Blastomyces dermatitidis causes a systemic disease called blastomycosis that occurs primarily in the southeastern and south central United States. The portal of entry for blastomycosis is the lung; ocular and CNS involvement is 176 The Neurologist • Volume 10, Number 4, July 2004 FIGURE 6. Focal retinochoroiditis in AIDS patient with acquired, disseminated histoplasmosis. caused by dissemination from a primary pulmonary infection. Unlike most fungi, Blastomyces dermatitidis is capable of producing disease in immune competent patients. Virtually every structure of the eye can be affected though the organism has a predilection for the uveal tract. Ocular findings include anterior uveitis with hypopyon, iridocyclitis, posterior uveitis, multifocal choroidal and peripapillary scarring, and panuveitis.46,47 CNS blastomycosis may cause pyogranulomas or meningitis.48,49 Immunocompromised patients may develop invasive Aspergillus infections with organisms spreading to the CNS either directly from a paranasal sinus or hematogenously from distant sites of infection. Intraocular infection by aspergillus is uncommon with the majority of cases resulting in endogenous endophthalmitis with chorioretinitis.50,51 Orbital infections, manifesting as eye pain, proptosis, optic neuropathy, and ophthalmoparesis, are far more common than intraocular infections. Acute or chronic aspergillus meningitis is rare but may result from spread of infection from the paranasal sinuses, middle or inner ear, or orbit.52 The diagnosis of a specific fungal etiology generally requires identification of fungal elements in a tissue specimen, fungal stain or positive culture. Appropriate antifungal therapy should be directed at the causative agent in fungal uveo-meningeal disease. PARASITIC AND PROTOZOAL INFECTIONS These are uncommon causes for uveo-meningeal disease in the United States. Ocular toxoplasmosis, caused by Toxoplasma gondii, is the most common cause of posterior segment inflammation. Most cases of toxoplasmosis retinochoroiditis are because of reactivation of prior infection. © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 Acquired toxoplasmosis is unusual in immunocompetent individuals and more commonly occurs as an acute disseminated infection in immunocompromised persons, especially patients immunosuppressed after organ transplantation. Acquired toxoplasmosis causes an acute choroiditis, retinitis, or chorioretinitis that is usually unilateral, as opposed to the bilateral disease that occurs in patients with congenital toxoplasmosis.53 The intense, white inflammatory focus is generally adjacent to a prior chorioretinal scar and is associated with a severe vitritis (“headlight in the fog”) (Fig. 7). A neuroretinitis can also occur.54 Necrotizing retinitis may be seen especially in immunodeficient patients. Disseminated toxoplasmosis with CNS involvement is a common cause of neurologic disease in patients with AIDS. Clinical manifestations include encephalitis, meningoencephalitis, meningitis, or expanding mass lesions. Appropriate antitoxoplasmosis therapy with daraprim, sulfasalazine, clindamycin, or trimethaprim/sulfamethoxazole is usually administered and is especially important in immunocompromised patients. Gnathostomiasis is a helminth infection resulting from Gnathostoma spinigerum. This organism may rarely cause anterior, posterior, or pan-uveitis and may also cause meningitis or encephalitis.55 Other less common parasitic and protozoan causes of uveo-meningeal syndrome should be considered in patients from endemic areas. VIRAL INFECTIONS Many viruses may cause uveitis; Herpes simplex and varicella-zoster virus may often cause blepharitis, conjunctivitis, scleritis, keratitis, iridocyclitis, vitritis, and retinitis. The FIGURE 7. Focal recurrent toxoplasma retinochoroiditis with overlying vitritis (“headlight in the fog” appearance). © 2004 Lippincott Williams & Wilkins Uveo-Meningeal Syndromes iridocyclitis caused by these organisms can vary from a mild anterior chamber inflammatory reaction to severe exudative infection with hypopyon. Various viruses are known to infect the retina or choroid resulting in posterior uveitis. Common viral agents causing posterior uveitis include cytomegalovirus (CMV), herpes simplex, and varicella-zoster; rare viral agents causing posterior uveitis include herpes B virus, Epstein-Barr virus, coxsackie virus, and rubella. Rift Valley fever, because of infection via Phlebovirus species endemic in Africa, may also cause retinitis, retinal vasculitis, and, occasionally, severe encephalitis.56 CMV retinitis is the most common ocular infection in patients with AIDS though its incidence has dramatically decreased with the development of highly active antiretroviral therapy (HAART). Patients who develop CMV retinitis typically