* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Clinical trial wikipedia , lookup
Epinephrine autoinjector wikipedia , lookup
Bad Pharma wikipedia , lookup
Biosimilar wikipedia , lookup
























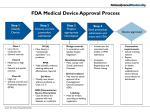



![FDA Regulatory Process for Premarket [510(k)] Submission: General and](http://s1.studyres.com/store/data/008269843_1-6bd3477559b8f78259d8368a9a95629a-150x150.png)