complain of blurred vision in one or both eyes, although when the disease occurs in the periphery, it may be asymptomatic. The infection often starts with white granular foci of infection in a perivascular distribution with or without associated vitritis (Figs. 8, 9). The lesions typically grow and spread over weeks to months eventually resulting in fullthickness necrosis of the retina often with associated hemorrhage.56 CMV-induced meningoencephalitis is not uncommon in immunodeficient patients with other CNS manifestations of CMV infection including encephalitis, ventriculo-encephalitis, and CNS vasculitis.56 Herpes simplex virus may cause retinitis and chorioretinitis. Acute retinal necrosis syndrome is a diffuse uveitis characterized by a peripheral necrotizing retinitis and retinal vasculitis associated in many cases with anterior chamber inflammation, vitritis, papillitis, or a combination of these FIGURE 8. Peripapillary CMV retinitis with mild hemorrhages in AIDS patient. 177 Brazis et al The Neurologist • Volume 10, Number 4, July 2004 FIGURE 9. The CMV retinitis appears as a granular, nearconfluent retinal infiltration with varying amounts of hemorrhage at its leading edge. FIGURE 11. Dendritic ulcer in patient with Herpes simplex keratitis. manifestations (Fig. 10). This syndrome may be a result of herpes simplex infection and may occur in isolation, or it may occur after, or concurrent with, primary herpes simplex infection, including herpes simplex keratitis (Fig. 11) and encephalitis. Herpes simplex virus is one of the most common causes of viral encephalitis in the United States and many other countries, particularly in children, and intraocular inflammation affecting the retina, choroid, optic nerve, or a combination of these occasionally occurs concurrently with this encephalitis.56 Varicella-zoster infection often may cause corneal, scleral, and conjunctival infection often with anterior uveitis. Progressive outer retinal necrosis (PORN) may occur with zoster infection, especially in AIDS patients, and is very difficult to treat. It consists of infection of the outer layers of the retina unassociated with vitritis and with little or no clinical evidence of retinal vasculitis (Fig. 12).56 The syndrome of acute retinal necrosis, particularly in older individuals, is often because of varicella-zoster infection. Aseptic meningitis, encephalitis, and myelitis are relatively rare in patient with varicella-zoster infection.56 Appropriate antiviral therapy should be directed against the causative agent in viral uveo-meningeal disease. Many patients with viral retinitis can be effectively treated with intraocular drug therapy. FIGURE 10. Acute retinal necrosis (ARN) in patient with coexisting Herpes simplex encephalitis. Photo reveals diffuse, necrotic retinal whitening with moderate intraretinal hemorrhage. 178 Inflammatory or autoimmune diseases are probably the most common clinically recognized causes of true uveo-meningeal syndromes. © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 Uveo-Meningeal Syndromes FIGURE 13. Noninfectious, peripheral corneal ulceration in patient with Wegener granulomatosis. FIGURE 12. PORN in AIDS patient with Herpes zoster infection. Photo shows numerous foci of outer retinal whitening. INFLAMMATORY ETIOLOGIES OF THE UVEO-MENINGEAL SYNDROME Inflammatory or autoimmune diseases are probably the most common clinically recognized causes of true uveomeningeal syndromes. These entities often cause granulomatous inflammation of various tissues in the body, including ocular structures and the meninges. Most of these inflammatory disorders are associated with systemic signs and symptoms. A complete review of systems should, therefore, be performed in all patients with a uveo-meningeal syndrome. The review should include assessment of the skin (eg, rash), mucous membranes (eg, oral ulcers), joints (eg, arthritis), heart (eg, pericarditis), lungs (eg, hilar adenopathy, interstitial disease), sinuses (eg, sinusitis), kidneys (eg, glomerulonephritis), gastrointestinal system (eg, diarrhea), and genitourinary (eg, genital ulcers) systems. Wegener granulomatosis is characterized by necrotizing granulomatous lesions of the respiratory tract, glomerulonephritis, and systemic vasculitis. In some patients, ocular, meningeal, or cerebral inflammation may be the earliest manifestation of Wegener and, indeed, may occur with no evidence of pulmonary, sinus, or renal involvement (“limited Wegener granulomatosis”).57 Ocular structures are affected in 30 to 60% of patients with both generalized and limited Wegener.58 The main categories of ocular involvement include orbital granuloma, scleritis with or without peripheral keratitis (Fig. 13) or iritis, and vascular complications from vasculitis. Retinal involvement includes asymptomatic cotton-wool spots or severe diffuse retinal vasculitis with widespread retinal vascular occlusions and perivascular venous sheathing, vitreous hemorrhages, and glaucoma. Anterior or © 2004 Lippincott Williams & Wilkins retrobulbar optic neuropathy may occur. Neurologic manifestations occur in 20 to 50% of patients and may include diffuse or focal meningeal involvement.59,60 Sarcoidosis is a systemic disease of unknown etiology characterized pathologically by the presence of noncaseating granulomas. Almost any tissue can be affected, but the lungs and thoracic lymph nodes are major sites of involvement. Ocular involvement may be the initial manifestation of sarcoidosis in approximately 20% of patients61 and approximately 25 to 80% of patients with sarcoidosis develop ocular manifestations at some time during the course of the disease.62 Anterior uveitis is the most common ocular manifestation of this disease. It is usually bilateral and granulomatous and is characterized by the formation of grayish-white “mutton-fat” keratic precipitates on the endothelial surface of the cornea. Occasional patients with sarcoidosis present with acute unilateral nongranulomatous or granulomatous anterior uveitis or iridocyclitis with sudden eye pain, redness, photophobia, and decreased vision. Patients with chronic anterior uveitis develop granulomatous nodules on the iris (Koeppe nodules and Busacca nodules). Although sarcoid uveitis is usually confined to the anterior chamber, some patients develop an isolated vitritis or even a panuveitis. Intermediate uveitis often presents with discrete gray-white spherical bodies that may occur in chains like a “string of pearls” or “snowballs” in the inferior vitreous. Retinal periphlebitis may occur with segments of retinal blood vessels showing an irregular waxy yellow coating described as “candle wax drippings.” Other posterior segment manifestations include multifocal or serpiginous chorioretinitis, choroidal nodules, retinal vein occlusions, retinal and vitreous hemorrhages, and optic nerve head granulomas (Fig. 14).63 The diagnosis can be suggested by chest imaging (eg, chest radiography, chest 179 Brazis et al FIGURE 14. Retinal vasculitis and optic nerve granuloma in patient with active sarcoidosis. computed tomography, Gallium scan) and laboratory testing (eg, serum or CSF angiotensin converting enzyme). Generally, however, a tissue diagnosis (eg, conjunctiva, bronchial, lymph node) is preferred. Neurosarcoidosis occurs in 5 to 10% of patients.64 In some cases the neurologic manifestations are the presenting features of the disease and in some of these, there is no overt evidence of sarcoidosis elsewhere in the body. Patients develop multifocal or diffuse leptomeningeal granulomas that may cause nonspecific meningeal symptoms (eg, headache, stiff neck), mentation changes, hydrocephalus, cranial neuropathies, hypothalamic dysfunction, and pituitary abnormalities.63,64 Sarcoid meningitis may be acute but is more often chronic. Single or multiple parenchymal granulomas may also develop. The treatment of sarcoidosis is usually corticosteroids but additional immunosuppressive therapy with methotrexate, azathioprine, or cyclosporine may be required in unresponsive cases. The prognosis is variable but generally good. Behçet disease is a chronic relapsing syndrome characterized by the clinical triad of oral ulcerations (essentially the sine qua non for diagnosis), genital ulcerations, and uveitis. Most cases are reported from Eastern Mediterranean countries and Japan. Although anterior uveitis (eg, acute bilateral nongranulomatous iridocyclitis) with hypopyon may occur, posterior uveitis develops in most patients.58,65 Although ocular inflammation is seldom the initial manifestation of the disease, bilateral uveitis ultimately develops in approximately 70% of patients. Retinal vasculitis, usually 180 The Neurologist • Volume 10, Number 4, July 2004 bilateral and affecting both arteries and veins, occurs frequently and may be the initial manifestation of Behçet disease. Retinal vasculitis may result in vascular occlusion, hemorrhagic retinal necrosis, neovascular glaucoma, and blindness.58 Other ocular manifestations include conjunctivitis, keratitis, vitritis, and scleritis. Neurologic manifestations of Behçet disease may be a result of meningitis, meningoencephalitis, or focal parenchymal involvement. Meningitis or meningoencephalitis occurs in 4 to 29% of patients with Behçet disease but rarely as the initial manifestation. However, approximately 5% of patients develop neurologic involvement weeks or months before other clinical features of the disease (“neuro-Behçet disease”).66 Focal or multifocal neurologic deficits occur with the brainstem being the most frequent site of focal involvement. The diagnosis is made clinically. Identifying neuroBehçet becomes important because of its prognostic value as it indicates severe and possibly fatal disease.67 The treatment is generally immunosuppression therapy with systemic corticosteroids, chlorambucil, cyclophosphamide, azathioprine, and cyclosporine. Vogt-Koyanagi-Harada (VKH) syndrome consists of 2 clinical entities: an anterior granulomatous iridocyclitis associated with poliosis, vitiligo, alopecia, and dysacusis (summarized by J. Lawton Smith’s pneumonic, “Too bad if you got PUVAD”) described by Vogt and Koyanagi and a posterior granulomatous choroiditis with multifocal serous retinal detachment, meningeal findings, and cerebrospinal fluid pleocytosis described by Harada. The 2 clinical entities combine to make a clinical syndrome characterized by panuveitis, neurologic impairment, and skin changes.68,69 The disease usually affects darkly pigmented Asian, American Indian, Hispanic, or Afro-American adults; it is only rarely seen in Caucasians. The syndrome can be divided into 4 clinical phases. The first phase or prodromal phase consists of a flu-like illness associated with meningeal involvement manifesting as meningismus, headaches, mental status changes, cerebrospinal fluid pleocytosis, tinnitus, and dysacusis.69,70 An autoimmune attack against melanin containing cells in the cochlea and meninges causes these symptoms.71 The second or uveitic phase is associated with acute granulomatous iridocyclitis, vitritis, optic disc edema, and multiple serous retinal detachments. The anterior iritis is usually bilateral with mutton-fat keratic precipitates, aqueous cell and flare, peripheral anterior synechiae, depigmentation of the perilimbus (Sugiura sign), iris nodules, cataracts, and glaucoma.68,69 Anterior and posterior uveitis may occur independently or simultaneously. Posterior segment findings include vitritis, disc swelling, and retinal hemorrhages and exudates. Recurrent bilateral serous retinal detachments, most commonly inferiorly and multiple, are essentially pathognomonic for the disease. The third or convalescent phase of the disease is associated with subsidence of the uveitis but with the onset of © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 skin manifestations and diffuse depigmentation of the choroid. Choroidal pigment decreases resulting in a mottled “sunset glow” depigmented fundus and multiple peripheral yellowish-white lesions (Dalen-Fuchs nodules) occur in the inferior peripheral retina. Cutaneous manifestations include alopecia, vitiligo, and poliosis (whitening) of the eyebrows, eyelashes, and hair of the head. The fourth phase of the disease consists of continuous recurrent inflammatory attacks characterized primarily by anterior uveitis. The major ophthalmic complications of the disease occur most often in this recurrent phase. Corticosteroid therapy is generally the first line of treatment but poorly responsive cases require azathioprine, cyclophosphamide and cyclosporine. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is a disease of unknown etiology typically affecting young adults 20 to 30 years of age without gender or racial predilection. It often begins with a prodromal phase of nonspecific upper respiratory complaints followed by sudden loss of central vision. Fundus examination shows multiple round, discrete, pale yellow-white lesions in the posterior pole at the level of the retinal pigment epithelium.72 Both eyes are usually affected and spontaneous recovery usually occurs within 3 weeks though mottling of the retinal pigment epithelium may be permanent. Older patients, however, may have a less favorable prognosis. Patients with APMPPE occasionally have other ocular findings including iridocyclitis, corneal thinning, retinal vasculitis with serous retinal detachment, and optic neuritis.58 Some patients with APMPPE have neurologic manifestations including headache, meningoencephalitis, cerebrospinal fluid pleocytosis, tinnitus, hearing loss, and cerebral vasculitis with stroke.58,73–75 Fortunately, cerebral vasculitis occurs rarely, but immunosuppressive treatment should be considered if this leads to strokes.76 NEOPLASTIC AND PARANEOPLASTIC ETIOLOGIES OF THE UVEO-MENINGEAL SYNDROME Lymphoma may affect both ocular structures and the meninges. Categories of intraocular lymphoma include: 1) Primary non-Hodgkin malignant lymphoma of the CNS with ocular involvement primarily of the vitreous or retina; 2) Secondary or metastatic intraocular involvement in systemic non-Hodgkin lymphomas, in which the uveal tract is most frequently affected; 3) Lymphoid hyperplasia (or lymphoid tumor) of uvea; 4) Angiotropic lymphoma (formerly neoplastic angioendotheliomatosis); and 5) Hodgkin disease and mycosis fungoides. Primary Ocular-CNS non-Hodgkin Lymphoma may present with neurologic or ocular findings.77–79 Twelve to 15% of primary CNS lymphoma patients present with associated ocular signs or symptoms while a further 10 to 20% of primary CNS lymphoma patients eventually develop ocular findings. On the other hand, 80 to 90% of primary ocular © 2004 Lippincott Williams & Wilkins Uveo-Meningeal Syndromes lymphoma patients concurrently or subsequently develop CNS or systemic involvement while only 10 to 20% stay ocular. Ocular lymphoma (usually a large B-cell lymphoma) appears to arise primarily in the vitreous and subretinal pigment epithelial space and only secondarily extends into the retina, choroid, and optic nerve. Patients with primary CNS large-cell lymphoma may present only with visual signs. Although any part of the visual system may be involved, the most common ocular presentation is uveitis. Both eyes are usually affected although the disease may begin monocularly. Patients typically complain of mild decreased vision or “floaters” in one or both eyes associated with mild to moderate unilateral or bilateral uveitis unassociated with eye pain or redness. Vitritis is always present but there may be cells in the anterior chamber as well. Some patients either concurrently or subsequently develop multifocal yellow-orange or white infiltrates beneath the retina or retinal pigment epithelium. Other patients develop exudative retinal detachments produced by subretinal pigment epithelium collections of tumor cells, retinal vasculitis, retinal artery occlusions, retinal or vitreous hemorrhages, or retinal necrosis. One should consider the diagnosis of primary ocularCNS lymphoma in patients 40 years or older with bilateral vitritis that fails to respond to treatment. Also consider this diagnosis if vitritis accompanies neurologic symptoms. Primary CNS lymphoma usually manifests as focal or multifocal parenchymal lesions but may be limited to the meninges.80 Other forms of lymphoma may cause an uveo-meningeal syndrome.77 Systemic non-Hodgkin lymphoma with secondary (metastatic) ocular involvement may present as anterior uveitis with hypopyon or hyphema but more commonly with diffuse choroidal infiltration (Fig. 15) or a choroidal mass. Systemic visceral or lymph node involvement precedes ocular involvement in approximately 57% of patients though bilateral uveal lesions may occasionally be the first manifestation of lymphoma. Angiotrophic large cell lymphoma or intravascular lymphomatosis may also present with decreased visual acuity, cortical blindness, small white retinal or choroidal infiltrates, retinal pigment epithelial changes, retinal artery occlusions, retinal hemorrhages, retinal vascular sheathing, vitritis, or iridocyclitis. Metastatic carcinoma to the eye is generally regarded as the most common intraocular malignant tumor. Because of the distribution of ocular blood flow, most intraocular metastases involve the choroid and present with yellowish-white, round or oval lesions. Exceptionally metastatic carcinoma to the eye may present with infiltration of the retina or even dispersed vitreous cells.81,82 Lung and breast carcinomas are the most common sources of metastatic tumors. A uveomeningeal syndrome may be produced by concomitant ocular metastasis and carcinomatous meningitis. A paraneoplastic visual disorder with combined optic neuritis (Fig. 16) and retinitis has been described, usually in 181 Brazis et al The Neurologist • Volume 10, Number 4, July 2004 FIGURE 16. Optic nerve swelling with mild vitritis in patient with CRMP-5-IgG paraneoplastic syndrome because of lung carcinoma. FIGURE 15. Diffuse creamy choroidal infiltrates in patient with systemic lymphoma. patients with small-cell lung cancer or, less often, thymoma or other malignancies. These cases are defined serologically by the presence of a paraneoplastic IgG autoantibody CRMP5-IgG.83,84 Neurologic findings include chorea, cranial neuropathy, loss of olfaction and taste, peripheral neuropathy, autonomic neuropathy, cerebellar ataxia, myelopathy, dementia, and neuromuscular junction disorders.83,84 Spinal fluid studies often show inflammatory changes. Cross has described a subset of 172 patients with neurologic disorders and CRMP-5-IgG.84 Fifteen patients had optic neuritis with retinitis documented in 5. Vitreous cells were noted in 9 cases and diagnostic vitrectomy revealed reactive lymphocytosis in 4 of 4 patients. APPROACH TO THE PATIENT WITH THE UVEO-MENINGEAL SYNDROME Our diagnostic approach to the uveo-meningeal syndromes is summarized as follows: 1. All patients should undergo extensive and complete ocular and medical histories and examinations to identify any systemic findings that might suggest a specific diagnosis. 2. The immune status should be determined. Immunocompromised individuals should be evaluated more aggressively for infectious etiologies (eg, Candida, Cryptococcus, Cytomegalovirus). 3. The eye should be scrutinized for distinctive signs of a specific etiology. Table 3 lists the distinctive signs in 182 selected uveo-meningeal processes. 4. Patients without a specific etiology identified by careful history and examination should undergo screening tests for treatable or common etiologies for uveo-meningeal disease (eg, sarcoidosis, tuberculosis, syphilis). Most patients with a uveo-meningeal process should at least undergo a magnetic resonance imaging (MRI) with gadolinium and a lumbar puncture. Patients from Lyme endemic areas or with a history of erythema chronica migrans should be considered for Lyme serology. More specific TABLE 3. Distinctive Presentations in Uveo-Meningeal Disease Optic disc edema with a macular star figure Focal chorioretinitis with massive vitritis (“headlight in the fog”) Acute retinal necrosis in peripheral retina Diarrhea and intermediate uveitis Hemorrhagic, perivascular necrotic retinitis Multifocal serous retinal detachments Hypopyon uveitis, genital or oral ulcers Acute, multifocal, placoid, pigment epitheliopathy Bilateral chronic uveitis in an elderly patient Cat scratch disease Toxoplasmosis Herpetic disease Whipple disease Cytomegalovirus Vogt-Koyanagi-Harada Behçet disease AMPPE Ocular lymphoma © 2004 Lippincott Williams & Wilkins The Neurologist • Volume 10, Number 4, July 2004 studies should be directed by the history and examination and we discourage a “shot-gun” approach to laboratory testing. 5. Special studies (eg, acid-fast-staining, polymerase chain reaction testing, cytology) may be necessary in cases where routine laboratory and radiographic tests are unremarkable. In some cases, the diagnosis requires vitreous or meningeal biopsy. The least invasive site for tissue diagnosis should be biopsied in patients with multisystem involvement. 6. Treatment should be directed at the causative agent and appropriate consultation should be obtained for inflammatory (eg, rheumatology), infectious (eg, infectious disease), neoplastic (eg, oncology) etiologies. Patients with uveo-meningeal processes typically require a multidisciplined approach to establish the diagnosis and formulate an effective treatment plan. It is incumbent upon the primary physician to involve appropriate consultants and direct the work-up of these complex patients. Utilizing a thoughtful, methodical approach, a correct diagnosis can be established in the majority of these cases. REFERENCES 1. Moorthy RS, Rao NA. Uveitis: introduction and classification. In: Wright KW, ed. Textbook of Ophthalmology. Baltimore, MD: Williams and Wilkins; 1997:477– 479. 2. Forster DJ. General approach to the Uveitis patient and treatment strategies. In: Yanoff M, Duker JS, eds. Ophthalmology. London: Mosby; 1999:3 3. Moorthy RS, Rao NA. Approach to the uveitis patient. In: Wright KW, ed. Textbook of Ophthalmology. Baltimore, MD: Williams and Wilkins; 1997:481– 486. 4. Henderly DE, Gensler AJ, Smith RE, et al. Changing patterns of uveitis. Am J Ophthalmol. 1987;103:131–136. 5. Rodriguez A, Calonge M, Pedroza-Seres M, et al. Referral patterns of uveitis in a tertiary eye care center. Arch Ophthalmol. 1996;114:593– 599. 6. Brazis PW, Lee AG. Optic disk edema with a macular star. Mayo Clin Proc. 1996;71:1162–1166. 7. Cohen SM, Davis JL, Gass DM. Branch retinal arterial occlusions in multifocal retinitis with optic nerve edema. Arch Ophthalmol. 1995;113: 1271–1276. 8. Golnik KC, Marotto ME, Fanous MM, et al. Ophthalmic manifestations of Rochalimaea species. Am J Ophthalmol. 1994;118:145–151. 9. Solley WA, Martin DF, Newman NJ, et al. Cat scratch Disease. Posterior segment manifestations. Ophthalmology. 1999;106:1546 –1553. 10. Wong MT, Dolan MJ, Lattuada CP Jr. , et al. Neuroretinitis, aseptic meningitis, and lymphadenitis associated with Bartonella (Rochalimaea) henselae infection in immunocompetent patients and patients infected with human immunodeficiency virus type I. Clin Infect Dis 1995;21: 352–360. 11. Knox DL, Bayless TM, Yardley JH, et al. Whipple’s disease presenting with ocular inflammation and minimal intestinal symptoms. Johns Hopkins Med J. 1968;123:175–182. 12. Finelli PF, McEntree WJ, Lessell S, et al. Whipple’s disease with predominantly neuro-ophthalmologic manifestations. Ann Neurol. 1977; 1:247–252. 13. Font RL, Rao NA, Issarescu S, et al. Ocular involvement in Whipple’s disease: light and electron microscopic observations. Arch Ophthalmol 1978;96:1431–1436. 14. Leland TM, Chambers JK. Ocular findings in Whipple’s disease. South Med J. 1978;71:335–338. © 2004 Lippincott Williams & Wilkins Uveo-Meningeal Syndromes 15. Avila MP, Jalkh AE, Feldman E, et al. Manifestations of Whipple’s disease in the posterior segment of the eye. Arch Ophthalmol. 1984;102: 384 –390. 16. Rickman LS, Freeman WR, Green WR, et al. Brief report: uveitis caused by Tropheryma Whippelii (Whipple’s Bacillus). New Engl J Med. 1995;332:363–366. 17. Schwartz MA, Harris JK, Campbell WW, et al. Oculomasticatory myorhythmia: a sign of Whipple’s disease. Ann Neurol. 1985;18:135– 136. 18. Schwartz MA, Selhorst JB, Ochs Al, et al. Oculomasticatory myorhythmia: a unique movement disorder occurring in Whipple’s disease. Ann Neurol 1986;20:677– 683. 19. Louis ED, Kusske JA, Rush JL, et al. Diagnostic guidelines in central nervous system Whipple’s disease. Ann Neurol. 1996;46:561–568. 20. Park SS, To KW, Friedman AH, et al. Infectious causes of posterior uveitis. In: Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders; 1994:453– 454. 21. Helm CJ, Holland GN. Ocular tuberculosis. Surv Ophthalmol. 1993;38: 229 –256. 22. Jabbour NM, Faris B, Trempe CL. A case of pulmonary tuberculosis presenting with a choroidal tuberculoma. Ophthalmology. 1985;92:834 – 837. 23. Kent SJ, Crowe SM, Yung A, et al. Tuberculous meningitis: a 30-year review. Clin Infect Dis. 1993;17:987–994. 24. Berile GR, Flynn TE. Syphilis exposure in patients with uveitis. Ophthalmology. 1997;104:1605–1609. 25. Ho AC, Guyer DR, Yannuzzi LA, et al. Ocular syphilis. Classic manifestations and recent observations. Semin Ophthalmol. 1993;8:53– 60. 26. Yagasaki T, Akiyama K, Nomura H, et al. Two cases of acquired syphilis with acute central choroiretinitis as initial manifestation. Jpn J Ophthalmol. 1992;36:301–309. 27. Anderson M. Management of cerebral infection. J Neurol Neurosurg Psychiatr. 1993;56:1243–1258. 28. Steere AC. Lyme disease. New Engl J Med 321;586 –596. 1989. 29. Jacobson DM, Frens DB. Pseudotumor cerebri syndrome associated with Lyme disease. Am J Ophthalmol. 1989;107:81– 82. 30. Aaberg TM. The expanding ophthalmologic spectrum of Lyme disease. Am J Ophthalmol. 1989;107:77– 80. 31. Breeveld J, Kuiper H, Spanjaard L, et al. Uveitis and Lyme borreliosis. Br J Ophthalmol. 1993;77:480 – 481. 32. Bialasiewicz AA, Ruprecht KW, Naumann GOH, et al. Bilateral diffuse choroiditis and exudative retinal detachment with evidence of Lyme disease. Am J Ophthalmol. 1988;105:419 – 420. 33. Kuiper H, Koelman JH, Jager MJ. Vitreous clouding associated with Lyme borreliosis. Am J Ophthalmol. 1989;108:453– 454. 34. Winterkorn JMS. Lyme disease: neurologic and ophthalmic manifestations. Surv Ophthalmol. 1990;35:191–204. 35. Brooks RG. Prospective study of Candida endophthalmitis in hospitalized patients with candidemia. Arch Int Med. 1989;149:2226 –2228. 36. Menezes AV, Sigesmund DA, Demajo WA, et al. Mortality of hospitalized patients with Candida endophthalmitis. Arch Int Med. 1994;154: 2093–2097. 37. Donohue SP, Eller AW. Intraocular Candida infection. Semin Ophthalmol. 1993;8:70 –76. 38. Moorthy RS, Sidikaro Y, Foos RY, et al. Coccidioidomycosis iridocyclitis. Ophthalmology. 1994;101:1923–1928. 39. Rodenbiker HT, Ganley JP. Ocular coccidioidomycosis. Surv Ophthalmol. 1980;24:263–290. 40. Crumo JR, Elner SG, Elner VM, et al. Cryptococcal endophthalmitis: case report and review. Clin Iinfect Dis. 1992;14:1069 –1073. 41. Garrity JA, Herman DC, Imes R, et al. Optic nerve sheath decompression for visual loss in patients with Acquired Immunodeficiency Syndrome and Cryptococcal meningitis with papilledema. Am J Ophthalmol. 1993;116:472– 478. 42. Denning DW, Armstrong RW, Fishman M, et al. Endophthalmitis in a patient with disseminated cryptococcosis and AIDS who was treated with itraconazole. Rev Infect Dis. 1991;13:1126 –1130. 43. Weingeist TA, Watzke RC. Ocular involvement by Histoplasma capsulatum. Int Ophthalmol Clin. 1983;23:33– 47. 183 Brazis et al 44. Specht CS, Mitchell KT, Bauman AE, et al. Ocular histoplasmosis with retinitis in a patient with acquired immune deficiency syndrome. Ophthalmology. 1991;98:1356 –1359. 45. Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseinated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis, and treatment, and review of the literature. Medicine. 1990;69:361–374. 46. Gottleib JL, McAllister IL, Guttman FA, et al. Choroidal blastomycosis: a report of two cases. Retina. 1995;15:248 –252. 47. Safneck JR, Hogg GR, Napier LB. Endophthalmitis due to Blastomyces dermatitidis. Case report and review of the literature. Ophthalmology. 1990;97:212–216. 48. Bradsher RW. Blastomycosis. Clin Infect Dis. 1992;14(Suppl 1):S82– S90. 49. Harding CV. Blastomycosis and opportunistic infections in patients with acquired immunodeficiency syndrome. Arch Path Lab Med. 1991;115: 1133–1166. 50. Graham DA, Kinyuon JL, George DP. Aspergillus endophthalmitis after lung transplantation. Am J Ophthalmol. 1995;119:107–109. 51. Valluri S, Moorthy RS, Rao NA. Endogenous Aspergillus endophthalmitis in an immunocompetent individual. Int Ophthalmol. 1993;17: 131–135. 52. Shirouzu K, Hasuo K, Yasumori J, et al. MR imaging in paranasal and intracranial aspergillosis. Nippon Acta Radiol. 1991;51:768 –774. 53. Ronday MJH, Luyendijk L, Baarsma GS, et al. Presumed acquired ocular toxoplasmosis. Arch Ophthalmol. 1995;113:1524 –1529. 54. Moreno RJ, Weisman J, Waller S. Neuroretinitis: an unusual presentation of ocular toxoplasmosis. Ann Ophthalmol. 1992;24:68 –70. 55. Odel JG, Moazami G. Disease caused by helminths. In: Miller NR, Newman NJ, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 5th Edition. Baltimore, MD: Williams and Wilkins; 1998:4500 – 4503. 56. Brazis PW, Miller NR. Viruses (except retroviruses) and viral diseases. In: Miller NR, Newman NJ, eds. Walsh and Hoyt’s Clinical NeuroOphthalmology, 5th Edition. Baltimore, MD: Williams and Wilkins; 1998:4945–5360. 57. Newman NJ, Slamovits TL, Friedland S, et al. Neuro-ophthalmic manifestations of meningocerebral inflammation form limited form of Wegener’s granulomatosis. Am J Ophthalmol. 1995;120:613– 621. 58. Galetta SL. Vasculitis. In: Miller NR, Newman NJ, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 5th Edition. Baltimore, MD: Williams and Wilkins; 1998:3725–3886. 59. Nishino H, Rubino FA, DeRemee RA, et al. Neurologic involvement in Wegener’s granulomatosis: an analysis of 324 consecutive patients at the Mayo Clinic. Ann Neurol. 1993;33:4 –9. 60. Specks U, Moder KG, McDonald TJ. Meningeal involvement in Wegener granulomatosis. Mayo Clin Proc. 2000;75:856 – 859. 61. Obenauf CD, Shaw HE, Sydnor CF, et al. Sarcoidosis and its ophthalmic manifestations. Am J Ophthalmol. 1978;86:648 – 655. 62. Silver MR, Messner LV. Sarcoidosis and its ocular manifestations. J Am Optom Assoc. 1994;65:321–327. 63. Hamilton SR. Sarcoidosis and idiopathic hypertrophic cranial pachymenigitis. In: Miller NR, Newman NJ, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 5th Edition. Baltimore, MD: Williams and Wilkins; 1998:5465–5538. 184 The Neurologist • Volume 10, Number 4, July 2004 64. Sharma OP, Sharma AM. Sarcoidosis of the nervous system: a clinical approach. Arch Int Med. 1991;151:1317–1321. 65. Wakefield D, McCluskey P. Behçet’s syndrome: ocular features in an Australian population. Aust NZ J Ophthalmol. 1990;18:129 –135. 66. Shimuzu T, Ehrlich GE, Inaba G, et al. Behçet’s disease. Semin Arth Rheum. 1979;8:223–260. 67. Kansu T. Neuro-Behçet’s Disease. The Neurologist. 1998;4:31–39. 68. Sugiura S. Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1978;22: 9 –35. 69. Ohno S, Char DH, Kimura SJ, et al. Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1977;83:735–740. 70. Perry HD, Font RL. Clinical and histopathological observations in severe Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 1977;83: 242–254. 71. Reed RW, Rao NA, Cunningham ET Jr. Vogt-Koyanagi-Harada disease. Curr Opin Ophthalmol. 2000;11:437– 442. 72. Gass JDM. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80:177–185. 73. Bullock JD, Fletcher RL. Cerebrospinal fluid abnormalities in Acute posterior multifocal placoid pigment epitheliopathy. Am J Ophthalmol. 1977;84:45– 49. 74. Comu S, Verstraeten T, Rinkoff JS, et al. Neurologic manifestations in Acute posterior multifocal placoid pigment epitheliopathy. Stroke. 1996; 27:996 –1001. 75. Kersten DH, Lessel S, Carlow TJ. Acute posterior multifocal placoid pigment epitheliopathy and late onset menigoencephalitis. Ophthalmology. 1987;84:393–396. 76. Comu S, Verstraeten T, Rinkoff JS, et al. Neurological manifestations of acute posterior multifocal placoid pigment epitheliopathy. Stroke. 1996; 27:996 –1001. Deleted in Proof. 77. Felton WL III. Tumors derived from hematopoietic cells and tissue. In: Miller NR, Newman NJ, eds. Walsh and Hoyt’s Clinical Neuro-Ophthalmology, 5th Edition. Baltimore, MD: Williams and Wilkins; 1998: 2329 –2436. 78. Whitcup SM, de Smet MD, Rubin BI, et al. Intraocular lymphoma. Clinical and histopathologic diagnosis. Ophthalmology. 1993;100: 1399 –1406. 79. Peterson K, Gordon KB, Heinemann MK, et al. The clinical spectrum of orbital lymphoma. Cancer. 1993;72:843– 849. 80. Lachance DH, O’Neill BP, Macdonald DR, et al. Primary leptomenigeal lymphoma: report of 9 cases, diagnosis with immunocytochemical analysis, and review of the literature. Neurology. 1991;41:95–100. 81. Leys AM, Van Eyck LM, Nuttin BJ, et al. Metastatic carcinoma to the retina. Clinicopathologic findings in two cases. Arch Ophthalmol. 1990; 108:1448 –1452. 82. Spraul CW, Martin DF, Hagler WS, et al. Cytology of metastatic cutaneous melanoma to the vitreous and retina. Retina. 1996;16:328 – 332. 83. Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49:146 –154. 84. Cross S. A paraneoplastic syndrome of combined optic neuritis and retinitis defined serologically by CRMP-5-IgG 关Abstract兴. J NeuroOphthalmol. 2002;22:169. © 2004 Lippincott Williams & Wilkins